

Abstract
The aim of this study is to determine whether primary over-expression of AβPP in skeletal muscle results in the development of features of inclusion body myositis (IBM) in a new lineage of the MCK-AβPP transgenic mouse. Quantitative histological, immunohistochemical and western blotting studies were performed on muscles from 3 to 18 month old transgenic and wild-type C57BL6/SJL mice. Electron microscopy was also performed on muscle sections from selected animals. Although western blotting confirmed that there was over-expression of full length AβPP in transgenic mouse muscles, deposition of amyloid-β and fibrillar amyloid could not be demonstrated histochemically or with electron microscopy. Additionally, other changes typical of IBM such as rimmed vacuoles, cytochrome C oxidase-deficient fibres, upregulation of MHC antigens, lymphocytic inflammatory infiltration and T cell fibre invasion were absent. The most prominent finding in both transgenic and wild-type animals was the presence of tubular aggregates which was age-related and largely restricted to male animals. Expression of full length AβPP in this MCK-AβPP mouse lineage did not reach the levels required for immunodetection or deposition of amyloid-β as in the original transgenic strains, and was not associated with the development of pathological features of IBM. These negative results emphasise the potential pitfalls of re-deriving transgenic mouse strains in different laboratories.
Keywords: amyloid-β, inclusion body myositis, MCK-AβPP transgenic mouse, muscle histology, tubular aggregates
Introduction
There are two alternative theories for the pathogenesis of inclusion body myositis (IBM), the most common inflammatory myopathy in individuals over the age of 50 years (Needham & Mastaglia 2008). The first proposes that IBM is primarily an immune-mediated inflammatory disorder which is initiated by the presentation of antigenic peptides by muscle fibres, and is associated with a number of characteristic myodegenerative changes (Dalakas 2005). The second theory proposes that IBM is caused by abnormal accumulation of amyloid-β (Aβ) and other misfolded proteins in intracellular inclusions, with associated impairment of proteasomal and mitochondrial function and increased oxidative stress, culminating in autophagic degeneration of muscle fibres (Askanas & Engel 2003). In this scenario, the T cell predominant lymphocytic inflammation typical of IBM may be regarded as a secondary feature.
One approach to elucidating the pathogenesis of IBM is the use of animal models such as the MCK-AβPP transgenic mouse. This C57BL6/SJL transgenic mouse strain, first reported by Sugarman et al. in 2002, carries the human amyloid β precursor protein (AβPP) cDNA transgene encoding the brain isoform (AβPP695) harbouring the Swedish double mutation, under control of the muscle creatine kinase (MCK) gene promoter, resulting in over-expression of AβPP in skeletal muscle, particularly in fast-twitch type IIB muscle fibres (Sugarman et al. 2006). Since Aβ accumulation is specifically caused by AβPP over-expression, the model provides an opportunity for investigating the potential role of AβPP and secondary downstream events in the pathogenesis of IBM. In the original study of the MCK-APP mouse, the predominant isoform of AβPP expressed in muscles after the age of 4–6 months was the C99 fragment which is a product of post-translational cleavage of AβPP by β-secretase (Sugarman et al. 2002), and only a small proportion of the Aβ42 peptide was produced, which is the predominant form in IBM (Vattemi et al. 2009). Histological studies confirmed the presence of AβPP/Aβ-reactive deposits in muscle fibres but other typical changes of IBM, such as rimmed vacuoles, muscle fibre necrosis and regeneration, or T cell myofibre invasion were not reported. Although there was an inflammatory response, it was predominantly neutrophilic rather than lymphocytic as in IBM. More recent studies in the MCK-APP mouse have reported only mitochondrial and other nonspecific abnormalities in muscle fibres (Beckett et al. 2010; Boncompagni et al. 2012).
Here we report the results of a more detailed quantitative histological, immunohistochemical and ultrastructural study of skeletal muscles at different ages in a new lineage of the MCK-AβPP mouse derived from the original transgenic strain. Our aim was to further investigate the spectrum of pathological changes and their comparability to human IBM.
Materials and methods
Transgenic mice and tissue preparation
The MCK-AβPP mouse colony was re-derived at the Animal Resources Centre (Murdoch University, WA, Australia) from a breeding pair obtained from the University of California, Irvine where the model was first developed (courtesy of Professor F LaFerla, University of California, Irvine, CA, USA). All experiments performed were approved by the University of Western Australia Animal Experimentation Committee. A total of 46 age-matched transgenic and wild-type mice were sacrificed at 3, 6, 9, 12 and 18 months of age (Table 1). The triceps brachii, quadriceps femoris, and tibialis anterior muscles were snap frozen in isopentane pre-cooled with liquid nitrogen and stored at −80 °C. Sections 8 μm thick for histological studies and immunoblotting were prepared using a Leica CM1900 cryostat (Leica Microsystems, North Ryde, NSW, Australia).
Table 1.
Mice used in the present study
| Genotype | ||||
|---|---|---|---|---|
| wt | tg | |||
| Age | Male | Female | Male | Female |
| 3 months | 2 | 1 | 2 | 3 |
| 6 months | 3 | 1 | 5 | 1 |
| 9 months | 3 | 2 | 3 | 2 |
| 12 months | 2 | 2 | 5 | 1 |
| 18 months | 3 | 2 | 3 | 0 |
MCK-AβPP mouse genotyping
PureLink Genomic DNA mini kits (Invitrogen, Mulgrave, SW, Australia) were used for DNA extraction. DNA was isolated and purified from approximately one hundred 7 μm thick cryostat muscle sections according to the manufacturer's instructions. The concentration of DNA was measured using a ND-1000 spectrophotometer (Thermo Scientific, Scoresby, Vic., Australia). A 25 μl amplification reaction was set up containing 100 ng genomic DNA, 10 mM Tris-HCl pH 6.8, 50 mM KCl, 2 mM MgCl2, 0.2 mM dNTPs, 0.5 U AmpliTaq DNA polymerase and 25 ng primers. Forward primer, AβPP gatgcagaattccgacatga; reverse primer, SV40 caaaccacaactagaatgcagtg. PCR cycling conditions were 94 °C for 6 min, 35 cycles of 94 °C for 30 s, 55 °C for 1 min, 72 °C for 2 min. Amplicons were electrophoresed on 2% agarose gels and imaged using a Chemi-Smart 3000 gel documentation system (Vilber Lourmat, Marne-la-Vallée, France). They were also sequenced on an Applied Biosystems 3730xl DNA Sequencer (Invitrogen).
Muscle histology and histochemistry
The numbers of necrotic and regenerating fibres and fibres with tubular aggregates per 1000 fibres were quantified in 10 randomly selected fields at ×400 in haematoxylin and eosin (H & E) stained sections. Necrotic fibres were identified as paler-staining fibres undergoing phagocytosis, and regenerating fibres as basophilic fibres with enlarged nuclei with prominent nucleoli. Sections were also stained using the modified Gomori trichrome, nicotinamide adenine dinucleotide-tetrazolium reductase (NADH), cytochrome C oxidase (COX), succinate dehydrogenase (SDH) and Congo red techniques. Slides were viewed under an Olympus BX41 microscope (Olympus, Mt Waverley, Vic., Australia) and polarised light.
Immunohistochemistry
Immunohistochemistry for AβPP/Aβ, tubular aggregates, MHC antigens and inflammatory cells was performed on 8 μm frozen muscle sections. The antibodies used are listed in Table 2. Detection of CD3, CD20, AβPP/Aβ (6E10 and 22C11) and SERCA 1 ATPase was performed using an Envision kit (Dako, Campbellfield, Vic., Australia). Endogenous peroxidase was blocked in 1% (v/v) H2O2 in methanol, followed by incubation with primary antibodies for 60 min at room temperature. Sections were then placed in horseradish peroxidase (HRP)-polymer labelled goat anti-mouse/rabbit immunoglobulin, followed by colouration with diaminobenzidine. Sections were dehydrated with ethanol, cleared with xylene and mounted with a synthetic resin-mounting medium. For immunolabelling of antibodies to CD68, neutrophils and MHC-I the endogenous peroxidase was blocked as described above followed by incubation in 10% (v/v) normal goat serum. Primary antibodies were applied for 1.5 h at room temperature followed by incubation in HRP-labelled goat anti-rat immunoglobulins (STAR72; AbD Serotec, Kidlington, UK). Diaminobenzidine was used as the detection colouring agent. The antibodies to MHC-II and AβPP/Aβ (6E10 and 22C11) were fluorescently labelled using a Zenon kit (Invitrogen) according to the manufacturer's instructions at a 4.5:1 molar ratio and slides were viewed on an Olympus IX70 fluorescence microscope (Olympus).
Table 2.
Details of antibodies used in the present study
| Antigen | Isotype | Host | Clonality | Clone/code | Source | Dilution |
|---|---|---|---|---|---|---|
| CD3 | IgG | Rabbit | polyclonal | ab5690 | Abcam, Cambridge, MA, USA | 1:50 |
| CD20 | IgG | Rabbit | monoclonal | EP459Y | Novus Biologicals, Littleton, CO, USA | 1:50 |
| CD68 | IgG2a | Rat | monoclonal | FA-11 | AbD Serotec | 1:50 |
| Neutrophil | IgG2b | Rat | monoclonal | NIMP-R14 | Abcam | 1:50 |
| MHC-I | IgG2a | Rat | monoclonal | ER-HR 52 | Abcam | 1:15 |
| MHC-II | IgG1 | Mouse | monoclonal | OX6 | Abcam | 1:225 |
| Serca1 ATPase | IgG1 | Mouse | monoclonal | IIH11 | Thermo Scientific | 1:15 |
| AβPP | IgG1 | Mouse | monoclonal | 22C11 | Millipore, Billerica, MA, USA | 1:300 |
| AβPP/Aβ | IgG1 | Mouse | mononclonal | 6E10 | Covance, Princeton, NJ | 1:200 |
Brain tissue from the tg2576 mouse that over-expresses human AβPP was used as a positive control for AβPP and Aβ42 (kindly provided by Professor Ralph Martins, Edith Cowen University).
Electron microscopy
Muscles were sliced into thin strips immediately after harvesting and fixed in 2.5% (v/v) buffered gluteraldehyde at 4 °C then post-fixed in 2% (v/v) osmium tetroxide for 60 min. After dehydrating in a graded series of ethanol concentrations, the strips were embedded in Fluka araldite resin and cured for 24 h at approximately 80 °C. Sections 90 nm thick were cut with an LKB 8800 ultramicrotome and grids were stained with uranyl acetate and lead citrate. Grids were viewed and photographed with a Phillips CM-10 transmission electron microscope and the negatives subsequently scanned to produce digital images.
Immunoblot analysis of AβPP expression
Cryostat muscle sections (total weight 4.5 mg) were dissociated in 150 μl treating buffer containing 125 mM Tris-HCl (pH 6.8), 15% (w/v) SDS, 10% (v/v) glycerol, 0.5 mM phenylmethylsulfonyl fluoride and 9 μl protease inhibitor (Sigma Aldrich, Castle Hill, NSW, Australia). After sonication the homogenates were stored at −80 °C. Before blotting they were reduced by adding 50 mM dithiothretol and mixed with 0.4 μg/ml bromophenol blue. The protein extracts were denatured at 95 °C and then centrifuged at 14,000 g. For myosin densitometry, 5 μl protein lysate was diluted with 45 μl treating buffer and 6 μl samples were loaded on NuPAGE 4–12% Bis-Tris gel (Invitrogen). After separation, gels were stained with Coomasie blue and destained with 7% acetic acid overnight. Equal amounts of protein were loaded for each sample based on myosin heavy chain densitometry and were separated on NuPAGE 4–12% Bis-Tris gels and electrotransferred to polyvinylidene fluoride membranes. After blocking in blocking solution (Novex WesternBreeze Immunodetection kit; Invitrogen), membranes were incubated with primary antibody to AβPP (6E10, dilution 1:600, Covance; 22C11, 1:200, Millipore) for 6 h. After washing, membranes were incubated with alkaline phosphatase labeled anti-mouse secondary antibody and were incubated with chemiluminescent substrate for 5 min. The membrane was imaged using the Chemi-Smart 3000 gel documentation system (Vilber Lourmat), with gel and band densitometry analysis carried out using Bio-1D software version 11.06.
Data analysis
Linear regression was used to test the correlation between the number of tubular aggregates and age in male mice. Independent-samples t-test was used to compare the difference in tubular aggregates between wild-type and transgenic mice. A P-value <0.05 was considered statistically significant.
Results
Confirmation of mouse genotype
Primers targeted to exon 14 of human AβPP gene and simian virus 40 polyadenylation signal (SV40 signal) were used for transgenic mouse genotyping. An approximately 800 base amplicon was amplified in transgenic mice, while it was absent in wild-type mice (Figure 1). Direct DNA sequencing confirmed that this amplicon consists of exon 14–16 of the human AβPP gene and the downstream SV40 signal.
Figure 1.

PCR demonstrates the amplification of part of the human AβPP gene together with SV40 signal in MCK-AβPP transgenic mice, but not in wild-type mice. Lane 1: 100 bp DNA ladder, the top high intensity band is 1000 bp; lanes 2 and 3: 6 and 3 month transgenics; lane 4: 12 month wild-type animal.
Light microscopy
The most prominent finding in both transgenic and wild-type animals was the presence of tubular aggregates in muscle fibres (see below). Other histological changes were relatively minor and nonspecific (Figure 2). Necrotic and regenerating fibres were sparse (0–1.3%) and were found in both transgenic and wild-type animals. Neither rimmed vacuoles nor congophilic inclusions under polarised light were observed in transgenic or wild-type animals. Immunohistochemistry did not demonstrate reactivity for AβPP (or Aβ-related products) with either the 6E10 antibody or the 22C11 antibody (Figure 2e). There were no inflammatory infiltrates in transgenic animals, but a single perivascular mononuclear cell infiltrate was found in two 18 month old animals, one wild-type and one transgenic. Immunohistochemistry showed no CD68-positive macrophages. Staining for MHC-I and MHC-II antigens was negative and there was no invasion of non-necrotic muscle fibres by mononuclear cells. Muscle histochemical stains were normal, except for rare COX-negative fibres in one transgenic animal.
Figure 2.
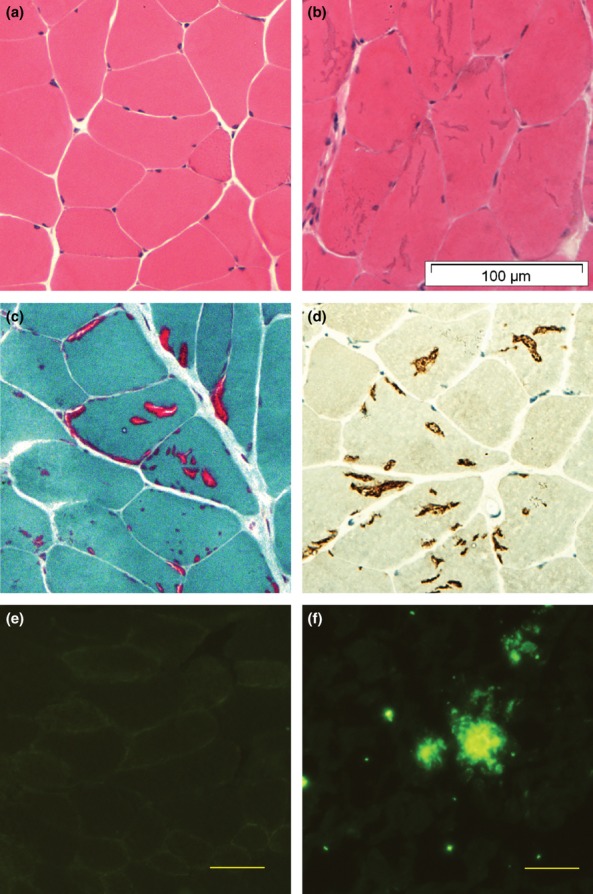
(a) Normal muscle from a 12 month female transgenic mouse (H&E); (b) Multiple tubular aggregates in muscle fibres from an 18 month old male transgenic mouse (H&E); (c) Tubular aggregates demonstrated on Gomori-Trichrome stain; (D) Tubular aggregates are positive for SERCA1-ATPase immunostaining; (e) Muscle fibres from a 12 month old transgenic animal does not show immuno-reactivity for AβPP/Aβ; (f) AβPP/Aβ-positive aggregates in tg2576 mouse brain tissue. Magnification: (a–d) ×400, (e,f) ×200. Antibody for (e,f): 6E10.
Tubular aggregates were detected in male animals after three months of age and only rarely in female animals, and were confined to type II muscle fibres. They were stained blue with H&E, bright red with Gomori, and also stained strongly with NADH and SERCA1 antibodies (Figure 2b,c,d). In H&E and Gomori stained sections the tubular aggregates had the appearance of cracks or vacuoles of various sizes, and were initially misinterpreted as rimmed vacuoles. In male mice the number of tubular aggregates increased with age in all muscles (P < 0.0001, Table 3) and there were no significant differences in their numbers between wild-type and transgenic animals (triceps brachii, P = 0.227; quadriceps femoris, P = 0.669; tibialis anterior, P = 0.358).
Table 3.
Fibres with TAs in male mice (per 1000 fibres, mean ± SE)
| Triceps brachii | Quadricpes femoris | Tibialis anterior | ||||
|---|---|---|---|---|---|---|
| Age group | wt | tg | wt | tg | wt | tg |
| 3 months | 9 ± 8 | 1 ± 1 | 7 ± 6 | 2 ± 2 | 11 ± 15 | 0 |
| 6 months | 90 ± 17 | 38 ± 26 | 50 ± 20 | 48 ± 18 | 30 ± 7 | 40 ± 17 |
| 9 months | 79 ± 28 | 77 ± 14 | 100 ± 22 | 116 ± 22 | 86 ± 25 | 82 ± 33 |
| 12 months | 148 ± 3 | 118 ± 30 | 181 ± 65 | 103 ± 23 | 63 ± 27 | 66 ± 32 |
| 18 months | 225 ± 121 | 105 ± 41 | 139 ± 63 | 209 ± 14 | 80 ± 51 | 181 ± 65 |
Electron microscopy
Electron microscopy confirmed the presence of tubular aggregates in muscle fibres in both wild-type and transgenic animals aged 6 and 9 months, with preservation of normal myofibrillar structure around them (Figure 3c,d). Other non-specific changes in transgenic animals included disruption of Z line alignment and occasional necrotic muscle fibres undergoing phagocytosis (Figure 3b). There were no intranuclear or sarcoplasmic filamentous inclusions and mitochondrial size and morphology was comparable in wild-type and transgenic animals. No fibrillar amyloid deposits were found.
Figure 3.
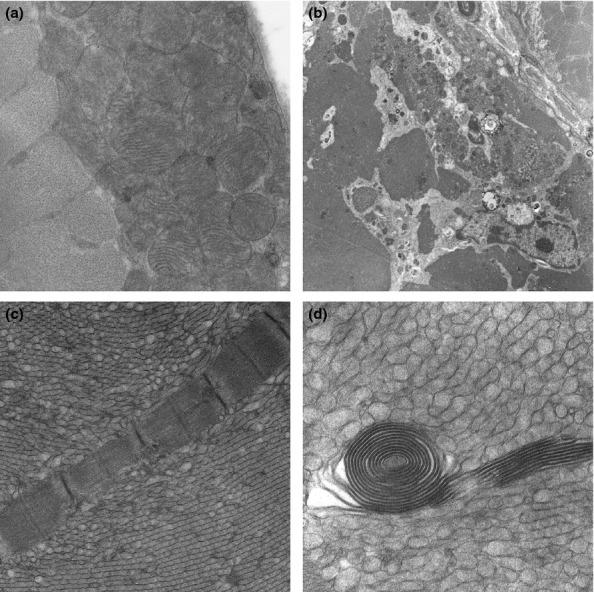
Electron microscopic findings: (a) Aggregates of subsarcolemmal mitochondria in a 9 month old wild-type male mouse. (b) A necrotic muscle fibre undergoing phagocytosis in a 6 month old transgenic male mouse. (c,d) Tubular aggregates in a 6 month old male transgenic mouse and a spiral membranous body in one tubular aggregate (d). Magnification: (a) ×6610; (a) ×1300; (c) ×5200; (d) ×12600.
AβPP immunoblots
Immunoblots demonstrated an approximately 100 kDa full-length (fl-) AβPP band in muscles from transgenic animals (6–18 months) and in the tg2576 mouse brain, but not in wild-type mouse muscles (Figure 4). Densitometry showed that the amount of muscle AβPP protein in MCK-APP transgenic animals was between 11 and 30% of the levels found in the tg2576 brain tissue. Neither Aβ42 nor C99 were found in muscles from transgenic animals even with repeated attempts using different levels of protein loading (data not shown).
Figure 4.
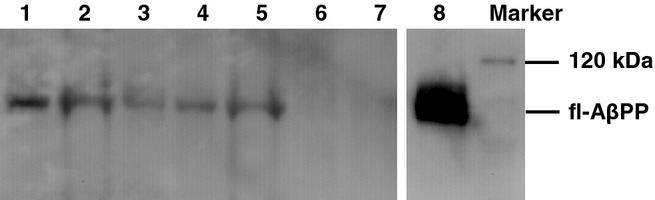
Western blots on transgenic and wild-type mouse muscles. Lanes 1 and 2: 9 month old transgenic; lanes 3 and 4:12 month old transgenic; lane 5: 18 month old transgenic; lane 6: 18 month old wild-type; lane 7: 12 month old wild-type; lane 8: tg2576 mouse brain. Antibody: 6E10.
Discussion
The MCK-AβPP mouse expresses the human AβPP695 isoform with the Swedish double mutation selectively in skeletal muscles. In the original model, accumulation of AβPP and Aβ with a diffuse or focal granular pattern of deposition in muscle fibres was demonstrated in animals older than 12 months (Sugarman et al. 2002). In addition, other histological changes, including central nucleation of muscle fibres and a neutrophilic inflammatory infiltrate were found. The authors concluded that selective over-expression of AβPP can lead to the development of a subset of IBM-like changes and that abnormal metabolism of AβPP may have a role in human IBM pathogenesis. In contrast, in this study of another MCK-AβPP colony with confirmed expression of fl-AβPP in muscle, we were unable to demonstrate accumulation of Aβ or of the C99 fragment, suggesting that the levels of AβPP expression and post-translational processing to the C99 and Aβ peptides were insufficient to reach the threshold required for immunodetection. In addition, we did not find any of the typical histological features of IBM. In particular, there was no evidence of an antigen-driven immune response against muscle fibre components, as shown by the absence of MHC antigen expression or T-cell invasion of muscle fibres. Although the breeding pair from which our colony was derived came from the original A6 line which had high AβPP expression levels as reported in the original study (M. Kitazawa, personal communication) (Sugarman et al. 2002), the level of AβPP expression in our animals was shown to be much lower than in brain tissue from the tg2576 mouse, which also carries the AβPP695 isoform with the Swedish double mutation (Hsiao et al. 1996).
The human AβPP gene produces three major isoforms through alternative splicing, AβPP770, AβPP751 and AβPP695 (Menéndez-González et al. 2005). The AβPP cDNA used in the development of the MCK-AβPP transgenic mouse encodes AβPP695, the predominant AβPP isoform in neurons, while AβPP751 is the isoform which is elevated in IBM and normal ageing brain (Oyama et al. 1991; Askanas et al. 1996). The structural difference between the two isofoms is the presence of an extra Kunitz-type protease inhibitor (KPI) domain in the extracellular region of AβPP751, which forms part of the secretory AβPP generated by the action of α- or β-secretase. Brain levels of AβPP with a KPI domain have been found to increase with ageing and in neurodegenerative diseases whereas levels of the AβPP isoform without a KPI domain are reduced with ageing and in AD (Konig et al. 1991; Zhan et al. 1995; Johnston et al. 1996; Beyer et al. 2004). AβPP with a KPI domain is involved in the formation of neuritic plaques and may interfere with the secretion of Aβ into the peripheral circulation and thereby increase intracellular levels of Aβ (Moir & Tanzi 2005). Thus the KPI domain seems to play an important role in post-translational processing of AβPP and amyloidogenesis, and it is likely that the AβPP695 isoform is associated with lower levels of Aβ formation.
There are a number of possible explanations for the lower levels of AβPP expression in our MCK-AβPP mouse line than in the original transgenic lines developed and investigated by Sugarman et al. 2002. The most likely explanation is that the founder animals from which our colony was derived carried a lower copy number of the transgene than the original lines. It is known that the expression of transgenes can be lost during prolonged culture in vitro and with repeated propagation in vivo due to various reasons. These include failure of incorporation into the host genome or incorporation into unstable sites in the genome. Alternatively, the transgene can be subjected to genome rearrangement, loss of copy number or unequal crossing-over during breeding, all of which can lead to a reduced expression of the transgene (Migliaccio et al. 2000; Barnes et al. 2003; Roy et al. 2009). We did show by sequencing the presence of the transgene in muscles from our transgenic animals, confirming that at least one copy was stably passed on through propagation. However, we were unsuccessful in trying to demonstrate the site of incorporation of the transgene. In addition, dietary and environmental differences may also influence AβPP processing and need to be considered. It is known that a number of dietary changes such as high fat, high cholesterol, high trans fatty acid diets and thiamine deficiency are amyloidogenic, while calorie restriction, increased dietary copper levels and docosahexanoic acid supplementation reduce Aβ production (Levin-Allerhand et al. 2002; Ho et al. 2004; Wang et al. 2005; Hooijmans et al. 2009; Zhao et al. 2011). Moreover, environmental enrichment that is associated with increased cognitive and physical stimulation is known to reduce Aβ plaque formation in the mouse (Calderon-Garcidueñas et al. 2004; Herring et al. 2011).
Our finding of tubular aggregates in both wild-type and transgenic animals are consistent with other reports showing age-related accumulation of tubular aggregates in various strains of wild-type and gene knockout mouse models (viz. CK−/−, Caveolin-1−/− and Caveolin-2−/− mice) (Novotová et al. 2002; Schubert et al. 2007). The absence of any differences in the numbers of tubular aggregates between wild-type and transgenic animals in the present study suggests that over-expression of AβPP is not a causal factor in their formation. However, the possibility cannot be excluded that the tubular aggregate formation in our animals is due to an unrelated gene defect accrued as a result of the re-derivation of the transgenic mouse strain, which is also present in the wild-type animals. None of the previous studies of muscle pathology in MCK-AβPP mice has reported the presence of tubular aggregates, except for one study which commented on the appearance of “punctate inclusions”, which may have been tubular aggregates (Beckett et al. 2010). A double transgenic mouse model combining MCK-AβPP and presenilin-1 noted “vacuole-like rimmed cracks” in muscle fibres, which were not characterised any further (Kitazawa et al. 2009) but probably represented tubular aggregates. Interestingly, tubular aggregates were also reported in the MLC-AβPP mouse, another possible model of IBM (Moussa et al. 2006) but this paper did not elaborate on their frequency or their presence in wild-type mice.
Studies in the MCK-AβPP mouse have reported increased resting calcium levels as well as a reduced sarcoplasmic reticulum calcium release in muscle fibres, leading to the conclusion that AβPP/Aβ overexpression leads to calcium dysregulation (Lopez & Shtifman 2010; Shtifman et al. 2010). Several of the component proteins of the sarcoplasmic reticulum, such as SERCA, have been identified in tubular aggregates, suggesting that they originate from the sarcoplasmic reticulum. SERCA, the calcium-ATP pump on the sarcoplasmic reticulum membrane, and the activity of one of its isoforms, has been shown to regulate Aβ production in vitro (Green et al. 2008). However, it is not clear if accumulation of Aβ in turn stimulates SERCA activity. The observation that tubular aggregates accumulate calcium ions indicates that they may still be functionally active in calcium uptake (Salviati et al. 1985) and may thereby contribute to disturbances in cytosolic calcium levels. A link between AβPP overexpression and calcium dyshomeostasis, as suggested by previous studies on MCK-AβPP mice (Lopez & Shtifman 2010), therefore needs to be viewed with caution given the presence of tubular aggregates in wild-type as well as transgenic strains. Similarly, any conclusions regarding the relationship between AβPP/Aβ and calcium homeostasis in the MCK-AβPP mouse and, by inference, in IBM need to be made with caution.
In conclusion, the level of expression of fl-AβPP in this transgenic mouse strain was much lower than in the original strains from which they were derived and was insufficient to reach the threshold for immunodetection or to be processed or assembled into Aβ. Moreover, neither the typical immunopathologic or myodegenerative changes of IBM developed in animals up to 18 months of age. The present findings emphasise the potential pitfalls when transgenic mouse models are re-derived in different laboratories, and illustrate the phenotypic changes that can occur in more distant lineages as a result of altered expression of the transgene.
Acknowledgments
The authors thank Dr Limbi Kanyenda and Dr Sherif Boulos for their helpful advice and Abbie Adams, Kane Greer, Loren Price for technical support. Professors Masashi Kitazawa and Michael P. Murphy are gratefully acknowledged for helpful discussion. We also thank Professor Norman Palmer for critical reading of the manuscript. This work was supported by the Neuromuscular Foundation of Western Australia. Dr Yue-Bei Luo was supported by a China Scholarship Council-University of Western Australia joint PhD scholarship.
Conflict of interests
The authors declare no conflict of interests.
References
- Askanas V, Engel WK. Proposed pathogenetic cascade of inclusion-body myositis: importance of amyloid-β, misfolded proteins, predisposing genes, and aging. Curr. Opin. Rheumatol. 2003;15:737–744. doi: 10.1097/00002281-200311000-00009. [DOI] [PubMed] [Google Scholar]
- Askanas V, McFerrin J, Baqué S, Alvarez RB, Sarkozi E, Engel WK. Transfer of β-amyloid precursor protein gene using adenovirus vector causes mitochondrial abnormalities in cultured normal human muscle. Proc. Natl Acad. Sci. USA. 1996;93:1314–1319. doi: 10.1073/pnas.93.3.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes LM, Bentley CM, Dickson AJ. Stability of protein production from recombinant mammalian cells. Biotechnol. Bioeng. 2003;81:631–639. doi: 10.1002/bit.10517. [DOI] [PubMed] [Google Scholar]
- Beckett TL, Niedowicz DM, Studzinski CM, et al. Effects of nonsteroidal anti-inflammatory drugs on amyloid-β pathology in mouse skeletal muscle. Neurobiol. Dis. 2010;39:449–456. doi: 10.1016/j.nbd.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer K, Lao JI, Carrato C, et al. Upregulation of amyloid precursor protein isoforms containing Kunitz protease inhibitor in dementia with Lewy bodies. Brain Res. Mol. Brain Res. 2004;131:131–135. doi: 10.1016/j.molbrainres.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Boncompagni S, Moussa CE, Levy E, et al. Mitochondrial dysfunction in skeletal muscle of amyloid precursor protein-overexpressing mice. J. Biol. Chem. 2012;287:20534–20544. doi: 10.1074/jbc.M112.359588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon-Garcidueñas L, Reed W, Maronpot RR, et al. Brain inflammation and Alzheimer's-like pathology in individuals exposed to severe air pollution. Toxicol. Pathol. 2004;32:650–658. doi: 10.1080/01926230490520232. [DOI] [PubMed] [Google Scholar]
- Dalakas MC. Inflammatory, immune, and viral aspects of inclusion-body myositis. Neurology. 2005;66:S33–S38. doi: 10.1212/01.wnl.0000192129.65677.87. [DOI] [PubMed] [Google Scholar]
- Green KN, Demuro A, Akbari Y, et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Gen. Physiol. 2008;132:1107–1116. doi: 10.1085/JGP1322OIA1. [DOI] [PubMed] [Google Scholar]
- Herring A, Lewejohann L, Panzer AL, et al. Preventive and therapeutic types of environmental enrichment counteract beta amyloid pathology by different molecular mechanisms. Neurobiol. Dis. 2011;42:530–538. doi: 10.1016/j.nbd.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Ho L, Qin W, Pompl PN, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB. J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- Hooijmans CR, Van der Zee CE, Dederen PJ, et al. DHA and cholesterol containing diets influence Alzheimer-like pathology, cognition and cerebral vasculature in APPswe/PS1dE9 mice. Neurobiol. Dis. 2009;33:482–498. doi: 10.1016/j.nbd.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Norgren S, Ravid R, et al. Quantification of APP and APLP2 mRNA in APOE genotyped Alzheimer's disease brains. Brain Res. Mol. Brain Res. 1996;43:85–95. doi: 10.1016/s0169-328x(96)00161-1. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Vasilevko V, Cribbs DH, LaFerla FM. Immunization with amyloid-β attenuates inclusion body myositis-like myopathology and motor impairment in a transgenic mouse model. J. Neurosci. 2009;29:6132–6141. doi: 10.1523/JNEUROSCI.1150-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig G, Salbaum JM, Wiestler O, et al. Alternative splicing of theβA4 amyloid gene of Alzheimer's disease in cortex of control and Alzheimer's disease patients. Brain Res. Mol. Brain Res. 1991;9:259–262. doi: 10.1016/0169-328x(91)90010-u. [DOI] [PubMed] [Google Scholar]
- Levin-Allerhand JA, Lominska CE, Smith JD. Increased amyloid-β levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J. Nutr. Health Aging. 2002;6:315–319. [PubMed] [Google Scholar]
- Lopez JR, Shtifman A. Intracellular β-amyloid accumulation leads to age-dependent progression of Ca2+ dysregulation in skeletal muscle. Muscle Nerve. 2010;42:731–738. doi: 10.1002/mus.21745. [DOI] [PubMed] [Google Scholar]
- Menéndez-González M, Pérez-Pinera P, Martínez-Rivera M, Calatayud MT, Blázquez Menes B. APP processing and the APP-KPI domain involvement in the amyloid cascade. Neurodegener. Dis. 2005;2:277–283. doi: 10.1159/000092315. [DOI] [PubMed] [Google Scholar]
- Migliaccio AR, Bengra C, Ling J, et al. Stable and unstable transgene integration sites in the human genome: extinction of the Green Fluorescent Protein transgene in K562 cells. Gene. 2000;256:197–214. doi: 10.1016/s0378-1119(00)00353-x. [DOI] [PubMed] [Google Scholar]
- Moir RD, Tanzi RE. LRP-mediated clearance of Aβ is inhibited by KPI-containing isoforms of APP. Curr. Alzheimer Res. 2005;2:269–273. doi: 10.2174/1567205053585918. [DOI] [PubMed] [Google Scholar]
- Moussa CE, Fu Q, Kumar P, et al. Transgenic expression of β-APP in fast-twitch skeletal muscle leads to calcium dyshomeostasis and IBM-like pathology. FASEB J. 2006;20:2165–2167. doi: 10.1096/fj.06-5763fje. [DOI] [PubMed] [Google Scholar]
- Needham M, Mastaglia F. Sporadic inclusion body myositis: a continuing puzzle. Neuromuscul. Disord. 2008;18:6–16. doi: 10.1016/j.nmd.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Novotová M, Zahradník I, Brochier G, Pavlovicová M, Bigard X, Ventura-Clapier R. Joint participation of mitochondria and sarcoplasmic reticulum in the formation of tubular aggregates in gastrocnemius muscle of CK−/− mice. Eur. J. Cell Biol. 2002;81:101–106. doi: 10.1078/0171-9335-00230. [DOI] [PubMed] [Google Scholar]
- Oyama F, Shimada H, Oyama R, Titani K, Ihara Y. Differential expression of beta amyloid protein precursor (APP) and tau mRNA in the aged human brain: individual variability and correlation between APP-751 and four-repeat tau. J. Neuropathol. Exp. Neurol. 1991;50:560–578. doi: 10.1097/00005072-199109000-00004. [DOI] [PubMed] [Google Scholar]
- Roy B, Shukla S, Stoermann B, Kremmer E, Duber S, Weiss S. Loss of lambda2(315) transgene copy numbers influences the development of B1 cells. Mol. Immunol. 2009;46:1542–1550. doi: 10.1016/j.molimm.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Salviati G, Pierobon-Bormioli S, Betto R, et al. Tubular aggregates: sarcoplasmic reticulum origin, calcium storage ability, and functional implications. Muscle Nerve. 1985;8:299–306. doi: 10.1002/mus.880080406. [DOI] [PubMed] [Google Scholar]
- Schubert W, Sotgia F, Cohen AW, et al. Caveolin-1(−/−)- and Caveolin-2(−/−)-deficient mice both display numerous skeletal muscle abnormalities, with tubular aggregate formation. Am. J. Pathol. 2007;170:316–333. doi: 10.2353/ajpath.2007.060687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtifman A, Ward CW, Laver DR, et al. Amyloid-beta protein impairs Ca2+ release and contractility in skeletal muscle. Neurobiol. Aging. 2010;31:2080–2090. doi: 10.1016/j.neurobiolaging.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugarman MC, Yamasaki TR, Oddo S, et al. Inclusion body myositis-like phenotype induced by transgenic overexpression of βAPP in skeletal muscle. Proc. Natl Acad. Sci. USA. 2002;99:6334–6339. doi: 10.1073/pnas.082545599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugarman MC, Kitazawa M, Baker M, Caiozzo VJ, Querfurth HW, LaFerla FM. Pathogenic accumulation of APP in fast twitch muscle of IBM patients and a transgenic model. Neurobiol. Aging. 2006;27:423–432. doi: 10.1016/j.neurobiolaging.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Vattemi G, Nogalska A, Engel WK, D'Agostino C, Checler F, Askanas V. Amyloid-β42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion-body myositis. Acta Neuropathol. 2009;117:569–574. doi: 10.1007/s00401-009-0511-6. [DOI] [PubMed] [Google Scholar]
- Wang J, Ho L, Qin W, et al. Caloric restriction attenuates β-amyloid neuropathology in a mouse model of Alzheimer's disease. FASEB J. 2005;19:659–661. doi: 10.1096/fj.04-3182fje. [DOI] [PubMed] [Google Scholar]
- Zhan SS, Sandbrink R, Beyreuther K, Schmitt HP. APP with Kunitz type protease inhibitor domain (KPI) correlates with neuritic plaque density but not with cortical synaptophysin immunoreactivity in Alzheimer's disease and non-demented aged subjects: a multifactorial analysis. Clin. Neuropathol. 1995;14:142–149. [PubMed] [Google Scholar]
- Zhao J, Sun X, Yu Z, et al. Exposure to pyrithiamine increases Neuropathol Appl Neurobiol-amyloid accumulation, Tau hyperphosphorylation, and glycogen synthase kinase-3 activity in the brain. Neurotox. Res. 2011;19:575–583. doi: 10.1007/s12640-010-9204-0. [DOI] [PubMed] [Google Scholar]