

Abstract
Oxidative stress damages multiple cellular components including DNA, lipids, and proteins and has been linked to pathological alterations in nonalcoholic fatty liver disease (NAFLD). Reactive oxygen species (ROS) emission, resulting from nutrient overload and mitochondrial dysfunction, is thought to be a principal mediator in NAFLD progression, particularly toward the development of hepatic insulin resistance. In the context of insulin signalling, ROS has a dual role, as both a facilitator and inhibitor of the insulin signalling cascade. ROS mediate these effects through redox modifications of cysteine residues affecting phosphatase enzyme activity, stress-sensitive kinases, and metabolic sensors. This review highlights the intricate relationship between redox-sensitive proteins and insulin signalling in the context of fatty liver disease, and to a larger extent, the importance of reactive oxygen species as primary signalling molecules in metabolically active cells.
In humans, excessive storage of triglycerides in the liver, resulting from various etiologies including chronic over nutrition and physical inactivity, potentiates the development of nonalcoholic fatty liver disease (NAFLD). The first stage of the disease is simple steatosis, characterized by triglyceride deposits within lipid droplets in hepatocytes. Simple steatosis can progress to nonalcoholic steatohepatitis (NASH) following hepatocyte injury (hepatocyte ballooning, cell death), inflammation, and fibrosis. 20–40% of the population of industrialized countries have high levels of fat in their livers due to diet, sedentary lifestyle, and poor health [1–3]. Our understanding of the pathogenesis of nonalcoholic fatty liver disease (NAFLD) is limited by the difficulty of early detection and diagnosis [4]. Poor liver health affects many organ systems and impacts multiple arms of the metabolic syndrome, including cardiovascular health, insulin sensitivity, and circulating lipid levels [5, 6]. Half of obese humans (adults and children) have fatty livers and with the steadily increasing incidence of obesity, fatty liver disease will likely become a significant burden on our health care system [7]. NAFLD and NASH are also strongly correlated with insulin resistance, a major risk factor for type 2 diabetes [3]. Interestingly, the presence of liver fat is a better predictor of type 2 diabetes risk than obesity or BMI [8, 9]. In addition, NASH patients have a significantly higher risk of developing liver cancer (hepatocellular carcinoma) [10].
There is convincing evidence for a central role of mitochondrial dysfunction in the pathophysiology of NAFLD/NASH [11–18]. Mitochondrial dysfunction alters lipid metabolism, increases oxidative stress, and promotes proinflammatory cytokine production [11, 16]. Inflammatory cytokine signalling and stellate cell activation result in fibrosis and endoplasmic reticulum stress, stimulating apoptosis and necrosis [19, 20]. Altered oxidative phosphorylation and increased reactive oxygen species (ROS) levels are reported in patients with NASH [13–17] and are associated with structural abnormalities in liver mitochondria that appear swollen, rounded, and have decreased cristae density [16, 21]. Accordingly, decreases in the mitochondrial electron transport chain (ETC) complex activity and ATP synthase are reported in NAFLD patients [15]. Several studies in experimental animal models and humans indicate a strong association between the severity of NAFLD/NASH and degree of mitochondrial dysfunction and oxidative stress [12, 15, 22–24]. Increased serum oxidative markers (thioredoxin, oxidized LDL, thiobarbituric acid-reactive substances, malondialdehyde) are also observed in patients [22, 25]. Deficient antioxidant defenses are a major factor promoting oxidative stress and decreased coenzyme Q10, CuZn-superoxide dismutase, catalase activity, glutathione, and glutathione S-transferase correlate with the severity of liver disease [26, 27].
Oxidative stress occurs when there is an imbalance of pro-oxidant (ROS) formation and reduced antioxidant defenses [28]. ROS are free radicals derived from oxygen and include singlet oxygen (O•), superoxide anion (O−), and hydroxyl (HO• or HO−) radicals. ROS are highly reactive, resulting in a short life and limited diffusion radius (e.g., super- oxide anion has a half-life of 10−6 s) [29, 30]. ROS are formed through radical leakage from enzymes such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [31], cyclooxygenases [32], lipoxygenases [33], and the mitochondrial ETC [34]. The NOX family of NADPH oxidase enzymes are membrane-bound electron carriers, which use NADPH as electron donor and oxygen as acceptor. NADPH oxidases localize to cellular membrane compartments via targeting proteins, facilitating hydrogen peroxide (H2O2) production within spatially confined areas [31, 35]. There are various molecular mechanisms within a cell designed to counteract overproduction of ROS. Superoxide anion is dismutated by mitochondrial manganese superoxide dismutase (MnSOD) into H2O2, which is more chemically stable and has a potentially wider diffusion range. It is then detoxified into water by mitochondrial glutathione peroxidase-1 (GPx) [29, 36]. Mitochondrial catalase also has a detoxifying effect against overproduction of hydrogen peroxide [37].
Mitochondria are a principal source of cellular ROS due to electron leak along the ETC [37]. Oxidative reactions in the mitochondria (β-oxidation, Krebs' cycle) generate reduced cofactors (NADH and FADH2), which are then oxidized (NAD+ and FAD). Electrons formed by oxidation of these reduced cofactors are carried through the redox complexes of the respiratory chain (complexes I, III, and IV) to the final electron acceptor, molecular oxygen [37]. Mitochondrial ROS generation is governed by the redox state of the respiratory chain [38–40]. Electron transfer through the mitochondrial respiratory chain generates an electrochemical gradient and the energy of this gradient is used to generate adenosine triphosphate (ATP) by ATP synthase. Mitochondrial ROS generation can be modulated by bioenergetics substrates such as uncouplers, free fatty acids, or adenosine diphosphate (ADP) [34, 41, 42]. Mitochondria continually exposed to high levels of ROS can suffer deleterious consequences, such as oxidative damage to ETC complexes, mtDNA, or lipids [21], leading to mitochondrial dysfunction.
A master regulator of mitochondrial biogenesis and function is the transcriptional coactivator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) [43, 44]. PGC-1α coordinates the transcriptional activity of several nuclear transcription factors such as nuclear respiratory factors 1 and 2 (NRF-1 and -2) and transactivates genes involved in the respiratory chain, mitochondrial import machinery, and transcription factors of mtDNA (such as the mtDNA transcription factor A (TFAM)). Decreased mitochondrial biogenesis associated with impaired biological activity of PGC-1α or reductions in TFAM has been observed in fatty livers [45–47]. Moreover, chronic liver-specific deletion of PGC-1α, or its homologue PGC-1β, in mice leads to hepatic steatosis and insulin resistance [48, 49]. PGC-1α also regulates the induction of antioxidant defenses including SODs, catalase, and GPx [50] and increases expression of the metabolic sensor NAD+-dependent deacetylase sirtuin 3 (Sirt3), a key regulator of the mitochondrial antioxidant system [51]. Deficient Sirt3 activity in livers predisposes mice to NASH [52]. In addition, nuclear Sirtuin 1 (Sirt1) deacetylates PGC-1α to regulate mitochondrial biogenesis and function [53]. Sirt1 is decreased in a rat model of NAFLD [54] and Sirt1 hepatic deficiency leads to oxidative damage and insulin resistance [55]. Thus, there is a strong mechanistic link between deficiencies of proteins controlling mitochondrial biogenesis, function, and antioxidant capacity and the development of fatty liver disease, stimulating great interest in targeting these pathways for new therapeutics [56].
While there is a compelling correlation between mitochondrial dysfunction and fatty liver disease, the molecular pathways directly linking altered mitochondrial function, insulin resistance, and inflammation are still unclear. While increased ROS remains an attractive theory, there is still much debate and little direct evidence on whether accumulated ROS due to mitochondrial dysfunction is a significant causative factor in NAFLD/NASH, in particular hepatic insulin resistance. Houstis and colleagues [57] show that ROS production precedes insulin resistance in cultured 3T3-L1 adipocytes and ROS scavenging rescues insulin sensitivity, providing evidence toward ROS as a primary cause of insulin resistance. To address whether mitochondrial ROS contributes to insulin resistance in vivo, Anderson and colleagues administered a potent small molecule antioxidant peptide, SS31, that specifically targets mitochondria [58, 59]. They show that coadministration of SS31 to rats consuming a high-fat diet for six weeks reduces mitochondrial H2O2 emission by 50%, normalizes the mitochondrial H2O2-emitting potential, and prevents the development of insulin resistance [58, 60]. Moreover, palmitate-induced hepatic insulin resistance is dependant on the generation of mitochondrial ROS [61], demonstrating that the negative effect of ROS on insulin signalling is a phenomenon shared by a variety of metabolic tissues. Other approaches to target mitochondria with antioxidants, such as transgenic mice overexpressing mitochondrial peroxiredoxin 3 [62] or targeted mitochondrial overexpression of catalase [63], have established mitochondrial H2O2 as a primary signal linking metabolic imbalance to insulin resistance.
Conversely, some argue that insulin resistance, resulting from nutrient overload and mitochondrial exhaustion, is the initiating event leading to mitochondrial dysfunction and increased ROS in metabolic disease [64]. However, insulin receptor deficiency, at least in muscle, does not generate oxidative stress [65] and most studies using environmental and nutritional factors to induce insulin resistance, such as a high-fat diet, report increased ROS and oxidative damage prior to the development of insulin resistance [66–68]. Interestingly, mice with whole body deficiency of GPx1, a key enzyme in ROS detoxification, fail to become obese mice on a high-fat diet, do not develop hepatic steatosis, and have improved hepatic insulin signalling [69]. While this suggests increased ROS does not necessarily lead to hepatic insulin resistance and fatty liver disease, global GPx1 deletion and the general failure of these mice to gain fat mass makes it difficult to isolate direct effects of GPx1 loss on liver metabolism. Further investigation using a conditional knockout approach will help clarify the role of GPx1, if any, in fatty liver disease. It is still not clear whether increased ROS is a major player in decreased insulin signalling in fatty liver disease. Well-designed studies using tissue-specific and inducible genetic models of insulin resistance (e.g., insulin receptor or insulin receptor substrates proteins knockouts) and more advanced tools that can detect changes in ROS production in vivo are needed to decipher the precise temporal link between insulin resistance and ROS.
Evidence linking increased ROS to insulin resistance is strong, yet it is still not well understood how alterations in hormone signalling are explained by variations in ROS concentration. Irreversible oxidative damage to key signalling mediators has been proposed; however, the system is further complicated by the fact that ROS are also essential players in many hormone-regulated cellular processes. H2O2 is widely accepted as a crucial signalling molecule [70, 71]. Generation of H2O2 at concentrations that are not thought to promote oxidative stress or damage is involved in the regulation of redox signalling pathways [71]. Of all cysteine residues within the proteome, it is estimated that more than 10% are redox-sensitive [71]. The sulphur atom within cysteine can exist as a reduced thiol (SH) or in different oxidized states, such as thiolate anion (S−), sulfenate (SO−), disulfide (S-S) sulfinate (SO2 −), or sulfonate (SO3 −) [22]. Alterations in the redox state of these sulphur atoms induce changes in protein conformation affecting enzyme activity, protein interaction, trafficking, degradation, and transcription factor binding to DNA [71]. In fact, the intracellular redox circuit is a master regulator of phosphorylation/dephosphorylation events in the cell due to the presence of redox-sensitive cysteine residues within nearly all classes of protein phosphatase enzymes [72]. Thus, phosphatase activity can be reduced in response to an oxidative shift in the redox environment. For example, protein tyrosine phosphatases (PTPs) are deactivated by oxidation of a conserved redox-sensitive cysteine residue within their catalytic sites [73]. The phosphoprotein family of Ser/Thr phosphatases also appear susceptible to oxidative deactivation [74, 75]. Protein phosphatase 2A (PP2A) and protein phosphatase 1 (PP1), which account for the majority of all Ser/Thr phosphatase activity [76], are sensitive to oxidative inactivation due to the presence of a conserved CXXC motif within their catalytic domains [74, 77].
Under resting conditions, the redox environment is in a reduced state and phosphatase activity exceeds kinase activity by 10-fold [78]. This generates an intracellular “phosphatase tone” preventing inappropriate phosphorylation events in vivo [75]. H2O2 emission leads to deactivation of key phosphatases, potentiating kinase activity and altering signal propagation in the cell. Imbalances between pro-oxidants and antioxidants that provoke phosphatase inactivation have been linked to disease development. For example, oxidative changes in phosphatase enzymes, such as MAP kinase phosphatase (MKP-1), phosphatase and tensin homolog (PTEN), and mitochondrial matrix targeted PP2C (PP2Cm) are linked to aging [79], cancer [80], and apoptosis [81]. Kinase and phosphatase activity play an integral role in propagation and regulation of the insulin response in cells.
The insulin signalling cascade begins with insulin binding to the insulin receptor at the cell surface, activating intrinsic tyrosine kinase activity (Figure 1). Insulin receptor substrate (IRS) docking proteins become phosphorylated, leading to the recruitment and activation of phosphoinositide 3-kinase (PI3K). A major substrate for PI3K is the membrane lipid phosphatidylinositol-4,5-bisphosphate (PIP2), which is phosphorylated to produce phosphatidylinositol-3,4,5-trisphosphate (PIP3). Increases in PIP3 attract pleckstrin homology domain-containing signalling proteins, such as the Ser/Thr kinases phosphoinositide-dependent protein kinase-1 (PDK1) and protein kinase B (PKB/Akt). Phosphorylated PDK1 activates the PKB/Akt kinase, which potentiates glycogenesis through phosphorylation of glycogen synthase kinase 3, stimulates fatty acid synthesis via activation of ATP citrate lyase, and inhibits gluconeogenesis by inhibitory phosphorylation of Forkhead box protein O1. PKB/AKT also promotes mTORC1 signalling to regulate protein synthesis. To amplify the signal, insulin binding initiates deactivation of phosphatases such as PTP1B, PTEN, and SH2 domain-containing phosphatase (SHP2) [82–84]. Inactivation of these phosphatases due to an oxidative shift in the redox environment at the level of the plasma membrane potentiates downstream kinase signalling [82, 83]. The source of the insulin-induced oxidative shift at the plasma membrane appears to be extracellular H2O2 generated by NOX complexes [85, 86] (Figure 1). Consistently, exposure to H2O2 markedly reduces Ser/Thr phosphatase activity in skeletal muscle [75] and hepatocytes [84]. Furthermore, this mechanism may be independent of insulin binding to its receptor. Adiponectin, known to increase insulin sensitivity, acts through small GTPase Rac1 activation and 5-lipooxygenase stimulation. It creates an ROS burst that deactivates PTP1B and increases insulin signalling [87]. In addition, receptor tyrosine kinase activation leads to the transient phosphorylation and inactivation of membrane-associated peroxiredoxin 1, a major H2O2-detoxifying enzyme [88]. Thus, insulin-stimulated tyrosine kinase and NADPH oxidase activation coordinate generation of an oxidized environment localized along the plasma membrane to inhibit phosphatase activity and facilitate insulin signalling. Once insulin levels decline, NADPH oxidase activity decreases and the local redox environment at the plasma membrane returns to a reduced state due to activity of the antioxidant systems (catalase, peroxiredoxins), restoring phosphatase activity and resetting the insulin signalling cascade at a basal state. Thus, insulin signalling requires ROS to be effective and it seems paradoxical that evidence points to elevated ROS as a primary cause of decreased insulin signalling and insulin resistance [57, 58]. However, there is growing evidence that altered activities of redox-sensitive phosphatases enzymes are implicated in the detrimental effects of ROS on the insulin signal pathway.
Figure 1.
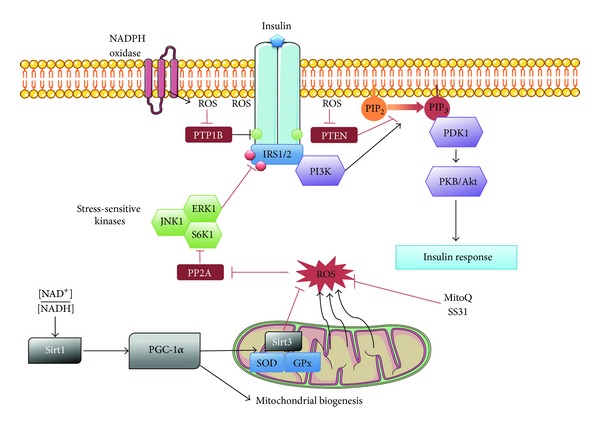
Molecular mechanisms linking ROS, antioxidants, and the insulin signalling pathway. To allow a response to insulin stimulation, ROS are relieving insulin receptor's inhibition by phosphatases such as PTP1B. When the cellular environment shifts to an oxidative one, because of increased mitochondrial respiration and ROS release for example, the stress-sensitive kinases are activated upon redox-sensitive phosphatases inhibition. Theses kinases are inhibiting signal amplification by inhibitory phosphorylation of IRS proteins. Mitochondrial ROS can be regulated by PGC-1α-dependent detoxification system (Sirt3, SOD, GPx) or by specific mitochondrial antioxidants (SS31, MitoQ).
Several studies show that activation of stress-sensitive Ser/Thr kinases causes inhibitory phosphorylation of insulin receptor and IRS1/2. An oxidized environment due to ROS accumulation inhibits phosphatase activity, and thus promotes stress-kinase activity, providing a potential mechanism for ROS-induced insulin resistance [89]. Among these Ser/Thr kinases, p70-S6 kinase 1 (S6K1), c-Jun N-terminal kinase-1 (JNK1), extracellular signal regulated kinase-1 (ERK1), and inhibitor of NF-κB kinase β (IKKβ) have all been implicated in insulin resistance [89]. Furthermore, mice deficient for JNK1 [90], ERK1 [91], S6K1 [92], and IKKβ [93] are protected from diet-induced insulin resistance. In addition, decreased activity of liver S6K1 [94], IKKβ, and JNK1 [95] improves hepatic insulin sensitivity. Reciprocally, S6K1 activation in fat-fed mouse liver or specific activation of hepatic ERK1 by C-reactive protein is linked to steatosis and insulin resistance [96, 97]. These stress kinases are targets of PP2A [98–100], a redox-sensitive protein phosphatase inactivated in an oxidized environment. In addition to enhancing activity of the stress kinases, oxidant-mediated inactivation of PP2A can cause persistent hyper-activation of NF-κB following TNFα stimulation [101], which may also have negative consequences on insulin sensitivity. These findings are consistent with evidence that insulin resistance develops as a consequence of aberrant Ser-phosphorylation events within key molecular components of the insulin signalling pathway [82, 102, 103]. However, although the redox sensitivity of Ser/Thr phosphatases is well established, it has not been thoroughly investigated within the context of insulin resistance associated with metabolic disease. In a similar manner to phosphatases, redox modification of kinases may also play a role in ROS-mediated insulin resistance. p38 MAPK is responsible for integrating ROS signals into a phosphorylative cascade [104] and has been shown to mediate palmitate-induced insulin resistance in hepatocytes [105]. The majority of studies did not investigate these pathways in the context of insulin resistance even though kinases involved in insulin signalling (PKB/AKT) are directly regulated by ROS [106]. Relatively few studies have investigated whether these mechanisms hold true for patients with fatty liver disease. Thus, it will be imperative to assess whether altered activity of phosphatase or kinase enzymes, as the result of changing ROS levels, plays a pathogenic a role in worsening insulin sensitivity in NAFLD/NASH.
Due to the intimate involvement of ROS in multiple aspects of insulin signalling, unraveling the timeline and dynamics of ROS production within the context of insulin resistance will help to determine whether or not ROS is a viable target for the treatment of metabolic disease. However, current methods to study ROS in cells and in vivo are limiting. The most common method is the redox-sensitive probe H2-DCFDA; however it is fairly insensitive at detecting subtle changes in ROS concentration and cannot differentiate ROS production from different cellular compartments. Moreover, endogenous esterases are required to generate the active probe, enzymes that are themselves affected by ROS-inducing treatment and pathological conditions. Probe-based methods allow only snapshots of the oxidative environment, whereas ROS are highly dynamic and transient molecules. Therefore, more sensitive and precise methods are needed to investigate the impact of ROS on cellular signalling if we are to truly understand their role, if any, on the relatively gradual development of insulin resistance in metabolic disease. New advances in electron paramagnetic resonance spectroscopy [107] and amperometry of hydrogen peroxide [108] will hopefully provide a more detailed understanding of the dynamic cellular environment, as both methods are extremely sensitive and can be applied in vivo. Single cell fluorescence also allows direct observation of ROS dynamics in different cell organelles [109] and can be applied to in vitro systems. Emerging technologies such as proteomic analysis of carbonyl-enriched samples [110–112] and the newly developed NOxICAT technology using a specific redox-sensitive cysteine trap are global approaches toward unraveling pathways impacted by ROS generation [113].
While the molecular mechanisms remain unclear, increased ROS is associated with multiple aspects of the metabolic syndrome including obesity, fatty liver disease, and diabetes, and is thus an attractive therapeutic target to improve insulin sensitivity. Moreover, elevated ROS and oxidative damage are also implicated in inflammation and endoplasmic reticulum stress, additional aggravating factors in the pathogenesis of type 2 diabetes. While inhibiting or reducing ROS seems as appealing strategies to treat metabolic disease, it is clear that ROS are necessary for effective and efficient propagation of intracellular signals, including the insulin signalling cascade. Thus, simple untargeted antioxidant therapy to treat insulin resistance may have multiple effects on signalling pathways independent of preventing oxidative damage. In animal models of metabolic disease, there is incomplete recovery of insulin sensitivity after an antioxidant treatment [57] and only partial prevention of NAFLD progression in rats after N-acetylcysteine administration [114]. In humans, intervention trials restricted to antioxidant supplementation have yielded little, if any, measurable effects on insulin sensitivity in type 2 diabetics [115, 116]. However, in small groups of NASH patients, insulin sensitivity seems to be improved following administration of vitamin E [117]. Mitochondria-targeted therapies such as SS31 or MitoQ may have better chances, as they do not interfere with ROS-potentiated insulin signalling, but prevent rising mitochondrial H2O2 concentrations. MitoQ administration improves hepatic steatosis, hyperglycemia, and liver damage in high fat-fed ApoE-deficient mice [118] and prevents ROS-induced liver damage in animal models and humans after HCV infection [119], suggesting that detoxification of mitochondrial ROS may have beneficial effects in NASH, which is characterized by both insulin resistance and uncontrolled inflammation. A phase 2 clinical trial was initiated to investigate the benefits of MitoQ for the treatment of fatty liver disease (ID: NCT01167088), but the trial was recently terminated due to poor enrollment.
Mitochondrial dysfunction is a hallmark of NAFLD progression. Subsequent H2O2 production and emission oxidize redox-sensitive cysteine residues that initially potentiate insulin action, yet potentially lead to insulin resistance via chronic inhibition of phosphatases and aberrant kinase activity. Proteins involved in enhancing mitochondrial function and limiting ROS accumulation, such as PGC-1α, Sirt1, and Sirt3, are of interest as specific therapeutic targets to improve insulin resistance observed in NAFLD progression. In addition, there is significant promise in using mitochondrial-targeted antioxidants to prevent or reverse liver damage in NAFLD patients, yet additional preclinical and clinical trials are needed to determine the effectiveness of these strategies. Unravelling the impact of ROS on global hepatocyte signalling and using recent advances in methodology for ROS quantification and localization will help decipher the importance of redox regulation in altered hepatic metabolism to possibly reveal novel, targeted therapeutics for the treatment of NAFLD and NASH.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Hui AY, Wong VW-S, Chan HL-Y, et al. Histological progression of non-alcoholic fatty liver disease in Chinese patients. Alimentary Pharmacology & Therapeutics. 2005;21(4):407–413. doi: 10.1111/j.1365-2036.2005.02334.x. [DOI] [PubMed] [Google Scholar]
- 2.Wong VW, Wong GL-H, Choi PC-L, et al. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010;59(7):969–974. doi: 10.1136/gut.2009.205088. [DOI] [PubMed] [Google Scholar]
- 3.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332(6037):1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunt EM. Pathology of nonalcoholic fatty liver disease. Nature Reviews Gastroenterology & Hepatology. 2010;7(4):195–203. doi: 10.1038/nrgastro.2010.21. [DOI] [PubMed] [Google Scholar]
- 5.Smith BW, Adams LA. Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment. Nature Reviews Endocrinology. 2011;7:456–465. doi: 10.1038/nrendo.2011.72. [DOI] [PubMed] [Google Scholar]
- 6.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. The New England Journal of Medicine. 2010;363(14):1341–1350. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 7.Schwimmer JB, Pardee PE, Lavine JE, Blumkin AK, Cook S. Cardiovascular risk factors and the metabolic syndrome in pediatric nonalcoholic fatty liver disease. Circulation. 2008;118(3):277–283. doi: 10.1161/CIRCULATIONAHA.107.739920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahn HS. The lipid accumulation product is better than BMI for identifying diabetes: a population-based comparison. Diabetes Care. 2006;29(1):151–153. doi: 10.2337/diacare.29.1.151. [DOI] [PubMed] [Google Scholar]
- 9.Musso G, Gambino R, Bo S, Cassader M. Should nonalcoholic fatty liver disease be included in the definition of metabolic syndrome? A cross-sectional comparison with adult treatment panel III criteria in nonobese nondiabetic subjects. Diabetes Care. 2008;31(3):562–568. doi: 10.2337/dc07-1526. [DOI] [PubMed] [Google Scholar]
- 10.Ascha MS, Hanouneh IA, Lopez R, Tamimi TA-R, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. 2010;51(6):1972–1978. doi: 10.1002/hep.23527. [DOI] [PubMed] [Google Scholar]
- 11.Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 12.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6(1):1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. Journal of the American Medical Association. 1999;282(17):1659–1664. doi: 10.1001/jama.282.17.1659. [DOI] [PubMed] [Google Scholar]
- 14.Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radical Biology and Medicine. 2000;28(10):1456–1462. doi: 10.1016/s0891-5849(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 15.Perez-Carreras M, del Hoyo P, Martín MA, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38(4):999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 16.Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. Journal of Hepatology. 2002;37(1):56–62. doi: 10.1016/s0168-8278(02)00073-9. [DOI] [PubMed] [Google Scholar]
- 17.Caldwell SH, Swerdlow RH, Khan EM, et al. Mitochondrial abnormalities in non-alcoholic steatohepatitis. Journal of Hepatology. 1999;31(3):430–434. doi: 10.1016/s0168-8278(99)80033-6. [DOI] [PubMed] [Google Scholar]
- 18.Vendemiale G, Grattagliano I, Caraceni P, et al. Mitochondrial oxidative injury energy metabolism alteration in rat fatty liver: effect of the nutritional status. Hepatology. 2001;33(4):808–815. doi: 10.1053/jhep.2001.23060. [DOI] [PubMed] [Google Scholar]
- 19.Marchesini G, Marzocchi R, Agostini F, Bugianesi E. Nonalcoholic fatty liver disease and the metabolic syndrome. Current Opinion in Lipidology. 2005;16(4):421–427. doi: 10.1097/01.mol.0000174153.53683.f2. [DOI] [PubMed] [Google Scholar]
- 20.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. American Journal of Physiology. 2006;291(2):E275–E281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 21.Ricci C, Pastukh V, Leonard J, et al. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. American Journal of Physiology. 2008;294(2):C413–C422. doi: 10.1152/ajpcell.00362.2007. [DOI] [PubMed] [Google Scholar]
- 22.Yesilova Z, Yaman H, Oktenli C, et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic fatty liver disease. American Journal of Gastroenterology. 2005;100(4):850–855. doi: 10.1111/j.1572-0241.2005.41500.x. [DOI] [PubMed] [Google Scholar]
- 23.Letteron P, Fromenty B, Terris B, Degott C, Pessayre D. Acute and chronic hepatic steatosis lead to in vivo lipid peroxidation in mice. Journal of Hepatology. 1996;24(2):200–208. doi: 10.1016/s0168-8278(96)80030-4. [DOI] [PubMed] [Google Scholar]
- 24.Yang SQ, Zhu H, Li Y, et al. Mitochondrial adaptations to obesity-related oxidant stress. Archives of Biochemistry and Biophysics. 2000;378(2):259–268. doi: 10.1006/abbi.2000.1829. [DOI] [PubMed] [Google Scholar]
- 25.Sumida Y, Nakashima T, Yoh T, et al. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. Journal of Hepatology. 2003;38(1):32–38. doi: 10.1016/s0168-8278(02)00331-8. [DOI] [PubMed] [Google Scholar]
- 26.Sreekumar R, Rosado B, Rasmussen D, Charlton M. Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis. Hepatology. 2003;38(1):244–251. doi: 10.1053/jhep.2003.50290. [DOI] [PubMed] [Google Scholar]
- 27.Leclercq IA. Antioxidant defence mechanisms: new players in the pathogenesis of non-alcoholic steatohepatitis? Clinical Science. 2004;106(3):235–237. doi: 10.1042/CS20030368. [DOI] [PubMed] [Google Scholar]
- 28.Robertson G, Leclercq I, Farrell GC. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. American Journal of Physiology. 2001;281(5):G1135–G1139. doi: 10.1152/ajpgi.2001.281.5.G1135. [DOI] [PubMed] [Google Scholar]
- 29.D'autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature Reviews Molecular Cell Biology. 2007;8(10):813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 30.Mittler R, Vanderauwera S, Suzuki N, et al. ROS signaling: the new wave? Trends in Plant Science. 2011;16(6):300–309. doi: 10.1016/j.tplants.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 31.Ushio-Fukai M. Compartmentalization of redox signaling through NaDPH oxidase-derived rOS. Antioxidants and Redox Signaling. 2009;11(6):1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryter SW, Tyrrell RM. Singlet molecular oxygen ((1)O2): a possible effector of eukaryotic gene expression. Free Radical Biology and Medicine. 1998;24(9):1520–1534. doi: 10.1016/s0891-5849(97)00461-9. [DOI] [PubMed] [Google Scholar]
- 33.Zuo L, Christofi FL, Wright VP, Bao S, Clanton TL. Lipoxygenase-dependent superoxide release in skeletal muscle. Journal of Applied Physiology. 2004;97(2):661–668. doi: 10.1152/japplphysiol.00096.2004. [DOI] [PubMed] [Google Scholar]
- 34.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Letters. 1997;416(1):15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 35.Chen K, Craige SE, Keaney JF. Downstream targets and intracellular compartmentalization in nox signaling. Antioxidants & Redox Signaling. 2009;11(10):2467–2480. doi: 10.1089/ars.2009.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turrens JF. Mitochondrial formation of reactive oxygen species. The Journal of Physiology. 2003;552(2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochimica et Biophysica Acta. 1998;1363(2):100–124. doi: 10.1016/s0005-2728(97)00091-1. [DOI] [PubMed] [Google Scholar]
- 39.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I) Journal of Biological Chemistry. 2004;279(38):39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 40.Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Quarterly Reviews of Biophysics. 1996;29(2):169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]
- 41.Okuda M, Lee H-C, Kumar C, Chance B. Comparison of the effect of a mitochondrial uncoupler, 2,4-dinitrophenol and adrenaline on oxygen radical production in the isolated perfused rat liver. Acta Physiologica Scandinavica. 1992;145(2):159–168. doi: 10.1111/j.1748-1716.1992.tb09351.x. [DOI] [PubMed] [Google Scholar]
- 42.Murphy MP, Echtay KS, Blaikie FH, et al. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: studies using a mitochondria- targeted spin trap derived from α-phenyl-N-tert-butylnitrone. Journal of Biological Chemistry. 2003;278(49):48534–48545. doi: 10.1074/jbc.M308529200. [DOI] [PubMed] [Google Scholar]
- 43.Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochimica et Biophysica Acta. 2011;1813(7):1269–1278. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rolo AP, Gomes AP, Palmeira CM. Regulation of mitochondrial biogenesis in metabolic syndrome. Current Drug Targets. 2011;12(6):872–878. doi: 10.2174/138945011795529056. [DOI] [PubMed] [Google Scholar]
- 45.Aharoni-Simon M, Hann-Obercyger M, Pen S, Madar Z, Tirosh O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Laboratory Investigation. 2011;91(7):1018–1028. doi: 10.1038/labinvest.2011.55. [DOI] [PubMed] [Google Scholar]
- 46.Wang S, Kamat A, Pergola P, Swamy A, Tio F, Cusi K. Metabolic factors in the development of hepatic steatosis and altered mitochondrial gene expression in vivo . Metabolism. 2011;60(8):1090–1099. doi: 10.1016/j.metabol.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 47.Nadal-Casellas A, Amengual-Cladera E, Proenza AM, Lladó I, Gianotti M. Long-term high-fat-diet feeding impairs mitochondrial biogenesis in liver of male and female rats. Cellular Physiology and Biochemistry. 2010;26(3):291–302. doi: 10.1159/000320552. [DOI] [PubMed] [Google Scholar]
- 48.Estall JL, Ruas JL, Choi CS, et al. PGC-1α negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erbα axis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(52):22510–22515. doi: 10.1073/pnas.0912533106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chambers KT, Chen Z, Crawford PA, et al. Liver-specific PGC-1beta deficiency leads to impaired mitochondrial function and lipogenic response to fasting-refeeding. PLoS ONE. 2012;7(12) doi: 10.1371/journal.pone.0052645.e52645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 51.Kong X, Wang R, Xue Y, et al. Sirtuin 3, a new target of PGC-1α, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011707.e11707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He J, Hu B, Shi X, et al. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Molecular and Cellular Biology. 2013;33(10):2047–2055. doi: 10.1128/MCB.01658-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Canto C, Auwerx J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current Opinion in Lipidology. 2009;20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng XQ, Chen LL, Li NX. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver International. 2007;27(5):708–715. doi: 10.1111/j.1478-3231.2007.01497.x. [DOI] [PubMed] [Google Scholar]
- 55.Wang RH, Kim H-S, Xiao C, Xu X, Gavrilova O, Deng C-X. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. The Journal of Clinical Investigation. 2011;121(11):4477–4490. doi: 10.1172/JCI46243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Serviddio G, Bellanti F, Sastre J, Vendemiale G, Altomare E. Targeting mitochondria: a new promising approach for the treatment of liver diseases. Current Medicinal Chemistry. 2010;17(22):2325–2337. doi: 10.2174/092986710791698530. [DOI] [PubMed] [Google Scholar]
- 57.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 58.Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. The Journal of Clinical Investigation. 2009;119(3):573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxidants & Redox Signaling. 2008;10(3):601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 60.Zhao K, Zhao G-M, Wu D, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. Journal of Biological Chemistry. 2004;279(33):34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 61.Nakamura S, Takamura T, Matsuzawa-Nagata N, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. Journal of Biological Chemistry. 2009;284(22):14809–14818. doi: 10.1074/jbc.M901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen L, Na R, Gu M, et al. Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell. 2008;7(6):866–878. doi: 10.1111/j.1474-9726.2008.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee HY, Choi CS, Birkenfeld AL, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metabolism. 2010;12(6):668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holloszy JO. ‘Deficiency’ of mitochondria in muscle does not cause insulin resistance. Diabetes. 2013;62(4):1036–1040. doi: 10.2337/db12-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’neill ED, Wilding JPH, Kahn CR, et al. Absence of insulin signalling in skeletal muscle is associated with reduced muscle mass and function: evidence for decreased protein synthesis and not increased degradation. Age. 2010;32(2):209–222. doi: 10.1007/s11357-009-9125-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsuzawa-Nagata N, Takamura T, Ando H, et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57(8):1071–1077. doi: 10.1016/j.metabol.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 67.Bonnard C, Durand A, Peyrol S, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. The Journal of Clinical Investigation. 2008;118(2):789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barazzoni R, Zanetti M, Cappellari GG, et al. Fatty acids acutely enhance insulin-induced oxidative stress and cause insulin resistance by increasing mitochondrial reactive oxygen species (ROS) generation and nuclear factor-κB inhibitor (IκB)-nuclear factor-κB (NFκB) activation in rat muscle, in the absence of mitochondrial dysfunction. Diabetologia. 2012;55(3):773–782. doi: 10.1007/s00125-011-2396-x. [DOI] [PubMed] [Google Scholar]
- 69.Merry TL, Tran M, Stathopoulos M, et al. High fat fed obese Gpx1-deficient mice exhibit defective insulin secretion but protection from hepatic steatosis and liver damage. Antioxidants & Redox Signaling. 2013 doi: 10.1089/ars.2013.5428. [DOI] [PubMed] [Google Scholar]
- 70.Forman HJ. Reactive oxygen species and α,β-unsaturated aldehydes as second messengers in signal transduction. Annals of the New York Academy of Sciences. 2010;1203:35–44. doi: 10.1111/j.1749-6632.2010.05551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jones DP. Radical-free biology of oxidative stress. American Journal of Physiology. 2008;295(4):C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chiarugi P. PTPs versus PTKs: the redox side of the coin. Free Radical Research. 2005;39(4):353–364. doi: 10.1080/10715760400027987. [DOI] [PubMed] [Google Scholar]
- 73.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121(5):667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 74.Foley TD, Petro LA, Stredny CM, Coppa TM. Oxidative inhibition of protein phosphatase 2A activity: role of catalytic subunit disulfides. Neurochemical Research. 2007;32(11):1957–1964. doi: 10.1007/s11064-007-9394-x. [DOI] [PubMed] [Google Scholar]
- 75.Wright VP, Reiser PJ, Clanton TL. Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. The Journal of Physiology. 2009;587(23):5767–5781. doi: 10.1113/jphysiol.2009.178285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annual Review of Biophysics and Biomolecular Structure. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 77.Fetrow JS, Siew N, Skolnick J. Structure-based functional motif identifies a potential disulfide oxidoreductase active site in the serine/threonine protein phosphatase-1 subfamily. FASEB Journal. 1999;13(13):1866–1874. doi: 10.1096/fasebj.13.13.1866. [DOI] [PubMed] [Google Scholar]
- 78.Maher P. Redox control of neural function: background, mechanisms, and significance. Antioxidants & Redox Signaling. 2006;8(11-12):1941–1970. doi: 10.1089/ars.2006.8.1941. [DOI] [PubMed] [Google Scholar]
- 79.Dasgupta J, Kar S, Liu R, et al. Reactive oxygen species control senescence-associated matrix metalloproteinase-1 through c-Jun-N-terminal kinase. Journal of Cellular Physiology. 2010;225(1):52–62. doi: 10.1002/jcp.22193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Covey TM, Edes K, Coombs GS, Virshup DM, Fitzpatrick FA. Alkylation of the tumor suppressor PTEN activates Akt and β-catenin signaling: a mechanism linking inflammation and oxidative stress with cancer. PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013545.e13545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu G, Ren S, Korge P, et al. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes and Development. 2007;21(7):784–796. doi: 10.1101/gad.1499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bashan N, Kovsan J, Kachko I, Ovadia H, Rudich A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiological Reviews. 2009;89(1):27–71. doi: 10.1152/physrev.00014.2008. [DOI] [PubMed] [Google Scholar]
- 83.Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxidants & Redox Signaling. 2005;7(7-8):1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iwakami S, Misu H, Takeda T, et al. Concentration-dependent dual effects of hydrogen peroxide on insulin signal transduction in H4IIEC hepatocytes. PLoS ONE. 2011;6(11) doi: 10.1371/journal.pone.0027401.e27401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mahadev K, Motoshima H, Wu X, et al. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Molecular and Cellular Biology. 2004;24(5):1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Y, Mouche S, Sajic T, et al. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. International Journal of Obesity. 2012;36(1):1503–1513. doi: 10.1038/ijo.2011.279. [DOI] [PubMed] [Google Scholar]
- 87.Fiaschi T, Buricchi F, Cozzi G, et al. Redox-dependent and ligand-independent trans-activation of insulin receptor by globular adiponectin. Hepatology. 2007;46(1):130–139. doi: 10.1002/hep.21643. [DOI] [PubMed] [Google Scholar]
- 88.Woo HA, Yim SH, Shin DH, Kang D, Yu D-Y, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell. 2010;140(4):517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 89.Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. American Journal of Physiology. 2009;296(4):E581–E591. doi: 10.1152/ajpendo.90437.2008. [DOI] [PubMed] [Google Scholar]
- 90.Hirosumi J, Tuncman G, Chang L, et al. A central, role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 91.Bost F, Aouadi M, Caron L, et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. 2005;54(2):402–411. doi: 10.2337/diabetes.54.2.402. [DOI] [PubMed] [Google Scholar]
- 92.Sung HU, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431(7005):200–205. doi: 10.1038/nature02866. Erratum in Nature, vol. 431, no. 7007, p. 485, 2004. [DOI] [PubMed] [Google Scholar]
- 93.Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ . Science. 2001;293(5535):1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 94.Xiao F, Huang Z, Li H, et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes. 2011;60(3):746–756. doi: 10.2337/db10-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang S, Messina JL. Role of inhibitory κB kinase and c-Jun NH2-terminal kinase in the development of hepatic insulin resistance in critical illness diabetes. American Journal of Physiology. 2011;301(3):G454–G463. doi: 10.1152/ajpgi.00148.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Y, Shi M, Fu H, et al. Mammalian target of the rapamycin pathway is involved in non-alcoholic fatty liver disease. Molecular Medicine Reports. 2010;3(6):909–915. doi: 10.3892/mmr.2010.365. [DOI] [PubMed] [Google Scholar]
- 97.Xi L, Xiao C, Bandsma RHJ, Naples M, Adeli K, Lewis GF. C-reactive protein impairs hepatic insulin sensitivity and insulin signaling in rats: role of mitogen-activated protein kinases. Hepatology. 2011;53(1):127–135. doi: 10.1002/hep.24011. [DOI] [PubMed] [Google Scholar]
- 98.Mumby MC, Walter G. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiological Reviews. 1993;73(4):673–699. doi: 10.1152/physrev.1993.73.4.673. [DOI] [PubMed] [Google Scholar]
- 99.Shearn CT, Reigan P, Petersen DR. Inhibition of hydrogen peroxide signaling by 4-hydroxynonenal due to differential regulation of Akt1 and Akt2 contributes to decreases in cell survival and proliferation in hepatocellular carcinoma cells. Free Radical Biology and Medicine. 2012;53(1):1–11. doi: 10.1016/j.freeradbiomed.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hahn K, Miranda M, Francis VA, Vendrell J, Zorzano A, Teleman AA. PP2A regulatory subunit PP2A-B′ counteracts S6K phosphorylation. Cell Metabolism. 2010;11(5):438–444. doi: 10.1016/j.cmet.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 101.Loukili N, Rosenblatt-Velin N, Rolli J, et al. Oxidants positively or negatively regulate nuclear factor κB in a context-dependent manner. Journal of Biological Chemistry. 2010;285(21):15746–15752. doi: 10.1074/jbc.M110.103259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Szypowska AA, Burgering BMT. The peroxide dilemma: opposing and mediating insulin action. Antioxidants & Redox Signaling. 2011;15(1):219–232. doi: 10.1089/ars.2010.3794. [DOI] [PubMed] [Google Scholar]
- 103.Tanti JF, Jager J. Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Current Opinion in Pharmacology. 2009;9(6):753–762. doi: 10.1016/j.coph.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 104.Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochimica et Biophysica Acta. 2008;1780(11):1325–1336. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 105.Gao D, Nong S, Huang X, et al. The effects of palmitate on hepatic insulin resistance are mediated by NADPH oxidase 3-derived reactive oxygen species through JNK and p38 MAPK pathways. Journal of Biological Chemistry. 2010;285(39):29965–29973. doi: 10.1074/jbc.M110.128694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Corcoran A, Cotter TG. Redox regulation of protein kinases. The FEBS Journal. 2013;280(9):1944–1965. doi: 10.1111/febs.12224. [DOI] [PubMed] [Google Scholar]
- 107.Khan N, Swartz H. Measurements in vivo of parameters pertinent to ROS/RNS using EPR spectroscopy. Molecular and Cellular Biochemistry. 2002;234-235(1):341–357. [PubMed] [Google Scholar]
- 108.Drougard A, Duparc T, Brenachot X, et al. Hypothalamic apelin/reactive oxygen species signaling controls hepatic glucose metabolism in the onset of diabetes. Antioxidants & Redox Signaling. 2013;20(4):557–573. doi: 10.1089/ars.2013.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lu YY, Chen T-S, Wang X-P, Li L. Single-cell analysis of dihydroartemisinin-induced apoptosis through reactive oxygen species-mediated caspase-8 activation and mitochondrial pathway in ASTC-a-1 cells using fluorescence imaging techniques. Journal of Biomedical Optics. 2010;15(4) doi: 10.1117/1.3481141.046028 [DOI] [PubMed] [Google Scholar]
- 110.Valle A, Catalán V, Rodríguez A, et al. Identification of liver proteins altered by type 2 diabetes mellitus in obese subjects. Liver International. 2012;32(6):951–961. doi: 10.1111/j.1478-3231.2012.02765.x. [DOI] [PubMed] [Google Scholar]
- 111.D’alessandro A, Rinalducci S, Zolla L. Redox proteomics and drug development. Journal of Proteomics. 2011;74(12):2575–2595. doi: 10.1016/j.jprot.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 112.Shearn CT, Fritz KS, Reigan P, Petersen DR. Modification of Akt2 by 4-hydroxynonenal inhibits insulin-dependent Akt signaling in HepG2 cells. Biochemistry. 2011;50(19):3984–3996. doi: 10.1021/bi200029w. [DOI] [PubMed] [Google Scholar]
- 113.Lindemann C, Leichert LI. Quantitative redox proteomics: the NOxICAT method. (Methods in Molecular Biology).Quantitative Methods in Proteomics. 2012;893:387–403. doi: 10.1007/978-1-61779-885-6_24. [DOI] [PubMed] [Google Scholar]
- 114.Baumgardner JN, Shankar K, Hennings L, Albano E, Badger TM, Ronis MJJ. N-acetylcysteine attenuates progression of liver pathology in a rat model of nonalcoholic steatohepatitis. The Journal of Nutrition. 2008;138(10):1872–1879. doi: 10.1093/jn/138.10.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen H, Karne RJ, Hall G, et al. High-dose oral vitamin C partially replenishes vitamin C levels in patients with type 2 diabetes and low vitamin C levels but does not improve endothelial dysfunction or insulin resistance. American Journal of Physiology. 2006;290(1):H137–H145. doi: 10.1152/ajpheart.00768.2005. [DOI] [PubMed] [Google Scholar]
- 116.Liu S, Lee I-M, Song Y, et al. Vitamin E and risk of type 2 diabetes in the women’s health study randomized controlled trial. Diabetes. 2006;55(10):2856–2862. doi: 10.2337/db06-0456. [DOI] [PubMed] [Google Scholar]
- 117.Dufour JF, Oneta CM, Gonvers J-J, et al. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin E in nonalcoholic steatohepatitis. Clinical Gastroenterology and Hepatology. 2006;4(12):1537–1543. doi: 10.1016/j.cgh.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 118.Mercer JR, Yu E, Figg N, et al. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free Radical Biology and Medicine. 2012;52(5):841–849. doi: 10.1016/j.freeradbiomed.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 119.Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Annals of the New York Academy of Sciences. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]