

Abstract
Background & Aims:
The Hedgehog signaling pathway contributes to cholangiocarcinoma biology. However, canonical Hedgehog signaling requires cilia, and cholangiocarcinoma cells often do not express cilia. To resolve this paradox, we examined non-canonical (G-protein coupled, pertussis toxin sensitive) Hedgehog signaling in cholangiocarcinoma cells.
Methods:
Human [non-malignant (H69), malignant (HuCC-T1 and Mz-ChA-1)] and rat [non-malignant (BDE1 and NRC), and malignant (BDEneu)] cell lines were employed for this study. A BDEΔLoop2 cell line with the dominant-negative receptor Patched-1 was generated with the Sleeping Beauty transposon transfection system.
Results:
Cilia expression was readily identified in non-malignant, but not in malignant cholangiocarcinoma cell lines. Although the canonical Hh signaling pathway was markedly attenuated in cholangiocarcinoma cells, they were chemotactic to purmorphamine, a small-molecule direct Smoothened agonist. Purmorphamine also induced remodeling of the actin cytoskeleton with formation of filopodia and lamellipodia-like protrusions. All these biological features of cell migration were pertussis toxin sensitive, a feature of G-protein coupled (Gis) receptors. To further test the role of Hedgehog signaling in vivo, we employed a syngeneic orthotopic rat model of cholangiocarcinoma. In vivo, genetic inhibition of the Hedgehog signaling pathway employing BDEΔLoop2 cells or pharmacological inhibition with a small-molecule antagonist of Smoothened, vismodegib, was tumor and metastasis suppressive.
Conclusions:
Cholangiocarcinoma cells exhibit non-canonical Hedgehog signaling with chemotaxis despite impaired cilia expression. This non-canonical Hedgehog signaling pathway appears to contribute to cholangiocarcinoma progression, thereby, supporting a role for Hedgehog pathway inhibition in human cholangiocarcinoma.
Keywords: Dominant-negative Ptch1, Biliary tract cancer, G-protein coupled receptor, Patched-1, Smoothened
Introduction
Cholangiocarcinoma (CCA) is a lethal primary hepatobiliary malignancy [1]. Therapeutic advances for CCA will require a deeper understanding of the molecular pathways driving progression of this neoplasm. However, knowledge regarding the pivotal signaling pathways responsible for CCA biology is incomplete. Several studies suggest activation of the Hedgehog (Hh) signaling pathway as a key feature of CCA progression [2,3].
In mammalian cells, Hh signaling comprises complex relationships between the Hh ligands [Sonic Hh (Shh), Indian Hh, and Desert Hh] and two plasma membrane proteins, Patched-1 (Ptch1) and Smoothened (Smo) [4] Canonically, binding of the Hh ligand to Ptch1 leads to derepression and translocation of Smo to cilia resulting in activation of the glioma-associated transcriptional factors (Gli1, Gli2, and Gli3) [4]. Smo translocation into the cilia membrane appears to be requisite for Gli activation [5-9]. This facet of Hh signaling results in a conundrum. Aberrant activation of Hh pathway signaling is a characteristic of many malignancies [10], and tumor progression is often Hh signaling responsive despite the lack of cilia expression by many cancers. This puzzle is particularly relevant to CCA cell biology. For example, Gli activation can be identified in the KMCH CCA cell line, which variably forms rudimentary or incomplete cilia and robustly expresses Gli1 [11,12]. However, most CCA cell lines appear to be Hh responsive despite the absence of cilia and minimal Gli expression.
Recently, a non-canonical pathway for Hh signaling was identified in drosophila cells lacking cilia, namely, a non-Gli, inhibitory G-protein (Gis) coupled pathway [7]. The identified downstream effect of this non-canonical Hh signaling activation is cytoskeleton remodeling and cell migration via involvement of the small Rho GTPases Rac1 and RhoA [13]. This pathway is pertussis toxin (PTX) sensitive, a feature of G-protein coupled receptors involving Gis. A role for this non-canonical Hh signaling pathway in mammalian, especially CCA, cell biology remains plausible, but has yet to be examined.
Herein, we identify Hh signaling in CCA cells despite failure to express cilia. The pathway is inhibitable by the small-molecule Smo inhibitor vismodegib, is PTX sensitive, blocked by forced expression of a dominant-negative Ptch1 construct, and is not associated with Gli activation. The pathway appears to be critical for CCA migration in vivo. These observations provide further insight regarding Hh signaling in cancer biology, and support a role for Hh signaling inhibition in the treatment of human CCA.
Materials and methods
Cell lines and cell culture
H69 is a non-malignant SV40-immortalized human cholangiocyte cell line [14]. Malignant, patient-derived CCA cell lines were: KMCH, HuCC-T1, and Mz-ChA-1 [15,16]. The BDE1 and NRC are immortalized non-tumorigenic rat cholangiocyte cell lines [14,17]. The BDEneu cell line was generated by genetic transformation of BDE1 cells with the mutationally activated rat neu oncogene [18]. All cell lines were cultured as previously described by us in detail [11,19].
Immunofluorescence
Cells were cultured and incubated at 37 °C in an atmosphere containing 5% CO2 at 100% confluency for 5 days with media exchange daily to stimulate cilia expression. In an experiment examining Smo translocation from the cell interior to the plasma membrane, cells were cultured and treated with either vehicle, recombinant mouse Shh ligand (6 μM; rm-Shh-N; R&D Systems, Minneapolis, MN), or a direct small-molecule agonist of Smo, purmorphamine (2 μM; Calbiochem, Billerica, MA, USA) with and without PTX (200 μg/ml; Sigma-Aldrich) for 16 h. In an experiment examining Gli2 translocation to the cell nuclei, cells were cultured and treated with either vehicle or purmorphamine (2 μM; Calbiochem) for 8 h. For immunofluorescence, cells were washed with phosphate-buffered saline (PBS) and fixed with either ice cold methanol (5 min) or 4% paraformaldehyde (10 min) for cilia and Gli2 or Smo immunofluorescence, respectively. All subsequent washes were performed using PBS with (cilia and Gli2 immunofluorescence) or without (Smo immunofluorescence) 0.1% Triton X-100 (Fisher Scientific, Pittsburg, PA, USA). Cells were incubated for 1 h at room temperature in blocking serum [5% fetal bovine serum (FBS) with 1% bovine serum albumin in PBS for cilia and Gli2 immunofluorescence; and 1% bovine serum albumin (BSA) with 10% goat serum, and 0.3 M glycine in PBS for Smo immunofluorescence], and then with primary antiserum (Supplementary Table 1) at 4 °C overnight. Cells were washed, incubated for 1 h with secondary antiserum (Supplementary Table 1) at room temperature, washed again, and mounted using Prolong Gold Antifade with DAPI (Invitrogen, Carlsbad, CA, USA). Cells were examined with confocal microscopy (LSM 510, Carl Zeiss, Jena, Germany) in at least 5 high power fields for Gli2 translocation to the cell nuclei, for percent of ciliated cells, or cells with Smo translocation to the plasma membrane.
To study actin cytoskeleton remodeling and expression of paxillin [22], we treated cultured cells either with vehicle or purmorphamine (2 μM; Calbiochem) with and without PTX (200 μg/ml; Sigma-Aldrich). Cell were washed with PBS, fixed with 4% paraformaldehyde, permeabilized with the 0.1% of Triton X-100 (Fisher Scientific), incubated in blocking serum (5% goat serum and 5% glycerol in PBS) and then with primary antibodies (Supplementary Table 1) for 2 h at 37 °C. Cells were subsequently washed with PBS and incubated with secondary antibodies (Supplementary Table 1) and phalloidin-FITC (Sigma-Aldrich; dilution of 1:300) for 1 h at 37 °C. Slides were mounted with Prolong Gold Antifade with DAPI (Invitrogen) and examined with fluorescence microscopy (Carl Zeiss).
Cell migration assay
The lower well of the modified Boyden chamber (Neuro Probe, Gaithersburg, MD, USA) was filled with the growth medium containing either vehicle, rm-Shh-N (6 μM, R&D), purmorphamine (2 μM, Calbiochem) with or without PTX (200 μg/ml; Sigma-Aldrich), or GANT61 (20 μM; Selleck, Randor, PA, USA) with or without purmorphamine (2 μM; Calbiochem). The polycarbonate membrane with 10 lm pores (Neuro Probe) covered by 0.01% collagen was placed on top of the lower well, and cells (at 105 density) suspended in a reduced-serum media (5% FBS) were added to the upper well. After incubation for the desired time period, the chamber was carefully disassembled; the membrane was washed with PBS; fixed with 4% paraformaldehyde; washed again; and mounted with Prolong Antifade with DAPI (Invitrogen). Both sides of the membrane were examined via fluorescence microscopy using excitation and emission wavelengths of 358 and 461 nm, respectively, to identify labeled cells. At least 5 high power fields per experimental condition were examined, and migrated cells were expressed as a percentage of total cells.
Supplementary materials and methods
Details regarding the assays for generation of the BDEneu cell line expressing dominant-negative Ptch1, quantitative real time PCR (qRT-PCR), cell proliferation assay, cell surface protein biotinylation, animal experiments, and statistical analysis are described in detail within the Supplementary materials and methods section.
Results
CCA cells have impaired cilium expression
We initially examined cellular cilia expression using immunocytochemistry for acetylated α-tubulin [11]. Human CCA cells minimally expressed cilia (0.4% of HuCC-T1, and none of the Mz-ChA-1 cells) as compared to non-malignant cholangiocytes which abundantly expressed cilia (40.3% of H69 cells; p <0.001; Fig. 1A). Similar findings were observed in rat cell lines where BDEneu cells completely lacked cilia expression unlike non-malignant cholangiocytes in which 18.5% of BDE1 and 15.6% of NRC cells expressed cilia (p <0.001; Fig. 1B). Thus, we confirmed prior findings that human and rat CCA cells minimally express cilia [11]. As cilia expression is highly cell-cycle dependent [23], cilia expression by cultured proliferating non-malignant cholangiocytes was less than 50% as expected.
Fig. 1. CCA cells have impaired cilium expression and do not display canonical Hh signaling activation.
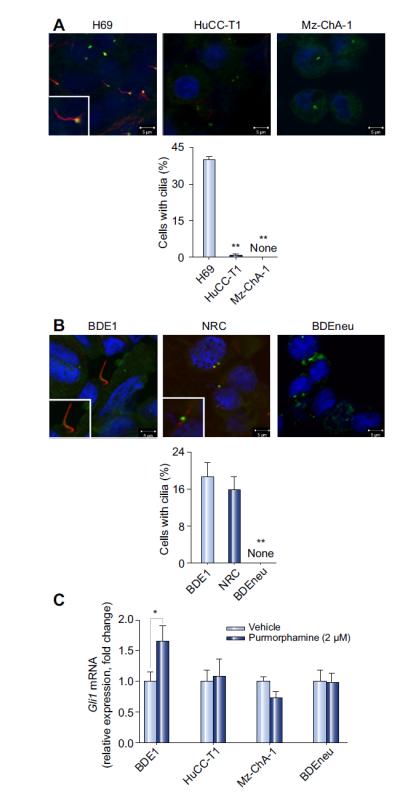
(A) Human non-malignant (H69) and CCA (HuCC-T1 and Mz-ChA-1) cells and (B) rat non-malignant (BDE1 and NRC) and CCA (BDEneu) cells were examined by confocal microscopy for immunofluorescence for acetylated α- and γ-tubulin, markers for cilia (red) and centromere (green) expression (A and B, top) respectively. Slides were analyzed under direct visualization, and results are presented as a percent of cells possessing cilia from the total number of cells in a high power microscopy field (A and B, bottom; mean ± SEM; **p <0.01). (C) Non-malignant (BDE1) and malignant (HuCC-T1, MzChA-1, and BDEneu) cell lines were cultured and treated with a small-molecule agonist of Smo, purmorphamine, at 2 μM for 72 h. Total RNA was then subjected to qRT-PCR for Gli1 as well as 18S (internal control) mRNA expression. Relative expression was determined (Δ-Δ CT compared to 18S), and results are presented as a fold change in Gli1 mRNA expression in the purmorphamine treated cells as compared to the vehicle treated cells (mean ± SEM; *p <0.05). (This figure appears in colour on the web.)
CCA cells do not display canonical Hh signaling pathway activation
Increase in Gli1 mRNA expression and translocation of the transcriptional factor Gli2 from the cell cytoplasm to the nuclei are indicators of canonical Hh signaling [4]. Therefore, we initially measured basal expression of Gli1 mRNA in non-malignant and malignant cholangiocytes (Supplementary Fig. 1A). We observed that in the human cell lines relative basal expression of Gli1 is increased in the non-malignant H69 cells and malignant KMCH cells expressing cilia as compared to the malignant HuCC-T1 and MzChA-1 cells. Among the rat cell lines, non-malignant NRC and BDE1 cell lines had lower expression of Gli1 as compared to the malignant BDEneu cells where overexpression of neu oncogene likely upregulates Gli1 in a non-canonical manner (Supplementary Fig. 1A). We next examined BDE1, HuCC-T1, Mz-ChA-1, and BDEneu cells for Gli1 mRNA expression and translocation of Gli2 into the cell nuclei following stimulation with the direct Smo agonist, purmorphamine [7]. We observed that all studied human and rat CCA cell lines (e.g., Mz-ChA-1, HuCC-T1, and BDEneu) treated with purmorphamine failed to increase Gli1 mRNA expression or translocate Gli2 protein into the cell nuclei. However, the non-malignant cholangiocyte cells, BDE1, demonstrated a 1.7-fold increase in Gli1 mRNA expression (p <0.05; Fig. 1C) and efficient nuclear translocation of Gli2 protein (Supplementary Fig. 1B). Thus, canonical Hh signaling pathway activation is disabled in HuCC-T1, Mz-ChA-1, and BDEneu CCA cells likely consistent with their impaired cilia expression.
Hh signaling is non-canonical in CCA cells
Next, we sought to explore the potential non-canonical Hh signaling in CCA cells. To investigate this signaling pathway via a loss of function paradigm, we used a genetic approach and generated a BDEneu cell line expressing dominant-negative Ptch1 (BDEΔLoop2 cells) [20,21]. We confirmed expression of the dominant-negative Ptch1ΔL2 in the transfected BDEneu cells (BDEΔLoop2, Fig. 2A). We also confirmed that the basal expression of Gli1 in the BDEΔLoop2 cells, which was equivalent to the parental cell line (Supplementary Fig. 1A). Because the non-canonical Hh signaling pathway is associated with Smo translocation to the plasma membrane [7], we examined Smo translocation from the cell interior to the plasma membrane upon Hh signaling stimulation in the BDEneu and BDEΔLoop2 cells by immunocytochemistry. rm-Shh-N ligand induced Smo translocation from the cell interior to the plasma membrane in BDEneu, but not BDEΔLoop2 cells as anticipated due to their expression of the dominant-negative Ptch1ΔL2 construct (Fig. 2B and C). However, Smo translocation to the plasma membrane was observed in the BDEΔLoop2 cells following direct Smo stimulation with purmorphamine (Fig. 2D and E). Smo translocation to the plasma membrane upon Hh signaling pathway stimulation with purmorphamine was also observed in BDEneu cells by demonstrating an increase in cell surface Smo biotinylation (Fig. 2F). Smo translocation was abrogated by PTX (Fig. 2D-F). The incompleteness of PTX inhibition is likely secondary to partial pharmacological antagonization. These findings are consistent with activation of the Hh signaling pathway in CCA cells in a Ptch1-dependent and PTX sensitive manner by a process associated with Smo plasma membrane translocation.
Fig. 2. The Hh signaling pathway is activated in CCA cells.

(A) BDEneu cells were genetically modified to express dominant-negative Ptch1. Genomic DNA was isolated from BDEneu and BDEΔLoop2 cells and examined on an agarose gel for presence of Ptch1 lacking second extracellular loop (PtchΔLoop2). The apparent DNA size is indicated in number of base pairs (bp). (B) The BDEneu and BDEΔLoop2 cells were plated and treated with either vehicle or rm-Shh-N (6 μM). Cells were examined by confocal microscopy for Smo immunofluorescence (green). (C) Slides were analyzed and cells counted using ImageJ software; results are presented as percent of cells with Smo expression at the plasma membrane from the total number of cells per a high power field (mean ± SEM; ***p <0.001). (D) The BDEneu and BDEΔLoop2 cells were plated and treated with either vehicle or purmorphamine (2 μM) with and without PTX (200 μg/ml) for 16 h. Cells were examined by confocal microscopy for Smo immunofluorescence (green) and Smo localization to the plasma membrane. (E) Slides were analyzed and cells counted using ImageJ software; results are presented as percent of cells with Smo expression at the plasma membrane from the total number of cells per a high power field (mean ± SEM; ***p <0.001). (F) BDEneu cells were cultured and treated with either vehicle or purmorphamine (2 μM) with and without PTX (200 μg/ml) for 16 h. Surface proteins were biotinylated, purified, and analyzed for Smo protein expression with immunoblot. Apparent molecular weight is indicated in kDa. (This figure appears in colour on the web.)
We next determined if the Hh signaling pathway stimulation in CCA cells leads to chemotaxis and if this process is PTX sensitive. Both human CCA cell lines revealed purmorphamine-induced chemotaxis (61% for HuCC-T1, and 62% for Mz-ChA-1 cells, p <0.01 and p <0.01, respectively; Fig. 3A). This chemotaxic response to purmorphamine stimulation was abrogated by PTX (Fig. 3A). Purmorphamine treatment in HuCC-T1 cells was also associated with actin cytoskeleton remodeling with the formation of filopodia- and lamellipodia-like protrusions; the focal adhesion-associated adaptor protein paxillin also co-localized to these protrusions confirming their status as bona fide focal adhesion complexes (Fig. 3B, arrows) [22]. PTX prevented these observed cytoskeleton rearrangements (Fig. 3B). In both human CCA cell lines, chemotaxis by the Smo agonist, purmorphamine, was unaffected by Gli inhibition with GANT61 (Fig. 3C). After affirming that the BDEneu and BDEΔLoop2 cells have similar proliferation rates (Fig. 3D), we examined these cell lines for chemotaxis in response to Hh signaling agonists. In response to rm-Shh-N ligand only the BDEneu, but not the BDEΔLoop2 cells, exhibited an increase in migratory behavior as compared with vehicle treated cell groups (21% increase for BDEneu cells, p <0.001; Fig. 3E). However, both, BDEneu and BDEΔLoop2, cell lines demonstrated chemotaxis to purmorphamine (Fig. 3F). The later effect was again blunted by PTX (Fig. 3F). Collectively, these observations suggest that both human and rat CCA cells respond to Hh signaling activation by displaying chemotaxis in a G-protein dependent manner.
Fig. 3. In CCA cells, activation of the Hh signaling pathway requires Ptch1 and leads to cell migration with cytoskeleton rearrangements in a G-protein dependent manner.

Modified Boyden chambers were employed to study cell migration (A, C, E, and F). Membranes were prepared, and cell nuclei were marked with DAPI. Cell migration across the membrane was assessed with direct visualization using fluorescence microscopy. Due to differences in migratory biology, human and rat CCA cell lines were treated over different time periods. Results are presented as a percent of migrated cells from the total number of cells. (A) The human CCA (HuCC-T1 and Mz-ChA-1) cells were studied for chemotaxis by vehicle and purmorphamine (2 μM) with and without PTX (200 μg/ml) for 8 h (mean ± SEM; **p <0.01). (B) The CCA cell line was studied for the actin cytoskeleton remodeling and localization of the focal adhesion-associated protein, paxillin. The HuCC-T1 cells were treated with either vehicle or purmorphamine (2 μM) with and without PTX (200 μg/ml) for 8 h and examined under the fluorescence microscope for phalloidin (red) marking F-actin expression and paxillin (green) marking focal adhesions (arrows). Results are shown as representative images taken at resolution of 630×. (C) The human CCA (HuCC-T1 and Mz-ChA-1) cells were studied for chemotaxis by vehicle or GANT61 (20 μM) with and without purmorphamine (2 μM) for 8 h (mean ± SEM; **p <0.01). (D) The rat CCA cells (BDEneu and BDEΔLoop2) were cultured and examined daily with colorimetric assay for the cell proliferation rate. Results are presented as an absorbance at 480 nm wavelength (reference wavelength is 630 nm). (E, F) The rat CCA cells (BDEneu and BDEΔLoop2) were studied for chemotaxis by vehicle, rm-Shh-N (6 μM; E), or purmorphamine (2 μM; F) with or without PTX (200 μg/ml; F) for 24 h (mean ± SEM; *p <0.05; ***p <0.001). (This figure appears in colour on the web.)
Genetic and pharmacological Hh signaling suppression inhibits tumor engraftment and metastases in vivo
We also sought to verify the importance of the Hh signaling pathway in CCA biology in vivo. Therefore, we employed a “patient-like” syngeneic orthotopic rat CCA model [24]. We first implanted two groups of animals with either BDEneu or BDEΔLoop2 cells. At day 21, none of the animals in the group implanted with cells with dominant-negative Ptch1 (BDEΔLoop2 cells) developed tumors; in contrast, 100% of animals implanted with the BDEneu cells developed aggressive tumors (p <0.05; Fig. 4A and B). Moreover, 89% of animals injected with BDEneu cells had tumors associated with extrahepatic metastases (Fig. 4B). To verify these genetic studies, we employed a pharmacological approach using the Smo inhibitor, vismodegib; a clinically approved small-molecule inhibitor of Smo [25]. After orthotopic implantation of BDEneu cells, animals were treated with either vehicle or vismodegib starting day 0 through day 6. On day 7 animals were sacrificed and examined for the presence of tumor and tumor burden. We observed that only 54.4% of animals in the vismodegib treated group developed tumors as compared with 100% of animals in the vehicle treated group (p <0.05, Fig. 4C). The tumor burden measured by tumor to liver weight ratio was also lower in animals treated with vismodegib as compared with animals treated with vehicle (3.2% vs. 0.7%, p = 0.15; Fig. 4D). Finally, we examined the in vivo efficacy of vismodegib on advanced tumor progression and metastases. In our orthotopic rodent CCA model, we injected the animals with BDEneu cells and started treatment with either vehicle or vismodegib on day 7, when the tumors are moderately advanced [24]. After 14 days of treatment we observed that animals treated with vismodegib had the significantly lower tumor to liver weight ratio (54% vs. 25%, p <0.05; Fig. 4E) and metastases burden (10 vs. 4 per animal, p <0.005; Fig. 4F). Collectively, these results demonstrate that genetic and pharmacological inhibition of Hh signaling impairs tumor implantation and progression.
Fig. 4. In vivo, genetic and pharmacological inhibition of the Hh pathway is tumor and metastases suppressive.

A syngeneic orthotopic rat CCA model was employed for the experiments (Fisher 344 rats; A–D). (A, B) 21 days after CCA cells (BDEneu and BDEΔLoop2) implantation, animals were euthanized and examined for the presence of tumor and tumor metastases. (A) Depicted are representative explanted livers from the animals implanted with either BDEneu (n = 9; left) or BDEΔLoop2 (n = 9; right) cells. (B) A stacked column plot represents a percent of animals with tumor with and without metastases (mean ± SEM; *p <0.05). (C, D) Animals were implanted with BDEneu cells and treated with either vehicle (n = 10) or vismodegib (n = 11; 25 mg/kg, intraperitoneally; day 0–6). Animals were euthanized on day 7 and examined for the presence of tumors (C) and liver and tumor weight (D). Results are presented as a percent of animals that developed tumor (C) and as a percent of tumor weight from the total liver weight (D; mean ± SEM; *p <0.05; n.s., non significant). (E, F) Animals were implanted with BDEneu cells and treated with either vehicle (n = 9) or vismodegib (n = 10; 25 mg/kg; intraperitoneally; day 7–21). Animals were euthanized on day 22 and examined for tumor (E) and metastases burden (F). Results for tumor burden are presented as a percent of tumor weight from total liver weight (E; mean ± SEM; *p <0.05). Metastases burden is represented as an average metastases burden [size of metastases for each examined site (omentum, peritoneum, and diaphragm) on a scale from 0 to 4] per animal in vehicle and vismodegib treated groups (mean ± SEM; **p <0.01). (G) A schematic diagram illustrating the role of the non-canonical Hh signaling pathway in CCA. (This figure appears in colour on the web.)
Discussion
The results of the current study provide mechanistic insights regarding the role of Hh signaling in CCA biology. The results indicate that in both human and rat CCA cell lines lacking cilia, Hh signaling agonists: (i) fail to enhance Gli1 expression or Gli2 nuclear translocation but do stimulate Smo plasma membrane translocation; and (ii) induce cell migration and cytoskeleton remodeling in a PTX sensitive manner. Furthermore, Hh signaling inhibition is tumor suppressive in an animal model of CCA. Collectively, these data implicate a non-canonical Hh signaling pathway in CCA tumor progression (Fig. 4G).
Cancer cell implantation and dissemination are essential steps in cancer metastasis. The cell implantation process is complex but one of the initial steps is the establishment of productive connections within the microenvironment [26]. Prevention of cancer cell interactions with the tumor microenvironment, and especially migration to and lodging of the cancer cells at metastatic sites may be therapeutic for CCA. Although the Hh signaling pathway has been implicated in CCA progression and metastasis [3,27], the precise role of Hh signaling activation in CCA remains elusive. The canonical Hh signaling pathway involves regulation of the transcriptional factors of the Gli family as first described in the context of Drosophila melanogaster morphogenesis. Intriguingly, canonical Hh signaling involves cilia as Smo receptor translocation to the cilia is requisite for Glis activation [4]. However, CCA cells frequently do not express cilia [11], and in the current study did not respond to the Hh signaling agonist by increasing Gli1 mRNA expression or Gli2 nuclear translocation. Hence canonical Hh signaling does not appear to be very robust in CCA cells. Smo signaling, thus, likely is pleiotropic. Smo represents a seven-membrane spanning domain protein, able to couple to the inhibitory G-proteins (Gis). Gis proteins promote actin cytoskeleton remodeling likely via activation of small GTPases, RhoA, and Rac [4]. Indeed, we observed that CCA cells respond to Hh signaling agonists by displaying cytoskeleton remodeling and cell migration by a PTX sensitive mechanism. Thus, Smo appears to function as a classic G-protein receptor promoting cell implantation and migration in CCA cells. Further work will be required to dissect the mechanisms, by which Smo activation in the context of cilia differs from that in the absence of these organelles. The precise G-protein mediating Smo signaling cascades also need to be elucidated but are beyond the scope of the current work.
Inhibition of the Hh signaling pathway was found to be tumor suppressive for several solid organs and hematological malignancies in experimental and clinical settings [28,29]. Therefore, we examined the effect of Hh signaling disruption in an in vivo pre-clinical model of CCA. First, we tested cell engraftment in the implantation model of CCA. CCA cells displayed impaired engraftment after the interruption of the Hh pathway either genetically with dominant-negative Ptch1 or pharmacologically with vismodegib. Next, we examined the efficacy of vismodegib in more advanced stage of cancer. The CCA cell migratory and dissemination potential was attenuated by vismodegib after allowing for initial tumor implantation in vivo. Finally, vismodegib also reduced the tumor burden in an advanced stage of CCA in this rodent model. Hence direct Smo inhibition was quite tumor suppressive in the syngeneic orthotopic model of CCA, likely secondary to blocking a non-canonical Hh signaling pathway; while other non-canonical cell survival signals mediated via Smo receptor can also be contributors to these effects. In addition, as cancer-associated fibroblasts (CAFs) in tumor microenvironment are important for tumor progression and sensitive to canonical Hedgehog signaling [30], a partial effect of the Smo inhibitor on CCA suppression via effects on CAFs in vivo cannot be excluded.
Collectively, our findings suggest the dominance of a Gli- and cilia-independent non-canonical Hh signaling pathway in CCA cell lines. The present work for the first time provides mechanistic insight regarding the involvement of the G-protein coupling property of Hh pathway in mammalian tumor cell engraftment, chemotaxis, and migration in in vitro and in vivo models. It also demonstrates that genetic and pharmacological inhibition of the Hh pathway is tumor suppressive in a preclinical model of CCA. Further studies are warranted to elucidate the role of non-canonical Hh signaling in other malignancies and perhaps taking this preclinical data to the CCA patient bedside.
Supplementary Material
Acknowledgements
Financial Support
This project was supported by NIH grants DK59427 (GJG), CA83650 (AES), T32 DK007198 (NR), and R21CA166635 (SAG). Additional support was received from the Mayo Clinic and the NIDDK funded Genetics and Optical Microscopy Cores of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567).
The authors want to thank Dr. Harmeet Malhi for kindly contributing to the discussion of the methodology of the study, and Ms. Courtney Hoover for her outstanding secretarial support.
Abbreviations
- CAFs
cancer-associated fibroblasts
- CCA
cholangiocarcinoma
- DAPI
4′-6-diamidino-2-phenylindole
- FBS
fetal bovine serum
- Hh
Hedgehog
- Gli
glioma-associated transcriptional factor
- Ptch1
Patched-1
- PTX
pertussistoxin
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- PBS
phosphate-buffered saline
- Shh
Sonic Hh
- Smo
Smoothened
Footnotes
Conflict of interest
The underlying research reported in the study was funded by the NIH Institutes of Health.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jhep.2013.11.005.
References
- 1.Razumilava N, Gores GJ. Classification, diagnosis, and management of cholangiocarcinoma. Clin Gastroenterol Hepatol. 2013;11:13–21. doi: 10.1016/j.cgh.2012.09.009. [e11; quiz e13–14] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 3.El Khatib M, Kalnytska A, Palagani V, Kossatz U, Manns MP, Malek NP, et al. Inhibition of hedgehog signaling attenuates carcinogenesis in vitro and increases necrosis of cholangiocellular carcinoma. Hepatology. 2013;57:1035–1045. doi: 10.1002/hep.26147. [DOI] [PubMed] [Google Scholar]
- 4.Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal. 2012;5:re6. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 6.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bijlsma MF, Damhofer H, Roelink H. Hedgehog-stimulated chemotaxis is mediated by Smoothened located outside the primary cilium. Sci Signal. 2012;5:ra60. doi: 10.1126/scisignal.2002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med. 2009;15:1062–1065. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 10.McMillan R, Matsui W. Molecular pathways: the hedgehog signaling pathway in cancer. Clin cancer Res. 2012;18:4883–4888. doi: 10.1158/1078-0432.CCR-11-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gradilone SA, Radtke BN, Bogert PS, Huang BQ, Gajdos GB, Larusso NF. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013;73:211–217. doi: 10.1158/0008-5472.CAN-12-2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurita S, Mott JL, Almada LL, Bronk SF, Werneburg NW, Sun SY, et al. GLI3-dependent repression of DR4 mediates hedgehog antagonism of TRAIL-induced apoptosis. Oncogene. 2010;29:4848–4858. doi: 10.1038/onc.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polizio AH, Chinchilla P, Chen X, Kim S, Manning DR, Riobo NA. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem. 2011;286:19589–19596. doi: 10.1074/jbc.M110.197111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grubman SA, Perrone RD, Lee DW, Murray SL, Rogers LC, Wolkoff LI, et al. Regulation of intracellular pH by immortalized human intrahepatic biliary epithelial cell lines. Am J Physiol. 1994;266:G1060–G1070. doi: 10.1152/ajpgi.1994.266.6.G1060. [DOI] [PubMed] [Google Scholar]
- 15.Knuth A, Gabbert H, Dippold W, Klein O, Sachsse W, Bitter-Suermann D, et al. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J Hepatol. 1985;1:579–596. doi: 10.1016/s0168-8278(85)80002-7. [DOI] [PubMed] [Google Scholar]
- 16.Miyagiwa M, Ichida T, Tokiwa T, Sato J, Sasaki H. A new human cholangiocellular carcinoma cell line (HuCC-T1) producing carbohydrate antigen 19/9 in serum-free medium. In Vitro Cell Dev Biol. 1989;25:503–510. doi: 10.1007/BF02623562. [DOI] [PubMed] [Google Scholar]
- 17.Yang L, Faris RA, Hixson DC. Long-term culture and characteristics of normal rat liver bile duct epithelial cells. Gastroenterology. 1993;104:840–852. doi: 10.1016/0016-5085(93)91021-9. [DOI] [PubMed] [Google Scholar]
- 18.Lai GH, Zhang Z, Shen XN, Ward DJ, Dewitt JL, Holt SE, et al. ErbB-2/neu transformed rat cholangiocytes recapitulate key cellular and molecular features of human bile duct cancer. Gastroenterology. 2005;129:2047–2057. doi: 10.1053/j.gastro.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Razumilava N, Bronk SF, Smoot RL, Fingas CD, Werneburg NW, Roberts LR, et al. MiR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology. 2012;55:465–475. doi: 10.1002/hep.24698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parra LM, Zou Y. Sonic hedgehog induces response of commissural axons to Semaphorin repulsion during midline crossing. Nat Neurosci. 2010;13:29–35. doi: 10.1038/nn.2457. [DOI] [PubMed] [Google Scholar]
- 21.Hackett PB, Ekker SC, Largaespada DA, McIvor RS. Sleeping beauty transposon-mediated gene therapy for prolonged expression. Adv Genet. 2005;54:189–232. doi: 10.1016/S0065-2660(05)54009-4. [DOI] [PubMed] [Google Scholar]
- 22.Schaller MD. Paxillin: a focal adhesion-associated adaptor protein. Oncogene. 2001;20:6459–6472. doi: 10.1038/sj.onc.1204786. [DOI] [PubMed] [Google Scholar]
- 23.Plotnikova OV, Pugacheva EN, Golemis EA. Primary cilia and the cell cycle. Methods Cell Biol. 2009;94:137–160. doi: 10.1016/S0091-679X(08)94007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sirica AE, Zhang Z, Lai GH, Asano T, Shen XN, Ward DJ, et al. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology. 2008;47:1178–1190. doi: 10.1002/hep.22088. [DOI] [PubMed] [Google Scholar]
- 25.Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171–2179. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shibue T, Brooks MW, Inan MF, Reinhardt F, Weinberg RA. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov. 2012;2:706–721. doi: 10.1158/2159-8290.CD-11-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song Q, Li Y, Zheng X, Fang Y, Chao Y, Yao K, et al. MTA1 contributes to actin cytoskeleton reorganization and metastasis of nasopharyngeal carcinoma by modulating Rho GTPases and Hedgehog signaling. Int J Biochem Cell Biol. 2013;45:1439–1446. doi: 10.1016/j.biocel.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 28.Blotta S, Jakubikova J, Calimeri T, Roccaro AM, Amodio N, Azab AK, et al. Canonical and non-canonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood. 2012;120:5002–5013. doi: 10.1182/blood-2011-07-368142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104:8455–8460. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.