

Abstract
Repairing or disposing of a malfunctioning object is an everyday dilemma. Replacing an item may be quicker than repairing it, but may also be more costly. Cells are faced with the same options when their organelles are challenged. Ensuring the health of the mitochondrial network is of utmost importance for cellular health and, not surprisingly, mitochondrial quality control can take both the repair and disposal routes. Spectacular advances have been made in recent years and a picture is starting to emerge of what drives a cell to take one or the other path. Interestingly, mitochondrial quality control seems to be deficient in various medically relevant conditions, such as neurodegeneration and aging.
Introduction
Mitochondria are double-membraned organelles of endosymbiotic origin. They are not only the “powerhouses of the cell”, they are also major triggers for the intrinsic pathway of apoptosis, regulators of calcium homeostasis and providers of various chemicals for the cell, such as phospholipids and iron-sulfur clusters (indispensable for membrane biogenesis and for the assembly of many catalytic centers, respectively). Mitochondrial homeostasis is ensured by a balance between biogenesis and turnover. This balance is achieved through coordination of several pathways. For instance, mitochondrial proteins are encoded within two genomes. The vast majority of mitochondrial proteins are encoded in the nuclear genome and are imported post-translationally, while a dozen proteins are directly encoded in the mitochondrial genome [1]. As both genomes contribute subunits to the respiratory chain complexes, coordination of the expression of the nuclear and the mitochondrial genome, as well as the rate of protein import, is crucial for the proper stoichiometric assemblies of these complexes. There are a number of ways that this can go wrong. Facing such problems, the cell is left with two options: repair the faulty mitochondria or get rid of them. This decision depends on several criteria. How bad is the damage and is it fixable? Are the damaged mitochondria doing any harm to the rest of the cell? Are there enough mitochondria to compensate for the loss of the damaged ones?
Interestingly, mitochondrial quality control is tightly linked to medically important phenomena, such as neurodegeneration and aging. While Parkinson’s disease is a mostly sporadic disease characterized by the cytoplasmic accumulation of plaques containing aggregates of the protein alpha-synuclein in dopaminergic neurons, a small percentage of Parkinson’s disease is strictly genetically inherited and its onset is earlier than sporadic Parkinson’s disease [2]. Besides pathological dominant alleles of the alpha synuclein-gene itself, most of these genetic cases of Parkinson’s disease are due to recessive mutations in genes encoding phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) and Parkin. These two proteins are part of a recently identified mitochondrial quality control pathway [2]. The activation of another mitochondrial quality control pathway extends the lifespan of worms and mice [3,4]. Therefore the study of mitochondrial quality control holds promise to boost our understanding of these medically relevant conditions.
Mitophagy: trash it
Autophagy allows the degradation and recycling of cytoplasmic components. This pathway can be triggered by a sudden need for energetic resources, for instance in the case of a starvation response [5]. In this case, random bits of the cytoplasm are engulfed in a double membraned organelle (autophagosome) and delivered to the lysosome for degradation and recycling. In other conditions, select parts of the cytoplasm, such as organelles, are sorted for engulfment in a selective autophagic process [6]. This happens to damaged mitochondria in a process called mitophagy. Treating cells with harsh mitochondria damaging agents can cause the virtually complete mitophagic disappearance of mitochondria (e.g. [7]). In physiological conditions, faulty mitochondria need to be flagged. This flag is then used to recruit the mitophagy machinery. In this sense, mitophagy not only is specific for mitochondria but it also only targets a subset of mitochondria, that is, the terminally damaged ones. PINK1 and Parkin are the two main components of this flagging/recruiting process. PINK1 is a serine/threonine kinase bearing a mitochondrial targeting sequence and a transmembrane domain. In the absence of damage, PINK1 is quickly turned over and does not accumulate. However, PINK1 is selectively stabilized on the surface of faulty mitochondria [8-10]. PINK1 accumulation then causes the recruitment of Parkin, which is a ring-domain containing E3-ubiquitin ligase. Parkin broadly ubiquitinylates proteins on the cytosolic side of the outer mitochondrial membrane (OMM) [11]. Ubiquitin is then used as a flag to degrade OMM proteins and to recruit the autophagic machinery.
Mitochondrial dynamics: break it
Mitochondria are highly dynamic, in that they undergo cycles of fusion and fission [12]. As a result, the number of mitochondria in a cell is not fixed, but fluctuates depending on whether mitochondria are rather elongated or fragmented. The physiological role of mitochondrial dynamics is largely mysterious, but one of its important functions appears to be quality control [13].
On the one hand, a damaged mitochondrion may be able to segregate its damaged components into subcompartments and divide, giving rise to one healthy mitochondrion and one that presumably carries the damage load of the original mitochondrion [14]. This damaged mitochondrion has an opportunity to repair itself, or is targeted to mitophagy by the PINK1-Parkin pathway.
On the other hand, fusion is inhibited in damaged mitochondria. The OPA1 protein (mutated in dominant optic atrophy) is involved in the fusion of the inner mitochondrial membrane (IMM) during mitochondrial fusion [15]. OPA1 exists in various forms that are produced by proteolytic cleavage [16,17]. Among the proteases that process OPA1 is OMA1. Mitochondrial damage appears to regulate OPA1 cleavage by OMA1 [18,19]. This leads to an overall inhibition of fusion in faulty mitochondria. Damaged organelles are therefore prevented from contaminating healthy ones. Moreover, preventing fusion also leads to a shortening of mitochondria, which are thereafter more easily engulfed in mitophagosomes [14,20]. Another way to prevent fusion is the selective PINK1-Parkin-dependent proteasomal degradation of the mitofusins [21,22]. Mitofusins are dynamin-related GTPases essential for OMM fusion. Their turnover may thus contribute to the fragmentation of the mitochondrial network.
Mitochondrial unfolded protein response: fix it
Cells undergoing stress homeostatically adapt by changing their transcription program to cope with and overcome the stress. Such transcriptional stress responses include the cytoplasmic heat-shock response and the endoplasmic reticulum unfolded protein response (UPR) [23]. In both of these cases, the accumulation of misfolded proteins is sensed by one or more sensor proteins in the respective compartments, causing the activation of a nuclear transcription factor, which turns on a transcriptional program aimed at fixing the problem. A more recently discovered pathway is the mitochondrial UPR [24,25], which has been most extensively studied in the worm Caenorhabditis elegans [26]. At the core of this pathway is activating transcription factor associated with stress-1 (ATFS-1). ATFS-1 is a basic leucine zipper (bZip) transcription factor, which bears a mitochondria targeting sequence at its N-terminus [27]. In healthy cells, ATFS-1 is efficiently imported into the mitochondrial matrix, where it is quickly degraded. Upon mitochondrial dysfunction, the import efficiency of ATFS-1 drops and ATFS-1 starts accumulating outside of mitochondria. Because ATFS-1 bears a nuclear localization sequence (NLS) close to its bZip domain, it is imported into the nucleus where it starts a transcription program. Among the transcriptional targets of ATFS-1 are mitochondrial chaperones, which are upregulated to cope with the stress in mitochondria [27]. Therefore, mitochondrial UPR helps damaged mitochondria to restore their function.
Sensing damage
The pathways we have just described have almost completely opposite outcomes. Both mitochondrial fragmentation and mitophagy aim at getting rid of the problem, while mitochondrial UPR tries to fix it. When and why does the cell choose one over the other? One possibility is that qualitatively different types of stresses elicit different responses. This explanation, however, does not hold as the stresses eliciting mitophagy, mitochondrial UPR and fusion inhibition appear to be identical: it’s a loss of electrochemical potential in the IMM, which is the cause of PINK1 accumulation in the OMM [9,10], of ATFS-1 failure to enter the mitochondria [27] and possibly also of OMA1 activation [18,19]. The mechanisms underlying these various phenomena are actually very similar and relate to the fact that the electrochemical potential of the IMM is crucial for the translocation of proteins across the IMM [28]. In healthy mitochondria, the mitochondrial targeting sequence of PINK1 precursor crosses both translocases of the outer and inner membranes (TOM and TIM), to reach the IMM, where its transmembrane domain is inserted. IMM insertion of PINK1 triggers a degradation reaction initiated by the intra-membrane cleavage of the transmembrane domain by the presenilin-associated rhomboid-like protease (PARL) [29,30] (Fig. 1, left). The final degradation reaction is pursued in the cytosol (likely after retrotranslocation through the OMM) by the cytosolic proteasome [31] because the N-terminal amino acid of cleaved PINK1 is targeted by the N-end rule pathway [32]. Compromised mitochondria are depolarized, therefore PINK1 can reach TOM but does not efficiently translocate into the TIM complex. As a result, it is most likely released from the TOM complexes and inserted laterally into the OMM, where it can recruit Parkin and trigger mitophagy [9,10,29] (Fig. 1, right).
Figure 1. Scheme of the various mitochondria quality control pathways operative in a healthy mitochondrion (left) or a depolarized one (right).
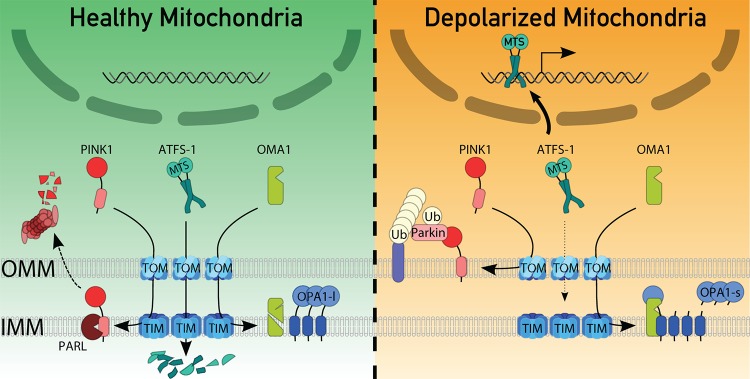
In healthy mitochondria, PTEN-induced kinase 1 (PINK1) is transported to the inner mitochondrial membrane (IMM) where it is processed by the intramembrane presenilin-associated rhomboid-like protease (PARL). This leads to the retrotranslocation of PINK1 to the cytosol, where it is degraded by the proteasome via the N-end rule pathway. In depolarized mitochondria, PINK1 fails to reach the IMM and instead inserts in the outer mitochondrial membrane (OMM), where it recruits the ubiquitin (Ub) ligase Parkin to ubiquitinylate OMM resident proteins. In healthy cells, ATFS-1 is imported into the mitochondrial matrix, thanks to its mitochondrial targeting sequence (MTS). In depolarized mitochondria, ATFS-1 import is inhibited. As a result, ATFS-1 translocates to the nucleus where it acts as a transcription factor to induce the mitochondrial UPR. The metalloprotease OMA1 inserts in the IMM. In healthy cells it is likely in an inactive form. As a consequence, a long non-processed form of its substrate – the dynamin-related protein OPA1 – accumulates (OPA1-l). In depolarized cells, OMA1 is activated and cleaves OPA1 to a short form (OPA1-s), thereby inhibiting mitochondrial fusion.
The case of ATFS-1 is more straightforward: a simple kinetic competition appears to take place between its mitochondrial targeting sequence and its NLS. Since the electrochemical gradient across the IMM is crucial for mitochondrial protein import [28] and, since depolarized mitochondria do not import proteins efficiently, loss of electrochemical potential will shift the equilibrium to favor nuclear import of ATFS-1 and trigger mitochondrial UPR [27] (Fig. 1).
The mechanism of OMA1 activation is less clear. It is proposed that OMA1 inserts in the OMM, where it is rendered inactive by proteolytic cleavage (Fig. 1, left). Upon depolarization, OMA1 might insert into the IMM in a way that prevents its proteolytic cleavage and instead accumulates as an active precursor [19] (Fig. 1, right).
Thus, if the type of insult that triggers mitophagy or mitochondrial UPR is not qualitatively different, then what decides between the two cellular pathways? The difference may just be quantitative. A single damaged mitochondria will be sufficient to expose PINK1 on its surface and trigger its own degradation. However a single faulty mitochondrion will likely not be able to release enough ATFS-1 to mount a mitochondrial UPR, as enough healthy mitochondria can compete with the nucleus for ATFS-1 import. In genetics terms, one could say that mitophagy is dominant, while mitochondrial UPR is recessive. As a result, mitophagy is an organelle-specific response while mitochondrial UPR is a systemic response.
Therefore, the choice of one pathway over the other will depend on the overall degree of damage of the mitochondrial network: a few highly damaged mitochondria will trigger their selective elimination by mitophagy, while a low general level of damage to most mitochondria will cause mitochondrial UPR. Of course, any intermediate situation could be dealt with by a combination of mitophagy and mitochondrial UPR activation. These two dominant and recessive types of response are actually a very neat way of fine tuning the cellular response to mitochondrial damage. On the one hand, a single faulty mitochondrion might not necessitate the induction of a costly systemic program and can be dealt with mostly by mitophagy. On the other hand, eating up the whole mitochondrial network is a bad idea. Repair is probably the way to go when too many mitochondria are damaged.
Mitochondrial quality control in health and disease
All of the above pathways have intricate connections with human health. PINK1 and Parkin are the most prevalent recessive mutations found in patients with early onset Parkinson’s disease [2], suggesting that a specific malfunction in the machinery that gets rid of damaged mitochondria can cause the disease. The aetiology of the disease is not clear in PINK1 and Parkin-related early onset Parkinson’s disease. Sporadic Parkinson’s disease is characterized by the appearance of amyloid plaques of alpha-synuclein, which is not a mitochondrial protein. Moreover, most patients suffering from Parkin- or PINK1-related Parkinson’s disease do not show plaques. Thus the connection between Parkinson’s disease and mitochondria still needs to be clarified.
Another spectacular health-related aspect of mitochondrial quality control is aging. Since the original observations that calorie restriction could extend lifespan, a close connection exists between energy metabolism and aging. Mitochondria, as an important source of reactive oxygen species, have long been considered a prime suspect for causing cellular aging. However, a different picture emerges from recent studies. In a screen for long-lived worm mutants, Andrew Dillin’s lab found that creating imbalance in the assembly of respiratory chain subunits caused increased lifespan [3]. That phenomenon was soon related to an activation of the mitochondrial UPR. Indeed, this increase in lifespan did not correlate to a reduced activity of the mitochondria, but to an activation of mitochondrial UPR, since increase in lifespan is blunted in mutants incapable of mounting a mitochondrial UPR. Thus, although direct causality between mitochondrial UPR induction and lifespan extension is not clearly established, it seems that mitochondrial quality control, more than mitochondrial activity, contributes to aging regulation. These observations, originally made in worms, could be extended to mice, where a large-scale genomic association study found that longevity was associated with variations in genes encoding mitochondrial proteins [4].
Finally, besides aging and Parkinsonism, other medically relevant conditions seem to involve mitochondrial biogenesis and turnover, and mitochondrial dynamics and quality control genes are often mutated in neurological diseases. These include dominant optical atrophy (OPA1), Charcot-Marie-Tooth type 2a (Mitofusin 2), Leber hereditary optical neuropathy (various mutations in mitochondrial DNA) among others (reviewed in [33]).
The unexpected relationship between mitochondrial quality control, neurodegeneration and aging thus stresses the importance of the global and detailed understanding of these pathways for human health.
Acknowledgments
I would like to thank Koji Yamano, Thomas Langer, Alexander Lang and Agnès H. Michel for critical comments on the manuscript. This work is supported by the Swiss National Science Foundation (grant # PP00P3_13365). I would like to thank the band Daft Punk for their song “Technologic”, which was the inspiration for part of the title of this report.
Abbreviations
- ATFS-1
activating transcription factor associated with stress-1
- bZip
basic leucine zipper
- IMM
inner mitochondrial membrane
- NLS
nuclear localization sequence
- OMA1
overlaps with mitochondrial AAA ATPase
- OMM
outer mitochondrial membrane
- OPA1
dominant optical atrophy 1
- OPA1-l/s
OPA1 long form/short form
- PARL
presenilin-associated rhomboid-like protease
- PINK1
PTEN-induced kinase 1
- PTEN
phosphatase and tensin homolog
- TIM
translocase of the inner membrane
- TOM
translocase of the outer membrane
- UPR
unfolded protein response
Disclosures
I hereby declare that I have no disclosures.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/b/6/15
References
- 1.Dolezal P, Likic V, Tachezy J, Lithgow T. Evolution of the molecular machines for protein import into mitochondria. Science. 2006;313:314–8. doi: 10.1126/science.1127895. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718265910
- 2.Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718265911
- 3.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/8487957
- 4.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–7. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718012749
- 5.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Priault M, Salin B, Schaeffer J, Vallette FM, Di Rago J, Martinou J. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 2005;12:1613–21. doi: 10.1038/sj.cdd.4401697. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718265912
- 7.Narendra D, Tanaka A, Suen D, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1132883
- 8.Vives-Bauza C, Zhou C, Huang Y, Cui M, Vries RLA de, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–83. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1836974
- 9.Narendra DP, Jin SM, Tanaka A, Suen D, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/2111956
- 10.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou Y, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–21. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/3099963
- 11.Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–6. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718077487
- 12.van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718265914
- 15.Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, Nunnari J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–95. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1053737
- 16.Herlan M, Vogel F, Bornhovd C, Neupert W, Reichert AS. Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem. 2003;278:27781–8. doi: 10.1074/jbc.M211311200. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1013197
- 17.Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson N, Neupert W, Reichert AS. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–9. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- 18.Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou J, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023–36. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718265916
- 19.Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–66. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718265917
- 20.Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta. 2008;1777:860–6. doi: 10.1016/j.bbabio.2008.05.442. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718265919
- 21.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen D, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–80. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/7548956
- 22.Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, Glickman MH, Weissman AM. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. 2012;47:547–57. doi: 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718265921
- 23.Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. 2010;123:3849–55. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–9. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718265922
- 25.Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–66. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- 26.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–40. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–90. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717952882
- 28.Schleyer M, Schmidt B, Neupert W. Requirement of a membrane potential for the posttranslational transfer of proteins into mitochondria. Eur J Biochem. 1982;125:109–16. doi: 10.1111/j.1432-1033.1982.tb06657.x. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718265924
- 29.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–42. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718265925
- 30.Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SHY, Renton AEM, Harvey RJ, Whitworth AJ, Martins LM, Abramov AY, Wood NW. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet. 2011;20:867–79. doi: 10.1093/hmg/ddq526. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/7519969
- 31.Takatori S, Ito G, Iwatsubo T. Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett. 2008;430:13–7. doi: 10.1016/j.neulet.2007.10.019. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718265926
- 32.Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9:1758–69. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718173981
- 33.Finsterer J. Inherited mitochondrial neuropathies. J Neurol Sci. 2011;304:9–16. doi: 10.1016/j.jns.2011.02.012. [DOI] [PubMed] [Google Scholar]