

Summary
The centromeric histone H3 variant centromeric protein A (CENP-A), whose sequence is the least conserved among all histone variants, is responsible for specifying the location of the centromere. Here, we present a comprehensive study of CENP-A nucleosome arrays by cryo-electron tomography. We see that CENP-A arrays have different biophysical properties than canonical ones under low ionic conditions, as they are more condensed with a 20% smaller average nearest-neighbor distance and a 30% higher nucleosome density. We find that CENP-A nucleosomes have a predominantly crossed DNA entry/exit site that is narrowed on average by 8°, and they have a propensity to stack face to face. We therefore propose that CENP-A induces geometric constraints at the nucleosome DNA entry/exit site to bring neighboring nucleosomes into close proximity. This specific property of CENP-A may be responsible for generating a fundamental process that contributes to increased chromatin fiber compaction that is propagated under physiological conditions to form centromeric chromatin.
Introduction
In eukaryotes, chromosomes are accurately segregated during mitosis to their daughter cells by connecting each chromosome to a highly dynamic microtubule structure called the spindle. To faithfully segregate sister chromatids during cell division, thereby preserving the integrity of the genome, a complex of proteins called the kinetochore mediates connections to the spindle. Kinetochores assemble specifically at the centromere, a chromosomal region that is epigenetically marked by the histone H3 variant centromeric protein A (CENP-A) (1). Whereas the centromere in budding yeasts is defined genetically by a specific ∼125 bp DNA sequence, this sequence is not conserved in other eukaryotes (2). Rather, in most eukaryotes, including mammals, a model of epigenetic centromere inheritance has emerged wherein an array of CENP-A nucleosomes are crucial for establishing and maintaining centromere identity independently of the DNA sequence (3). A key biochemical demonstration of this concept involved reconstituted CENP-A nucleosome arrays that were sufficient to assemble centromere and kinetochore proteins, to bind and stabilize microtubules, and to facilitate a mitotic checkpoint function in vitro using Xenopus laevis cell-free extracts (4). In this context, an important question arises regarding the biophysical properties of CENP-A that facilitate the differentiation of the centromere from the remaining chromatin landscape.
Over the last 6 years, researchers have investigated the specific properties of CENP-A by using a multitude of structural and biochemical approaches to explore the stoichiometry, composition, size, and topology of DNA wrapped around the histone core and the flexibility of the entry/exit site DNA (5–15). These extensive studies led to proposals favoring either 1), a right-handed hemisome model with a nucleosome composed of one copy each of CENP-A, H4, H2A, and H2B (16); or 2), a model with a more compact, left-handed octameric nucleosome whose entry/exit site DNA is looser compared with canonical H3 nucleosomes (8,9,11–14). The latter model has recently gained support since the major form of CENP-A nucleosomes in humans was shown to be octameric (13). Moreover, studies on reconstituted recombinant CENP-A nucleosome arrays confirmed that CENP-A nucleosomes are octameric and revealed that the height difference between CENP-A- and H3-containing nucleosomes commonly measured by atomic force microscopy (AFM) is not related to alterations in the composition of histone subunits as invoked in the hemisome model (17).
Here, we used reconstituted 24-mer human CENP-A and X. laevis H3 arrays, and subjected them to cryo-electron tomography (CET) to analyze their biophysical properties. CET is the only technique that can investigate structurally individual pleiomorphic objects (e.g., chromatin) with a resolution in the nanometer range. Although the individual micrographs provide a global view of arrays, more detailed information can be mined by subtomogram averaging and classification techniques that enable one to discern the various conformational associations among individual arrays and nucleosomes in solution. The results revealed that CENP-A nucleosomes are distinct from their canonical counterparts. They are more condensed and have a narrower angle of the DNA entry/exit site. CENP-A nucleosomes have a propensity to stack face to face even under low ionic conditions.
Materials and Methods
Nucleosome array preparation of recombinant human and X. laevis histones
Human (CENP-A/H4)2 subnucleosomal heterotetramer was purified as previously described (18). Human H2A was purified with a 6x-His tag that was cleaved by Precission protease (GE Healthcare) before dimer reconstitution with untagged human H2B as previously described (11). Canonical histones from X. laevis H2A, H2B, H3, and H4 (GenBank IDs X03018.1, X03018.1, X72949.1, and X00224, respectively) were produced as previously described (19). The DNA template used consists of 24 tandem head-to-tail repeats of a 207 bp nucleosome repeat length that is composed of the 601 Widom nucleosome positioning sequence (20) and a 60 bp internucleosomal linker sequence. The 24-mer 601 DNA was prepared as described previously (21). Subsequently, the 24-mer 601 DNA was excised by XbaI/HindIII double-restriction digestion and separated from the three cleavage by-products by gel filtration through a Sephacryl S-1000, eluting from TE (10 mM Tris pH 8.0, 1 mM EDTA). The three pUC19 cleavage by-products (∼700, 800, and 1100 bp) were used as carrier DNA for the reconstitution of canonical nucleosome arrays.
Human canonical histones H2A, H2B, and H4, and centromeric H3 variant CENP-A (GenBank IDs CAA58539.1, CAB02542.1, CAA58538.1, and AAH02703.1, respectively) were utilized directly from ∼2.5 μM glycerol stocks of H2A/H2B dimer and CENP-A/H4 tetramer. Nucleosome arrays were reconstituted at 25 μg/ml 24-mer 601 DNA by salt gradient dialysis according to the protocol established by the Rhodes laboratory (22), with slight modifications. First, CENP-A octamers were formed from human H2A/H2B dimers and CENP-A/H4 tetramers in histone buffer (10 mM Tris, 0.6 M NaCl, 0.5 mM EDTA, pH 7.5) on ice before application to the reconstitution reaction. Second, no carrier DNA was used for the human nucleosome array reconstitution, because histone octamers unfavorably assembled equally on both the 601 DNA array and carrier DNA, which led to underloaded 601 DNA arrays. Third, overnight dialysis was split into two dialysis steps, first dialyzing against 250 mM NaCl and then against 5 mM NaCl low-salt buffer. The molar input ratio of octamer that was required to obtain full saturation of the 601 DNA array was determined empirically by titration. In the case of glutaraldehyde fixation before electron microscopy (EM) analysis, the conventional Tris-HCl buffer system of the last dialysis step was substituted for HEPES ((4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) to avoid quenching of the cross-linker reaction by the primary amine group of the buffer molecule. Upon dialysis, the X. laevis H3 reconstituted nucleosome arrays were purified from carrier DNA by MgCl2 precipitation (23). For this reason, an equal volume of MgCl2 solution was added to the reaction to 2.5 mM, incubated at 4°C for 10 min, and pelleted at 13,000 rpm for 15 min at 4°C. The supernatant containing the carrier DNA was removed and the pellet was gently resuspended in 5 mM NaCl low-salt buffer. To examine whether nucleosome arrays had been fully reconstituted, 5 μl of the reaction was digested with EcoRI to produce mononucleosomes and analyzed by native agarose gel electrophoresis (0.7% agarose gel in 0.2× TBE buffer, post-staining with ethidium bromide).
Analysis of nucleosome arrays by EM and CET
To assess regular loading of the nucleosome arrays by negative-stain EM, the sample was mildly fixed by 30 min incubation in a low-salt buffer containing 5 mM NaCl, 20 mM HEPES, and 0.5% glutaraldehyde (Carl-Roth, Karlsruhe, Germany). Then 4 μl of the sample was applied to glow-discharged 400 mesh, 5–10 nm extra-carbon supported grids (Plano, Wetzlar, Germany), incubated for 2 min at room temperature, washed three times with distilled water, and finally incubated for 2 min at room temperature in 2% uranyl-acetate (Ted Pella, Redding, CA) aqueous solution before final blotting and air drying. The negative-stained grids were analyzed with a 200 kV F-20 transmission electron microscope (FEI, Eindhoven, The Netherlands) equipped with a 4k FEI Eagle CCD camera. All images were taken at 200 kV with a defocus between −6 and −12 μm and exposure times ranging from 0.25 to 1.0 s at a magnification of 29,000–50,000×.
Before CET analysis, a 3 μl sample was diluted 1:4 with Protein A 10 nm fiducial markers (Aurion, Wageningen, The Netherlands) that had been washed three times with 5 mM NaCl low-salt buffer to remove residual MgCl2 in the storage buffer. Further, the diluted sample was applied to glow-discharged 300 mesh formvar-carbon supported lacey grids (Plano, Wetzlar, Germany) and plunge-frozen with a Vitrobot Mark IV (FEI) with zero blotting force and 1.5 s blotting time.
Tomograms were recorded using DigitalMicrograph (Gatan, Pleasanton, CA) on a 300 kV Titan Krios transmission electron microscope (FEI) with a GATAN GIF Quantum SE postcolumn energy filter and 4k UltraScan 4000 multichannel CCD camera. All tomograms were acquired at 300 kV with the GIF operating in zero loss peak mode, with tilt series ranging from −66° to +66° with an angular increment of 1°, defocus set between −6 and −8 μm, and an electron dosage not exceeding 100 e-/Å−2. Tilt series were obtained at a nominal magnification of 26,000× or 33,000×, which corresponds to a pixel size of 0.48 or 0.38 nm, respectively, at the specimen level. Tilted images were aligned using gold beads as fiducial markers and reconstructed by weighted back-projection with a Gaussian image filter to the first zero crossing of the contrast transfer function (CTF).
Image analysis of nucleosome arrays
Data were acquired from multiple chromatin array preparations that were followed by several cryo preparations to carry out CET. After data mining, the technically best tomograms from several recording sessions (resulting in four tomograms for each nucleosome type) were used for data analysis.
Three-dimensional (3D) reconstructions were analyzed by the EM package in Amira (24) and by custom image processing scripts and principal component analysis (PCA) classification (25) performed in MATLAB (The MathWorks, Natick, MA) that are available at http://www.biophys.uni-frankfurt.de/frangakis. In general, the particles in the cryo-tomograms were cropped out by the watershed labeling/subtomogram averaging detection procedure (26), and the resulting 643 voxel subtomograms with a voxel size of either 0.78 for CENP-A or 0.96 nm for H3 were aligned by a template-free procedure until the average reached convergence. The missing wedge was taken into account in all alignment and averaging steps.
To analyze the higher compaction of CENP-A arrays, we utilized two strategies measuring 1), the nucleosome density (number of nucleosomes per tomogram); and 2), the average nearest-neighbor distance. For the first measure, we quantified the labels obtained by the above-mentioned labeling technique (26) for the CENP-A and canonical H3 nucleosomes, each for four tomograms, and set them into the context of the label volume encompassing all nucleosomes found in each tomogram. For the second measure, we converted the labels for the CENP-A and canonical H3 nucleosomes into Cartesian coordinates and calculated the distance to each nucleosome, where the minimal distance corresponds to the closest neighbor. Subsequently, sample points that were five times higher than the standard deviation (SD; z = 5) were defined as outliers and removed from both data sets.
To investigate the nucleosome stacking motif, the particles were aligned using a rotationally symmetrized nucleosome derived from the PDB structure 1ID3. The properly aligned particles (exceeding a cross-correlation value of 0.6) were then classified by 3D PCA (27) with a spherical mask that was four times the radius of a nucleosome that was capable of discerning the conformational differences.
To determine the molecular weight and dimensions of the nucleosomes, we used 30% of the data set with the highest cross-correlation value for alignment. The nucleosomes were thereafter mean-normalized and masked with a 20 pixel spherical mask that corresponds in radius 1.5 for CENP-A and 2 times the size for H3 nucleosomes to set a threshold for segmentation (assuming a radius of 5nm for an ideal nucleosome adapted to the pixel size of 0.38 nm for CENP-A and 0.48 nm for H3 nucleosomes at the specimen level). The criterion for setting a threshold that distinguished background (noise) from foreground (nucleosome) was evaluated to be the mean value of the subtomogram histograms multiplied by a global factor that was manually screened for CENP-A and H3 nucleosomes. The ideal threshold for segmentation was determined in the screening process to be consistent when the segmented nucleosome matched the dimensions of the corresponding unsegmented nucleosome upon visual inspection. The resulting volume size of the segmented nucleosome was then multiplied by a protein average density of 1.3 g/cm3 and converted to Dalton (corresponding to 0.8 kDa per nm3) to determine the molecular weight. The dimension of the segmented nucleosome was measured by computing the difference between the maximum and minimum value in each x, y, z dimension. The diameter of the nucleosomes was calculated by the mean value of the x-y dimensions.
To determine the nucleosome-to-nucleosome distances and the ω angle, 170 trinucleosomes for CENP-A and 171 for canonical H3 were manually selected. Subtomograms of 1283 voxels containing the trinucleosomes were extracted and rotated to bring the nucleosomes onto a plane. The positions between the central and neighboring nucleosomes were assumed to be connected by straight linker DNA. To investigate the configuration of the entry/exit site DNA of the central nucleosomes, the trinucleosomes were low-pass filtered, 10-nm-thick tomographic slices along the z-axis were computed, and the linker DNA arms together with the central nucleosome were manually segmented and binarized. The binarized central nucleosome with its linker DNA arms was then subjected to subtomogram averaging until the average reached convergence. The trinucleosomes were then extracted from tomograms filtered to the first zero of the CTF and aligned by the angles and translational shifts that had been determined by the subtomogram averaging procedure of the respective binarized nucleosome with its linker DNA. To distinguish open and crossed entry/exit site DNA, the aligned particles were classified by 3D PCA using a spherical mask of 10 pixels in diameter to encompass the DNA entry/exit site.
The model of the centromeric CENP-A fiber under physiological conditions was derived from in situ measurements of chicken erythrocyte chromatin (28). To model the CENP-A chromatin fiber, the pitch of the canonical two-start helix fiber was kept constant while the angle between consecutive nucleosomes and the rise was varied until the neighboring distance of consecutive nucleosomes and the ω angle approximately reached the measured values derived from the CENP-A arrays.
Results
Unfolded CENP-A arrays are more condensed than canonical arrays
To investigate the biophysical differences between canonical and centromeric chromatin, equimolar 24-mer 601 DNA tandem repeat arrays were reconstituted with either X. laevis canonical (H3) or human centromeric histone octamers (CENP-A) (Fig. S1, A and B, in the Supporting Material) under low monovalent salt conditions (5 mM NaCl; Fig. S1, C and D), plunge frozen, and analyzed by CET.
The arrays under this low monovalent salt condition formed patches of well-dispersed polynucleosomes (Fig. 1, A and B) in a thin vitreous ice layer of holey carbon support grids. The cryo preparation was carried out with equimolar nucleosome array solutes, but CENP-A nucleosome arrays tend to be compacter associated, resulting in more nucleosomes per tomogram. To quantify the difference in condensation, four tomograms of CENP-A and four tomograms of canonical H3 nucleosomes were automatically labeled (26), which resulted in ∼11,200 CENP-A and ∼8600 canonical H3 nucleosomes (Table S1).
Figure 1.

Unfolded CENP-A arrays are more condensed. (A) A tomographic slice (0.76 nm thick) through an area of unfolded CENP-A 24x207bp-mer 601 DNA nucleosome arrays, visualizing disperse polynucleosomes with a distinct population of nucleosomes in which the entry/exit site DNA is apparent (white arrowheads) and nucleosomes stack face to face (boxes). The dashed line frames a carbon support bar. The inset represents the histogram of the nearest-neighbor distance distribution for 6600 CENP-A nucleosomes, with mean distance above the peak in nanometers and curve fitting superimposed with a black dashed line. NN-Distance, nearest-neighbor distance. (B) As in A, but visualizing canonical H3 24x207bp-mer 601 DNA nucleosome arrays. The inset represents the same histogram as the inset in A, but for the canonical H3 nucleosomes. The nucleosome model inset schematically depicts the population of nucleosomes in which the entry/exit site DNA is visible. NN1 is the distance between consecutive nucleosomes, whereas NN2 is equal to the distance of N+2 neighboring nucleosomes.
The analysis yielded an average nearest-neighbor distance of 16.2 nm (SD = 7 nm) for CENP-A (Fig. 1 A, inset) and 19.8 nm (SD = 5.9 nm) for H3 arrays (Fig. 1 B, inset). Thus, the average nearest-neighbor distance is 20% smaller in the CENP-A tomograms. Next, we investigated the extent to which the consecutive nucleosome-to-nucleosome (N+1) distance (NN1) contributes to the nearest-neighbor distance. For this purpose, we selected all nucleosomes for which the linker DNA, connecting consecutive nucleosomes, was visible (Fig. 1 A and B, white arrowheads), and measured the consecutive nucleosome-to-nucleosome distance. The consecutive nucleosome-to-nucleosome distance was found to be 29 nm for both nucleosome types (SD = 7 nm; Fig. S2). This indicates that both nucleosome types are homogeneously aligned on the 601 DNA tandem repeat array, as 29 nm corresponds to ∼60 bp, which coincidences with the spacing (linker length) between consecutive nucleosomes positioned in sequence of the 601 DNA template employed in this work (21). Since the distance between the majority (∼93%) of the neighboring nucleosomes is smaller than 29 nm, we conclude that these distances correspond primarily to N+2 distances (NN2) or distances between nucleosomes originating from different arrays. In both cases, NN2 distances and distances between nucleosomes from different arrays, the average nearest-neighbor distance indicates a higher condensation rate of CENP-A arrays. In particular, 34% of the neighboring distance in the nonnormally distributed CENP-A histogram is in the range of 7–13 nm, whereas this range is only covered by 7% in the normally distributed H3 histogram (Fig. 1). Measuring the absolute number of nucleosomes per tomogram in a statistically significant sample (equimolar concentrations) also showed that the average density of the CENP-A nucleosomes was ∼2800 nucleosomes/tomogram, whereas the canonical nucleosome density was only ∼2160 nucleosomes/tomogram (Table S1). Thus, we found that the nucleosome density was 30% higher in the CENP-A tomograms.
Unfolded CENP-A arrays exhibit more juxtaposed nucleosomes
Next, we performed subtomogram averaging to reveal arrangement motifs that neighboring nucleosomes have in common. We extracted subtomograms around nucleosomes from the tomographic reconstructions depicted in Fig. 1 and aligned them using a rotationally symmetrized nucleosome template generated from the crystal structure of the nucleosome. To discern the various motifs, we subsequently classified the subtomograms by PCA. PCA is a standard technique in the field of cryo-EM to distinguish molecules that differ from each other in their conformational state, and has been successfully used to discern the dynamics of many macromolecular structures (29). Technically, template-based alignment is prone to bias; however, newly arising densities surrounding the aligned nucleosome are considered to be independent of the template used (26).
After the alignment of the CENP-A and canonical H3 nucleosomes, only the nucleosomes with appropriate alignment (cross-correlation value higher than 0.6), resulting in 1810 CENP-A and 1286 canonical H3 nucleosomes, were used for classification. The classification resulted in class averages with a nucleosome arrangement in which CENP-A nucleosomes were preferably juxtaposed with a face-on orientation and distance of ∼10 nm, reminiscent of nucleosome stacking (Fig. 2).
Figure 2.
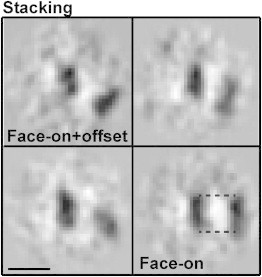
Unfolded CENP-A arrays exhibit more juxtaposed nucleosomes. CENP-A nucleosomes, a few of which are boxed in Fig. 1A, were classified. The entire pool of neighboring nucleosomes (n = 40) show a stacking motif that is distinguished by face-on and face-off positioning of nucleosomes. Face-on juxtaposition is indicated by the dashed lines between the nucleosomes. The scale bar is 15 nm.
The analysis of 1810 nucleosomes of the CENP-A data set showed that 2.1% contributed to the specific stacking motif. The remaining class averages did not contain nucleosomes with a specific arrangement or conformation, but only mononucleosomes (Fig. S3 A). The control experiment, in which 1286 subtomograms of the canonical H3 data set were analyzed, did not show any evidence of classes with stacked nucleosomes (Fig. S3 B), thereby rendering it as a significant motif in the CENP-A arrays (z-test, p-value < 0.01).
Both CENP-A and H3 arrays have a higher ratio of crossed versus open DNA entry/exit sites
In many cases, the linker DNA between nucleosomes could be visualized in our tomographic reconstructions (Fig. 1, A and B). We therefore selected ∼520 nucleosomes out of four tomograms for each nucleosome type (Table S1) for which the entry/exit sites of the DNA and the linker DNA could be unambiguously traced to their consecutive nucleosomes. Nucleosome arrays with three nucleosomes account for the majority of traceable polynucleosomes and are referred to as trinucleosomes (Fig. 3, A and C, left). Traceable nucleosome arrays composed of more than four nucleosomes exist, but are less frequently found in the data sets (Fig. 3, A and C, right).
Figure 3.
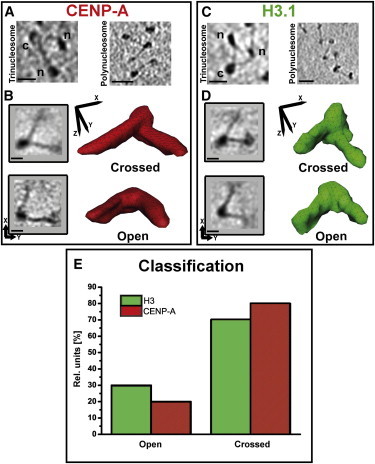
Both CENP-A and H3 arrays show a higher ratio of crossed versus open entry/exit DNA site. (A) Representative tomographic slice (9 nm thick) of a CENP-A trinucleosome (left) and polynucleosome (right) as they were regularly found in CENP-A tomograms. (C) Representative tomographic slice (12 nm thick) of a canonical H3 trinucleosome (left) and polynucleosome (right) as they were regularly found in canonical H3 tomograms. In A and C, left, the central nucleosome is labeled with (c) and the neighboring nucleosomes are labeled with (n). The scale bars are 15 nm for A and C, left, and 30 nm for A and C, right. (B) 2D projections of subtomograms show trinucleosomes in the crossed/open state for CENP-A arrays as classified by PCA (left). The orientation of the projections is indicated by a coordinate axis to the left. The corresponding subtomogram average of these classes is illustrated by a red isosurface representation of the subtomogram average volume (right). The orientation of the red isosurface representation is indicated by superimposing the Cartesian coordinate axis and also applies to the orientation of the subtomogram average shown below (open). (D) As in A, but for the classification of trinucleosomes of the canonical H3 arrays. The scale bar for both panels is 10 nm. (E) Bar chart of the relative ratio of open/crossed state of CENP-A and canonical H3 trinucleosomes as determined by PCA. The sample size was 170 for CENP-A and 171 for canonical H3 trinucleosomes, with a percentage of ambiguous trinucleosomes (i.e., nucleosomes with a low signal/noise ratio that could not be properly classified) of 27% and 15%, respectively.
To assess the structural effect of CENP-A on the DNA entry/exit site, subtomograms of CENP-A and canonical H3 nucleosomes with visible entry/exit site DNA were aligned according to their linker DNA segments connected to the central nucleosome, and classified by the positions of the DNA entry/exit site. The classification showed that both the CENP-A and canonical H3 nucleosomes exhibit two classes for an open and a crossed DNA entry/exit site (Fig. 3). We observe that the central nucleosome is clearly resolved, whereas the neighboring nucleosomes are undefined due to flexibility in the length and angle of the linker DNA arms. This effect is observed for crossed trinucleosomes (Fig. 3, B and D, crossed) as well as for open ones (Fig. 3, B and D, open).
The class averages for the crossed nucleosomes showed the particles to be left-handed and octameric (Fig. S4) with negatively crossed entry/exit site DNA for both the CENP-A and canonical H3 nucleosomes (Fig. 3, B and D, crossed). The open class averages do not verify whether the DNA is wrapped around the nucleosome core particle in a left-handed manner, as the resolution is not sufficient to discern a difference in the entry/exit points of the planar DNA linker arms (Fig. 3, A and B, open).
Regarding the proportion of open- to crossed-state nucleosomes, the class averages yielded a ratio of 30–70% open/crossed DNA entry/exit sites for canonical H3 and 20–80% for CENP-A arrays (Fig. 3 E). The 10% difference between the open/crossed configuration of CENP-A and canonical H3 nucleosomes was verified to be statistically insignificant (z-test: p-value > 0.05). Thus, we conclude that both CENP-A and canonical H3 nucleosomes indicate a similar proportion of open to crossed entry/exit site DNA under low ionic conditions, and thus the predominant crossing of the DNA entry/exit site must be responsible for the higher compaction of unfolded CENP-A arrays.
The DNA entry/exit angle is 8° narrower in CENP-A versus H3 arrays
With the ∼175 trinucleosomes selected from four tomograms for each nucleosome type (Table S1), we measured the planar angle (ω angle) between the flanking DNA arms that protrude from the central nucleosome (Fig. 4 A, inset).
Figure 4.
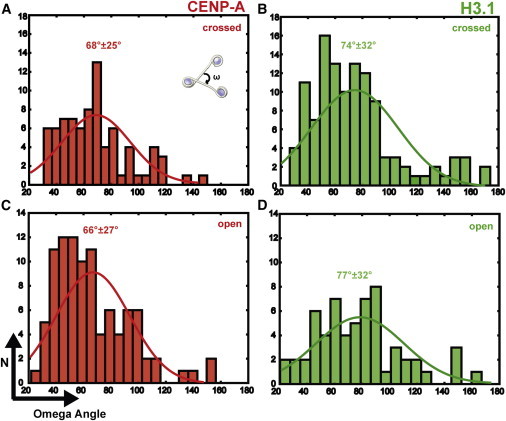
The ω angle between entry/exit sites of DNA is 8° smaller in CENP-A than in H3 arrays. (A and C) ω Angle histogram of CENP-A with open (A) and crossed DNA entry/exit site (C). (B and D) As in A and C, but for canonical H3 trinucleosomes. The mean ω angle and its SD are indicated above the histogram peaks. The mean ω angles are 74° (SD = 32°) and 77° (SD = 32°) for canonical H3, and 68° (SD = 25°) and 66° (SD = 27°) for CENP-A nucleosomes with crossed/open DNA entry/exit site. The values for the ω angles of crossed and open DNA entry/exit sites of CENP-A and canonical H3 data sets differ significantly from each other according to an unpaired two-sample t-test (p-value < 0.05). The sample size is 175 for CENP-A (open: 96; crossed: 79) and 176 trinucleosomes for canonical H3 (open: 60; crossed: 116). In each panel the curve fitting is superimposed on its corresponding color.
The mean ω angle is 74° (SD = 32°) and 77° (SD = 32°) for canonical H3 and 67° (SD = 25°) and 66° (SD = 27°) for CENP-A nucleosomes with a crossed/open DNA entry/exit site. The ω angle for CENP-A is consistently narrower throughout the open/crossed DNA entry/exit site configuration in comparison with canonical H3 nucleosomes (Fig. S5). The measured angles for the canonical H3 arrays are consistent with in situ measurements of the ω angle of chicken erythrocyte chromatin (26), and the variance observed is in agreement with previous studies in which the nucleosome arrays did not contain the H1 linker histone (30). The difference in ω angle obtained between the pooled crossed/open DNA entry/exit site of CENP-A and canonical H3 arrays was verified to be statistically significant by a t-test (p-value < 0.05). Thus, the 8° smaller ω angle observed in the CENP-A arrays has a direct effect on condensation by bringing the neighboring nucleosomes into closer proximity, thereby achieving an increased compactness of the nucleosome fiber.
Discussion
Centromeric chromatin harboring the histone H3 variant CENP-A is present within a very condensed region of the chromosome (31–33). CENP-A is needed to maintain the centromere (reviewed in Allshire and Karpen (34)) and serves as anchoring platform for the kinetochore microtubule network during mitosis (3). Our data show that CENP-A arrays are 30% more compact than canonical H3 arrays. This deviates from previous analytical ultracentrifugation (AUC) interpretations, which on average show that CENP-A and canonical arrays sediment mainly as individual arrays with similar levels of compaction at low ionic strength. However, in the same AUC profiles, a trend toward larger sedimenting CENP-A species compared with canonical arrays could be detected ((14); compare Fig. 2, A and B), which confirms our findings. Therefore, our experiments adequately complement the AUC experiments by enabling the investigation of potential dynamic larger array complexes. Since these larger array complexes can be discerned in individual arrays by CET, it also enables direct analysis of interactions between nucleosomal DNA and the histone core. Thus, we were able to measure that CENP-A nucleosomes have a significantly narrower DNA entry/exit angle compared with canonical H3 nucleosomes. The narrowing of the DNA entry/exit angle leads to a decreased nearest-neighbor distance, which consequently brings nucleosomes into closer proximity, raising the probability for stacking (Fig. 5 A).
Figure 5.
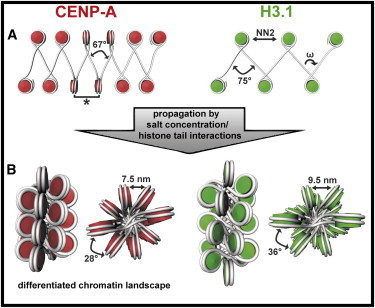
Model of the impact of CENP-A’s specific biophysical properties on forming centromeric chromatin. (A) Depiction of CENP-A and canonical H3 fibers under low ionic conditions in which CENP-A-containing nucleosome fibers are more condensed, as they have a more narrowed angle of the DNA entry/exit site (ω), thereby bringing N and N+2 nucleosomes into close proximity (NN2) and positioning neighboring nucleosomes in an arrangement similar to stacking (∗). (B) Under physiological salt conditions, the condensation of the CENP-A fibers is propagated as histone tail interactions become operational, so that CENP-A nucleosomes are brought into closer proximity. The narrower ω angle and enhanced condensation yields a differentiated chromatin landscape for the recognition of centromere- and kinetochore-related proteins. The canonical H3 model originates from in situ measurements of chicken erythrocyte chromatin (28) and was used to model the two-start helix CENP-A fiber model (for details, refer to Materials and Methods).
The stacked nucleosomes have a distance of ∼10 nm, which we do not attribute to the juxtaposition by histone tail interactions. This is supported by previous studies that showed that histone tails are tightly bound to the nucleosome core particle DNA under low ionic conditions (35–37). It is questionable whether the increased density is a factor in the formation of nucleosome pairs, but if we take into account that the average ω angle is narrower for CENP-A arrays, it is more likely that this conformation of the DNA entry/exit site promotes the arrangement of nucleosome pairs by bringing nucleosomes into proximity. Although this effect might pertain only to 2.1% of the CENP-A nucleosome arrays, we can clearly detect this motif in the CENP-A, but not in the canonical H3 data set. Notably, cryo-EM is capable of taking snapshots of molecular-dynamic motions. Thus, it might be conceivable that nucleosome pairs arrange in a stacking motif at low abundance because only a fraction of the nucleosome population adopts this distinct (transient) orientation at the time of rapid freezing.
Many studies on CENP-A have investigated structural differences at the DNA entry/exit site that make CENP-A nucleosomes different from canonical H3 nucleosomes (8,9,11–14). We show that an alteration of the DNA entry/exit angle is a core biophysical feature of CENP-A-containing nucleosome arrays. Further, this geometrical constraint resembles a unique packaging mechanism in low ionic buffer that may be propagated at physiological salt conditions when internucleosomal interactions are also operational (Fig. 5 B). We thus propose that CENP-A confers a narrower DNA entry/exit angle that in turn minimizes the repulsive forces of the linker DNA arms, allowing CENP-A nucleosomes to come into closer proximity than is observed for canonical H3 arrays.
However, there may be processes other than a more geometrically constrained DNA entry/exit site that govern the compaction. Previous studies have suggested that lysine 49 in the alpha-N helix of CENP-A is important at the DNA entry/exit site, since lysine confers more flexibility to the terminal nucleosome DNA contacts compared with the corresponding arginine in canonical H3 nucleosomes (14). Our results indicate that changes at the DNA entry/exit site of CENP-A and canonical H3 nucleosomes are not due to an intrinsic preference of steady-state nucleosomal arrays for the open versus crossed entry/exit site DNA configuration. The increased flexibility, as measured by more rapid backbone exchange on the alpha-N helix of CENP-A, has only been measured under conditions where there are topological constraints that accompany chromatin folding (13,14), which does not occur under the low ionic conditions used in this study. Therefore, it seems likely that steady-state flexibility or a more open configuration of the entry/exit site DNA for CENP-A nucleosomes only occurs when internucleosomal DNA is relaxed by ions. Our findings show that under low ionic conditions, unfolded CENP-A arrays have a similar steady-state open/crossed configuration at the entry/exit site DNA compared with their canonical counterparts assembled with conventional H3. Therefore, an increased opening of the DNA entry/exit site cannot be responsible for the higher compaction of CENP-A arrays under low ionic conditions. However, the extent to which a geometrical constrained DNA entry/exit site contributes to a higher compaction at physiological salt concentrations remains to be tested by other methods.
In conclusion, we hypothesize that CENP-A generates chromatin that is especially compacted, and that this feature is crucial for its mitotic function. Propagation of the specific CENP-A properties observed here under physiological salt conditions would result in a more compact fiber, thereby restricting access of remodeling factors and other chromatin-binding proteins, but enabling recognition by centromere- and kinetochore-related proteins (Fig. 5 B). This in turn would result in a centromere that is differentiated from the rest of the chromatin landscape, thereby providing a means of epigenetic inheritance that is independent of DNA sequence. In the future, it would be interesting to perform CET on reconstituted centromere-specific/kinetochore assembly proteins and on centromeric chromatin in situ to continue unraveling the structural determinants that enable CENP-A to epigenetically mark the centromere.
Acknowledgments
We thank Dr. S. Grigoryev for kindly providing the 601 Widom DNA template, Dr. Karolin Luger for the X. Laevis histone plasmids, and Dr. Maria Hondele for helpful discussions. We are also very grateful to Dr. Don W. Cleveland for providing us with the bicistronic human CENP-A:H4, H2A, and H2B plasmids.
This work was also supported by National Institutes of Health grant GM082989 (B.E.B.), a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund (B.E.B.), a Rita Allen Foundation Scholar Award (B.E.B.), and a postdoctoral fellowship from the American Cancer Society (N.S.). D.K. was supported by a Long-Term EMBO Fellowship. This work was supported by SFB 902 and an ERC starting grant to A.S.F.
Supporting Material
References
- 1.Cleveland D.W., Mao Y., Sullivan K.F. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 2003;112:407–421. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 2.Henikoff S., Ahmad K., Malik H.S. The centromere paradox: stable inheritance with rapidly evolving DNA. Science. 2001;293:1098–1102. doi: 10.1126/science.1062939. [DOI] [PubMed] [Google Scholar]
- 3.Sekulic N., Black B.E. Molecular underpinnings of centromere identity and maintenance. Trends Biochem. Sci. 2012;37:220–229. doi: 10.1016/j.tibs.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guse A., Carroll C.W., Straight A.F. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature. 2011;477:354–358. doi: 10.1038/nature10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalal Y., Wang H., Henikoff S. Tetrameric structure of centromeric nucleosomes in interphase Drosophila cells. PLoS Biol. 2007;5:e218. doi: 10.1371/journal.pbio.0050218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dimitriadis E.K., Weber C., Dalal Y. Tetrameric organization of vertebrate centromeric nucleosomes. Proc. Natl. Acad. Sci. USA. 2010;107:20317–20322. doi: 10.1073/pnas.1009563107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aravamudhan P., Felzer-Kim I., Joglekar A.P. The budding yeast point centromere associates with two Cse4 molecules during mitosis. Current biology: CB. 2013;23:770–774. doi: 10.1016/j.cub.2013.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dechassa M.L., Wyns K., Li M., Hall M.A., Wang M.D., Luger K. Structure and Scm3-mediated assembly of budding yeast centromeric nucleosomes. Nature communications. 2011;2:313. doi: 10.1038/ncomms1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kingston I.J., Yung J.S., Singleton M.R. Biophysical characterization of the centromere-specific nucleosome from budding yeast. J. Biol. Chem. 2011;286:4021–4026. doi: 10.1074/jbc.M110.189340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Padeganeh A., Ryan J., Boisvert J., Ladouceur A.M., Dorn J.F., Maddox P.S. Octameric CENP-A nucleosomes are present at human centromeres throughout the cell cycle. Current biology: CB. 2013;23:764–769. doi: 10.1016/j.cub.2013.03.037. [DOI] [PubMed] [Google Scholar]
- 11.Sekulic N., Bassett E.A., Black B.E. The structure of (CENP-A-H4)(2) reveals physical features that mark centromeres. Nature. 2010;467:347–351. doi: 10.1038/nature09323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tachiwana H., Kagawa W., Kurumizaka H. Crystal structure of the human centromeric nucleosome containing CENP-A. Nature. 2011;476:232–235. doi: 10.1038/nature10258. [DOI] [PubMed] [Google Scholar]
- 13.Hasson D., Panchenko T., Black B.E. The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat. Struct. Mol. Biol. 2013;20:687–695. doi: 10.1038/nsmb.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panchenko T., Sorensen T.C., Black B.E. Replacement of histone H3 with CENP-A directs global nucleosome array condensation and loosening of nucleosome superhelical termini. Proc. Natl. Acad. Sci. USA. 2011;108:16588–16593. doi: 10.1073/pnas.1113621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall M.A., Shundrovsky A., Wang M.D. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 2009;16:124–129. doi: 10.1038/nsmb.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuyama T., Henikoff S. Centromeric nucleosomes induce positive DNA supercoils. Cell. 2009;138:104–113. doi: 10.1016/j.cell.2009.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miell M.D., Fuller C.J., Allshire R.C. CENP-A confers a reduction in height on octameric nucleosomes. Nat. Struct. Mol. Biol. 2013;20:763–765. doi: 10.1038/nsmb.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Black B.E., Foltz D.R., Cleveland D.W. Structural determinants for generating centromeric chromatin. Nature. 2004;430:578–582. [Google Scholar]
- 19.Dyer P.N., Edayathumangalam R.S., Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- 20.Lowary P.T., Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 21.Grigoryev S.A., Arya G., Schlick T. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl. Acad. Sci. USA. 2009;106:13317–13322. doi: 10.1073/pnas.0903280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huynh V.A., Robinson P.J., Rhodes D. A method for the in vitro reconstitution of a defined “30 nm” chromatin fibre containing stoichiometric amounts of the linker histone. J. Mol. Biol. 2005;345:957–968. doi: 10.1016/j.jmb.2004.10.075. [DOI] [PubMed] [Google Scholar]
- 23.Schwarz P.M., Felthauser A., Hansen J.C. Reversible oligonucleosome self-association: dependence on divalent cations and core histone tail domains. Biochemistry. 1996;35:4009–4015. doi: 10.1021/bi9525684. [DOI] [PubMed] [Google Scholar]
- 24.Pruggnaller S., Mayr M., Frangakis A.S. A visualization and segmentation toolbox for electron microscopy. J. Struct. Biol. 2008;164:161–165. doi: 10.1016/j.jsb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Förster F., Pruggnaller S., Frangakis A.S. Classification of cryo-electron sub-tomograms using constrained correlation. J. Struct. Biol. 2008;161:276–286. doi: 10.1016/j.jsb.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Scheffer M.P., Eltsov M., Frangakis A.S. Nucleosomes stacked with aligned dyad axes are found in native compact chromatin in vitro. J. Struct. Biol. 2012;178:207–214. doi: 10.1016/j.jsb.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 27.Geladi P., Isaksson H., Esbensen K. Principal component analysis of multivariate images. Chemometr. Intell. Lab. Syst. 1989;5:209–220. [Google Scholar]
- 28.Scheffer M.P., Eltsov M., Frangakis A.S. Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proc. Natl. Acad. Sci. USA. 2011;108:16992–16997. doi: 10.1073/pnas.1108268108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frank J. Story in a sample-the potential (and limitations) of cryo-electron microscopy applied to molecular machines. Biopolymers. 2013;99:832–836. doi: 10.1002/bip.22274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bednar J., Horowitz R.A., Woodcock C.L. Chromatin conformation and salt-induced compaction: three-dimensional structural information from cryoelectron microscopy. J. Cell Biol. 1995;131:1365–1376. doi: 10.1083/jcb.131.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan B.A., Karpen G.H. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat. Struct. Mol. Biol. 2004;11:1076–1083. doi: 10.1038/nsmb845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schroeder-Reiter E., Sanei M., Wanner G. Current SEM techniques for de- and re-construction of centromeres to determine 3D CENH3 distribution in barley mitotic chromosomes. J. Microsc. 2012;246:96–106. doi: 10.1111/j.1365-2818.2011.03592.x. [DOI] [PubMed] [Google Scholar]
- 33.Blower M.D., Sullivan B.A., Karpen G.H. Conserved organization of centromeric chromatin in flies and humans. Dev. Cell. 2002;2:319–330. doi: 10.1016/s1534-5807(02)00135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allshire R.C., Karpen G.H. Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat. Rev. Genet. 2008;9:923–937. doi: 10.1038/nrg2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mangenot S., Leforestier A., Livolant F. Salt-induced conformation and interaction changes of nucleosome core particles. Biophys. J. 2002;82:345–356. doi: 10.1016/S0006-3495(02)75399-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leuba S.H., Bustamante C., Zlatanova J. Linker histone tails and N-tails of histone H3 are redundant: scanning force microscopy studies of reconstituted fibers. Biophys. J. 1998;74:2830–2839. doi: 10.1016/S0006-3495(98)77990-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng C., Lu X., Hayes J.J. Salt-dependent intra- and internucleosomal interactions of the H3 tail domain in a model oligonucleosomal array. J. Biol. Chem. 2005;280:33552–33557. doi: 10.1074/jbc.M507241200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.