

Abstract
Introduction:
The polyelectrolyte Complexes (PECs) are based on ionic cross-linking. They have been employed to prepare a sustained release matrix tablets. These systems are based upon the fact that their structure can entrap the drug within them. Isosorbide Mononitrate (ISMN) is an anti-anginal organic nitrate vasodilator used in the treatment of various cardiovascular disorders and prophylaxis of angina Pectoris, which is poorly absorbed from the upper GIT, hence CR formulation is desirable.
Materials and Methods:
Chitosan (CH)/Sodium alginate (SA), Guar gum (GG), and Xanthan gum (XG) were used as PECs, and were prepared using different proportions i.e., in 1:1 and 1:2 ratio. The optimum ratio of CH: SA, CH: GG and CH: XG was in the ratio was 1:2; these are formed due to electrostatic interaction between oppositely charged poly ions. These normally employ a hydrophilic matrix system. Matrix tablet of ISMN was formulated by using PECs as matrix forming agent by wet granulation technique.
Results:
The tablets were evaluated for hardness, wt variation, drug content, and in-vitro dissolution studies and found to be within limits. Release kinetics data indicated that ISMN released from the PECs-based matrix tablets of CH-SA, CH-GG and CH-XG CP in 1:1 and 1:2 ratio, followed Fickian and non-Fickian diffusion mechanism respectively. Thus, the drug release rate was extended for over a period of more than 12 h stability studies. There is no significant difference in the mean % drug released from formulation CH-X2 after storing for 3 months at 40°C/75% RH. The FT-IR spectra revealed that there was no interaction between polymers and drug, Statistical analysis showed a significant differences (P < 0.05) for the amount of ISMN released from the formulations (MXG) and formulations (CH-X2).
Conclusion:
Formulation CH-XG2 (1:2) showed better sustained release of highly water-soluble ISMN with the desired release rate. Thus, the formulated PECs-based matrix tablets seems to be a potential candidate for sustained drug delivery of highly soluble drug ISMN in the symptomatic therapy of angina pectoris.
Keywords: Chitosan, isosorbide mononitrate, polyelectrolyte complex, sustained release, Xanthan gum
INTRODUCTION
Polyelectrolyte complexes (PECs) are the association complexes formed between oppositely charged particles (e.g., polymer-polymer, polymer-drug, and polymer-drug-polymer). These are formed due to electrostatic interaction between oppositely charged polyions.[1] The term polyelectrolyte denotes a class of macromolecular compounds, which when dissolved in a suitable polar solvent (generally water), spontaneously acquires or can be made to acquire a large number of elementary charges distributed along the macromolecular chain.[2] In its uncharged state, a polyelectrolyte behaves like any other macromolecules, but the dissociation of even a small fraction of its ionic (side) groups leads to dramatic changes of its properties.[3] Polyelectrolyte or polysalt complexes are formed, when macromolecules of opposite charge are allowed to interact. The interaction usually involves a polymeric acid or its salt with a polymeric base or its salt. Depending on a variety of factors, it may cause the system to separate into a dilute phase and a concentrated complex coacervate phase, or it may result in a more-or-less compact precipitate or gel. The complexes can also remain in solution. Electrostatic interactions constitute the main attractive forces, but hydrogen bonding, ion dipole forces, and hydrophobic interactions frequently play a significant role in determining the ultimate structures.[4,5]
Polymer complexation leads to a loss of translational and conformational entropy of the polymer chain, which has to be counterbalanced if complexation is to occur. The loss in entropy (per bond formed) is largest for the first bond formed between the two polymers, but is much smaller for subsequent (neighboring) bonds. The enthalpic change (per bond) due to the interaction of the monomeric units however, is nearly constant, and it is easily understood that at a certain critical chain (or sequence) length, complexation becomes energetically favorable.[6,7] The short range of these interactions (Vander Waals forces) makes a good sterical fit between the polymers essential if complexation is to occur, leading to very high demands on the polymers chemical structure and tacticity. The complexes formed show a very high degree of ordering and crystal like properties, and have quite compact structures.[8,9] These are formed due to electrostatic interaction between oppositely charged polyions which avoids the use of covalent cross linkers. In general, these polymeric networks are well tolerated, biocompatible and are more sensitive to changes in environmental conditions.[10,11] Chitosan is a deacetylated derivative of chitin, which is a naturally occurring polysaccharide comprising copolymers of glucosamine and N-acetylglucosamine. Chitosan is biocompatible and biodegradable cationic polymer which is most widely used due to its reduced toxicity and better patient compliance. The cationic amino groups on the C2 position of the repeating glucopyranose units of chitosan can interact electrostatically with the anionic groups (usually carboxylic acid groups) of other polyions to form polyelectrolyte complexes. Many different polyions from natural origin (e.g., pectin, alginate, carrageenan, xanthan gum, carboxymethyl cellulose, chondroitin sulfate, dextran sulfate, hyaluronic acid) or synthetic origin (e.g., poly (acrylic acid)), polyphosphoric acid, poly(L-lactide) have been used to form polyelectrolyte complexes with chitosan in order to provide the required physiochemical properties for the design of specific drug delivery systems.[12]
Isosorbidemononitrate (ISMN) is an anti-anginal organic nitrate vasodilator used in the treatment of various cardiovascular disorders and prophylaxis of Angina Pectoris. It is highly soluble in water. It has been classified as a class I substance according to the bio-pharmaceutics classification system (BCS), highly soluble in water (1g/20mL). The drug is readily and completely absorbed throughout the intestinal tract. After a single oral dose, peak plasma concentrations occur after 1-2 hours.[11,12] The drug is eliminated within 3-4 hours, which depending on therapeutic intention, makes it necessary to administer simple formulation of ISMNupto four times daily.[13] Developing oral sustained release tablets for water-soluble drugs with constant release rate has always been a challenge to the pharmaceutical technologist. Most of these water-soluble drugs if not formulated properly, may readily release the drug at a faster rate and produce a toxic concentration of drug on oral administration. Hence, it is a challenging task to formulate a suitable tablet dosage form for prolonged delivery of highly water-soluble drugs. In the present study an attempt has been carried out to evaluate the affect of PECs in the matrix core component, which is formed with natural polymers i.e., sodium alginate (SA), guar gum (GG), and xanthan gum (XG) as a cross-linking agent with cationic chitosan to from PECs. The aim of designing the formulation is to sustained the drug delivery of Isosorbide 5-mononitrate by using polyelectrolyte complexes in the formulation to reduce the dosing frequency and to improve patient compliance.
MATERIALS AND METHODS
Isosorbide mononitrate was obtained as gift sample from JP fine chemicals Ltd., India, Bangalore. Chitosan from Kerala state co-operative federation for Fisheries development Ltd, Kerala, India, Guar gum from H.B Gum, Kalol, Gujarat, India, Sodium Alginate SD Fine Chemicals, Mumbai, Xanthan gum from Raj Enterprises Mumbai, India was used. All other materials were of analytical or reagents grade.
Calculation of required first order release rate constant
kr1 = Ke (exp (-ke × Ti) was the equation used to calculate first-order rate constant, (kr1) of ISMN from tablets formulation, where ke is the elimination rate constant (0.142h-1 ) and Ti, crossing time at which the blood level profiles produced by administration, the value of Ti = h-Tp (where ‘h’ is the duration of therapy, i.e., 12h in the present study and Tp the time taken for maximum plasma concentration at second hour. This is based on the mean pharmacokinetic parameters of drug in humans.[14]
Preparation of PECs
Chitosan (CH) solution was prepared in 1% v/v acetic acid. Solutions of SA, XG and GG were separately prepared by hydrating them in distilled water. The dissolved chitosan was slowly added with stirring into SA, XG and GG aqueous solutions, to give CH-SA, CH-XG and CH-GG in 1:1 and 1:2 ratios, respectively, and is represented in the Table 1. The complexes were left at room temperature for 12h. The complexation will have interaction. The solid complexes were washed with distilled water and resulting mixture was poured into Petri plates and dried in hot air oven at 40°C for 24 h. The dried complex was grounded and made into fine powder by was passing through sieve #100. The interaction between the polymers and cationic chitosan is depicted below.
Table 1.
Composition of polyelectrolyte complexes of chitosan (CH) with SA, GG and XG

Polymer − COO- + CH − NH3 + Polymer − COO- + NH3 − CH
Preparation of ISMN matrix tablets by using natural polymer as matrix forming agent
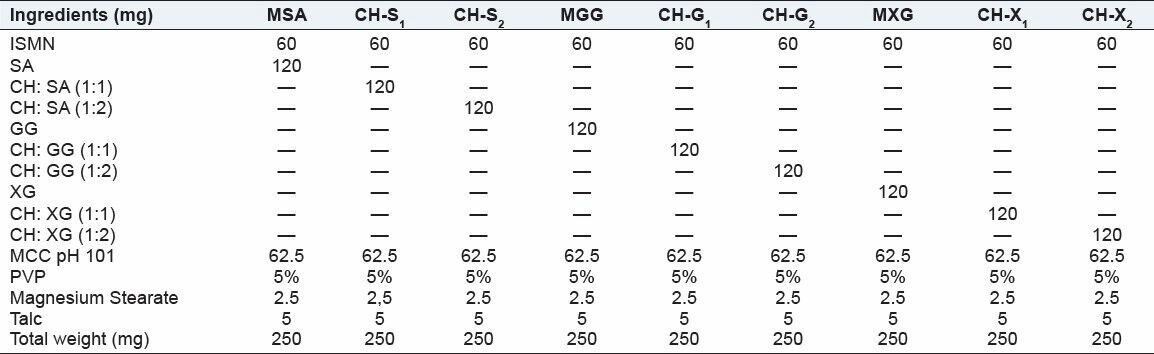
Matrix tablets were prepared by using natural polymers in the matrix core and different combinations of drug and natural polymer (SA, XG, GG) loaded in the matrix core, the composition is shown in Table 2, wet granulation technique is used to prepare the granules. The ratio of ISMN-SA, XG, and GG is 1:2 and were mixed properly in a mortar and pestle. MCC pH 101 was added and mixed well, it act as diluents, and the mixture is passed through sieve #44. Granules were prepared by utilizing PVP K-30-5% w/v. Then the wet mass was passed through sieve #16. The wet granules were air dried for 2 hours. The granules were then sized by sieve #22 and then mixed with talc and magnesium stearate in the ratio of 2:1. The formulation is shown in Table 2. Tablets were compressed using a multistation rotary punching machine using 9-mm diameter punch. The hardness of the tablet and was adjusted to 6-7kg/cm2.
Table 2.
Formulae of matrix and matrix and PECs-based matrix tablets of isosorbide mononitrate
Preparation of ISMN matrix core granules by using PECs as matrix forming agent
Matrix tablets were prepared by wet granulation method. Drug and polyelectrolyte complex in the ratio 1:2 were mixed in a mortar and pestle. MCC pH 101 was added and mixed well, it act as diluents, and the mixture is passed through sieve #44. Granules were prepared by utilizing PVP K-30-5% w/v. Then the wet mass was passed through sieve #16. The wet granules were air dried for 2 h. The granules were then sized by sieve #22 and then mixed with talc and magnesium stearate in the ratio of 2:1. The formulation is shown in Table 2. Tablets were compressed using a multistation rotary punching machine. The hardness of the tablet and was adjusted to 6-7 kg/cm2.
Physical tests for the prepared PEC-based ISMN matrix tablets
Ten tablets from each formulation were taken for measurement of diameter and crown thickness with VernierCalipers and an average of 10 determinations was carried out. Hardness of the PECs-based matrix tablets was evaluated by using hardness tester (Pfizer) and mass determination was performed for 20 tablets from each batch and average values were calculated. Friability of the PECs-based matrix tablets was determined by first weighing 10 tablets after de dusting and placing in a friability tester (Roche friabilator, Pharma labs, Ahmedabad, India), which was rotated for 4 min at 25 rpm. After dedusting, the total remaining weight of the tablets was recorded and the percent friability was calculated. The drug content of the prepared tablets of each batch was determined in triplicate.
In vitro drug release studies
In vitro dissolution studies for the prepared matrix tablet and triple-layered matrix tablets were conducted for a period of 12h using a six station (1) USP XXII type II apparatus (Lab India Disso 2000 system, India.) at 37 ± 0.5°C and 50 rpm speed. The dissolution studies were carried out in triplicate for 2 h in pH 1.2 medium (900 ml) and then phosphate buffer of pH 7.2. The drug content was determined by using HPLC method. The experiments were repeated thrice and the results were taken as average of three test readings with standard deviations.
High performance liquid chromatographic determination of ISMN from dissolution sample
The dissolution media consisted of 0.1N HCI, (pH1.2) for 2 h and then phosphate buffer pH7.2. At a specific time interval samples of ISMN were withdrawn, filtered and analyzed using a HPLC (Shimadzu HPLC Class VP series) system. Chromatographic separation was performed on a RP C-18 column (250 mm 34.6 mm i.d., particle size 5 μm;) was used at 25°C. The optimized mobile phase composition was water-methanol (80:20, v/v) at flow rate of 1.5ml/min. Loop size: 20 μL Stock solutions of ISMN were prepared in phosphate buffer in pH 7.2 as 1mg/mL. Calibration curve was prepared for each of the analytes after appropriate dilution of stock solutions to obtain final concentrations of 0.1, 0.5, 1, 2, 5, 10, and 20 μg/mL, detection was performed at 220 nm using a UV detector. The calibration curve was prepared taking the peak area of the analytes (ISMN) versus the concentration (μg/mL) using a weighted (1/concentration2 ) linear least squares regression as the mathematical model. The regression equation of the calibration curve was then used to calculate the drug content and in vitro drug release. The lowest limit of quantitation for ISMN was determined from the peak signal to noise level (S/N) as 10. Results is given as mean ± standard deviation.
Characterization of release data
The description of dissolution profiles has been attempted using different release models. The data were evaluated according to the following equations.
Zero order: Mt = Mo + Ko t
First order: ln Mt = ln Mo + K1 t
Higuchi model: Mt = KH√t
Korsmeyer — Peppas model: Mt /Mo = Kk tn
Where Mt is the amount of drug dissolved in time t, Mo the initial amount of drug, K1 is the first-order release constant, K0 the zero-order release constant, KH the Higuchi rate constant, Kk the release constant and n is the diffusional release exponent indicative of the operating release mechanism. The correlation coefficient (r2 ) was used as an indicator of the best fitting, for each of the models considered.
The dissolution parameters used for comparing the different formulations was MDT and DE8 %. The following equation was used to calculate the mean dissolution time (MDT) from the mean dissolution data.

Where i is the dissolution sample number, n is the number of dissolution sample time, t mid is the time at the midpoint between i and i-1 and ΔM is the additional amount of drug dissolved between i and i-1.[15] MDT, which is calculated from the amount of drug released to the total cumulative drug. MDT is a measure of the rate of the dissolution process: The higher the MDT, the slower the release rate.
Dissolution efficiency (DE) after 8hr of release test was used to compare the results of dissolution tests of different formulations.[16]

FT-IR spectroscopy studies
FTIR studies were performed on drug and the optimized formulation by using the following method, an approximately minimum quantity (less than 4 mg) of sample was thoroughly blended with adequate quantity of IR grade KBr (less than 100 mg) in mortar. The mix was then made into KBr pellets by hydraulic compression lever. The samples were analysed in a double beam IR Spectrometer using KBr film as negative control (blank). The samples were analyzed between wave numbers 400 and 4000 cm-1 . The samples of pure drug and formulated PECs-based matrix tablets CH-S2, CH-G2, CH-X2 were scanned individually.
Stability studies
Stability studies were conducted on ISMN release from the PEC-based matrix formulation of xanthan gum (CH-X2) to assess their stability with respect to their physical appearance, drug content and drug release characteristics after storing at 40°C/75% RH for 6 months was studied .[17]
Statistical analysis
In vitro release data of ISMN release from the formulations (MXG) and PEC-based formulations (CH-X2) were subjected to the 1-way analysis of variance (ANOVA) at different time intervals of drug released up to 12h, by Newman-Keulus multiple comparison test Graph pad prism version 5. (Graph pad prism Software, Inc).
RESULTS AND DISCUSSION
The objective of the present work is development of sustained release formulations of ISMN, for management of angina pectoris and hypertension, the usual dose is 40-60 mg in divided doses. In the present study, 60 mg of ISMN was incorporated in the formulations. In order to retard the highly soluble drug ISMN (1 g/20 mL) from the matrix core, to achieve the desired release rate. The first-order rate constant was found to be 0.034h-1 . The formulations developed till the required first-order rate constant of ISMN was obtained. The sustained release matrix tablets of Isosorbide 5-Mononitrate (ISMN) were prepared by using Polyelectrolyte Complexes (PECs) technique. Natural polymers Sodium alginate (SA), Guar gum (GG) and xanthan gum (XG) were used for cross linking with cationic chitosan to from PECs in the matrix core. Table 1 shows the product yield of PEC formed when the CH-SA, CH-GG and CH-XG ratio increases.
Physicochemical characterization of formulated PECs-based ISMN matrix tablets
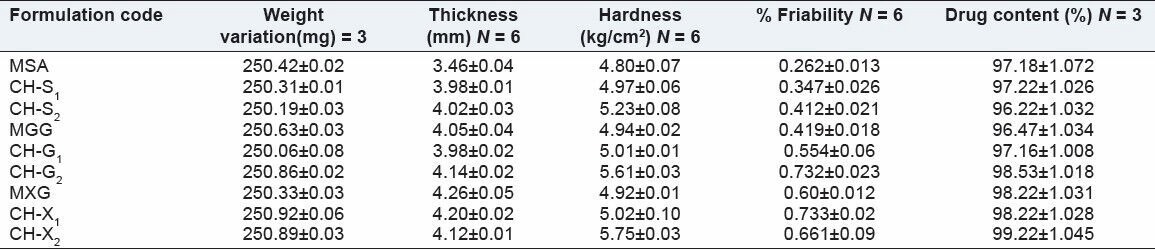
The prepared tablets were evaluated for physical parameters such as hardness, thickness, friability, weight variation, and drug content. The results are shown in Table 3. The mean values for hardness of the matrix tablets and PECs-based matrix tablets were in the range of 4.94 ± 0.02 to 5.75 ± 0.03 kg/cm2 . All the tablets passed the friability test as the loss the tablet material was less than 1%, indicating that the tablets prepared were of sufficient strength. The thickness of the prepared tablets is ranged from 3.46 ± 0.02 to 4.26 ± 0.05 mm. The matrix tablets and PECs-based matrix tablets also satisfied the drug content as they contained 96.22% ± 1.34% to 99.62% ± 2.65% of ISMN indicating the uniform mixing of the drug, polymer and PECs complexes along with the other formulation excipients. Hence, it attributes that the prepared tablets were found be practically within the limits.
Table 3.
Physical parameters of isosorbide mononitrate matrix tablets and PECs-based matrix tablets
In vitro drug release studies of sa matrix and PECs-based matrix tablets
The in-vitro release studies were carried out for formulations in both pH1.2 and pH 7.2 media and the release profile is shown in the Figure 1. The amount of drug release at the first hour for matrix tablets of SA and PECs (CH-S1 CH-S2) based matrix tablets was 21.71 ± 0.17%, 13.09 ± 0.15% and 12.23 ± 0.23% respectively, with more than 7.87 ± 0.11% getting released by end of 1hrs and 10.85 ± 0.11% of more release at the end of 12 hrs for matrix tablets, whereas around 82.42 ± 0.06% was released in case of PECs-based (CH-S2) matrix tablets till the end of 12 h. The correlation coefficient (r2 ) of the SA matrix tablet for first-order release kinetics was found to be higher (0.986), when compared to that of zero-order kinetics (0.948) indicating that the drug release from the matrix tablets followed first-order kinetics [Table 4]. PECs-based matrix tablets (CH-S2) the ‘r2 ’ values for zero-order kinetics were found to be higher when compared than that of first-order kinetics. It indicates that the drug released from PECs-based matrix tablets (CH-S2) follows a zero-order release. The first-order release rate constants obtained from MSA, CH-S1 and CH-S2 formulations were 0.077, 0.063 and 0.052 h, respectively. It signifies that with increasing the amount of SA in the chitosan; there is decrease in the first-order rate constant. Hence the release rate of formulation MSA is higher than PECs-based formulations, indicating that the formulation (MSA) shows higher release rate. Formulation CH-S2 follows a constant release of 0.133 mgh-1 in case of zero-order release. It is evident from this data that there is decrease in the release rate from formulation MSA to CH-S2.
Figure 1.
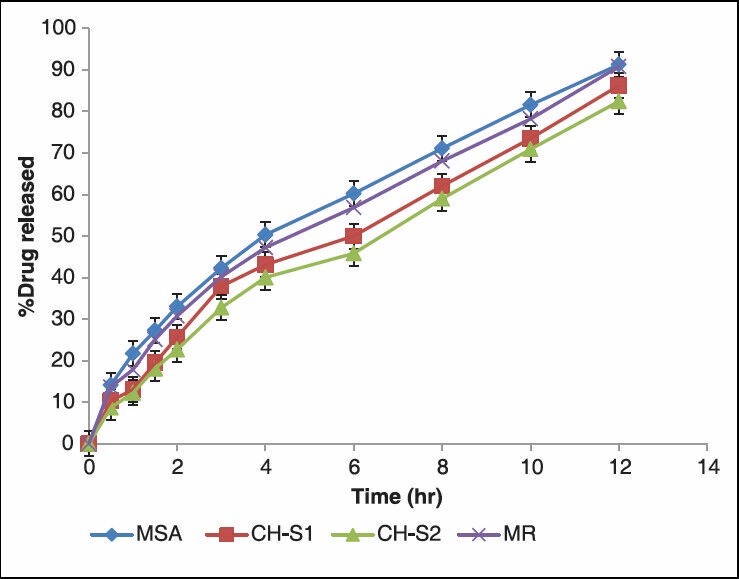
In-vitro characterization of release profiles of ISMN from (MR), plain matrix and (CH-SA) PEC-based matrix tablets
Table 4.
In-vitro dissolution kinetics, MDT and DE8% of Isosorbide Mononitrate (ISMN) released from matrix and PECs-based matrix tablets (n = 3)
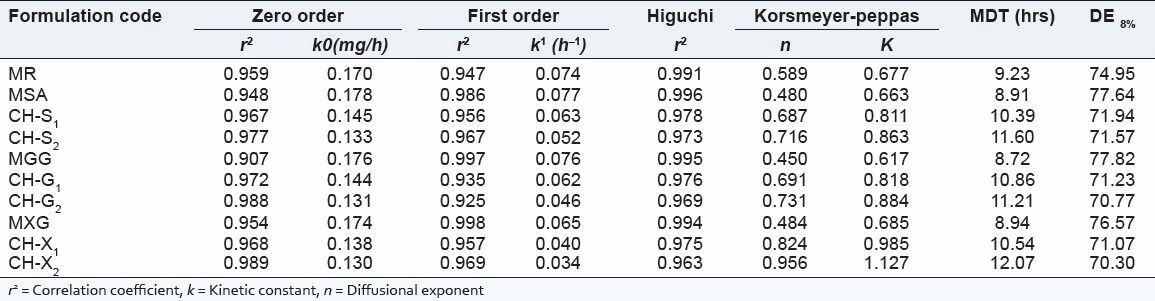
From this data [Table 4] shows that, the ‘r2 ’ values of Higuchi plot, for SA and PECs-based (CH-S1 CH-S2) matrix tablets were 0.996, 0.978 & 0.973, it can be said that the prepared formulation shows diffusion mechanism. This can be attributed to the higher viscosity of the formed PECs to retard the drug release. The diffusional coefficient values obtained according to the model developed by Korsemeyer et al., showed that matrix tablet followed Fickian diffusion and the PECs-based (CH-S1 CH-S2) matrix tablets followed non-Fickian diffusion, as the diffusion coefficient ‘n’ value was found to be less than 0.5 and greater than 0.5. It is evident from this data that, as SA is added in cationic chitosan there was retardation of the drug release from the matrix core; improved zero order of the ISMN release from the matrix core is achieved. From the above study, we may infer that SA along with cationic chitosan provided better release to achieve zero-order release than SA alone. In case of the PEC (CH-S2), the drug release was sustained with delay as the time prolonged.
In vitro drug release studies of GG matrix and PECs-based matrix tablets
The in vitro release studies were carried out for formulations in both pH1.2 and pH 7.2 media and the release profile is shown in the Figure 2. The amount of drug release at the first hour for matrix tablets of GG and PECs (CH-G1, CH-G2) based matrix tablets was 20.52 ± 0.02%, 12.97 ± 0.39% and 11.55 ± 0.24% respectively, with more than 8.87 ± 0.12% getting released by end of 1hrs and 9.85 ± 0.13% for more release at the end of 12h for matrix tablets, whereas around 82.42 ± 0.06% was released in case of PECs-based (CH-G2) matrix tablets till the end of 12h. The correlation coefficient (r2 ) of the GG matrix tablet for first-order release kinetics was found to be higher (0.997), when compared to that of zero-order kinetics (0.907) indicating that the drug release from the (GG) matrix tablets followed first-order kinetics. Data shown in Table 4. However, in case of PECs-based matrix tablets (CH-G2) the ‘r2 ’ values for zero-order kinetics were found to be higher when compared than that of first-order kinetics. It indicates that the drug released from PECs-based matrix tablets (CH-G2) follows a zero-order release. The first-order release rate constants obtained from MGG, CH-G1 and CH-G2 formulations were 0.076, 0.062 and 0.046h, respectively. It signifies that with increasing the amount of GG in the chitosan, there is decrease in the first-order rate constant. Hence the release rate of formulation MGG is higher than PECs-based formulations, indicating that the formulation (MGG) shows higher release rate. Formulation CH-G2 follows a constant release of 0.131mgh-1 in case of zero-order release. It is evident from this data that there is decrease in the release rate from formulation MGG to CH-G2.
Figure 2.
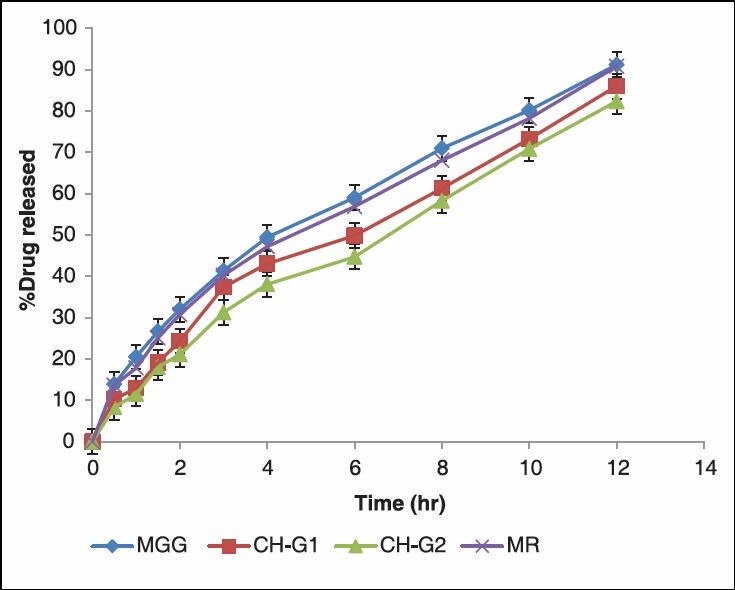
In-vitro Characterization of Release Profiles of ISMN from MR, Plain Matrix and (CH-GG) PEC matrix Tablets
From this data Table 4 show that, the ‘r2 ’ values of Hugchi plot, for GG and PECs-based (CH-G1 CH-G2) matrix tablets were 0.995, 0.976 and 0.969. From this data, it can be said that the prepared formulation shows diffusion mechanism. The diffusional coefficient values obtained according to the model developed by Korsemeyer et al., showed that matrix tablet (MGG) followed Fickian diffusion and the PECs-based (CH-G1 CH-G2) matrix tablets followed non-Fickian diffusion. It is evident from this data that, as GG is added in cationic chitosan there was retardation of the drug release from the matrix core; hence improved zero order of the ISMN release from the matrix core is achieved. In case of the PEC (CH-G2), the drug release was sustained with delay as the time prolonged.
In vitro drug release studies of XG matrix and PECs-based matrix tablets
The in-vitro release studies were carried out for formulations in both pH1.2 and pH 7.2 media and the release profile is shown in the Figure 3. The amount of drug release at the first hour for matrix tablets of XG and PECs (CH-X1 CH-X2) based matrix tablets was 19.66 ± 0.33%, 12.94 ± 0.50% and 11.23 ± 0.34%, respectively, with more than 9.87% getting released by end of 1h and 8.85% for more release at the end of 12hrs for matrix tablets, around 81.56 ± 0.49% was released in case of PECs-based (CH-X2) matrix tablets till the end of 12 h. The correlation coefficient (r2 ) of the XG matrix tablet for first-order release kinetics was found to be higher (0.998), when compared to that of zero-order kinetics (0.954) indicating that the drug release from the (GG) matrix tablets followed first-order kinetics. [Table 4], in case of PECs-based matrix tablets (CH-X2) the ‘r2 ’ values for zero-order kinetics were found to be higher when compared than that of first-order kinetics. It indicates that the drug released from PECs-based matrix tablets (CH-X2) follows a zero-order release. The first-order release rate constants obtained from MXG, CH-X1, and CH-X2 formulations were 0.065h-1, 0.040 h-1 and 0.034h-1, respectively. It signifies that with increasing the amount of XG in the chitosan, there is decrease in the first-order rate constant. Hence the release rate of formulation MXG is higher than PECs-based formulations, indicating that the formulation (MXG) shows higher release rate. Formulation CH-X2 follows a constant release of 0.130mgh-1 in case of zero-order release. It is evident from this data that there is decrease in the release rate from formulation MGG to CH-G2 . The PECs-based formulation CH-G2 showed the calculated first release rate constant of 0.034h-1 Hence the CH-G2 is an optimized formulation for the release of ISMN.
Figure 3.
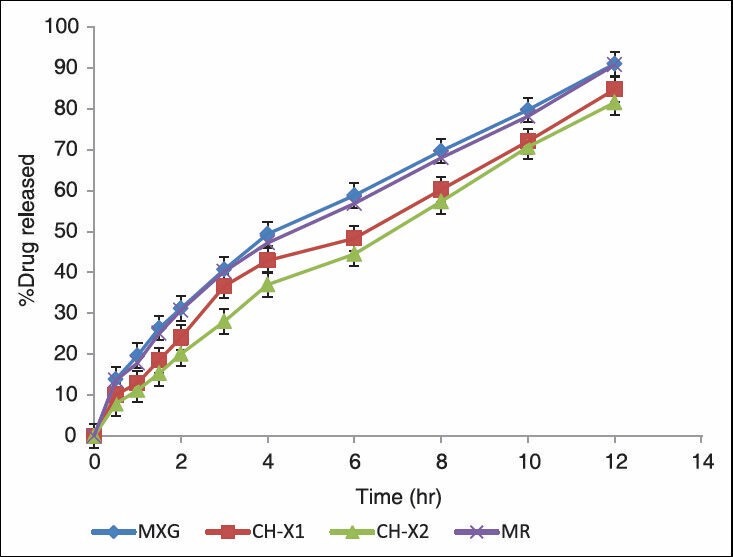
In-vitro dissolution release profiles of ISMN from MR, plain matrix and (CH-XG) PEC matrix tablets
From this data [Table 4] show that, the ‘r2 ’ values of Higuchi plot, for XG and PECs-based (CH-X1 CH-X2) matrix tablets were 0.994, 0.975 & 0.963. From this data, it can be said that the prepared formulation shows diffusion mechanism. The diffusional coefficient values obtained according to the model developed by Korsemeyer et al., showed that matrix tablet (MXG) followed Fickian diffusion and the PECs-based (CH-X1 CH-X2) matrix tablets followed non-Fickian diffusion, as the diffusion coefficient ‘n’ value was found to be less than 0.5 and greater than 0.5 respectively [Table 4]. It is evident from this data that, as XG is added in cationic chitosan there was retardation of the drug release from the matrix core; improved zero order of the ISMN release from the matrix core is achieved. From the above study, we may infer that XG along with cationic chitosan provided better release to achieve zero-order profile than XG alone. In case of the PEC (CH-X2), the drug release was sustained with delay as the time prolonged. Formulation (CH-X2) were found swollen and retain their physical integrity till the end of 12h dissolution study. This was because it involves formation of new bonds or correction of the distortions of the polymer chains with the resulting difference in electrostatic interactions, between the blend of polymer compared to the single polymer accounting for the above release pattern. The formulations were further optimized by varying the ratio of polymers (CH-XG, CH-GG and CH-SA) in the PECs as crosslinking agent. The results have shown that CH-S2, CH-G2 and CH-X2 showed was the best formulation, showed slowest release rate of 82.42 ± 0.06%, 82.35 ± 0.24% and 81.56 ± 0.49% respectively which showed sustained release than that of the marketed formulation MR (the drug release was 90.78% at 12 hrs), indicating that the drug release was more sustained with the formulation CH-X2, when compared with marketed tablets and the prepared tablets. This release pattern was anticipated and can be very well explained based on previous such similar studies.
The MDT and DE8% of the prepared formulations were calculated shown in Table 4 and it was found that as the MDT was increased, the DE8% was found to decrease. The MDT and DE8% valued for MXG and CH-X2 were found in the range of 8.92h, 12.07h and 76.57%, 70.30% respectively. Thus the formulation CH-X2 showed drug release that was extended for over a period of more than 12 h.
FT-IR studies
The interaction study between the pure drug (ISMN) and formulations CH-S2 CH-G2 and CH-X2 was evaluated using FT-IR spectrophotometer. The bands present in ISMN spectrum were at 3211.70, 2913.88, 1651.05, 1282.25cm-1],[ due to the formation of O-H, C-H, C = C and N-O linkage respectively, The wave numbers was also detected and identified in the spectrum of the formulations is shown in Figure 4. Hence the study indicates that there was no drug- polymer interaction.
Figure 4.
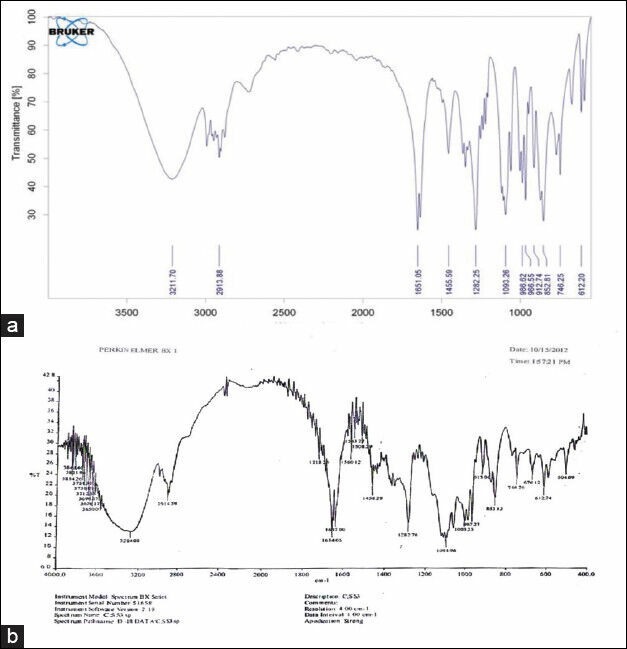
FT-IR spectra of pure Isosorbite mononitrate (a) and formulation X2 with PEC (CH: X2) 1: 2 (b)
Stability studies
At the end of testing period, the matrix tablets were observed for changes in physical appearance, analyzed for drug content and subjected to in vitro drug release studies. The resulted is shown in Figure 5. The drug content was found to be 96.75% ± 0.12. The % of ISMN released from the formulation CH-X2 before storage was 81.56 ± 0.49%, where as the released after storage was 82.45 ± 0.44%. There is no significant difference in the mean % drug released from formulation CH-X2 after storing for 6 months at 40°C/75% RH.
Figure 5.
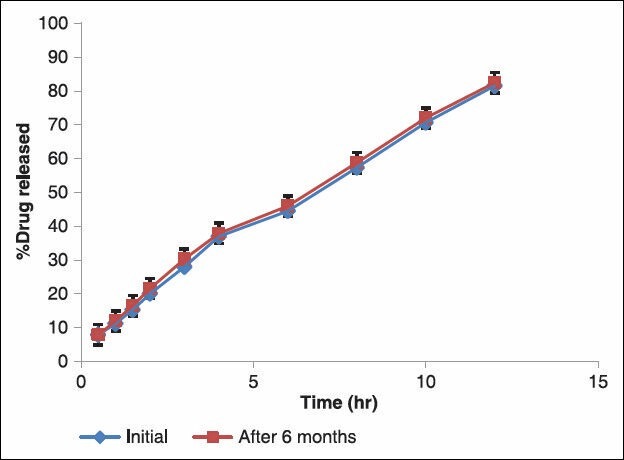
Comparison of In vitro drug release of formulation CH: X2 after 6 months storage
Statistical analysis
Analysis of variance (single factor ANOVA) showed a significant differences (P < 0.05) for the amount of ISMN released from the formulations (MXG), and formulations (CH-X2).
CONCLUSIONS
The present study was concerned with the formulation of oral sustained release matrix tablets of IsosorbideMononitrate, using different natural polymers and (cationic) chitosan (PECs) as matrix forming agent to obtain prolonged drug release. Increase in the ratio of Polymer (SA, GG and XG) to cationic Chitosan shows to decrease in the drug release from the Matrix tablets. The desired release rate constant of ISMN is achieved from Chitosan-Xanthan gum (1:2) PECs-based formulation. Thus, the formulated PECs-based Matrix tablets seems to be a potential candidate for sustained drug delivery of highly soluble drug ISMN in the symptomatic therapy of angina pectoris.
ACKNOWLEDGMENTS
The authors are thankful to JP fine chemicals Ltd., India, Bangalore., India, Chitosan from Kerala state Co-operative federation for Fisheries development Ltd, Kerala, India, H.B Gum, Kalol, Gujarat, India, and Raj Enterprises Mumbai, India, for generously giving the samples of Isosorbide Mononitrate, Chitosan, Guar gum and Xanthan gum, respectively. We are also thankful to the Principal and Management of SR College of Pharmacy, Warangal, for his constant encouragement.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Roy K, Mao HQ, Huang SK, Leong KW. Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat Med. 1999;5:387–91. doi: 10.1038/7385. [DOI] [PubMed] [Google Scholar]
- 2.Mark HF, Bikales NM, Overberger CG, Menges G. Encyclopedia of polymer science. 2nd ed. Vol. 2. New York: John Wiley and Sons, A Willey Interscience Publishers; 1987. p. 739. [Google Scholar]
- 3.Encyclopedia of polymer science and technology. 2nd ed. Vol. 2. Chichester: Wiley; 1988. pp. 739–829. [Google Scholar]
- 4.Philipp B, Doutzenberg H, Linow KJ, Koetz J, Dowydoff W. Polyelectrolyte-recent development and open problems. Prog Polym Sci. 1989;14:91. [Google Scholar]
- 5.Kabanov VA, Zezin AB, Ragacheva VB, Guskina N, Goethals EJ, Valde MV. Properties of polyelectrolyte complexes containing poly (N-tertbutylaziridine) Makromol Chem. 1986;187:1151. [Google Scholar]
- 6.Brinkhuis RH, Schouten AJ. Thin-film behaviour of poly (methyl methacrylates): Characteristics of the poly (methyl methacrylate) monolayer stereocomplexation process. Macromol. 1992;25:2732–8. [Google Scholar]
- 7.Kabanov VA, Papisov IM. Formation of complexes between complimentary synthetic polymers and oligomers in dilute solution review. Polym Sci. 1979;21:261–307. [Google Scholar]
- 8.Kikuchi Y, Kubota N. Structure properties and Na + transport membrane of polyelectrolyte complexes consisting of glycol chitosan and poly (vinyl sulfate) Bull Chem Soc J. 1987;60:375. [Google Scholar]
- 9.Smolen VF, Hahman DE. A water membrane hypothesis behavior of hydrated polycation-polyanion salt complexed membranes as appeared lipoidal barriers to solute transport. J Colloid Interface Sci. 1973;42:70–8. [Google Scholar]
- 10.Krone V, Magerstadt M, Walch A, Groner A, Hoffmann D. Pharmacological composition containing polyelectrolyte complexes in microparticulate form and at least on active agent. United States patent. 1997;5:700, 459. [Google Scholar]
- 11.Schaumann W. Pharmacokinetics of isosorbide dinitrate and isosorbide-5-mononitrate. Int J Clin Pharmacol Ther Toxicol. 1989;27:445–53. [PubMed] [Google Scholar]
- 12.Abshagen U, Sporl-Radun S. First data on the effects and pharmacokinetics of isosorbide-5-mononitrate in normal man. Eur J Clin Pharmacol. 1981;19:423–9. doi: 10.1007/BF00548586. [DOI] [PubMed] [Google Scholar]
- 13.Hutt V, Bonn R, Fritschi E, Jaeger H. Evaluation of the pharmacokinetics and absolute bioavailability of three isosorbide-5-mononitrate preparations in healthy volunteers. Arzneimittelforschung. 1995;45:142–5. [PubMed] [Google Scholar]
- 14.Lordi NG, Lachman L, Lieberman HA, Kanig JA. The Theory and Practice of Industrial Pharmacy. 2nd ed. Philadelphia: Lea and Febiger; 1987. pp. 430–56. [Google Scholar]
- 15.Gohel MC, Panchal MK. Novel use of similarity factors f2 and Sd for the development of diltiazem HCl modified-release tablets using a 3(2) factorial design. Drug Dev Ind Pharm. 2002;28:77–87. doi: 10.1081/ddc-120001488. [DOI] [PubMed] [Google Scholar]
- 16.Banakar UV. Pharmaceutical dissolution testing. 1st ed. New York: Marcel Dekker Inc; 1999. pp. 191–4. [Google Scholar]
- 17.Mathews BR. Regulatory aspects of stability testing in Europe. Drug Dev Ind Pharm. 1999;25:831–56. doi: 10.1081/ddc-100102245. [DOI] [PubMed] [Google Scholar]