

Summary
FSGS is a lesion, not a disease. The separation into primary FSGS (a result of immunologic-mediated injury) versus secondary FSGS (related to a variety of causes) is often difficult. Even when this particular issue is carefully evaluated, the therapeutic implications are not always apparent. Newer literature on both biomarker discovery and on the genetic basis of FSGS is reviewed in this context. In addition, the thorny implications of obesity as it relates to the FSGS lesion are discussed. An overall practical algorithmic approach to the management and treatment of the FSGS lesion that integrates these controversial overlap areas is suggested.
Introduction
FSGS is the most common primary glomerular histologic lesion associated with high-grade proteinuria and with ESRD (1). FSGS is a pattern of histologic injury rather than a disease and can be either primary or secondary to a variety of underlying processes. Separation into these two categories of FSGS is not always easy. There is a significant overlap of clinical and histologic features making the assignment of each patient to a single specific category difficult (Figure 1). Some FSGS patients are known to have significant genetic mutations but still respond to immunosuppressive treatment (2,3). This failure to appreciate the potential for overlap raises the concern that the finding of such abnormalities will automatically lead to avoidance of immunotherapy. This conundrum of primary versus secondary FSGS is critical not only for diagnostic but also therapeutic purposes because this decision virtually drives all subsequent aspects of patient management. Currently, the best method of separation is based on pathology (electron microscopy that demonstrates >80% diffuse foot process effacement is classically associated with primary FSGS) but the correlation with the clinical and laboratory parameters, response to therapy and eventual outcome is imprecise. A recent detailed, evidence-based FSGS treatment guideline was published under the auspices of the Kidney Disease Improving Global Outcomes (KDIGO) organization and the KDIGO guideline on GN remains a major reference text (4). However, the guideline begins only after confirmation that the lesion (and patient) under discussion represents primary FSGS.
Figure 1.

Overlap of different causal categories of FSGS. There is a complex interaction between different causes of FSGS. The overlap area conceptually indicates patients who may respond to treatment within the overlapping areas of primary and secondary causations.
Primary FSGS is usually a progressive disorder with <5% spontaneous remission and a 50% ESRD rate over a period of 5–8 years from the time of biopsy in patients that are either unresponsive to treatment or not treated (5). Nephrotic-range proteinuria with or without other features of the nephrotic syndrome is the classic pattern of presentation of primary FSGS and is seen in 75%–90% of children and 50%–60% of adults. Although there is evidence of improvement in long-term outcomes with treatment, large-scale observational renal replacement registries that focus on causation indicate a continuing increase in the number designated as having FSGS (6). It is likely that these divergent observations result from the imprecise classification of primary versus secondary causes of the lesion. Although primary FSGS is the major focus of this article, the recognition of the secondary variants and the potential overlap of the two processes are intrinsic to understanding the approach to the current management and treatment of patients with the FSGS lesion.
Histologic Variants
The most current histologic classification includes the following variants: classic FSGS (also called FSGS not otherwise specified [FSGS NOS]), collapsing, tip, perihilar, and cellular types (7). However, even this approach, designated the Columbia classification, has had a variable and at times poor correlation with both the natural history and therapeutic responsiveness of FSGS patients (5,8). Classic FSGS (NOS variant) is the most common variant observed. Prognosis of the cellular variant is intermediate between collapsing and a classic FSGS (9). Although the collapsing variant of FSGS is most often associated with HIV infection, other causes (including idiopathic causes) exist. Of all of the variants, it still carries the worse prognosis. Patients with the tip lesion have the most favorable prognosis and may be more responsive to corticosteroid therapy than the other types (10). The perihilar variant is more commonly associated with secondary FSGS and is considered to be mediated by an adaptive response to increased glomerular capillary pressures and flow rates.
There is an alternative taxonomy proposed for the podocytopathies that classifies them along two dimensions: histopathology, including podocyte phenotype and glomerular morphology (minimal change nephropathy, FSGS, diffuse mesangial sclerosis, and collapsing glomerulopathy), and etiology (11). According to this system, FSGS can be classified as idiopathic, nonsyndromic (genetic), and postadaptive. Despite some distinct advantages, this system has not been as widely accepted or validated compared with the Columbia classification.
Relationship between Steroid-Resistant Minimal Change Disease and Primary FSGS
Primary FSGS and minimal change disease (MCD) have many clinical as well as histologic similarities at presentation, making separation into these two categories difficult. Compounding the clinical complexity is the distinct possibility of sampling error (i.e., missing the lesion on biopsy given that the FSGS lesions are by definition focal and segmental in nature). The common target of injury in both is the podocyte. The distinction between the two disorders is important given the marked difference in terms of response to steroid treatment and long-term outcome. Currently, in the absence of seeing the FSGS lesion, response to corticosteroid therapy remains the clinical gold standard. The problem in distinguishing the two is accentuated by the age of onset of the process. Nephrotic children, given the commonness of the MCD diagnosis, are most commonly treated on a preemptive basis with steroids, whereas nephrotic adults almost always have a biopsy before initiating treatment. This creates a potential bias to fewer cases of FSGS in children compared with adults because the steroid-responsive child with FSGS will not have a biopsy and thus will not be counted in this histologic category. An earlier, more specific, and sensitive method of separating primary FSGS from MCD is an area of active investigation, with the objective to not only identify the underlying specific pathology but also to gain insight into the pathophysiology of the process. These inquiries have focused on biomarker discovery as well as intensive research into the genetic basis of these processes.
New Biomarkers.
Recurrent primary FSGS can be associated with massive proteinuria within hours of a kidney transplant. This has given rise to the concept of a permeability factor (or factors) playing a key role in podocyte injury. Potential permeability factors include hemopexin, vascular endothelial growth factor, and cardiotrophin-like cytokine-1 (12–14). Most of these factors have been found to be neither specific nor sensitive for separating primary FSGS from MCD. Soluble urokinase plasminogen-type activator receptor (suPAR) was recently found in association with FSGS. SuPAR is a glycosyl-phosphatidylinositol–anchored three-domain membrane protein that can bind to several ligands, including urokinase-type plasminogen activator, vitronectin, or integrins. SuPAR can be further cleaved to smaller fragments DI and DIIDIII (15). Wei et al. found that two thirds of patients with primary FSGS had increased suPAR and those with the highest levels had a greater chance of recurrence of the disease after transplantation (16). On the basis of both in vitro and in vivo studies, these investigators determined that suPAR binds and activates β3 integrin in cultured podocytes. Because this process is involved in foot process effacement, these results suggested a potential mechanistic link to the proteinuria seen in humans with FSGS.
Elevated suPAR concentrations are known to be increased in other conditions, including infection, sepsis, and solid tumors, as well as in states of systemic inflammation (17). SuPAR concentrations are also known to be inversely proportional to renal function, further complicating interpretation of their levels in patients with renal impairment (18). The clinical studies carried out to date are summarized in Table 1. Current testing reports only the sum of suPAR fragments, which potentially obfuscates the importance of the more active fragments. Currently, whether suPAR plays a functional role in the immunologic activity in primary FSGS and whether suPAR is a nonspecific marker remain open questions and major areas of investigation.
Table 1.
Clinical studies of suPAR in humans
| Study Parameter | Wei et al. (2011)16 | FSGS CT (2012)50 | Podonet (2012)50 | Huang (2012)51 |
| Patients with underlying kidney disease (n) | ||||
| Primary FSGS | 78 | 70 (35 patients received CSA and 35 patients received MMF/dexamethasone for 26 wk) |
94 | 74 |
| Secondary FSGS | 14 | |||
| MCD | 14 | |||
| MN | 25 | |||
| Preeclampsia | 11 | 40 | ||
| Controls | 7 | 110 (healthy) | 56 (healthy) | |
| Mean age (yr) | 27 | 19 | <18 | 29 |
| Race (%) | n/a | |||
| White | 60 | |||
| African American | 17 | 33 | ||
| Hispanic | 17 | |||
| Asian | 6 | 100 | ||
| Renal function, creatinine (mg/dl) | 54 in ESRD, 23 in CKD (mean 1.9) | Mean 1.1 | 0.69–0.91 | Median 1.1 |
| suPAR (pg/ml) | 71% with >3000 | 4588±203 | 3497±195 | 2923 (2205–4360) |
| Study conclusions | (1) suPAR is elevated in two thirds of patients with primary FSGS, but not in people with other glomerular diseases; (2) the higher concentration of suPAR before transplantation implies an increased risk for recurrence of FSGS after transplantation | (1) suPAR was markedly elevated in the majority of FSGS patients; (2) CRP levels were normal, suggesting that primary FSGS was not an inflammatory condition; (3) MMF therapy associated with lower suPAR levels; (4) a sustained decline in suPAR over 26 wk of treatment was associated with a reduction in proteinuria and greater odds for complete remission | (1) suPAR was markedly elevated in the majority of FSGS patients; (2) CRP levels were normal, suggesting that primary FSGS was not an inflammatory condition; (3) suPAR levels were higher in familial cases of FSGS, including those with a defined podocin mutation | (1) suPAR levels were higher in patients with FSGS compared with patients with MCD or MN and healthy controls; (2) there was no significant difference in suPAR levels between primary and secondary FSGS patients; (3) suPAR levels increased in the order of tip variant, to not otherwise specified variant, and a cellular variant; (4) soluble urokinase receptor levels were significantly but negatively correlated with creatinine clearance at presentation but positively correlated with crescent formation in patients with primary FSGS |
CSA, cyclosporine A; MMF, mycophenolate mofetil; MCD, minimal change disease; MN, membranous nephropathy; suPAR, soluble urokinase plasminogen-type activator receptor; CRP, C-reactive protein.
Genetics and FSGS
Genetic defects have been identified in up to two thirds of patients with FSGS who present in the first year of life and are responsible for the subsequent clinical phenotype in this age group (19). However, the direct causal relationship to the disease process (i.e., proteinuria and renal failure) is not so apparent in older children and adults with FSGS and an associated genetic mutation. It has been suggested that a second hit may be required in this setting and speculations on the origin of these triggers are wide ranging and include additional genetic and/or external environmental factors.
Specific Mutations
One of the major components of the slit membrane is the transmembrane protein nephrin (NPHS1). This gene was found mutated in patients with Finnish-type congenital nephrotic syndrome and has been directly implicated in kidney tissue injury (20). Podocin (NPHS2) is exclusively expressed in podocytes and localizes at the insertion of the slit membrane (21).
Patients with major mutations, NPHS 1 and NPHS 2 are resistant to immunotherapy. They have low rates of recurrence after transplantation, further supporting the concept that these mutations lead to the FSGS lesion and the clinical phenotype.
Many other genetic mutations have been recognized in both older children and adults with FSGS. These have been important discoveries that have led to significant advances in our understanding of the pathobiology of the podocyte and its relationship to the integrity of the glomerular basement membrane. These include R229Q, CD2AP, α-actinin-4, transient receptor potential cation channel, phospholipase Cε1, and laminin β-2, and others (3,22–24). Their relationship to treatment response and recurrence after transplantation is not as clear.
APOL1.
Recent studies have described polymorphisms in the APOL1 gene that are associated with the FSGS lesion and appear to be expressed exclusively in individuals of African descent (25,26). Its variants are associated with a 17-fold higher odds for FSGS and 29-fold higher odds for HIV-associated nephropathy. In the African American Study of Kidney Disease and Hypertension (AASK), patients with polymorphisms in APOL1 on average had higher baseline proteinuria and unrelenting kidney disease progression compared with those without the polymorphisms. This occurred despite equal BP control and angiotensin-converting enzyme inhibitor blockade, supporting the notion that additional mechanisms were in play in this cohort (27). In the AASK study, kidney biopsies of these patients showed extensive focal global glomerulosclerosis (FGGS), suggesting a strong association between APOL1 and FGGS (28). This polymorphism may help to explain the disproportion percentage of the patients of African ancestry in ESRD registries. Currently, most of these patients do not have a biopsy; thus, they are categorized solely based on their clinical features and potentially carry the (mis)label of hypertensive nephrosclerosis.
It is currently unclear how to integrate the information on these genetic mutations into the management and treatment strategies of patients with the FSGS lesion. The recent KDIGO guideline on GN does not recommend genetic testing on patients with FSGS beyond the age of 2 or 3 years and suggests that further genetic evaluation should only be performed after failure to respond to corticosteroid therapy. An alternative but earlier approach to this evaluation was recently suggested (29). The uncertainty of the clinical relevance of the underlying mutations increases potential error in assuming that these patients will not respond to treatment. Certainly, this has not proven to be the case, especially in the patient with older-onset FSGS. A large part of this confusion and controversy is related to our current lack of understanding of the specific factors that may modify the pathobiology of the mutation and lead to a delay in the clinical manifestations of the process to much later in life.
Obesity and FSGS
Obesity remains an important element related to the FSGS lesion. It seems unlikely that weight per se is the single factor in this association, given the current major difference in the number of people who are obese versus those with the diagnosis of FSGS.
A patient with the obesity-associated FSGS lesion (O-FSGS) typically presents with subnephrotic or nephrotic-range proteinuria but without other features of nephrotic syndrome. The classic pathology features include glomerulomegaly and lesions of focal and segmental sclerosis involving the perihilar regions with associated hyalinosis and mesangial changes but with little tubulointerstitial damage. There tends to be less diffuse foot process effacement on electron microscopy than that seen in patients with primary FSGS (30–32). Risk factors identified by physiological studies that may contribute to the development of the lesion include elevations of renal plasma flow and GFR, insulin resistance leading to an increased transcapillary pressure gradient and increased synthesis of growth factors promoting glomerular hypertrophy (33,34). Elevated plasma levels of leptin through upregulation of TGF-β1 in obesity may also predispose to glomerulosclerosis (35).
However, how or whether these specific pathophysiologic factors are different in the vast percentage of obese patients who do not develop the clinical phenotype or presumably the FSGS lesion is currently unknown. Hence, the therapeutic implications, beyond the obvious one of weight reduction, remain a conundrum as discussed in the section on overlap.
Treatment of FSGS
How to integrate this new information into management of the patient with the FSGS lesion is a challenge. However, once the decision is made to proceed to immunotherapy, a guideline is available. The goal of therapy is to induce a complete remission of proteinuria that in turn will lead to better long-term preservation of renal function. Achieving partial remission, although not optimal, does slow the progression of kidney disease and substantially improve renal survival (36). Regardless of the underlying causative process, the signs and symptoms of nephrotic syndrome should be managed with renin-angiotensin system inhibitors, statins, a low-salt diet, and diuretics, because even low-level persistent proteinuria has been associated with an increased risk of cardiovascular disease and the potential for long-term organic kidney damage (37,38).
Immunosuppressive Treatment of Primary FSGS.
As the presumed origin of primary FSGS is a dysregulated autoimmune response, the use of immunosuppressive agents is advocated in its treatment. Recently, direct effects of some of these agents on the podocyte have been determined that potentially augments or supplements their immunosuppressive action. Calcineurin inhibitors (CNIs) have been shown to stabilize the podocyte actin cytoskeleton by blocking the calcineurin-mediated dephosphorylation of synaptopodin, a protein critical for actin filament reformation (39). Rituximab, a chimeric mAb against CD20 on the surface of B cells and a well established B cell–depleting immunosuppressive agent, may have a direct antiproteinuric effect by preventing actin cytoskeleton disruption (40).
The current KDIGO guideline on GN recommends initial treatment of primary FSGS with high-dose prednisone given for between 4 and 16 weeks or until complete remission (4). CNIs are recommended for patients with FSGS who are resistant or intolerant to glucocorticoids and are continued for a minimum of 1 year if the patient is responsive. The above treatments are effective but side effects are significant, and rates of treatment failure and relapse are high. Steroid resistance can be seen in up to 50% of patients and a prolonged course is associated with significant side effects, including diabetes, increased infection rates, osteoporosis, and weight gain. Prophylactic strategies should be initiated to minimize the toxic effects of a prolonged course of steroids as per the KDIGO guideline on GN (4). CNI treatment is also associated with significant but different adverse effects, including nephrotoxicity and hypertension. This therapy has been associated with a relapse rate of up to 50%.
The current guideline suggests that patients who relapse should be treated with the same agent and duration that resulted in their initial remission.
Other therapeutic options have less evidence but warrant discussion given the above limitations.
The current available data do not support the general use of alkylating agents in the treatment of FSGS in adults (4). Pilot studies in resistant patients using mycophenolate mofetil (MMF) alone showed a low but significant response rate of approximately15%–20%. More recently, a randomized controlled trial in 138 children and adults with FSGS compared MMF plus oral pulse dexamethasone to cyclosporine A (CSA) for 1 year (41). At the end of the 52 weeks, there was no statistical difference in remissions (complete plus partial: 46% CSA versus 33% MMF/dexamethasone) but the 95% confidence interval was wide (0.3 to 1.18), leaving open the possibility of missing a substantial benefit of CSA. Part of this problem in interpretation is related to the study reaching only 30% of its prerequisite sample size.
Rituximab therapy has been tried in small and uncontrolled studies in FSGS (42). The pediatric literature suggests a significant response rate but the adult literature is more mixed.
Plasma exchange focused on removing the permeability factor has also been advocated as adjunctive therapy.
Although the KDIGO guideline on GN indicates that there is insufficient evidence to support the use of alkylating agents, MMF, or rituximab in the treatment of FSGS, these drugs may have a role in patients who are resistant or intolerant to conventional treatment. A practical algorithm for consideration in the treatment of FSGS is provided (Figure 2). The newer but more experimental agents discussed below eventually may also play a role in resistant or intolerant patients.
Figure 2.
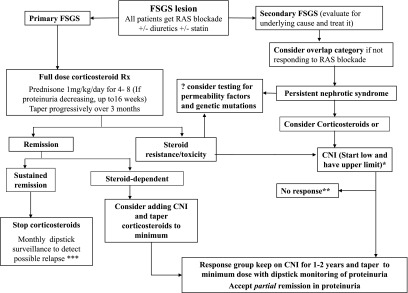
Treatment algorithm for FSGS. *CNI dose as per the KDIGO guidelines on GN. **See the text on options to consider in the management of nonresponders. ***See the text on how to manage relapse. RAS, renin-angiotensin system; CNI, calcineurin inhibitor; MMF, mycophenolate mofetil.
Galactose has a high affinity for permeability factors and could theoretically abolish plasma permeability activity in vivo. There are case reports in which oral galactose in combination with CNIs has achieved remission in FSGS patients who were resistant to currently available treatment (43). Certainly, a significant benefit to this approach is its benign side effect profile.
Pirfenidone is an oral antifibrotic agent. In an open-label trial involving 18 patients with primary or presumed secondary FSGS who received pirfenidone, participants showed an improved monthly change in GFR from a median of −0.61 ml/min per 1.73 m2 per month during the baseline period to −0.45 ml/min per 1.73 m2 per month. However, no effect on BP or proteinuria was observed after a median 13 months of treatment (44).
In a phase I trial, patients treated with adalimumab (a TNF inhibitor with antifibrotic properties) and rosiglitazone (an antidiabetic medication but with antifibrotic properties) showed improvement in GFR over time; however, this study was only 16 weeks and efficacy remains untested (45).
Deoxyspergualin derivative LF15-0195 ameliorated proteinuria in both a native kidney and a post-transplant animal model of FSGS by inducing regulatory T cells (46). No comparable human studies have been done.
Sirolimus, a mammalian target of rapamycin inhibitor, has been cited in anecdotal case reports to be effective in the treatment of FSGS; however, this drug can impair podocyte integrity and predispose patients to glomerular injury and is currently not recommended (47).
Adrenocorticotropic hormone (ACTH) therapy has been studied in the treatment of FSGS. In a pilot study of 11 FSGS patients treated with 16 weeks of subcutaneous ACTH, 6 patients had a reduction in proteinuria, with 2 patients achieving partial remission and 1 achieving complete remission (R.A.R.C. Lafayette and K. Mehta, unpublished observations). An open-label nonrandomized clinical trial is currently underway to investigate whether ACTH therapy is effective in primary FSGS (ClinicalTrials.gov identifier: NCT01155141).
Treatment of Secondary FSGS.
Management of secondary causes of the FSGS lesion involves treating the underlying condition. In large part, this is focused on reducing systemic and intraglomerular pressure using renin-angiotensin system inhibitors as first-line treatment. Their benefit on both proteinuria and disease progression in FSGS is quite variable; however, the risks are low, hence their recommendation. Obese patients need to lose weight to help control their proteinuria. Case series have shown that bariatric surgery tends to improve proteinuria in patients with obesity-related FSGS lesions (48).
Management of FSGS in the Overlap Group of Primary and Secondary FSGS.
Which patients with recognized factors associated with secondary FSGS should be considered for immunosuppressive therapy remains an important question. Unfortunately, there is little evidence on which to base the answer. This is in part because of the multiple unknown factors affecting the initiation and progression of the process. Perhaps a short course of immunosuppression should be considered for obese patients and for adults and older children with FSGS and a known mutation who, despite ideal BP and weight control and maximum renin-angiotensin system blockade for 3–6 months, continue to have nephrotic range proteinuria. Another scenario that may provide a clue to an underlying dysregulated autoimmune process is the rapid onset of full-blown nephrotic syndrome despite a documented potential secondary cause. Immunosuppressive treatment may be effective under these conditions.
If there is a good clinical response in proteinuria, the course could be continued similar to patients with more clear-cut primary FSGS. However, if there is no response, the treatment should be terminated early instead of the prolonged duration currently recommended. The choice of which drug and the duration of treatment should be determined by the specific clinical scenario (49).
Disclosures
None.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Haas M, Meehan SM, Karrison TG, Spargo BH: Changing etiologies of unexplained adult nephrotic syndrome: A comparison of renal biopsy findings from 1976-1979 and 1995-1997. Am J Kidney Dis 30: 621–631, 1997 [DOI] [PubMed] [Google Scholar]
- 2.Fuchshuber A, Gribouval O, Ronner V, Kroiss S, Karle S, Brandis M, Hildebrandt F: Clinical and genetic evaluation of familial steroid-responsive nephrotic syndrome in childhood. J Am Soc Nephrol 12: 374–378, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nürnberg G, Garg P, Verma R, Chaib H, Hoskins BE, Ashraf S, Becker C, Hennies HC, Goyal M, Wharram BL, Schachter AD, Mudumana S, Drummond I, Kerjaschki D, Waldherr R, Dietrich A, Ozaltin F, Bakkaloglu A, Cleper R, Basel-Vanagaite L, Pohl M, Griebel M, Tsygin AN, Soylu A, Müller D, Sorli CS, Bunney TD, Katan M, Liu J, Attanasio M, O’toole JF, Hasselbacher K, Mucha B, Otto EA, Airik R, Kispert A, Kelley GG, Smrcka AV, Gudermann T, Holzman LB, Nürnberg P, Hildebrandt F: Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 38: 1397–1405, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Kidney DiseaseImproving Global Outcomes (KDIGO) Glomerulonephritis Work Group: KDIGO Clinical Practice Guideline for Glomerulonephritis. Kidney Int Suppl 2: 139–274, 2012 [Google Scholar]
- 5.Korbet SM: Clinical picture and outcome of primary focal segmental glomerulosclerosis. Nephrol Dial Transplant 14[Suppl 3]: 68–73, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Kitiyakara C, Eggers P, Kopp JB: Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am J Kidney Dis 44: 815–825, 2004 [PubMed] [Google Scholar]
- 7.D’Agati VD, Fogo AB, Bruijn JA, Jennette JC: Pathologic classification of focal segmental glomerulosclerosis: A working proposal. Am J Kidney Dis 43: 368–382, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Deegens JK, Steenbergen EJ, Borm GF, Wetzels JF: Pathological variants of focal segmental glomerulosclerosis in an adult Dutch population—epidemiology and outcome. Nephrol Dial Transplant 23: 186–192, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Stokes MB, Valeri AM, Markowitz GS, D’Agati VD: Cellular focal segmental glomerulosclerosis: Clinical and pathologic features. Kidney Int 70: 1783–1792, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Stokes MB, Markowitz GS, Lin J, Valeri AM, D’Agati VD: Glomerular tip lesion: A distinct entity within the minimal change disease/focal segmental glomerulosclerosis spectrum. Kidney Int 65: 1690–1702, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Barisoni L, Schnaper HW, Kopp JB: A proposed taxonomy for the podocytopathies: A reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol 2: 529–542, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Cattran D, Neogi T, Sharma R, McCarthy ET, Savin VJ: Serial estimates of serum permeability activity and clinical correlates in patients with native kidney focal segmental glomerulosclerosis. J Am Soc Nephrol 14: 448–453, 2003 [DOI] [PubMed] [Google Scholar]
- 13.McCarthy ET, Sharma M, Savin VJ: Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 5: 2115–2121, 2010 [DOI] [PubMed] [Google Scholar]
- 14.McCarthy ET, Sharma M, Sharma R, Falk RJ, Jennette JC: Sera from patients with collapsing focal segmental glomerulosclerosis increase albumin permeability of isolated glomeruli. J Lab Clin Med 143: 225–229, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Gladson CL, Cheresh DA: Glioblastoma expression of vitronectin and the alpha v beta 3 integrin. Adhesion mechanism for transformed glial cells. J Clin Invest 88: 1924–1932, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, Maiguel D, Karumanchi SA, Yap HK, Saleem M, Zhang Q, Nikolic B, Chaudhuri A, Daftarian P, Salido E, Torres A, Salifu M, Sarwal MM, Schaefer F, Morath C, Schwenger V, Zeier M, Gupta V, Roth D, Rastaldi MP, Burke G, Ruiz P, Reiser J: Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med 17: 952–960, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Backes Y, van der Sluijs KF, Mackie DP, Tacke F, Koch A, Tenhunen JJ, Schultz MJ: Usefulness of suPAR as a biological marker in patients with systemic inflammation or infection: A systematic review. Intensive Care Med 38: 1418–1428, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koch A, Voigt S, Kruschinski C, Sanson E, Dückers H, Horn A, Yagmur E, Zimmermann H, Trautwein C, Tacke F: Circulating soluble urokinase plasminogen activator receptor is stably elevated during the first week of treatment in the intensive care unit and predicts mortality in critically ill patients. Crit Care 15: R63, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu J, Hasselbacher K, Hangan D, Ozaltin F, Zenker M, Hildebrandt F; Arbeitsgemeinschaft für Paediatrische Nephrologie Study Group: Nephrotic syndrome in the first year of life: Two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 119: e907–e919, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Putaala H, Soininen R, Kilpeläinen P, Wartiovaara J, Tryggvason K: The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet 10: 1–8, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Pereira AC, Pereira AB, Mota GF, Cunha RS, Herkenhoff FL, Pollak MR, Mill JG, Krieger JE: NPHS2 R229Q functional variant is associated with microalbuminuria in the general population. Kidney Int 65: 1026–1030, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Löwik M, Levtchenko E, Westra D, Groenen P, Steenbergen E, Weening J, Lilien M, Monnens L, van den Heuvel L: Bigenic heterozygosity and the development of steroid-resistant focal segmental glomerulosclerosis. Nephrol Dial Transplant 23: 3146–3151, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB: A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Zenker M, Aigner T, Wendler O, Tralau T, Müntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wühl E, Cochat P, Bouvier R, Kraus C, Mark K, Madlon H, Dötsch J, Rascher W, Maruniak-Chudek I, Lennert T, Neumann LM, Reis A: Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet 13: 2625–2632, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Genovese G, Tonna SJ, Knob AU, Appel GB, Katz A, Bernhardy AJ, Needham AW, Lazarus R, Pollak MR: A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int 78: 698–704, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR: Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipkowitz MS, Freedman BI, Langefeld CD, Comeau ME, Bowden DW, Kao WH, Astor BC, Bottinger EP, Iyengar SK, Klotman PE, Freedman RG, Zhang W, Parekh RS, Choi MJ, Nelson GW, Winkler CA, Kopp JB; SK Investigators: Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int 83: 114–120, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fogo A, Breyer JA, Smith MC, Cleveland WH, Agodoa L, Kirk KA, Glassock R; AASK Pilot Study Investigators: Accuracy of the diagnosis of hypertensive nephrosclerosis in African Americans: A report from the African American Study of Kidney Disease (AASK) Trial. Kidney Int 51: 244–252, 1997 [DOI] [PubMed] [Google Scholar]
- 29.Rood IM, Deegens JK, Wetzels JF: Genetic causes of focal segmental glomerulosclerosis: Implications for clinical practice. Nephrol Dial Transplant 27: 882–890, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Adelman RD, Restaino IG, Alon US, Blowey DL: Proteinuria and focal segmental glomerulosclerosis in severely obese adolescents. J Pediatr 138: 481–485, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Chen HM, Liu ZH, Zeng CH, Li SJ, Wang QW, Li LS: Podocyte lesions in patients with obesity-related glomerulopathy. Am J Kidney Dis 48: 772–779, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Kambham N, Markowitz GS, Valeri AM, Lin J, D’Agati VD: Obesity-related glomerulopathy: an emerging epidemic. Kidney Int 59: 1498–1509, 2001 [DOI] [PubMed] [Google Scholar]
- 33.Juncos LA, Ito S: Disparate effects of insulin on isolated rabbit afferent and efferent arterioles. J Clin Invest 92: 1981–1985, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verani RR: Obesity-associated focal segmental glomerulosclerosis: Pathological features of the lesion and relationship with cardiomegaly and hyperlipidemia. Am J Kidney Dis 20: 629–634, 1992 [DOI] [PubMed] [Google Scholar]
- 35.Wolf G, Hamann A, Han DC, Helmchen U, Thaiss F, Ziyadeh FN, Stahl RA: Leptin stimulates proliferation and TGF-beta expression in renal glomerular endothelial cells: potential role in glomerulosclerosis. Kidney Int 56: 860–872, 1999 [DOI] [PubMed] [Google Scholar]
- 36.Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC; Toronto Glomerulonephritis Registry Group: Focal and segmental glomerulosclerosis: Definition and relevance of a partial remission. J Am Soc Nephrol 16: 1061–1068, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Ordoñez JD, Hiatt RA, Killebrew EJ, Fireman BH: The increased risk of coronary heart disease associated with nephrotic syndrome. Kidney Int 44: 638–642, 1993 [DOI] [PubMed] [Google Scholar]
- 38.Abbate M, Zoja C, Remuzzi G: How does proteinuria cause progressive renal damage? J Am Soc Nephrol 17: 2974–2984, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P: The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14: 931–938, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, Li J, Mattiazzi A, Ciancio G, Chen L, Zilleruelo G, Abitbol C, Chandar J, Seeherunvong W, Ricordi C, Ikehata M, Rastaldi MP, Reiser J, Burke GW, 3rd: Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med 3: 85ra46, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gipson DS, Trachtman H, Kaskel FJ, Greene TH, Radeva MK, Gassman JJ, Moxey-Mims MM, Hogg RJ, Watkins SL, Fine RN, Hogan SL, Middleton JP, Vehaskari VM, Flynn PA, Powell LM, Vento SM, McMahan JL, Siegel N, D’Agati VD, Friedman AL: Clinical trial of focal segmental glomerulosclerosis in children and young adults. Kidney Int 80: 868–878, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernandez-Fresnedo G, Segarra A, González E, Alexandru S, Delgado R, Ramos N, Egido J, Praga M; Trabajo de Enfermedades Glomerulares de la Sociedad Española de Nefrología (GLOSEN): Rituximab treatment of adult patients with steroid-resistant focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 4: 1317–1323, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savin VJ, McCarthy ET, Sharma R, Charba D, Sharma M: Galactose binds to focal segmental glomerulosclerosis permeability factor and inhibits its activity. Transl Res 151: 288–292, 2008 [DOI] [PubMed] [Google Scholar]
- 44.Cho ME, Smith DC, Branton MH, Penzak SR, Kopp JB: Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2: 906–913, 2007 [DOI] [PubMed] [Google Scholar]
- 45.Peyser A, Machardy N, Tarapore F, Machardy J, Powell L, Gipson DS, Savin V, Pan C, Kump T, Vento S, Trachtman H: Follow-up of phase I trial of adalimumab and rosiglitazone in FSGS: III. Report of the FONT study group. BMC Nephrol 11: 2, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Berre L, Bruneau S, Naulet J, Renaudin K, Buzelin F, Usal C, Smit H, Condamine T, Soulillou JP, Dantal J: Induction of T regulatory cells attenuates idiopathic nephrotic syndrome. J Am Soc Nephrol 20: 57–67, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cho ME, Hurley JK, Kopp JB: Sirolimus therapy of focal segmental glomerulosclerosis is associated with nephrotoxicity. Am J Kidney Dis 49: 310–317, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Ramírez J, Carpio D, Mezzano S, Mukdsi J, Ardiles L: [Bariatric surgery in patients with focal segmental glomerulosclerosis secondary to obesity]. Nefrologia 29: 266–269, 2009 [DOI] [PubMed] [Google Scholar]
- 49.Radhakrishnan J, Cattran DC: The KDIGO practice guideline on glomerulonephritis: Reading between the (guide)lines—application to the individual patient. Kidney Int 82: 840–856, 2012 [DOI] [PubMed] [Google Scholar]
- 50.Wei C, Trachtman H, Li J, Dong C, Friedman AL, Gassman JJ, McMahan JL, Radeva M, Heil KM, Trautmann A, Anarat A, Emre S, Ghiggeri GM, Ozaltin F, Haffner D, Gipson DS, Kaskel F, Fischer DC, Schaefer F, Reiser J; PodoNet and FSGS CT Study Consortia: Circulating suPAR in two cohorts of primary FSGS. J Am Soc Nephrol 23: 2051–2059, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang J, Liu G, Zhang YM, Cui Z, Wang F, Liu XJ, Chu R, Chen Y, Zhao MH: Plasma soluble urokinase receptor levels are increased but do not distinguish primary from secondary focal segmental glomerulosclerosis. Kidney Int 84: 366–372, 2013 [DOI] [PubMed] [Google Scholar]