

Abstract
Background and objectives
The genetic cause of medullary cystic kidney disease type 1 was recently identified as a cytosine insertion in the variable number of tandem repeat region of MUC1 encoding mucoprotein-1 (MUC1), a protein that is present in skin, breast, and lung tissue, the gastrointestinal tract, and the distal tubules of the kidney. The purpose of this investigation was to analyze the clinical characteristics of families and individuals with this mutation.
Design, setting, participants, & measurements
Families with autosomal dominant interstitial kidney disease were referred for genetic analysis over a 14-year period. Families without UMOD or REN mutations prospectively underwent genotyping for the presence of the MUC1 mutation. Clinical characteristics were retrospectively evaluated in individuals with the MUC1 mutation and historically affected individuals (persons who were both related to genetically affected individuals in such a way that ensured that they could be genetically affected and had a history of CKD stage IV or kidney failure resulting in death, dialysis, or transplantation).
Results
Twenty-four families were identified with the MUC1 mutation. Of 186 family members undergoing MUC1 mutational analysis, the mutation was identified in 95 individuals, 91 individuals did not have the mutation, and111 individuals were identified as historically affected. Individuals with the MUC1 mutation suffered from chronic kidney failure with a widely variable age of onset of end stage kidney disease ranging from 16 to >80 years. Urinalyses revealed minimal protein and no blood. Ultrasounds of 35 individuals showed no medullary cysts. There were no clinical manifestations of the MUC1 mutation detected in the breasts, skin, respiratory system, or gastrointestinal tract.
Conclusion
MUC1 mutation results in progressive chronic kidney failure with a bland urinary sediment. The age of onset of end stage kidney disease is highly variable, suggesting that gene–gene or gene–environment interactions contribute to phenotypic variability.
Introduction
There are three types of autosomal dominant interstitial kidney disease (ADIKD). Medullary cystic kidney disease (MCKD) type 2 (Online Mendelian Inheritance in Man [OMIM] 162000) is caused by mutations in the UMOD gene and results in juvenile hyperuricemia, frequent gout, and progressive CKD (1). Mutations in the REN gene (OMIM 613092) encoding renin cause childhood anemia, hyperuricemia, mild hyperkalemia, and slowly progressive CKD (2). MCKD1 (OMIM 174000) is characterized by a bland urinary sediment and no other associated features other than progressive CKD (3). Linkage of MCKD1–1q21 was first shown in the work by Christodoulou et al. (4) in 1998, with refinement of the genetic locus shown in the works by Fuchshuber et al. (5), Wolf et al. (6), and Wolf et al. (7). Kirby et al. (8) identified a mutation in MUC1 (OMIM 158340) encoding mucoprotein-1 (MUC1) as the cause of MCKD1. MUC1 is a membrane-anchored mucoprotein expressed in the skin, breast, lung, gastrointestinal tract, salivary glands, and renal distal tubular cells (9). The mucoprotein provides a protective layer to cell surfaces and may be involved in signaling (10).
Before identification of the genetic cause of MCKD1, it was difficult to clinically characterize this disorder. Identification of the MUC1 etiology for MCKD1 permits us, for the first time, to provide a clinical characterization of individuals with known MCKD1, including individuals who have mild disease and in whom diagnosis could not be ascertained in the past. The only prior detailed clinical characterizations of this condition were in six families from Cyprus (11) and one Native American family (12) linked to chromosome 1. In this manuscript, we describe 24 families with an MUC1 mutation. Our results show a diverse clinical presentation within and between families with regards to the progression of CKD.
The identification of a mutation in MUC1 as the cause of MCKD1 was surprising, because MUC1 is widely expressed. With identification of a mutation in MUC1 as a cause of this condition, we were able to more closely examine if occult clinical expression in other organ systems was present.
Materials and Methods
A registry of families with inherited interstitial kidney disease had been constructed by A.J.B. and V.R. over 14 years, with the referral of approximately 400 families by physicians and/or family members. Families were screened by A.J.B., and medical records were obtained from these families. Information collected included demographics, family pedigree, age of ESRD, laboratory values, and ultrasound results. If appropriate, DNA was obtained and analyzed for UMOD and REN gene mutations by S.K. and P.S.H. If mutations were not found in these genes, linkage analysis was performed. In this manner, six large families with linkage to chromosome 1 were identified (Table 1, families L1–L6), including a family (L3) linked to chromosome 1 in the work by Kiser et al. (12), another family (L5) linked to chromosome 1 in the work by Kimmel et al. (13), and four kindreds (L1, L2, L4, and L6) linked by P.S.H. Samples were sent to the Broad Institute in Boston, Massachusetts for genetic analysis (8). Southern blots were used to identify mutations in the variable number of tandem repeat (VNTR)–coding region of MUC1 in the initial six families linked to chromosome 1. All families were found to have an additional cytosine insertion to a string of seven cytosines. A probe extension assay capable of distinguishing reference and mutant MUC1 VNTR repeat units was then used to specifically identify this same mutation (a cytosine insertion after a string of seven cytosines) in other families (8). Because of the extreme technical difficulties in the mutational analysis of this gene (8), we did not test for other mutations in MUC1, and we were not able to identify the particular VNTR subunit in which the cytosine insertion occurred.
Table 1.
Characteristics of families with MUC1 mutation
| Family Numbera | Ethnicity | MUC1 Mutation (n) | Historically Affected (n) | No MUC1 Mutation (n) | Median Age of ESRD (yr) | Kidney Biopsy (n) | Kidney Ultrasound Available for Review (n) | Prior Linkage to Chromosome 1 |
|---|---|---|---|---|---|---|---|---|
| L1 | Middle Eastern | 11 | 11 | 16 | 42 | 1 | 0 | Yes |
| L2 | African American | 11 | 12 | 20 | 57 | 0 | 6 | Yes |
| L3 | Native American | 22 | 5 | 36 | 45.5 | 8 | 7 | Yes |
| L4 | European American | 7 | 9 | 6 | 60 | 3 | 2 | Yes |
| L5 | European American | 9 | 6 | 0 | 34 | 2 | 0 | Yes |
| L6 | European American | 4 | 5 | 6 | 41 | 0 | 2 | Yes |
| 1 | European American | 3 | 0 | 1 | 28.5 | 1 | 2 | Yes |
| 2 | European American | 3 | 2 | 0 | 67.5 | 2 | 3 | Yes |
| 3 | European American | 1 | 4 | 1 | 42 | 2 | 1 | No |
| 4 | European American | 1 | 5 | 0 | 31.5 | 2 | 1 | No |
| 5 | African American | 2 | 6 | 1 | 29 | 0 | 2 | No |
| 6 | Finnish | 1 | 19 | 0 | 52.5 | 1 | 1 | No |
| 7 | European American | 1 | 9 | 1 | 33.5 | 0 | 1 | No |
| 8 | European American | 1 | 1 | 0 | 31.5 | 0 | 0 | No |
| 9 | European American | 1 | 1 | 0 | 39 | 1 | 1 | No |
| 10 | European American | 2 | 0 | 0 | No members yet with ESRD | 1 | 0 | No |
| 11 | Australian | 2 | 1 | 0 | 31 | 2 | 0 | No |
| 12 | Eastern European | 1 | 0 | 0 | 29 | 1 | 0 | No |
| 13 | Eastern European | 1 | 0 | 0 | 44 | 1 | 0 | No |
| 14 | European American | 6 | 6 | 3 | 43.5 | 2 | 1 | No |
| 15 | Australian | 1 | 2 | 0 | 25 | 2 | 2 | No |
| 16 | Russian | 2 | 1 | 0 | 55 | 1 | 1 | No |
| 17 | Australian | 1 | 1 | 0 | 38 | 0 | 0 | No |
| 18 | Israeli | 1 | 5 | 0 | 39 | 1 | 2 | No |
Family number: families L1–L6 refer to the first six families that were linked to chromosome 1 and had mutational analysis performed. Family numbering corresponds to numbering in ref. 8.
Samples from 21 additional families with UMOD- and REN-negative ADIKD were analyzed for this specific MUC1 mutation. Two of these families (Table 1, families 1 and 2) had previously been linked to chromosome 1 (14) (Scheinman SJ et al., unpublished data). Also, more samples from family members from the first six families were analyzed. Individuals were considered affected (genetically affected) if they had an MUC1 mutation. Individuals were considered to be unaffected (genetically unaffected) if they were negative for an MUC1 mutation. Individuals were considered historically affected if genetic analysis could not be done and the individuals fulfilled two requirements: (1) they were related to genetically affected individuals in such a way that ensured that they could be genetically affected, and (2) they had an estimated GFR (eGFR; calculated throughout this work according to the Chronic Kidney Disease Epidemiology Collaboration equation) (15) that placed them in CKD stage IV or they had a history of kidney failure resulting in death, dialysis, or transplantation. The age of ESRD was calculated as the age of dialysis, kidney transplantation, or death from kidney failure.
Statistical analysis was performed with SAS, version 9.3 (Cary, NC). To determine change in eGFR over time for patients with eGFR≥50 or <50 ml/min per 1.73 m2, the difference in eGFR between the two measurements with the greatest time interval between them was used, with an interval of at least 1 year between measurements; 50 ml/min per 1.73 m2 was chosen arbitrarily as a cutoff point after visual inspection of the data showed a steep decline in eGFR in these individuals.
To determine if sex was associated with age of onset of ESRD, a univariate model was created with age of ESRD as the dependent variable and sex as the independent variable.
To compare the age of starting dialysis between affected parents and their children, mixed models (using PROC MIXED) were used to fit a regression between parent and child age of onset of ESRD (controlling for within-family clustering).
This study was approved by the Wake Forest School of Medicine Institutional Review Board, and it adhered to the Declaration of Helsinki. Informed consent was obtained from all individuals.
Results
Eight of eight families linked to chromosome 1 (L1–L6, 1, and 2) were found to have an MUC1 mutation resulting in a cytosine insertion after a string of seven cytosines; 16 of 19 families with UMOD- and REN-negative ADIKD who had not been linked to chromosome 1 were found to have this same type of MUC1 mutation.
Of 186 individuals undergoing MUC1 genetic analysis, the MUC1 mutation was identified in 95 individuals, and 91 individuals did not have the MUC1 mutation. There were 111 individuals who were historically affected. There was one genetically unaffected individual with ESRD: this African American patient had a long history of nephrotic proteinuria and was considered a priori not to have MCKD1. Table 1 lists characteristics of the individual families.
Of 147 individuals who developed ESRD, ESRD developed in 1 individual (0.68%) at age<20 years, 24 (16.3%) individuals between 20 and 30 years, 72 (49.0%) individuals between 30 and 50 years, 37 (25.2%) individuals between 50 and 70 years, and 13 (8.8%) individuals at ≥70 years.
Figure 1 shows the age of onset of ESRD according to family. There seemed to be two groups of families: one group with a younger median age of ESRD and all family members on dialysis before age 50 years, and one group with a highly variable age of ESRD for individual family members and a higher median age of ESRD. In three families (5, 9, and 11), clinical information strongly suggested a de novo mutation (Figure 2, A–C).
Figure 1.
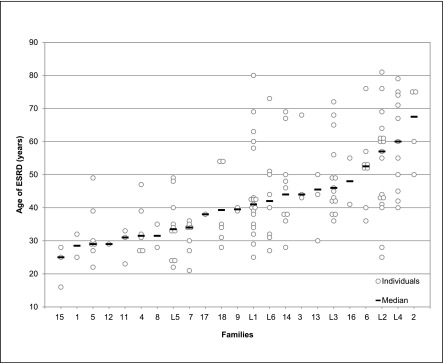
Age of onset of ESRD for individuals from families with the MUC1 mutation. (This figure does not include family members who have not developed ESRD. One family has no members with ESRD and, thus, is not represented here.)
Figure 2.

Representative pedigrees from families with the MUC1 mutation. Individuals with the MUC1 mutation are represented with black symbols. Historically affected individuals are denoted with dark gray symbols. Individuals with unknown status are shown with light gray symbols. Age of ESRD (years) is given below affected individuals. The proband for each family is indicated with an arrow. A–C show families with new mutations. The age (years) of death of individuals in generation I is given below the symbol. D shows a family with earlier onset of ESRD in succeeding generations.
We then analyzed eGFR values for affected and unaffected family members. Figure 3 shows the highest available measurement of eGFR at any time point for affected individuals and the lowest available eGFR measurement for unaffected individuals from families with MUC1 mutation to determine how well eGFR differentiated between the groups. For individuals in whom only the ESRD age and no serum creatinine values were available, the eGFR at the start of dialysis was approximated as 5 ml/min per 1.73 m2. Like in Figure 1, Figure 3 shows a wide variation in eGFR over time for affected individuals; some individuals had relatively preserved kidney function into their eighth decade, and other individuals required dialysis in their 20s. There is clear overlap between affected individuals and unaffected individuals caused by the presence of stages II and III kidney failure in a number of affected and unaffected individuals.
Figure 3.
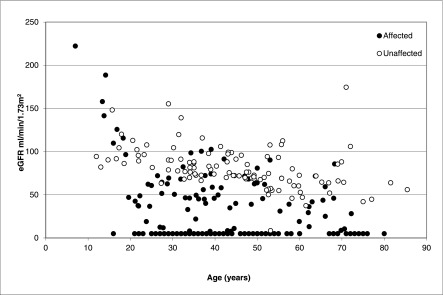
Estimated GFR (eGFR) versus age for individuals in families with the MUC1 mutation. This graph shows the highest available eGFR measurement for affected individuals and the lowest available eGFR measurement for unaffected individuals to determine overlap. Patients in whom only the age of ESRD is available are depicted with an eGFR value of 5 ml/min per 1.73 m2. There is considerable overlap between groups.
We analyzed the rate of decline in eGFR in affected individuals. In examination of the data, we noted that there were many measurements obtained by the patients’ nephrologists when the eGFR was below 50 ml/min per 1.73 m2, and the decline seemed quite rapid. We decided to arbitrarily analyze values ≥50 and <50 ml/min per 1.73 m2. Figure 4 displays progression of kidney disease in all 11 affected individuals in whom we had at least 10 measurements of eGFR<50 ml/min per 1.73 m2. In available data, 22 affected individuals with an eGFR≥50 ml/min per 1.73 m2 had a mean eGFR change of 0.99±6 ml/min per 1.73 m2 per year versus a mean eGFR change of −6.7±4.2 ml/min per 1.73 m2 per year for 29 affected individuals with an eGFR≤50 ml/min per 1.73 m2 (P<0.001).
Figure 4.

Decline in eGFR over time for individuals with more than 10 eGFR measurements. Decline was rapid when eGFR was <50 ml/min per 1.73 m2. The family to which each individual belongs is identified (family numbers correspond to Table 1).
We next evaluated whether ESRD developed at younger ages for successive generations. Figure 2D depicts a family in which this development seems to have occurred. To further evaluate the possibility, a graph was created (Figure 5), with parental age of ESRD on the y axis and the age of ESRD for the child on the x axis. There were 63 of 111 family pairs (57%) in which the parents were older at the start of ESRD, and there were 45 of 111 family pairs (41%) in which the child started at an older age than the parent. Using mixed models that accounted for clustering within families, the mean difference between parent and child age of starting ESRD was estimated at 4.9 years (SEM=2.0 years; parents being older when ESRD developed: 48.0±2.1 versus 43.1±2.1 years, P=0.02).
Figure 5.

Age of onset of ESRD for affected parents versus children. Parental age of ESRD is plotted versus the age of ESRD for the affected child. The line represents where parental age of ESRD would equal the child’s age of ESRD. Points that are above the line represent parental ESRD age being older than the child ESRD age. Points below the line represent parental ESRD age being lower than the child ESRD age.
We then determined whether sex was associated with variation in the age of ESRD. In a univariate model with age of ESRD as the outcome variable, sex was not associated with the age of onset of ESRD (P=0.62). Only two affected individuals also suffered from diabetes mellitus. Their rate of progression seemed consistent with other affected individuals, with ESRD at 43 and 45 years.
Table 2 shows clinical characteristics of individuals with MCKD1. Gout was identified in 30 of 127 individuals (24%) in whom sufficient clinical information was available to ascertain the presence of gout. Of 27 patients who eventually required dialysis and had gout, 3 (11%) patients had gout more than 10 years before kidney failure, 7 (26%) patients had gout 6–9 years before kidney failure, and 17 (63%) patients had gout occurring within 5 years of dialysis or after starting dialysis. The eGFR was less than 50 ml/min per 1.73 m2 in seven of eight (88%) patients in whom eGFR data were available at the time of gout.
Table 2.
Characteristics of affected individuals
| Characteristic | Number of Individuals with the Characteristic Evaluated | Value |
|---|---|---|
| N | 206 | |
| Sex (% men) | 206 | 100 (48.5%) |
| Age of ESRD (yr)a | 147 | 44.9±15.4 |
| Range of age of ESRD (yr) | 147 | 16–81 |
| Age of ESRD for women (yr) | 79 | 44.4±16.1 |
| Age of ESRD for men (yr) | 68 | 45.6±14.8 |
| Receiving antihypertensive therapy (%) | 70 | 43 (61.4%) |
| Systolic BP (mmHg) | 83 | 126±18.7 |
| Diastolic BP (mmHg) | 83 | 76.0±11.1 |
| Gout (%) | 127 | 30 (24%) |
| Serum uric acid level for individuals not on allopurinol | 43 | 7.4±1.89 |
| Renal cysts on imaging (%) | 27 | 48.15 |
| Medullary cysts on imaging (%) | 27 | 0 |
| Urinalysis dipstick for protein negative | 29 | 23 (79%) |
Continuous variables are represented as mean±SD.
In urinalyses from 29 individuals with MUC1 mutation, urine protein was negative in 23 (79%) individuals, trace in 2 individuals, 1+ in 3 individuals, and 2+ in 1 individual. The urine dipstick was negative for blood in 22 individuals and trace positive in 7 individuals. In ultrasounds from 35 individuals, kidneys ranged from normal size to small as CKD progressed. Twenty-one patients (60%) had no cysts. Of the total 70 kidneys, 49 (70%) kidneys had no cysts, 9 (13%) kidneys had 1 cyst, and 12 (17%) kidneys had 2 or more cysts. There were no medullary cysts identified in any ultrasounds. MCKD was not suspected in any ultrasounds. There were 34 renal biopsies performed that all showed tubulointerstitial kidney disease. In four (12%) individuals, microcystic dilation of the tubules was noted; in two of these cases, MCKD was suggested as a cause; in one case, polycystic kidney disease was suggested as a cause, and in one case, a toxic injury to the tubules was suggested as a cause. There was no increased incidence of polyuria, renal calculi, or urinary tract infections in affected individuals.
Review of records did not reveal an increased frequency of any clinical pathophysiology of the lungs, breasts, or gastrointestinal tract—tissues in which MUC1 is highly expressed. There were no instances of chronic obstructive pulmonary disease or other lung disease, lung cancer, gastric ulcers, or gastric cancer. Records were obtained on 17 women who had undergone mammograms. Of these women, 7 women had negative results, and 10 women were called back for additional evaluation. Of these 10 women, there were five biopsies; of these five biopsies, two individuals had cancer. Of 99 affected women for whom we have records, only 2 women were identified as having breast cancer.
Discussion
One cannot fully appreciate the clinical spectrum of a genetic disease until the mutation causing the disease has been identified. Identification of an MUC1 mutation in MCKD1 enables us to considerably increase our knowledge about the clinical manifestations of MCKD1, particularly for mild manifestations.
The mutation causing MCKD1 is in MUC1. MUC1 has a repetitive sequence of 60 nt that occurs 20–125 times (VNTR domain). This sequence encodes for a corresponding serine- and threonine-rich amino acid domain of MUC1. Glycosylation of the VNTR determines the mucinous properties of the protein. In all families identified in this work, the MUC1 mutation originates from the insertion in 1 of 60 nucleotide repeat units of a single cytosine within a consecutive sequence of seven cytosines. The mutation encodes within the VNTR domain for a shift in MUC1 transcript translation and results in proteosynthesis of an MUC1 neoprotein that contains a novel repeat and a C-terminal amino acid sequence lacking characteristic protein post-translational processing, transmembrane anchoring, and intracellular signaling domains of the wild-type MUC1.
For the first six families investigated, full mutational analysis of MUC1 was performed. Because of the intrinsic, laborious difficulties of screening the entire VNTR, the remaining families were only examined for mutations that would have included an extra cytosine residue (8). A high proportion (16 of 19; 84%) of families with ADIKD and no mutations in UMOD and REN was found to have a cytosine insertion in the VNTR of MUC1. Thus, it is important to screen for this mutation in families with UMOD- and REN-negative ADIKD. The three families that tested negative for the MUC1 insertion were too small for linkage analysis. It is possible that their condition results from a different mutation in MUC1 or mutation of another gene. However, the vast majority of individuals with this condition have the cytosine insertion in the VNTR of MUC1. These findings suggest that the neoprotein that is created because of the cytosine insertion is central to the pathogenesis of the disease. Moreover, the presence of clinical changes only in the kidney is suggestive that this neoprotein has a specific deleterious effect in renal tubular cells.
It was also not possible to determine in which particular VNTR subunit the mutation occurred. It is likely that the mutation occurs in different subunits for each family, although each family will have the mutation in the same VNTR subunit for all affected family members.
The most important findings of this study were the variable age of onset of ESRD within and between families, and the rapid progression to dialysis in the third decade of life in several families. Previously, families with early onset kidney failure had not been identified, because these families are smaller (because of limited time for child-bearing as a result of ESRD) and could not provide definitive genetic linkage. The site of the mutation (in an earlier versus later portion of the VNTR) might be responsible for this difference, although, at this time, it has not yet been possible to determine the precise position of the mutation for the individual families.
In some genetic conditions, the phenotype can worsen with succeeding generations. It was difficult to determine if ESRD occurred at younger ages with succeeding generations. In general, children proceeded to dialysis earlier than their affected parent. However, this finding was not always the case. The work by Christodoulou et al. (4) was the first to suggest early age of ESRD in succeeding generations in a study of six large Cypriot families that had shown DNA linkage to the MCKD1 locus. There is no biologic mechanism that has been identified that would suggest that the mutation under study would lead to this phenomenon. The temporal trends of starting patients on dialysis earlier or receiving transplantation earlier could be potentially responsible for these findings.
We found that the rate of eGFR decline seemed to be accelerated when the eGFR declined below 50 ml/min per 1.73 m2. This finding must be interpreted with caution. We arbitrarily chose to evaluate values ≥50 and <50 ml/min per 1.73 m2 after visual inspection of the data, and we did not have a large number of patients in our sample. Additional analyses will be required with a larger number of patients and evaluation in a prospective manner. However, clinicians may consider following laboratory values more closely when patients reach this level of eGFR until more data are available.
Like other investigators (11), we found that MCKD1 has a very limited clinical presentation. Urinalyses showed a bland sediment and minimal protein. Although MUC1 is produced in many tissues, we could not find evidence of clinical alterations of organs or tissues besides the kidney. The recall rate for mammograms was 58%, which was similar to the lifetime recall rate of 61.3% in a large study of mammogram screening (16). Renal ultrasounds showed occasional cysts, and medullary cysts were not identified in any patients. As initially described in the work by Hildebrandt and Omram (17), MCKD is a poor name for this condition, because cysts are often absent, and the misnomer may impede diagnosis of this condition. Kidney biopsy specimens did show tubular microcystic dilation in a small proportion (4 of 34) of biopsies.
There was considerable overlap in eGFR values between unaffected and affected members of families with MUC1 mutations. The high prevalence of stage III CKD in unaffected individuals may have been caused by a significant number of African Americans with stage III CKD, the use of the lowest available eGFR for unaffected individuals, and interlaboratory variation in serum creatinine determinations. These results show that it is not yet possible to clinically differentiate individuals without genetic testing, and there is no clinical method to diagnose MCKD1 or determine if families or family members are affected or unaffected.
Given the different characteristics of MCKD1, MCKD2, and REN mutations (Table 3), we propose a stepwise approach to genetic testing. In individuals with ADIKD, genetic testing for REN mutations should be performed if patients also suffer from anemia, mild hyperkalemia, and low normal BPs. If gout is a significant component of disease, mutational analysis of the UMOD gene should be performed. If these tests are negative or if gout, anemia, and mild hyperkalemia are absent, MUC1 testing should be done. In the future, tandem genetic analyses for all three conditions will likely become available. Currently, genetic testing is only available in a research setting, and we would be happy to discuss testing for families that may potentially be affected. Please contact A.J.B. with any questions in this regard.
Table 3.
Clinical characteristics of autosomal dominant interstitial kidney disease
| Characteristic | Medullary Cystic Kidney Disease Type 1 (Caused by Mutations in the MUC1 Gene) | Medullary Cystic Kidney Disease Type 2 (Caused by Mutations in the UMOD Gene) | Mutations in the REN Gene Encoding Renin |
|---|---|---|---|
| Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant |
| Urinary sediment | Bland | Bland | Bland |
| Urinary protein | Absent/minimal | Absent/minimal | Absent/minimal |
| Medullary cysts | Rare | Rare | Absent |
| Age of ESRD (yr) | Variable: late 20s to >70 | Variable: 30–60 | Variable: 40–70 |
| Gout in many affected family members (often occurring early in life) | Absent | Present | Present |
| Hyperkalemia and low BP (symptoms of low renin state) | Absent | Absent | Present |
| Anemia in childhood | Absent | Absent | Present |
In summary, MCKD1 is caused by a mutation in the MUC1 gene. Onset of ESRD is very variable between and within families. Although MUC1 is produced in many tissues, we could only find clinical abnormalities in the kidney.
Disclosures
None.
Acknowledgments
The authors thank Fabio Aglieco and Eric Brown for referral of families.
This research was supported, in part, by the Intramural Research Program of the US National Institutes of Health, National Human Genome Research Institute (NHGRI). S.K. was funded by Charles University Institutional Programs PRVOUK-P24/LF1/3, UNCE 204011, and SVV2013/266504. This work was specifically supported by Ministry of Education of the Czech Republic Grant LH12015 and Ministry of Health of the Czech Republic NT13116-4/2012.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
This article contains supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.06380613/-/DCSupplemental.
See related editorial, “Variable Presentations of Rare Genetic Renal Interstitial Diseases,” on pages 437–439.
References
- 1.Bleyer AJ, Zivná M, Kmoch S: Uromodulin-associated kidney disease. Nephron Clin Pract 118: c31–c36, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Zivná M, Hůlková H, Matignon M, Hodanová K, Vylet’al P, Kalbácová M, Baresová V, Sikora J, Blazková H, Zivný J, Ivánek R, Stránecký V, Sovová J, Claes K, Lerut E, Fryns JP, Hart PS, Hart TC, Adams JN, Pawtowski A, Clemessy M, Gasc JM, Gübler MC, Antignac C, Elleder M, Kapp K, Grimbert P, Bleyer AJ, Kmoch S: Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, and chronic kidney failure. Am J Hum Genet 85: 204–213, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bleyer AJ, Hart PS, Kmoch S: Hereditary interstitial kidney disease. Semin Nephrol 30: 366–373, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christodoulou K, Tsingis M, Stavrou C, Eleftheriou A, Papapavlou P, Patsalis P, Iounnou P, Pierides A, Constantinou Deltas C: Chromosome 1 localization of a gene for autosomal dominant medullary cystic kidney disease. Hum Mol Genet 7: 905–911, 1998 [DOI] [PubMed] [Google Scholar]
- 5.Fuchshuber A, Kroiss S, Karle S, Berthold S, Huck K, Burton C, Rahman N, Koptides M, Deltas C, Otto E, Rüschendorf F, Feest T, Hildebrandt F: Refinement of the gene locus for autosomal dominant medullary cystic kidney disease type 1 (MCKD1) and construction of a physical and partial transcriptional map of the region. Genomics 72: 278–284, 2001 [DOI] [PubMed] [Google Scholar]
- 6.Wolf MTF, Karle SM, Schwarz S, Anlauf M, Anlauf M, Glaeser L, Kroiss S, Burton C, Feest T, Otto E, Fuchshuber A, Hildebrandt F: Refinement of the critical region for MCKD1 by detection of transcontinental haplotype sharing. Kidney Int 64: 788–792, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Wolf MTF, van Vlem B, Hennies HC, Zalewski I, Karle SM, Puetz M, Panther F, Otto E, Fuchshuber A, Lameire N, Loeys B, Hildebrandt F: Telomeric refinement of the MCKD1 locus on chromosome 1q21. Kidney Int 66: 580–585, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Kirby A, Gnirke A, Jaffe DB, Barešová V, Pochet N, Blumenstiel B, Ye C, Aird D, Stevens C, Robinson JT, Cabili MN, Gat-Viks I, Kelliher E, Daza R, DeFelice M, Hůlková H, Sovová J, Vylet’al P, Antignac C, Guttman M, Handsaker RE, Perrin D, Steelman S, Sigurdsson S, Scheinman SJ, Sougnez C, Cibulskis K, Parkin M, Green T, Rossin E, Zody MC, Xavier RJ, Pollak MR, Alper SL, Lindblad-Toh K, Gabriel S, Hart PS, Regev A, Nusbaum C, Kmoch S, Bleyer AJ, Lander ES, Daly MJ: Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet 45: 299–303, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patton S, Gendler SJ, Spicer AP: The epithelial mucin, MUC1, of milk, mammary gland and other tissues. Biochim Biophys Acta 1241: 407–423, 1995 [DOI] [PubMed] [Google Scholar]
- 10.Pemberton LF, Rughetti A, Taylor-Papadimitriou J, Gendler SJ: The epithelial mucin MUC1 contains at least two discrete signals specifying membrane localization in cells. J Biol Chem 271: 2332–2340, 1996 [DOI] [PubMed] [Google Scholar]
- 11.Stavrou C, Koptides M, Tombazos C, Psara E, Patsias C, Zouvani I, Kyriacou K, Hildebrandt F, Christofides T, Pierides A, Deltas CC: Autosomal-dominant medullary cystic kidney disease type 1: Clinical and molecular findings in six large Cypriot families. Kidney Int 62: 1385–1394, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Kiser RL, Wolf MTF, Martin JL, Zalewski I, Attanasio M, Hildebrandt F, Klemmer P: Medullary cystic kidney disease type 1 in a large Native-American kindred. Am J Kidney Dis 44: 611–617, 2004 [PubMed] [Google Scholar]
- 13.Kimmel RJ, Kovacs I, Vrabel C, Wood B, Schalling M, Kelsoe JR: Cosegregation of bipolar disorder and autosomal-dominant medullary cystic kidney disease in a large family. Am J Psychiatry 162: 1972–1974, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Auranen M, Ala-Mello S, Turunen JA, Järvelä I: Further evidence for linkage of autosomal-dominant medullary cystic kidney disease on chromosome 1q21. Kidney Int 60: 1225–1232, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J, CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) : A new equation to estimate glomerular filtration rate. Ann Intern Med 150: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braithwaite D, Zhu W, Hubbard RA, O'Meara ES, Miglioretti DL, Geller B, Dittus K, Morre D, Wernli KJ, Mandelblatt J, Kerlikowske K, Breast Cancer Surveillance Consortium : Screening outcomes in older US women undergoing multiple mammograms in community practice: Does interval, age, or comorbidity score affect tumor characteristics or false positive rates. J Natl Cancer Inst 105: 334–341, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hildebrandt F, Omram H: New insights: Nephronophthisis-medullary cystic kidney disease. Pediatr Nephrol 16: 168–176, 2001 [DOI] [PubMed] [Google Scholar]