

Abstract
Tanjungides A (1) (Z isomer) and B (2) (E isomer), two novel dibrominated indole enamides, have been isolated from the tunicate Diazona cf formosa. Their structures were determined by spectroscopic methods including HRMS, and extensive 1D and 2D NMR. The stereochemistry of the cyclised cystine present in both compounds was determined by Marfey’s analysis after chemical degradation and hydrolysis. We also report the first total synthesis of these compounds using methyl 1H-indole-3-carboxylate as starting material and a linear sequence of 11 chemical steps. Tanjungides A and B exhibit significant cytotoxicity against human tumor cell lines.
Keywords: bromoindole, tunicate, Diazona cf formosa, cytotoxicity, isolation, structure elucidation, total synthesis
1. Introduction
Ascidians [1] are a rich source of bromoindole derived metabolites such as eudistomin [2], didemnimide [3], meridianin [4], coscinamide [5], rhopaladin [6], kottamide [7,8] and aplicyanin [9]. Most of these compounds exhibit antiviral, antibacterial and anti-inflammatory activity as well as cytotoxicity against tumor cell lines. The diazonamides isolated from Diazona angulata (originally misidentified as Diazona chinensis) [10] and Diazona sp. [11] provide a further example of secondary metabolites from ascidians. Strong cytotoxic activity has been reported for these compounds with IC50 values in the nanomolar range. As part of work to study marine organisms from Indonesia, we have examined the constituents of the tunicate Diazona cf formosa collected off the coast of Tanjung Liarua and Toro Doro (Timor Island). In this paper we report the isolation, structure elucidation and synthesis of two new indole alkaloids Tanjungides A and B (1 and 2). Tanjungides are novel alkaloids containing a dibromoindole joined to a disulfide dipeptide by an enamide bond.
2. Results and Discussion
2.1. Isolation and Structure Elucidation
Cytotoxicity bioassay-guided fractionation of an organic extract of the organism, including VLC RP-18 chromatography followed by reverse-phase preparative HPLC of selected active fractions, led to the isolation of Tanjungides A and B.
Compound 1 was isolated as an optically active pale yellow amorphous solid with a pseudomolecular ion in the (+)-HRESIMS at m/z 518.9142 and an isotopic cluster consistent with the presence of two bromine atoms. The presence of 16 signals in the 13C NMR spectrum (Table 1) was also in agreement with the molecular formula C16H1679Br2N4O2S2 (m/z 518.9142 [M + H]+, calcd. for C16H1779Br2N4O2S2, 518.9154). The presence of a 3,5,6-trisubstituted indole in 1 (Figure 1) was inferred by the existence of four characteristic signals in the low field region of the 1H NMR spectrum in DMSO-d6, two doublets at δH 7.78 (d, H-2, J = 2.4 Hz) and 11.78 (d, NH-1, J = 2.6 Hz) and two singlets at δH 7.81 (s, H-7) and δH 8.01 (s, H-4). In addition, the two bromine atoms contained in the molecular formula were located at C-5 and C-6 based on their 13C chemical shifts. The intense 3-bond long range couplings between H-4 and C-6 at δC 115.7 ppm and between H-7 and C-5 at δC 113.5 ppm observed in the HMBC spectrum further confirmed the chemical shifts of these two quaternary carbons. The nature of the substituent at C-3 was deduced from analysis of additional signals in the low field region of the 1H NMR spectrum and correlations observed in the COSY, HSQC and HMBC spectra. A spin system comprising two olefinic signals at δH 6.06 ppm (H-8) and 6.68 ppm (H-9), and an interchangeable proton at δH 9.60 ppm (NH-10) established the presence of an enamide. A coupling constant of 9.4 Hz between H-8 and H-9 confirmed a Z geometry for this double bond. Finally, HMBC correlations from H-9 to C-3 (δC 109.2 ppm) and from H-8 to C-2 (δC 126.8 ppm), and C-3a (δC 127.6 ppm) indicated that the indole moiety was substituted at C-3 with a Z geometry enamide fragment. The remaining atoms, C6H9N2O2S2, comprised two carbonyl (δC 169.9 and 167.1 ppm), two methine, (δC 52.5/δH 5.02 ppm and δC 51.2/δH 4.65 ppm) and two methylene groups (δC 41.6/δH 3.40 and 2.86 ppm and δC 39.7/δH 3.17 and 2.94 ppm) with three degrees of unsaturation being required for this molecular formula, including the two carbonyls mentioned previously. Analysis of the bidimensional spectra revealed the presence of a two spin system corresponding to two consecutive cysteine residues. Cross-peaks observed in the HMBC experiment between H-12 and H-16 and carbon C-14 at δC 169.9 ppm (Figure 2) confirmed this structural proposal. Furthermore, correlations observed in the HMBC experiment between H-9, NH-10, H-12 and H-17 to C-11, and a ROESY correlation between NH-10 and H-12, connected these cysteines residues to the enamide group through C-11. Finally, linkage of the two cysteine amino acids by a S–S bond to form a cyclic cystine explained the remaining unsaturation present and established the complete structure of Tanjungide A.
Table 1.
1H and 13C NMR (500 and 125 MHz) assignments for Tanjungide A (1) (DMSO-d6) and Tanjungide B (2) (CD3OD).
| Position | Tanjungide A (1) | Tanjungide B (2) | ||
|---|---|---|---|---|
| δH (m, J in Hz) | δC, mult. | δH (m, J in Hz) | δC, mult. | |
| 1 | 11.78 (d, 2.6) | - | - | - |
| 2 | 7.78 (d, 2.4) | 126.8, d | 7.35 (s) | 126.7, d |
| 3 | - | 109.2, s | - | 112.9, s |
| 3a | - | 127.6, s | - | 127.4, s |
| 4 | 8.01 (s) | 123.0, d | 8.04 (s) | 124.6, d |
| 5 | - | 113.5, s | - | 117.5, s |
| 6 | - | 115.7, s | - | 115.5, s |
| 7 | 7.81 (s) | 116.3, d | 7.72 (s) | 117.4, d |
| 7a | - | 135.4, s | - | 138.3, s |
| 8 | 6.06 (d, 9.4) | 103.8, d | 6.50 (d, 14.8) | 109.2, d |
| 9 | 6.68 (dd, 9.4, 9.8) | 118.9, d | 7.34 (d, 14.8) | 120.7, d |
| 10 | 9.60 (d, 9.8) | - | - | - |
| 11 | - | 167.1, s | - | 167.8, s |
| 12 | 5.02 (ddd, 11.8, 11.5, 3.6) | 52.5, d | 4.93 (m) | 54.2, d |
| 13 | 8.44 (br s) | - | - | - |
| 14 | - | 169.9, s | - | 169.0, s |
| 15 | 4.65 (m) | 51.2, d | 4.60 (m) | 53.7, d |
| 16 | 2.94 (dd, 14.2, 11.5) 3.17 (m) | 39.7, t | 3.11 (m) | 41.8, t |
| 17 | 2.86 (dd, 13.3, 11.6) 3.40 (m) | 41.6, t | 2.90 (dd, 12.4, 12.4) 3.47 (m) | 43.2, t |
Figure 1.

Chemical Structures of Tanjungides.
Figure 2.
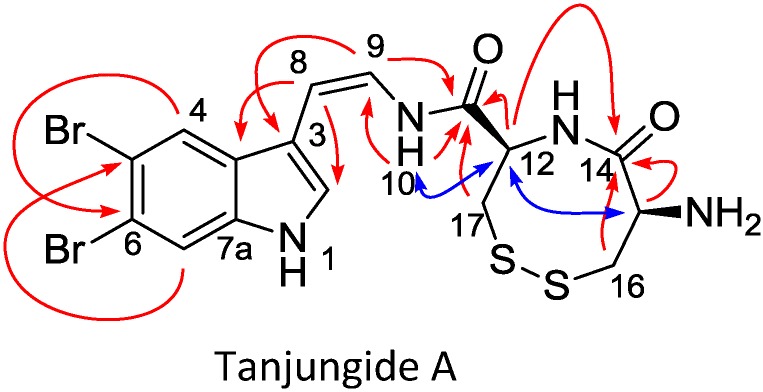
Selected HMBC (H → C, red) and ROESY (blue) correlations for Tanjungide A.
The absolute configuration of 1 was determined by converting the cyclized cystine into two alanines by Raney®-Nickel desulfurization [12]. The absolute configuration of the resulting Ala amino acids was determined to be R by comparing the hydrolysis products of 1 with appropriate amino acid standards using HPLC-MS chromatography and after derivatization with Marfey’s reagent l-FDAA (Nα-(2,4-dinitro-5-fluorophenyl)-l-alaninamide) [13].
Compound 2 (Figure 1) was isolated as an optically active pale yellow amorphous solid with the same molecular formula as 1 [(+)-HRESIMS m/z 518.9142 [M + H]+ (Calcd. for C16H1779Br2N4O2S2, 518.9154)]. After examination of the 1D and 2D NMR spectra we concluded that Tanjungide B (Table 1) was very similar to Tanjungide A, and the major difference found in the 1H NMR was the value of the coupling constant of the ∆8 olefin signals. Thus, the coupling constant JH8-H9 had a value of 14.6 Hz corresponding to a E geometry for the double bond. The absolute configuration of the Cys residues was not determined due to the small amount of compound isolated and was assumed to be the same as in Tanjungide A (1). The validity of this assumption was later confirmed by total synthesis of the molecule.
2.2. Biological Activities of Tanjungides A and B
The cytotoxic activity of the new compounds (Table 2) was tested against three human tumour cell lines, lung (A549), colon (HT29), and breast (MDA-MB-231), following a published procedure [14]. Tanjungide A (1) exhibited strong activity with GI50 values in the range 0.19 to 0.33 μM, whereas Tanjungide B (2) displayed only mild cytotoxicity, with GI50 values ranging from 1.00 to 2.50 μM.
Table 2.
Cytotoxic Activity Data (μM) of Compounds 1 and 2.
| Compound | Lung-NSCLC | Colon | Breast |
|---|---|---|---|
| A549 | HT29 | MDA-MB-231 | |
| GI50 | GI50 | GI50 | |
| Natural Tanjungide A | 0.33 | 0.19 | 0.23 |
| Synthetic Tanjungide A | 0.33 | 0.25 | 0.19 |
| Natural Tanjungide B | 2.50 | 2.31 | 1.63 |
| Synthetic Tanjungide B | 1.00 | 1.15 | 1.11 |
2.3. Total Synthesis of Tanjungides A and B
In order to solve the supply problem for these two new marine chemical entities and progress pharmaceutical development and in vivo preclinical studies, we have completed the first total synthesis of Tanjungides A and B. This synthesis uses methyl 1H-indole-3-carboxylate as starting material and involves a linear sequence of 11 chemical steps. Key elements of our approach include selective dibromination of the indole, formylation by Vilsmeier reaction, Wittig olefination, stereoselective enamide formation and oxidation to create the disulfide bond (Figure 3). The strategy uses vinyl iodide indole 8 as a common precursor to give both Tanjungides.
Figure 3.

Retrosynthetic analysis of Tanjungides A and B.
The synthesis started, as outlined in Scheme 1, from the cheap commercially available methyl 1H-indole-3-carboxylate as this provided a high yielding route to a 5,6-dibrominated indole possessing an aldehyde moiety at C3, a highly versatile building block for the total synthesis of the two natural products. The slow addition of two equivalents of bromine to methyl 1H-indole-3-carboxylate in acetic acid at 23 °C yielded the corresponding 5,6-dibromo intermediate 3 as a single pure product in 66% yield [15]. Disappointingly, attempted methyl ester reduction of 3 to give aldehyde 6 directly was unsuccessful, and an alternative stepwise process to give aldehyde 6 was used involving hydrolysis and decarboxylation to give 5,6-dibromo-1H-indole 5 in good yield (90% over two steps) followed by Vilsmeier formylation using dimethylformamide and phosphorus oxychloride. After protection of the indole nitrogen as a tert-butyl carbamate, Wittig olefination with (iodomethyl)triphenylphosphonium iodide [16] gave the desired vinyl iodide indole 8 in 83% yield and as a 9:1 ratio of Z:E isomers [17].
Scheme 1.
Total synthesis of Tanjungides A (1) and B (2).

Regents and conditions: (a) Br2, AcOH, 23 °C, 2 h, 66%; (b) aq NaOH 2 M, CH3OH, reflux, 2.5 h, 95%; (c) Pyridine, reflux, 12 h, 95%; (d) i POCl3, DMF, 35 °C, 1 h then 65 °C, 1 h, ii aq NaOH 2 M, 110 °C, 5 min, 92%; (e) (Boc)2O, DMAP, 1,4-Dioxane, 23 °C, 2 h, 86%; (f) Iodomethyltriphpenylphosphonium iodide, NaHMDS 1.0 M, THF, −78 °C, 2 h, 83%; (g) for (Z)-10: 9, Cul, Cs2CO3, DMEDA, THF, 60 °C, 18 h, 50% + 13% (E)-10; for (E)-10: 9, Cul, K2CO3, DMEDA, THF, 80 °C, 18 h, 60% + 14% (Z)-10; (h) Pd(PPh3)4, PhSiH3, CH2Cl2, 23 °C, 30 min, 85% (Z)-11 and 72% (E)-11; (i) N-Boc-l-(S-trityl)-Cys, HATU, HOBt, DIPEA, CH2Cl2:DMF (4:1), 23 °C, 2 h, 83% (Z)-12 and 62% (E)-12; (j) I2, CH2Cl2:CH3OH (10:1), 23 °C, 40 min, 84% (Z)-13 and 73% (E)-13; (k) TFA, CH2Cl2, 0 °C, 3 h, 60% (Z)-Tanjungide A and 45% (E)-Tanjungide B.
With vinyl iodide 8 in hand, the next step involved coupling of the suitably-protected cysteine amino acids (Scheme 1). As described by Buchwald and co-workers [18], depending on the conditions used for the coupling reaction, vinyl iodide 8 provided access to both stereoisomers of enamide 10 and hence to both Tanjungide A and B. Specifically, copper-catalyzed reaction of 8 with N-allyloxycarbonyl-S-trityl-l-cysteine-amide 9, made in one step from commercially available S-trityl-l-cysteine-amide, gave enamide 10 in moderate yield (50%−60%) with use of Cs2CO3 as base affording mainly enamide (Z)-10, which could be readily separated from the corresponding (E)-isomer by column chromatography, and K2CO3 providing predominantly enamide (E)-10. Next, removal of the Alloc group of (Z)-10 or (E)-10 under neutral conditions using Pd(PPh3)4 and PhSiH3 and coupling of the resulting primary amine with (N-(tert-butoxycarbonyl)-S-trityl-l-cysteine) by treatment with HATU and HOBt yielded the corresponding amide (Z)-12 or (E)-12. After substantial experimentation, the trityl group proved to be the best thiol protecting group for each of the cysteine amino acid building blocks. To complete the synthesis, the key cyclization of 12 to form the disulfide bond was accomplished using I2 in CH2Cl2:CH3OH at high dilution to avoid undesired side-products [19,20] and subsequent simultaneously cleavage of both Boc protecting groups of 13 with TFA gave Tanjungides A (1) and B (2). All the spectroscopic data (1H and 13C NMR, optical rotation, IR, etc.), HPLC retention times and biological activities of the synthetic samples exactly matched those of the isolated natural products. The Supplementary Information provides more details.
3. Experimental Section
3.1. General
Dry solvents were purchased and used without any extra processing. All reagents were used as purchased without further purification unless otherwise stated. All reactions were performed under an atmosphere of nitrogen in flame dried or oven dried glassware. Routine monitoring of reactions was performed using silica gel TLC plates (Merck 60 F254, Merck KGaA, Darmstadt, Germany). Spots were visualized by UV and/or dipping the TLC plate into an ethanolic phosphomolybdic acid solution and heating with a hot plate. Flash chromatography was carried out on silica gel 60 (200–400 mesh). 1H and 13C NMR were recorded on a Varian Unity 300 or 500 spectrometer at 300 or 500, and 75 or 125 MHz, respectively. Chemical Shifts (δ) are reported in parts per millions (ppm) referenced to CHCl3 at 7.26 ppm for 1H and CDCl3 at 77.0 ppm for 13C, to CH3OH at 3.30 ppm for 1H and CD3OD at 49.0 ppm; and to (CH3)2SO at 2.50 ppm for 1H and (CD3)2SO at 39.5 ppm for 13C. Coupling constants are reported in Hertz (Hz), with the following abbreviations used: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. When appropriate, the multiplicities are preceded with br, indicating that the signal was broad. Optical rotations were determined using a Jasco P-1020 polarimeter (Jasco Inc., Easton, MD, USA) with a sodium lamp and are reported as follows: [α]25D (c g/100 mL, solvent). (+)-HRESIMS was performed on an Applied Biosystems QStar pulsar Analyzer spectrometer (Applied Biosystems Inc., Foster City, CA, USA) employing 0.1% of formic acid in methanol as an ionic mobile phase. (+)-ESIMS were recorded using an Agilent 1100 Series LC/MSD spectrometer (Agilent Technologies, Santa Clara, CA, USA). UV spectra were performed using an Agilent 8453 UV-VIS spectrometer (Agilent Technologies). IR spectra were obtained with a Perkin Elmer Spectrum 100 FT-IR spectrometer (PerkinElmer Inc., Waltham, MA, USA) with ATR sampling.
3.2. Animal Material
The tunicate Diazona cf formosa (Order Phlebobranchia, Family Diazonidae, Genus Diazona) was collected by hand using a rebreather diving system in East Timor (08°25.637′S/126°22.849′E) at depths ranging between 6 and 80 m in June 2009. A sample of the specimen was deposited in the Center for the Advanced Studies of Blanes in Girona, Spain, with the reference code TISM-763.
3.3. Extraction and Isolation
A specimen of Diazona cf formosa (128 g) was triturated and exhaustively extracted with CH3OH:CH2Cl2 (50:50, 3 × 200 mL). The combined extracts were concentrated to yield a crude mass of 5.05 g that was subjected to VLC on Lichroprep RP-18 (Merck KGaA) with a stepped gradient from H2O to CH3OH and then CH3OH:CH2Cl2 (50:50). Fraction eluted with CH3OH:H2O (75:25, 63.7 mg) were subjected to preparative HPLC (Symmetry C18, 7 μm, 19 × 150 mm, gradient H2O + 0.1% TFA:CH3CN + 0.1% TFA from 32% to 49% CH3CN + 0.1% TFA in 14 min and then from 49% to 100% in 1 min, flow: 13.6 mL/min, UV detection) to yield Tanjungide A (31.8 mg, retention time: 10.1 min) and Tanjungide B (1.6 mg, retention time: 9.2 min). Fraction eluted with CH3OH (100.7 mg), was also subjected to the same conditions of preparative HPLC to yield additional amounts of Tanjungides A and B.
Tanjungide A (1): Pale yellow amorphous solid. [α]25D +114.9° (c 0.1, CH3OH); UV (CH3OH) λmax 201, 236, 288 nm; IR (KBr) νmax 3324, 1674, 1534, 1494, 1450, 1203, 1144, 803, 723 cm−1; (+)-HRESIMS m/z 518.9142 [M + H]+ (Calcd. for C16H1779Br2N4O2S2, 518.9154). 1H NMR (500 MHz) and 13C NMR (125 MHz) in DMSO-d6, see Table 1.
Tanjungide B (2): Pale yellow amorphous solid. [α]25D +46.2° (c 0.1, CH3OH); UV (CH3OH) λmax 201, 236, 291 nm; IR (KBr) νmax 3296, 1676, 1571, 1447, 1203, 1141, 803, 726 cm−1; (+)-HRESIMS m/z 518.9142 [M + H]+ (Calcd. for C16H1779Br2N4O2S2, 518.9154). 1H NMR (500 MHz) and 13C NMR (125 MHz) in CD3OD, see Table 1.
3.4. Absolute Configuration of Cysteine Residues
Approximately 100 μL of Raney®-Nickel (50% slurry in H2O, excess) was added to Tanjungide A (1.02 mg) in methanol (1.2 mL), N2 was bubbled through the solution to remove O2. The resulting suspension was heated at 65 °C for 4 h under nitrogen and then the reaction mixture was left overnight at 23 °C. The disappearance of starting material was monitored by HPLC. The resulting solution was purified on a C18 SPE cartridge using methanol as eluent, yielding desthiotanjungide A. Desthiotanjungide A (200 μg) was dissolved in 6 N HCl (500 μL) and heated in a sealed glass vial at 110 °C overnight. The solvent was removed in a stream of dry N2. To the acid hydrolysate of desthiotanjungide A, a solution of l-FDAA (Nα-(2,4-dinitro-5-fluorophenyl)-l-alaninamide, 700 μg) in acetone (160 μL), H2O (100 μL) and NaHCO3 1 N (50 μL) were added. The vials were heated at 40 °C for 1 h, and the contents neutralized with 2N HCl (20 μL) after cooling to 23 °C. H2O (800 μL) was added to each reaction and the resulting mixture filtered and analyzed by RP18-HPLC-MS (Symmetry C18, 5 μm, 4.6 × 150 mm; linear gradient from 20% to 50% CH3CN (0.04% TFA) in H2O (0.04% TFA) over 20 min, flow rate: 0.8 mL/min). The amino acid standards (R)- and (S)-Ala (200 μg) were derivatized in a similar manner, and the retention times were compared with those of the alanines of desthiotanjungide A hydrolysate. The retention times of the authentic FDAA-Ala used as standards were as follows: (S)-Ala (13.9 min) and (R)-Ala (16.3 min). The hydrolysate of Tanjungide A contained: (S)-Ala (13.7 min).
3.5. Total Synthesis of Tanjungides A (1) and B (2)
3.5.1. 5,6-Dibromo-1H-indole-3-carboxylic Acid (4)
To a stirred solution of methyl 5,6-dibromo-1H-indole-3-carboxylate (3) (12.5 g, 37.8 mmol) in CH3OH (124 mL) was added an aqueous solution of NaOH (188 mL, 2 M, 376 mmol). The suspension was refluxed for 2.5 h. After this time, the brown solution was cooled to 23 °C and the volatiles were evaporated. The aqueous phase was acidified with a 1 M solution of HCl until reached pH 2 and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo to afford crude 4 (11.4 g, 95% yield) as a brown solid which was used in the next step without further purification. 1H NMR (300 MHz, CD3OD) δH ppm: 8.36 (s, 1H), 7.97 (s, 1H), 7.79 (s, 1H). 13C NMR (75 MHz, CD3OD) δC ppm: 166.9, 136.6, 134.0, 127.2, 125.2, 117.2, 116.7, 116.5, 107.5.
3.5.2. 5,6-Dibromo-1H-indole (5)
5,6-Dibromo-1H-indole-3-carboxylic acid (4) (11.7 g, 36.7 mmol) was dissolved in pyridine (20.5 mL) and refluxed overnight. The solvent was concentrated in vacuo, the crude obtained was dissolved in CH2Cl2, precipitated with hexane and left at 5 °C overnight. The solid was filtered to yield crude 5 (9.6 g, 95% yield) which was used in the next step without further purification. 1H NMR (300 MHz, CDCl3) δH ppm: 7.90 (d, J = 1.1 Hz, 1H), 7.70 (t, J = 1.1 Hz, 1H), 7.21 (ddd, J = 3.6, 2.5, 1.2 Hz, 1H), 6.48 (ddt, J = 3.3, 2.2, 1.1 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC ppm: 135.7, 129.0, 126.3, 125.1, 117.3, 115.9, 115.3, 102.6.
3.5.3. 5,6-Dibromo-1H-indole-3-carbaldehyde (6)
To a stirred solution of DMF (46.8 mL) at 0 °C was dropwise added POCl3 (12.0 mL, 131.3 mmol). The mixture was further stirred for 5 min at 0 °C and a solution of 5,6-dibromo-1H-indole (5) (7.22 g, 26.3 mmol) in DMF (70 mL) was slowly added. The reaction mixture was stirred 1 h at 35 °C, 1 h at 65 °C, and was left to reach 23 °C. An aqueous solution of NaOH (72.3 mL, 2 N) was added at 0 °C and the reaction mixture was stirred 5 min at 110 °C, left to reach 23 °C, and then added over an ice-water bath in order to precipitate 6. The reaction mixture was left overnight at 5 °C and filtered to obtain crude 6 (7.32 g, 92% yield) which was used in the next step without further purification. 1H NMR (300 MHz, DMSO-d6) δH ppm: 9.91 (d, J = 1.3 Hz, 1H), 8.51–8.20 (m, 2H), 7.91 (d, J = 1.3 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δC ppm: 185.9, 140.7, 137.4, 125.7, 125.4, 118.2, 118.1, 117.8, 117.4.
3.5.4. tert-Butyl 5,6-dibromo-3-formyl-1H-indole-1-carboxylate (7)
To a stirred solution of 5,6-dibromo-1H-indole-3-carbaldehyde (6) (9.2 g, 30.4 mmol) in 1,4-dioxane (152 mL) was added successively di-tert-butyldicarbonate (7.9 g, 36.4 mmol) and DMAP (370 mg, 3.0 mmol). After stirring for 2 h at 23 °C, the mixture was quenched with H2O and extracted with EtOAc. The combined organic phases were washed thoroughly with H2O, dried over Na2SO4, filtered and concentrated in vacuo to afford 7 (10.5 g, 86% yield) as a slightly brown solid that was used in the next steps without further purification. 1H NMR (300 MHz, CDCl3) δH ppm: 10.03 (s, 1H), 8.54 (s, 1H), 8.47 (s, 1H), 8.18 (s, 1H), 1.71 (s, 9H). 13C NMR (75 MHz, CDCl3) δC ppm: 185.2, 148.3, 137.2, 135.5, 126.7, 126.6, 122.2, 120.9, 120.6, 120.4, 86.9, 28.2.
3.5.5. (Z)-tert-Butyl 5,6-dibromo-3-(2-iodovinyl)-1H-indole-1-carboxylate (8)
To a suspension of iodomethyltriphpenylphosphonium iodide (14.6 g, 27.5 mmol) in anhydrous THF (157 mL) was added a solution of sodium bis(trimethylsilyl)amide (NaHMDS) (27.5 mL, 1.0 M in THF, 27.5 mmol) dropwise at 23 °C. After stirring for 2 min, the yellow mixture was cooled to −78 °C and a solution of 7 (7.9 g, 19.6 mmol) in anhydrous THF (98 mL) was then added. The reaction mixture was stirred at −78 °C for 2 h, at 23 °C for 5 min, diluted with hexane, and filtered through a plug of Celite®. The plug was rinsed with hexane, the combined filtrates were evaporated under reduced pressure affording 8 (8.62 g, 83% yield) as a brown solid that was used in the next steps without further purification. 1H NMR (300 MHz, CDCl3) δH ppm: 8.56 (s, 1H), 8.48 (s, 1H), 7.80 (s, 1H), 7.43 (d, 1H, J = 8.7 Hz), 6.67 (d, 1H, J = 8.7 Hz), 1.69 (s, 9H). 13C NMR (75 MHz, CDCl3) δC ppm: 149.1, 135.5, 130.6, 128.5, 125.0, 124.0, 123.1, 120.5, 118.8, 116.4, 85.4, 81.0, 28.3.
3.5.6. (R)-Allyl (1-amino-1-oxo-3-(tritylthio)propan-2-yl)carbamate (9)
To a stirred solution of S-trityl-l-cysteine amide (500 mg, 1.38 mmol) in a mixture THF:H2O (2.5 mL:1.25 mL) at 0 °C was added solid NaHCO3 (232 mg, 2.76 mmol) followed by allyloxycarbonyl chloride (0.14 mL, 1.65 mmol). After stirring for 2 h at 0 °C, the mixture was quenched by slow addition of a 2 M solution of HCl until reached pH 2 and extracted with CH2Cl2. The combined organic layers were dried over Na2SO4, filtered and the solvent was removed under reduced pressure to afford 9 as a white solid (616 mg, 100% yield) that was used without further purification. 1H NMR (300 MHz, CDCl3) δH ppm: 7.43 (m, 5H), 7.33–7.19 (m, 10H), 5.88 (m, 1H), 5.81 (br s, 1H), 5.33 (br s, 1H), 5.29 (d, 1H, 16.0 Hz), 5.22 (d, 1H, J = 10.5 Hz), 5.06 (d, 1H, J = 7.2 Hz), 4.52 (dd, 2H, 5.7, J = 1.2 Hz), 3.87 (m, 1H), 2.76 (dd, 1H, J = 13.2, 7.2 Hz), 2.57 (dd, 1H, J = 13.2, 5.1 Hz). 13C NMR (75 MHz, CDCl3) δC ppm: 172.4, 156.0, 144.5, 132.5, 129.8, 128.3, 127.2, 118.2, 67.6, 66.3, 53.7, 33.9.
3.5.7. (R,Z)-tert-Butyl 3-(2-(2-(((allyloxy)carbonyl)amino)-3-(tritylthio)propanamido)vinyl)-5,6-dibromo-1H-indole-1-carboxylate (Z-10)
A Schlenk tube was charged with copper(I) iodide (39 mg, 0.20 mmol), cesium carbonate (667 mg, 2.05 mmol) and N-alloc-S-trityl-l-cysteine-amide (9) (455 mg, 1.02 mmol), evacuated and filled with N2. N,N′-dimethylethylenediamine (44 μL, 0.41 mmol), vinyl iodide 8 (360 mg, 0.68 mmol) and dry THF (4 mL) were added. The Schlenk tube was sealed, heated at 60 °C for 18 h and cooled to 23 °C. The resultant mixture was diluted with EtOAc and quenched with H2O. The organic layer was washed with H2O and dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (hexane:EtOAc, 4:1) to yield successively pure (Z)-10 (285 mg, 50% yield) and (E)-10 (77 mg, 13% yield) as brown solids. 1H NMR (300 MHz, CDCl3) δH ppm: 8.46 (s, 1H), 8.21 (d, 1H, J = 8.1 Hz), 7.73 (s, 1H), 7.61 (s, 1H), 7.34 (m, 5H), 7.22 (m, 10H), 6.96 (dd, 1H, J = 11.1, 9.3 Hz), 5.78 (m, 1H), 5.68 (d, 1H, J = 9.6 Hz), 5.21 (d, 1H, J = 17.1 Hz), 5.14 (d, 1H, J = 10.5 Hz), 4.90 (d, 1H, J = 7.2 Hz), 4.40 (m, 2H), 3.83 (m, 1H), 2.83 (dd, 1H, J = 13.2, 6.9 Hz), 2.58 (dd, 1H, J = 13.2, 5.7 Hz), 1.68 (s, 9H). 13C NMR (75 MHz, CDCl3) δC ppm: 168.1, 156.4, 149.0, 144.4, 134.7, 132.4, 130.4, 129.7, 128.3, 127.1, 124.7, 123.8, 123.3, 120.8, 120.5, 118.8, 118.2, 114.3, 100.2, 85.2, 67.7, 66.5, 54.1, 32.9, 28.3.
3.5.8. (R,E)-tert-Butyl 3-(2-(2-(((allyloxy)carbonyl)amino)-3-(tritylthio)propanamido)vinyl)-5,6-dibromo-1H-indole-1-carboxylate (E-10)
A Schlenk tube was charged with copper(I) iodide (11 mg, 0.06 mmol), potassium carbonate (78 mg, 0.57 mmol) and N-alloc-S-trityl-l-cysteine-amide (9) (127 mg, 0.284 mmol), evacuated and filled with N2. N,N′-dimethylethylenediamine (12 μL, 0.11 mmol), vinyl iodide 8 (100 mg, 0.19 mmol) and dry THF (5 mL) were added. The Schlenk tube was sealed, heated at 80 °C for 18 h and cooled to 23 °C. The resultant mixture was diluted with EtOAc and quenched with H2O. The organic layer was washed with H2O and dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (hexane:EtOAc, 4:1) to yield pure (E)-10 (96 mg, 60% yield) and (Z)-10 (22 mg, 14% yield) as brown solids. 1H NMR (300 MHz, CDCl3) δH ppm: 8.50 (s, 1H), 7.87 (s, 1H), 7.83 (d, 1H, J = 8.1 Hz), 7.50 (s, 1H), 7.45 (m, 5H), 7.35–7.21 (m, 11H), 6.10 (d, 1H, J = 15.3 Hz), 5.90 (m, 1H), 5.32 (d, 1H, J = 17.1 Hz), 5.25 (d, 1H, J = 10.2 Hz), 4.99 (d, 1H, J = 6.9 Hz), 4.56 (dd, 2H, J = 5.7, 2.7 Hz), 3.87 (q, 1H, J = 6.6 Hz), 2.85 (dd, 1H, J = 13.2, 6.9 Hz), 2.62 (dd, 1H, J = 13.2, 5.7 Hz), 1.66 (s, 9H). 13C NMR (75 MHz, CDCl3) δC ppm: 167.7, 156.2, 149.0, 144.5, 135.4, 132.4, 129.8, 129.4, 128.4, 127.2, 124.0, 123.5, 123.0, 120.6, 120.5, 118.8, 118.5, 116.2, 104.3, 85.0, 67.8, 66.6, 54.3, 33.6, 28.3.
3.5.9. (R,Z)-tert-Butyl 3-(2-(2-amino-3-(tritylthio)propanamido)vinyl)-5,6-dibromo-1H-indole-1-carboxylate (Z-11)
To a stirred solution of enamide (Z)-10 (1.53 g, 1.81 mmol) in CH2Cl2 (46 mL) was added successively PhSiH3 (4.46 mL, 36.2 mmol) and Pd(PPh3)4 (313 mg, 0.27 mmol). After stirring for 30 min at 23 °C all volatiles were evaporated and the crude mixture was purified by flash chromatography on silica gel (hexane:EtOAc, 2:1) to afford pure (Z)-11 (1.17 g, 85% yield) as a brown solid. 1H NMR (300 MHz, CDCl3) δH ppm: 9.57 (d, 1H, J = 12.0 Hz), 8.48 (s, 1H), 7.77 (s, 1H), 7.59 (s, 1H), 7.43 (m, 5H), 7.25 (m, 10H), 6.92 (dd, 1H, J = 11.0, 8.7 Hz), 5.69 (d, 1H, J = 9.3 Hz), 3.12 (m, 1H), 2.77 (dd, 1H, J = 9.0, 4.5 Hz), 2.66 (dd, 1H, J = 12.9, 8.4 Hz), 1.66 (s, 9H). 13C NMR (75 MHz, CDCl3) δC ppm: 170.5, 148.8, 144.4, 129.5, 128.0, 127.9, 126.9, 123.9, 123.8, 122.6, 122.5, 120.6, 120.2, 118.5, 114.8, 99.2, 84.7, 67.1, 53.7, 36.8, 28.1.
3.5.10. (R,E)-tert-Butyl 3-(2-(2-amino-3-(tritylthio)propanamido)vinyl)-5,6-dibromo-1H-indole-1-carboxylate (E-11)
To a stirred solution of enamide (E)-10 (265 mg, 0.31 mmol) in CH2Cl2 (8 mL) was added successively PhSiH3 (0.77 mL, 6.3 mmol) and Pd(PPh3)4 (54 mg, 0.05 mmol). After stirring for 25 min at 23 °C all volatiles were evaporated and the crude mixture was purified by flash chromatography on silica gel (hexane:EtOAc, 2:1) to obtain (E)-11 (170 mg, 72% yield) as a brown solid. 1H NMR (300 MHz, CDCl3) δH ppm: 9.14 (d, 1H, J = 11.4 Hz), 8.48 (s, 1H), 7.86 (s, 1H), 7.45 (m, 6H), 7.37–7.20 (m, 11H), 6.10 (d, 1H, J = 15.3 Hz), 3.14 (m, 1H), 2.78 (m, 1H), 2.67 (m, 1H), 1.68 (s, 9H). 13C NMR (75 MHz, CDCl3) δC ppm: 170.6, 149.0, 144.7, 135.4, 132.3, 129.8, 129.5, 128.2, 127.1, 124.0, 123.2, 120.5, 120.4, 118.7, 116.5, 103.6, 84.9, 67.4, 53.9, 37.2, 28.3.
3.5.11. tert-Butyl 5,6-dibromo-3-((6R,9R,Z)-2,2-dimethyl-4,7,10-trioxo-6,9-bis((tritylthio)methyl)-3-oxa-5,8,11-triazatridec-12-en-13-yl)-1H-indole-1-carboxylate (Z-12)
To a stirred solution of amine (Z)-11 (3.75 g, 4.92 mmol) and N-boc-l-(S-trityl)-cys (2.73 g, 5.91 mmol) in anhydrous CH2Cl2:DMF (4:1, 59 mL:15 mL) at 0 °C, were added diisopropylethylamine (DIPEA) (1.28 mL, 7.4 mmol), 1-hydroxybenzotriazole (HOBt) (730 mg, 5.41 mmol) and N,N,N′,N′-tetramethyl-O-(7-azabenzotriazol-1-yl)uronium hexafluorophosphate (HATU) (2.05 g, 5.41 mmol). After 30 min the cold bath was removed and the reaction mixture was stirred at 23 °C for 2 h, quenched with a saturated aqueous solution of NH4Cl, poured into H2O and extracted with CH2Cl2. The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash chromatography on silica gel (hexane:EtOAc, 4:1) to give pure (Z)-12 (4.97 g, 84% yield) as a yellow solid. 1H NMR (300 MHz, CDCl3) δH ppm: 8.44 (d, 1H, J = 11.3 Hz), 8.41 (s, 1H), 7.72 (s, 1H), 7.38 (m, 10H), 7.30 (s, 1H), 7.25 (m, 20H), 6.89 (dd, 1H, J = 11.4, 9.0 Hz), 6.02 (br s, 1H), 5.60 (d, 1H, J = 9.6 Hz), 5.09 (br s, 1H), 4.16 (m, 1H), 3.92 (m, 1H), 2.88 (m, 1H), 2.55 (dd, 1H, J = 12.9, 4.5 Hz), 2.49 (m, 1H), 2.37 (dd, 1H, J = 12.6, 6.3 Hz), 1.69 (s, 9H), 1.27 (s, 9H). 13C NMR (75 MHz, CDCl3) δC ppm: 171.3, 167.7, 155.4, 149.3, 144.5, 144.3, 134.6, 131.0, 129.7, 129.6, 128.3, 128.2, 127.2, 127.1, 126.6, 125.2, 123.6, 122.9, 120.4, 118.5, 114.1, 100.3, 85.1, 80.6, 67.6, 67.1, 53.5, 52.4, 34.1, 32.1, 28.4, 28.3.
3.5.12. tert-Butyl 5,6-dibromo-3-((6R,9R,E)-2,2-dimethyl-4,7,10-trioxo-6,9-bis((tritylthio)methyl)-3-oxa-5,8,11-triazatridec-12-en-13-yl)-1H-indole-1-carboxylate (E-12)
To a stirred solution of amine (E)-11 (170 mg, 0.22 mmol) and N-boc-l-(S-trityl)-cys (124 mg, 0.27 mmol) in anhydrous CH2Cl2:DMF (4:1, 2.6 mL:0.7 mL) at 0 °C, were added DIPEA (58 μL, 0.33 mmol), HOBt (33 mg, 0.24 mmol) and HATU (93 mg, 0.24 mmol). After 20 min the cold bath was removed and the reaction mixture was stirred at 23 °C for 2.4 h, quenched with a saturated aqueous solution of NH4Cl, poured into H2O and extracted with CH2Cl2. The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash chromatography on silica gel (hexane:EtOAc, 3:1) to afford (E)-12 (184 mg, 73% yield) as a yellow solid. 1H NMR (300 MHz, CDCl3) δH ppm: 8.86 (d, 1H, J = 10.5 Hz), 8.49 (s, 1H), 7.81 (s, 1H), 7.50–7.36 (m, 11H), 7.34–7.16 (m, 21H), 6.34 (d, 1H, J = 8.1 Hz), 6.25 (d, 1H, J = 14.4 Hz), 4.87 (br s, 1H), 4.40 (m, 1H), 3.65 (m,1H), 3.15 (dd, 1H, J = 12.3, 5.4 Hz), 2.70 (dd, 1H, J = 13.2, 4.5 Hz), 2.56 (m, 1H), 2.38 (dd, 1H, J = 12.9, 4.5 Hz), 1.66 (s, 9H), 1.26 (s, 9H).
3.5.13. tert-Butyl 5,6-dibromo-3-((Z)-2-((4R,7R)-7-((tert-butoxycarbonyl)amino)-6-oxo-1,2,5-dithiazocane-4-carboxamido)vinyl)-1H-indole-1-carboxylate (Z-13)
Over a solution of I2 (897 mg; 3.54 mmol) in CH2Cl2:CH3OH (10:1; 1416 mL) a solution of (Z)-12 (610 mg; 0.50 mmol) in CH2Cl2 (100 mL) was added at 23 °C. The reaction mixture was stirred over 40 min, and a 5% aqueous solution of Na2S2O4 was added. The aqueous layer was extracted with CH2Cl2, the combined organic layers were dried over Na2SO4, filtered, and concentrated under vacuum. The residue obtained was purified by flash chromatography on silica gel (hexane:EtOAc, 3:1) to give pure (Z)-13 (302 mg, 83% yield) as a white solid. 1H NMR (300 MHz, CDCl3) δH ppm: (mixture of conformers, signals for major conformer) 9.90 (d, 1H, J = 11.1 Hz), 8.45 (s, 1H), 7.78 (s, 1H), 7.62 (s, 1H), 7.08 (t, 1H, J = 9.9 Hz), 5.81 (d, 1H, J = 9.9 Hz), 5.65 (br s, 1H), 5.05 (br s, 1H), 4.93 (m, 1H), 4.48 (m, 1H), 3.67 (m, 1H), 3.37–2.87 (m, 3H), 1.70 (s, 9H), 1.25 (s, 9H). 13C NMR (75 MHz, CDCl3) δC ppm: (signals for major conformer) 172.3, 167.3, 154.8, 149.1, 130.3, 125.4, 125.1, 124.1, 123.7, 122.3, 120.7, 118.9, 114.0, 101.9, 85.2, 80.6, 57.1, 54.5, 43.2, 41.8, 28.6, 28.3.
3.5.14. tert-Butyl 5,6-dibromo-3-((E)-2-((4R,7R)-7-((tert-butoxycarbonyl)amino)-6-oxo-1,2,5-dithiazocane-4-carboxamido)vinyl)-1H-indole-1-carboxylate (E-13)
Over a solution of I2 (265 mg; 1.04 mmol) in CH2Cl2:CH3OH (10:1; 416 mL) a solution of (E)-12 (180 mg; 0.15 mmol) in CH2Cl2 (30 mL) was added at 23 °C. The reaction mixture was stirred over 40 min, and a 5% aqueous solution of Na2S2O4 was added. The aqueous layer was extracted with CH2Cl2, the combined organic layers were dried over Na2SO4, filtered, and concentrated under vacuum. The residue obtained was purified by flash chromatography on silica gel (hexane:EtOAc, 6:4) to obtain pure (E)-13 (65 mg, 62% yield) as a white solid. 1H NMR (300 MHz, CDCl3) δH ppm: (mixture of conformers, signals for major conformer) 9.40 (d, 1H, J = 10.5 Hz), 8.32 (s, 1H), 7.70 (s, 1H), 7.44 (s, 1H), 7.34 (m, 1H), 6.92 (d, 1H, J = 11.1 Hz), 6.21 (d, 1H, J = 15.0 Hz), 5.10 (br s, 1H), 5.01 (m, 1H), 4.80 (m, 1H), 3.78 (m, 1H), 3.45 (m, 1H), 3.05 (m, 1H), 2.87 (m, 1H), 1.63 (s, 9H), 1.42 (s, 9H). 13C NMR (75 MHz, CDCl3) δC ppm: (signals for major conformer) 173.3, 166.7, 155.6, 148.9, 135.1, 129.2, 123.7, 122.8, 120.4, 120.2, 118.5, 115.9, 115.7, 105.5, 85.0, 81.1, 53.8, 48.8, 42.8, 36.9, 28.6, 28.3.
3.5.15. Tanjungide A (1)
Over a solution of (Z)-13 (310 mg; 0.43 mmol) in CH2Cl2 (9.3 mL) was dropwise added TFA (2.8 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 8 h and a saturated aqueous solution of NaHCO3 was added until pH 8. The organic layer was extracted with EtOAc (×3), the combined organic layers were dried over Na2SO4, filtered and concentrated under vacuum. The residue obtained was purified by flash chromatography on silica gel (CH2Cl2:CH3OH, 30:1) to yield pure Tanjungide A (1) (135 mg, 60% yield) as a pale yellow solid and exhibited physical and spectroscopic characteristics (1H, 13C NMR and MS) equivalent to those reported in 3.3.
3.5.16. Tanjungide B (2)
Over a solution of (E)-13 (52 mg; 0.07 mmol) in CH2Cl2 (1.6 mL) was dropwise added TFA (0.47 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 3 h and a saturated aqueous solution of NaHCO3 was added until pH 8. The organic layer was extracted with EtOAc (×3), the combined organic layers were dried over Na2SO4, filtered and concentrated under vacuum. The residue obtained was purified by flash chromatography on silica gel (CH2Cl2:CH3OH, 20:1) to give pure Tanjungide B (2) (16 mg, 45% yield) as a pale yellow solid and exhibited physical and spectroscopic characteristics (1H, 13C NMR and MS) equivalent to those reported in section 3.3.
3.6. Biological Activity
A549 (ATCC CCL-185), lung carcinoma; HT29 (ATCC HTB-38), colorectal carcinoma and MDA-MB-231 (ATCC HTB-26), breast adenocarcinoma cell lines were obtained from the ATCC. Cell lines were maintained in RPMI medium supplemented with 10% fetal calf serum (FCS), 2 mM l-glutamine and 100 U/mL penicillin and streptomycin, at 37 °C and 5% CO2. Triplicate cultures were incubated for 72 h in the presence or absence of test compounds (at ten concentrations ranging from 10 to 0.0026 μg/mL). For quantitative estimation of cytotoxicity, the colorimetric sulforhodamine B (SRB) method was used [14]. Briefly, cells were washed twice with PBS, fixed for 15 min in 1% glutaraldehyde solution, rinsed twice in PBS, and stained in 0.4% SRB solution for 30 min at room temperature. Cells were then rinsed several times with 1% acetic acid solution and air-dried. Sulforhodamine B was then extracted in 10 mM trizma base solution and the absorbance measured at 490 nm. Results are expressed as GI50, the concentration that causes 50% inhibition in cell growth after correction for cell count at the start of the experiment (NCI algorithm). Doxorubicin and DMSO (solvent) were used as the positive and negative controls in this assay. Prism 3.03 from GraphPad was used for the statistical analysis of the cell growth inhibition results.
4. Conclusions
In summary, we have isolated, determined the structure and completed the first total synthesis of Tanjungides A and B, two new bromoindole enamides with interesting cytotoxic properties from the tunicate Diazona cf formosa. The total synthesis confirmed the structural assignment and provides rapid access to these new natural products and related analogues for biological evaluation and drug development.
Acknowledgments
The authors thank Susana González for recording the NMR experiments; Luis F. García-Fernández for performing the cytotoxicity assays; Santiago Bueno and Carlos de Eguilior for collection of the tunicate; Sebastiano Gulinello for logistical support with the diving expedition; Xavier Turon, Centre for Advanced Studies of Blanes (CEAB-CSIC), for the taxonomic identification of the sample; and Democratic Republic of Timor Leste, Ministry of Agriculture and Fisheries, Area of Agriculture, Fisheries and Livestock, Comoro-Dili for facilitating this work.
Supplementary Files
Supplementary Information (PDF, 2377 KB)
Conflicts of Interest
The authors declare that CM, LC, RF, MJM, AF, SM and CC are employees of PharmaMar. FR was an employee of PharmaMar.
References
- 1.Menna M., Fattorusso E., Imperatore C. Alkaloids from marine ascidians. Molecules. 2011;16:8694–8732. doi: 10.3390/molecules16108694. [DOI] [Google Scholar]
- 2.Blunt J.W., Copp B.R., Keyzers R.A., Munro M.H.G., Prinsep M.R. Marine natural products. Nat. Prod. Rep. 2013;30:237–323. doi: 10.1039/c2np20112g. [DOI] [PubMed] [Google Scholar]
- 3.Vervoort H.C., Richards-Gross S.E., Fenical W., Lee A.Y., Clardy J. Didemnimides A–D: Novel, predator-deterrent alkaloids from the caribbean mangrove ascidian Didemnum conchyliatum. J. Org. Chem. 1997;62:1486–1490. doi: 10.1021/jo961789s. [DOI] [Google Scholar]
- 4.Seldes A.M., Rodríguez M.F., Hernández L., Palermo J.A. Identification of two meridianins from the crude extract of the tunicate Aplidium meridianum by tandem mass spectrometry. Nat. Prod. Res. 2007;21:555–563. doi: 10.1080/14786410601133517. [DOI] [PubMed] [Google Scholar]
- 5.Bokesch H.R., Pannell L.K., McKee T.C., Boyd M.R. Coscinamides A, B and C, three new bis indole alkaloids from the marine sponge Coscinoderma sp. Tetrahedron Lett. 2000;41:6305–6308. doi: 10.1016/S0040-4039(00)01062-5. [DOI] [Google Scholar]
- 6.Sato H., Tsuda M., Watanabe K., Kobayashi J. Rhopaladins A–D, new indole alkaloids from marine tunicate Rhopalaea sp. Tetrahedron. 1998;54:8687–8690. doi: 10.1016/S0040-4020(98)00470-0. [DOI] [Google Scholar]
- 7.Appleton D.R., Copp B.R. Kottamide E, the first example of a natural product bearing the amino acid 4-amino-1,2-dithiolane-4-carboxylic acid (Adt) Tetrahedron Lett. 2003;44:8963–8965. doi: 10.1016/j.tetlet.2003.10.008. [DOI] [Google Scholar]
- 8.Parsons T.B., Spencer N., Tsang C.W., Grainger R.S. Total synthesis of kottamide E. Chem. Commun. 2013;49:2296–2298. doi: 10.1039/c3cc39062d. [DOI] [PubMed] [Google Scholar]
- 9.Reyes F., Fernández R., Rodríguez A., Francesch A., Taboada S., Ávila C., Cuevas C. Aplicyanins A–F, new cytotoxic bromoindole derivatives from the marine tunicate Aplidium Cyaneum. Tetrahedron. 2008;64:5119–5123. doi: 10.1016/j.tet.2008.03.060. [DOI] [Google Scholar]
- 10.Lindquist N., Fenical W. Isolation and structure determination of Diazonamides A and B, unusual cytotoxic metabolites from the marine ascidian Diazona chinensis. J. Am. Chem. Soc. 1991;11:2303–2304. doi: 10.1021/ja00006a060. [DOI] [Google Scholar]
- 11.Fernández R., Martín M.J., Rodríguez-Acebes R., Reyes F., Francesch A., Cuevas C. Diazonamides C–E, new cytotoxic metabolites from the ascidian Diazona sp. Tetrahedon Lett. 2008;49:2283–2285. doi: 10.1016/j.tetlet.2008.02.012. [DOI] [Google Scholar]
- 12.Whitson E.L., Ratnayake A.S., Bugni T.S., Harper M.K., Ireland C.M. Isolation, structure elucidation, and synthesis of Eudistomides A and B, lipopeptides from a fijian ascidian Eudistoma sp. J. Org. Chem. 2009;74:1156–1162. doi: 10.1021/jo8022582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marfey P. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984;49:591–596. [Google Scholar]
- 14.Skehan P., Storeng R., Scudiero D., Monks A., McMahon J., Vistica D., Warren J.T., Bokesch H., Kenney S., Boyd M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 15.Boyd E.M., Sperry J. Synthesis of the selective neuronal nitric oxide synthase (nNOS) inhibitor 5,6-dibromo-2′-demethylaplysinopsin. Synlett. 2011;6:826–830. [Google Scholar]
- 16.Seyferth S., Heeren J.K., Singh G., Grim S.O., Hughes W.B. Studies in phosphinemethylene chemistry: XIII. Routes to triphenylphosphine-halomethylenes and -dihalomethylenes. J. Organomet. Chem. 1966;5:267–274. [Google Scholar]
- 17.Stork G., Zhao K. A stereoselective synthesis of (Z)-1-iodo-1-alkenes. Tetrahedron Lett. 1989;30:2173–2174. doi: 10.1016/S0040-4039(00)99640-0. [DOI] [Google Scholar]
- 18.Jiang L., Job G.E., Klapars A., Buchwald S.L. Copper-catalyzed coupling of amides and carbamates with vinyl halides. Org. Lett. 2003;5:3667–3669. doi: 10.1021/ol035355c. [DOI] [PubMed] [Google Scholar]
- 19.Kamber B., Hartmann A., Eisler K., Riniker B., Rink H., Sieber P., Rittel W. The synthesis of cystine peptides by iodine oxidation of S-trityl-cysteine and S-acetamidomethyl-cysteine peptides. Helv. Chim. Acta. 1980;63:899–915. doi: 10.1002/hlca.19800630418. [DOI] [Google Scholar]
- 20.Boger D.L., Ichikawa S., Tse W.C., Hedrick M.P., Jin F.Q. Total synthesis of thiocoraline and BE-22179 and assessment of their DNA binding and biological properties. J. Am. Chem. Soc. 2001;123:561–568. doi: 10.1021/ja003602r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information (PDF, 2377 KB)