

Abstract
Membranoproliferative GN represents a pattern of injury seen on light microscopy. Historically, findings on electron microscopy have been used to further subclassify this pathologic entity. Recent advances in understanding of the underlying pathobiology have led to a proposed classification scheme based on immunofluorescence findings. Dysregulation of the complement system has been shown to be a major risk factor for the development of a membranoproliferative GN pattern of injury on kidney biopsy. Evaluation and treatment of this complex disorder rest on defining the underlying mechanisms.
Introduction
Membranoproliferative GN (MPGN), also termed mesangiocapillary GN, accounts for approximately 7%–10% of all cases of biopsy-confirmed GN (1,2). A light microscopic pattern of injury, MPGN occurs in both children and adults. The presentation is usually slowly progressive disease with hematuria and non-nephrotic proteinuria, but nephrotic syndrome and more severe presentations have been described as well. Among glomerular diseases, MPGN pathology has the most association with secondary causes, including viruses, autoimmune diseases, and paraproteins (Table 1) (3).
Table 1.
Immune complex-based conditions associated with membranoproliferative GN pattern of injury
| Autoimmune diseases |
| Systemic lupus |
| Sjogren’s syndrome |
| Rheumatoid arthritis |
| Mixed connective tissue disease |
| Infections |
| Hepatitis B |
| Hepatitis C |
| Endocarditis |
| Shunt infections |
| Visceral abscesses |
| Leprosy |
| Malaria |
| Schistosomiasis |
| Mycoplasma |
| Paraproteinemiasa |
| Monoclonal gammopathy of undetermined significance |
| Waldenstrom macroglobulemia |
| Chronic lymphocytic leukemia |
| Low-grade B-cell lymphoma |
| Cyroglobulinemia types 1 and 2 |
| Immunotactoid glomerulopathy |
| Monoclonal Ig deposition disease |
| Fibrillary glomerulopathy |
Some paraproteins (e.g., monoclonal globulins type I) can aggregate spontaneously and form nonimmune deposits (nonantigen–antibody complexes) in glomeruli.
Historical Classification
The term MPGN refers to the features noted on light microscopy (LM). It is usually characterized by mesangial hypercellularity, endocapillary proliferation, capillary wall remodeling, double contour formation, and duplication of basement membranes (Figure 1). Traditionally, MPGN has been further classified based on the findings of electron microscopy (EM) as primary MPGN type I (MPGN I), MPGN II, MPGN III, or secondary MPGN. MPGN I, the most common form, is characterized by subendothelial deposits, and MPGN III has both subepithelial and subendothelial deposits (4). MPGN II is characterized by dense deposits in the glomerular basement membrane (dense deposit disease [DDD]).
Figure 1.

Membranoproliferative GN (MPGN). Light microscopy hematoxylin & eosin stain. Reprinted from Glen Markowitz, with permission.
The newer classification (5–7) of MPGN is dependent on immunofluorescence (IF) staining. This pathophysiology-based classification is most useful to clinicians. Given recent advances in our understanding of the role of the alternative pathway of complement in MPGN, a practical approach is to view MPGN as either immune complex– or complement-mediated. Immune complex–mediated MPGN may occur when there are increased levels of circulating immune complexes, and complement-mediated MPGN may occur because of dysregulation of the alternative pathway of complement. If neither of these causes is present (null complement and null immune complex), then chronic thrombotic microangiopathy may be the cause of the MPGN (Figure 2). The older classification gave a diagnosis but not much insight into the pathophysiology of the disease (Table 2). Given the emergence of many secondary causes of MPGN, it is hard to quantify the true incidence of idiopathic MPGN in both children and adults.
Figure 2.
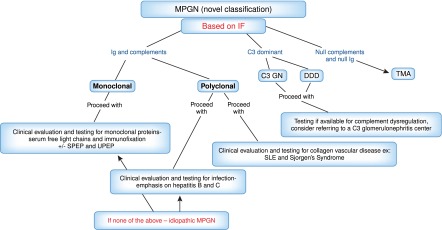
Simplistic breakdown of the new MPGN classification using immunofluorescence as the basis and an approach to evaluation when the kidney biopsy indicated MPGN. DDD, dense deposit disease; IF, immunofluorescence; SPEP, serum protein electrophoresis; TMA, thrombotic microangiopathy; UPEP, urine protein electrophoresis.
Table 2.
Older and newer classification of membranoproliferative GN
| Based on Electron Microscopy (Older) | Based on Immunofluorescence (Newer) | |
| Classification | MPGN 1: Subendothelial MPGN | Immune complex–mediated MPGN Igs and complements on IF (paraproteins, viruses, autoimmune) |
| MPGN 2: Dense subendothelial deposits (dense deposit disease) | Complement-mediated MPGN C3 dominant IF (C3 glomerulopathy) | |
| MPGN 3: Subendothelial membranoproliferative with intramembranous and subepithelial deposits | MPGN not related to complement or immune complex | |
| Negative IF (thrombotic microangiopathy) |
IF, immunofluorescence; MPGN, membranoproliferative GN.
Immune Complex–Mediated MPGN
Disease entities that are associated with MPGN are chronic or relapsing conditions, such as hepatitis B, hepatitis C (8), autoimmune diseases (9,10), and monoclonal gammopathy of undetermined significance (MGUS) (11). The immune complex form of MPGN is caused by deposition of immune complexes, which results from paraproteinemias, autoimmune diseases, or chronic infections. Complement is activated through the classic pathway, and on IF, there will be monoclonal or polyclonal Igs with complement components. Table 1 reviews known causes of MPGN that result from the above three main categories. MPGN has been noted with many autoimmune diseases (8). Although systemic lupus erythematosus may be the most common cause, Sjogren’s syndrome (SS) and rheumatoid arthritis (RA) are other common causes (9,10,12). Interestingly, a recent study from Mayo Clinic showed that the most common autoimmune causes of MPGN were SS and RA (13). Chronic infections are known to cause MPGN (Table 1). Hepatitis C and B are the most frequently described infections associated with MPGN (3,8), and MPGN I associated with type II cryoglobulinemia is the most common form of kidney disease associated with hepatitis C infection. Why only some patients develop kidney lesions has yet to be determined (8). Kidney Disease Improving Global Outcomes (KDIGO) established guidelines for management of hepatitis C-induced kidney diseases in 2008 (14). Given the high incidence of hepatitis C–associated glomerular diseases, it is suggested that patients be tested at least annually for proteinuria, hematuria, and serum creatinine.
Although prior reports had usually mentioned paraproteinemias as major causes of MPGN, recently, Sethi et al. (11) described the important association of MGUS and MPGN. Traditionally, MGUS was thought to be benign, although with some risk of progressing to multiple myeloma. However, it is now known that MGUS, even without progression to myeloma, may cause kidney injury. Particular pathologic B cell or plasma cell clones may be responsible and lead to an MPGN pattern of injury. Classically, all of these cases had monoclonal deposition findings on kidney biopsy (IF). Some cases were associated with low-grade B cell lymphoma, lymphoplasmacytic lymphoma, or chronic lymphocytic leukemia. Many studies have suggested calling it monoclonal gammopathy of renal significance (15).
C3 Glomerulopathy: DDD and C3GN
LM findings of MPGN, accompanied by C3 dominant staining on IF, point to dysregulation of the alternative pathway (AP) of the complement system as the underlying defect, which distinguishes C3 glomerulopathy from the more common immune complex–mediated forms of glomerular diseases, such as postinfectious GN. The term C3 glomerulopathy encompasses a spectrum of renal diseases highlighted by AP dysregulation. In other words, the glomerular morphology as shown on LM is heterogeneous. To date, the principal entities that represent C3 glomerulopathy are DDD, which is historically classified as MPGN II, and C3GN (14). EM aids in differentiating between these disorders; mesangial deposits accompanied by highly dense intramembranous deposits suggest a diagnosis of DDD (Figure 3), whereas the presence of deposits in mesangial, subendothelial, subepithelial, and/or intramembranous locations suggests C3GN. Of note, the composition of the deposits in C3 glomerulopathy is typically devoid of Ig and consists mainly of complement proteins, particularly C3 and terminal complement components (C5b-C9) (16).
Figure 3.

Dense deposit disease electron microscopy. Reprinted from Glen Markowitz, with permission.
The complement system represents a primary defense modality. Its activation results in recruitment of neutrophils and pathogen elimination by apoptosis and cell lysis. The complement system is activated through three primary pathways—classic pathway, lectin pathway, and AP. The classic pathway and lectin pathway are triggered by Igs and bacterial carbohydrates, respectively, whereas the AP maintains a constant, spontaneous low level of activity with mechanisms that exist to maintain tight control, including activating and inhibitory proteins. Genetic mutations or acquired antibodies to these proteins result in dysregulation of the complement system. It results in overdrive and ongoing amplification of the AP, leading to potential organ damage (17).
Activation of the AP begins with cleavage of C3 by C3 convertase, resulting in formation of C3a and C3b. Activators of the AP include factor B (FB) and factor D—both of which promote another generation of C3 convertase and lead to AP amplification (17,18).
C3b interacts with FB. This C3bFB complex then interacts with factor D and results in a complex of C3 convertase with C3bFB, which then promotes formation of C5 convertase; C5 convertase cleaves C5 into C5a and C5b. C5a (along with C3a) acts as a chemoattractant, recruiting neutrophils and promoting an inflammatory cascade. C5b leads to the final product of complement activation, C5b-C9 (otherwise known as soluble membrane attack complex). The membrane attack complex leads to the formation of transmembrane pores, resulting in osmotically-driven water entry, which causes cell lysis (17–19).
Inhibitors of the AP include factor H (FH) and its related proteins (FHRPs) factor I (FI), CD35, and membrane cofactor protein (MCP; also known as CD46). FH is the principal inhibitor, and mutations in FH or FHRP lead to significant AP dysregulation. Murine models support this pathobiology, because genetically altered mice deficient in FH develop a renal injury pattern similar to DDD (20). Figure 4 illustrates the general schematic of the AP.
Figure 4.

Alternate pathway (AP) schematic. Activation of the AP begins with C3 interacting with C3 convertase. C3 convertase is activated or inhibited by a variety of factors and proteins. C3b and C3 convertase promote formation of C5 convertase. C5 convertase, acting on C5, results in formation of C5a and C5b. C3a and C5a attract neutrophils. C5b interacts with C6–C9 to form membrane attack complex.
Depending on the environment, C3b, instead of interacting with FB, may be inactivated by FH and FI. FH binds to C3b and is the primary cofactor for FI-induced lysis of C3b. It results in the formation of inactive C3b, which can no longer participate in the formation of C3 convertase (18,19).
Properdin is a positive regulator of the AP and acts as a stabilizer of C3 convertase. In its absence, C3 convertase rapidly degrades, halting additional amplification of the AP. Deficiency of properdin is a risk factor for infections with encapsulated organisms because of eventual lack of C5b-9. Recent experimental studies of mice deficient in both FH and properdin surprisingly revealed increased C3 glomerular deposits and inflammation (21).
C3 nephritic factor (C3Nef) is an autoantibody that stabilizes the activity of C3 convertase, making it resistant to the degradation effects of FH, thereby increasing the half-life of C3 convertase and promoting AP amplification. Approximately 80% of DDD patients are positive for C3Nef. However, C3Nef presence has also been shown in ∼40% of C3GN patients, potentially nullifying its use as a specific marker for DDD (7).
Although C3Nef is common in DDD, other processes leading to AP dysregulation and DDD have been identified. Autoantibodies that bind to native FB and lead to stabilization of C3 convertase have been described, and patients with stabilizing autoantibodies to both FB and C3b have also been described. Blocking of FH activity by autoantibodies to FH has been shown in patients with DDD as well (22). DDD has also been also associated with monoclonal gammopathy. As a result, it is possible that not all MGUS patients have an immune complex–related MPGN pattern of injury in the kidney; some might have dominant C3 only on IF (23).
In 2010, Martínez-Barricarte et al. (16) first described a C3 mutation in two patients with DDD from the same family. The mutation made C3 resistant to cleavage by C3 convertase, consequently making the formation of C3b impossible. However, the mutated C3 did get cleaved by proteases and spontaneous hydrolysis, leading to formation of C3b and C3 convertase that were resistant to the breakdown efforts of FH. This case showed that DDD in this family was attributable to dysregulation of AP and not the typical mechanism of C3Nef/altered FH. It should be noted that the LM findings accompanying DDD are not limited to MPGN and can include mesangial proliferative as well as crescentric patterns of injury.
As stated earlier, the term C3 glomerulopathy represents a heterogeneous group of disorders; although the renal pathology is most commonly highlighted by MPGN on LM and C3 dominant deposits on IF, the underlying pathobiology can be quite variable (24). Servais et al. (24) first described C3 glomerulopathy in 19 patients. The patients were divided into two groups—patients with C3 dominant staining on IF with membranoproliferative features on LM (n=13) and patients with C3 dominant disease without mesangial proliferation (n=6). Servais et al. (24) showed a wide spectrum of disease, which was characterized combined or exclusively by mutations in FH, FI, MCP/CD46, and C3Nef. Of note, in those patients with findings of MPGN, 5 of 13 patients were positive for C3Nef compared with 2 of 6 patients in the C3GN group. Defects in complement regulatory proteins (primarily FH) were detected in 4 of 6 patients with C3GN compared with only 2 of 13 patients with MPGN. Importantly, serum C3 levels were variable, and the presence of a normal C3 did not preclude AP dysregulation (24).
Gale et al. (25) identified a gene mutation among two families from Cyprus that led to C3GN. A total of 26 patients were identified. The mutation results in defective co-FH–related protein 5 (CFHR5). Mutated CFHR5 shows a decrease in complement interaction compared with the wild type, potentially leading to AP dysregulation. A larger cohort of CFHR5 was identified by Athanasiou et al. (26). Expanding on the original cohort described by Gale et al. (25), Athanasiou et al. (26) described 91 patients from 16 different pedigrees. Approximately 30% of patients progressed with proteinuria/renal failure; 15% of patients developed ESRD. Of note, approximately 70% of those patients who developed ESRD were men. LM may show a mesangioproliferative or MPGN pattern of injury; on EM, there are typical subendothelial and mesangial deposits with occasional subepithelial deposits. At this point, CFHR5 nephropathy seems confined to those patients of Cypriot descent. All reported cases of CFHR5 to date have been negative for C3Nef (26).
Sethi et al. (27) described 12 cases of C3 glomerulopathy with varying LM features (MPGN, mesangial proliferative GN, and diffuse endocapillary GN) with isolated C3 deposits on IF. AP dysfunction was because of a range of acquired and genetic conditions. C3Nef was again found to be the most common acquired defect; FH allele polymorphisms were the most common genetic defect. FH autoantibodies, along with mutations in FH, FI, and FHRP, were noted as well.
Recently, Tortajada et al. (28) reported a genetic mutation affecting CFHR1 and resulting in C3GN. A single pedigree was studied with three members affected. Studies performed by Tortajada et al. (28) indicate that this particular mutation results in a CFHR1, which competes with the ability of FH to regulate the AP, leading to AP dysregulation and C3 deposition.
To further elucidate acquired and genetic abnormalities of the AP, Servais et al. (29) analyzed 134 patients across a spectrum of DDD, C3GN, and MPGN I. Less than one half of the patients presented with a low serum C3. Close to 50% of patients were C3Nef-positive, the majority of which were in the DDD category, confirming previous studies. Mutations in FH and FI were identified in ∼20% of patients, one half of whom were also C3Nef-positive. Unexpectedly, one half of the patients with MPGN I had evidence of AP dysfunction.
Taken together, the above case series represents a wide spectrum of disease under a rather simplified phenotype on renal biopsy. There is significant variability with regards to presentation and course of disease. The efforts to further elucidate the disease state will likely require a multicenter approach given its relatively low incidence. Table 3 summarizes all known mutations with C3 glomerulopathy. Although C3GN has been mostly found to have mutations and genetic abnormalities, an association with monoclonal gammopathy has been described as well (23).
Table 3.
Known mutations/causes associated with C3GN
| Autoantibodies to C3 convertase |
| Autoantibodies to factor H |
| Autoantibodies to factor B |
| Properdin deficiency |
| C3 nephretic factor |
| Complement factor H mutation screening identified the H402 allele and V62 allele |
| Heterozygous mutations in factor I gene |
| Heterozygous mutation in CD 46 gene (membrane cofactor protein) |
| Mutation in the gene for complement factor H-related protein 5 |
MPGN III is similar to MPGN I, except that subepithelial deposits are noted as well as subendothelial deposits. Based on the EM findings, MPGN III is further classified into two principal variants: the Strife and Anders variant and the Burkholder variant, which have different patterns of electron dense deposits and disorganization of the glomerular basement membrane (4). The majority of the cases that were diagnosed as MPGN III using the old classification are likely to be C3GN. Some of these cases were complement-positive but Ig-negative by IF microscopy. Some of these cases have been attributed to resolving postinfectious GN, even in the absence of a history of recent infection. In such cases, strong C3 staining and lack of significant Ig staining raise the possibility of C3GN. Consistent with this hypothesis, abnormalities in the alternative complement pathway have been found in many of these patients (30–32). In many cases of postinfectious GN, the glomerular disease persists in the absence of infection. This disease is called atypical postinfectious GN. Recent studies suggest that this variant might represent undiagnosed C3GN (32).
MPGN without Immune Complexes and Complement
When pathology reveals no immune complex deposits and no specific IF staining but the light microscope reveals an MPGN pattern of injury, thrombotic microangiopathy (TMA) is the most likely diagnosis. Injury to the endothelial cell can lead to a similar pattern without complement activation. Rennke (3) discussed this possibility as early as 1995 among secondary causes of MPGN. Table 4 lists all causes noted to cause TMA and an MPGN pattern of injury (10). With regards to transplant glomerulopathy, IF findings of null C3 and null Ig can aid in distinguishing from recurrent MPGN (33) (Table 4).
Table 4.
Thrombotic microangiopathies associated with MPGN pattern of injury
| Hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (resolving) |
| Antiphospholipid syndrome |
| Radiation nephropathy (bone marrow transplant-related nephropathy) |
| Sickle cell disease |
| Transplant glomerulopathy |
Recurrence Post-Transplantation
Post-transplant recurrence of MPGN has been reported with variable rates, in part depending on the underlying pathobiology. MPGN associated with monoclonal gammopathy seems to have an earlier recurrence with a more aggressive course. Interestingly, recurrence of MPGN in renal allografts is associated with the presence of crescents in the native kidneys as well as the allograft (34).
Given the lack of a proven beneficial therapeutic intervention, the aggressiveness of treatment should match the aggressiveness of the disease process. One large study using the older classification of MPGN looked at recurrence rates in 88 patients with MPGN I (35). The incidence of allograft loss at 10 years because of recurrent MPGN I was 14.4% (95% confidence interval, 7.6% to 26.4%), which was similar to the incidence of recurrent FSGS. In an analysis of nearly 190,000 renal transplant patients in the United Network for Organ Sharing database, the incidence of allograft loss at 10 years because of recurrent MPGN I was similarly 14.5%, which was higher than other glomerulonephritides combined (36). We must keep in mind that the histologic changes seen on transplant glomerulopathy have the MPGN pattern of injury, and many of the recurrence rates might be overestimated. Nevertheless, findings on EM (such as the absence of immune complex deposition in transplant glomerulopathy) may help distinguish between these two disorders (33).
With regards to C3 glomerulopathy, DDD recurrence is the norm, with a 50% graft failure rate; data regarding C3GN recurrence is limited, but recurrence has been reported (24,37,38). Ten patients with CFHR5 nephropathy are reported with successful kidney transplantation (26), and one other patient with disease recurrence after unrelated donor transplantation had successful kidney transplantation (39).
Evaluation of MPGN
The evaluation of MPGN usually begins with recognition of the clinical syndrome of GN, which leads to a kidney biopsy. In some cases, biopsy may be performed for other reasons, with MPGN being a surprise diagnosis. In either case, the pathologic diagnosis of MPGN leads to a defined diagnostic pathway driven by immunoflourescent pathology findings (Figure 2).
When renal pathology IF shows Igs and C3 (Ig+C3+), then, as discussed above, MPGN is considered to be immune complex–mediated. In these cases, a careful evaluation for infection is warranted. Specifically, serologies for evidence of infection with hepatitis B or C should be obtained. Evaluation for other infections should be based on a clinical constellation of suggestive findings. Another cause of immune complex–mediated MPGN is the presence of a monoclonal gammopathy. Of patients with MPGN without hepatitis B or C, 41% of patients have a monoclonal gammopathy of uncertain significance (11). Testing could include serum free monoclonal light chain analysis, immunofixation, and serum and urine protein electrophoresis. An additional cause of MPGN that is immune complex–mediated is SS. SS should be suspected if typical signs and symptoms are present.
The greatest advances in MPGN have been in the area of complement-mediated disease. As discussed above, when renal pathology shows predominant staining for C3, a C3 glomerulopathy is present. Pathologic features help to further define the disease as DDD or C3GN (40). With these findings, serologic testing of complement components should be performed with attention to activation of the alternative pathway. Most patients will have persistently low levels of C3 and may be positive for C3 nephritic factor (40). Ideally, testing to characterize the abnormalities leading to dysregulation of complement metabolism should be performed. Such testing is currently available only at select research laboratories. Tests of interest (Table 3) would include evaluation for antibodies associated with dysregulation of the alternative complement pathway, including C3Nef, anti–co-FB autoantibodies, anti-FH, and anti-FI. Genetic testing for key complement regulators, primarily co-FH, co-FI, MCP (CD46), and FH-related proteins, would be helpful if available (40).
Treatment
A review of the literature on the treatment of MPGN can be confusing, in that most published studies were conducted before current knowledge on the different causes of disease. As a result, it should be advised that earlier studies should probably not be considered when deciding on treatment for patients, because the study cohorts were an admixture of patients with different pathobiologic conditions. After evaluation, more targeted therapies based on current knowledge should be considered if a cause is identified (for example, hepatitis C). Otherwise, immunosuppressive therapy may often be required. In general, it might be said that treatment for MPGN syndromes, in general, is not well established.
The treatment of TMA-associated MPGN is to treat the underlying cause of the TMA, and it will not be discussed in this review. Therapy for Ig+C3+ forms of MPGN are varied and directed by the underlying pathobiology. Immune complex–mediated MPGN caused by SS, RA, and other rheumatologic diseases should prompt a rheumatology-driven treatment plan with immunosuppressive agents. Treatment of underlying chronic infection is required for MPGN associated with infections. Patients with MGUS and MPGN are probably the most difficult to treat, because there is a paucity of evidence-based medicine to guide therapy. These patients, however, likely have a very small clone of plasma cells producing offending Ig and may respond to novel myeloma based therapies. Treating the underlying clone of cells might be the solution (15). As per KDIGO recommendations from 2008, patients with acute flares of hepatitis C–associated cryoglobulinemia and MPGN should be treated with immunosuppressive drugs (steroids, cyclophosphamide, or rituximab), plasma exchange, and antiviral therapy. It is recommended that antiviral therapy with standard IFN or pegylated IFN plus ribavirin be given for at least 12 months. Patients with noncryoglobulinemic MPGN may be treated with standard IFN, pegylated IFN, or IFN plus ribavirin. Relapses of systemic cryoglobulinemia and MPGN may be treated with increment doses of rituximab. Patients with hepatitis C–associated glomerulopathy should receive therapy with antiproteinuric agents, such as angiotensin-converting enzyme inhibitors and/or ARBs, to reduce proteinuria and antihypertensive treatment to achieve target BP goals established for patients with CKD (14). Finally, for patients with MPGN and an Ig+C3+ IF pattern, in whom the evaluation has not determined a cause (idiopathic disease), treatment remains unclear. For less severe disease, conservative treatment aimed at BP control and proteinuria management with careful observation may be appropriate. For severe disease, immunosuppressive therapy is indicated. Although prednisone and cyclophosphamide may have grade 2C evidence for use in MPGN, those trials were done in an era preceding the novel classification (41). Nevertheless, treatment of rapidly progressive crescentic variant of the disease would favor using of cytotoxic agents earlier than later. A full discussion of this subject is beyond the scope of this article.
Discussion here on will focus on the recent evolution of treatment for the C3 dominant variant of MPGN or C3 glomerulopathy. The current literature of published studies on C3 dominant MPGN patients is scarce. Conservative therapy may be effective, but stabilization has also been found for various immune-modulating agents (26). In a family described by Habbig et al. (42), repletion of FH was effective. The two sisters described had a single deletion in the FH gene. There is currently no method for direct replacement of FH; instead, plasma exchange is required. The group described by Habbig et al. (42) was treated with fresh frozen plasma infusion for 36 months. This treatment will not be effective in all such patients because of the diversity of pathogenesis, which was shown by Martínez-Barricarte et al. (16). In the family described by Martínez-Barricarte et al. (16), a mutation in C3 convertase caused functional resistance to FH; replacement would not have been effective.
An advance has been the development of eculizimab for treatment of this disease and related diseases. This agent is a humanized monoclonal antibody that binds with great affinity to C5 proteins, inhibiting cleaving into C5a and C5b and blocking production of the C5b-9 membrane attack complex. Reports of individual cases showed that Ig−C3+ MPGN improved after treatment, with reduced serum creatinine and proteinuria (43–46). In an illustrative case, a 16-year-old girl with renal biopsy indicated MPGN and low C3 levels; plasma complement FH-related protein 1 was absent by immunoblotting. This finding suggested that plasma exchange could be effective, which is discussed above; however, in this case, anuria persisted, despite treatment. Treatment with eculizimab was initiated with dramatic results. Renal function normalized rapidly (43). In a series of six patients with DDD or C3GN, Bomback et al. (47) found eculizimab to result in either clinical or pathologic benefits in four subjects. Taken together, the early results with eculizimab in this disease are promising, but additional studies will be necessary, because the current literature is insufficient to assess the spectrum of benefit and risk of this agent. Herlitz et al. (48) reported that, after 1 year of therapy with eculizumab, there was reduction in active glomerular proliferation and neutrophil infiltration in three of five patients, consistent with effective C5 blockade, which prevents production of chemotactin C5a. Interestingly, all post-treatment biopsies showed de novo monoclonal staining for IgG-κ in the same distribution as C3 and C5b-9, mimicking monoclonal Ig deposition disease. Staining of the γ-heavy chain was restricted to the IgG2 and IgG4 subclasses, suggesting the binding of monoclonal eculizumab to C5 in renal tissues (48). The clinical relevance of this finding is unclear at this point.
In summary, MPGN is a pattern of disease that has undergone significant reclassification, leading to emergence of a new entity called C3 glomerulopathy. Nephrologists and pathologists should be aware of these recent changes, because there are novel clinical implications for the treatment for DDD and C3GN.
Disclosures
None.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Swaminathan S, Leung N, Lager DJ, Melton LJ, 3rd, Bergstralh EJ, Rohlinger A, Fervenza FC: Changing incidence of glomerular disease in Olmsted County, Minnesota: A 30-year renal biopsy study. Clin J Am Soc Nephrol 1: 483–487, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Briganti EM, Dowling J, Finlay M, Hill PA, Jones CL, Kincaid-Smith PS, Sinclair R, McNeil JJ, Atkins RC: The incidence of biopsy-proven glomerulonephritis in Australia. Nephrol Dial Transplant 16: 1364–1367, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Rennke HG: Secondary membanoproliferative glomerulonephritis. Kidney Int 47: 643–656, 1995 [DOI] [PubMed]
- 4.Jennette JC, Olson LJ, Schwartz MM: Heptinstall’s Pathology of the Kidney, 6th Ed., edited by Silva GF, Philadelphia, Lippincott Williams and Wilkins, 2007 [Google Scholar]
- 5.Sethi S, Fervenza FC: Membranoproliferative glomerulonephritis: Pathogenetic heterogeneity and proposal for a new classification. Semin Nephrol 31: 341–348, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Sethi S, Fervenza FC: Membranoproliferative glomerulonephritis—a new look at an old entity. N Engl J Med 366: 1119–1131, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Sethi S, Nester CM, Smith RJ: Membranoproliferative glomerulonephritis and C3 glomerulopathy: Resolving the confusion. Kidney Int 81: 434–441, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doutrelepont JM, Adler M, Willems M, Durez P, Yap SH: Hepatitis C infection and membranoproliferative glomerulonephritis. Lancet 341: 317, 1993 [DOI] [PubMed] [Google Scholar]
- 9.Bishof NA, Welch TR, Beischel LS, Carson D, Donnelly PA: DP polymorphism in HLA-A1,-B8,-DR3 extended haplotypes associated with membranoproliferative glomerulonephritis and systemic lupus erythematosus. Pediatr Nephrol 7: 243–246, 1993 [DOI] [PubMed] [Google Scholar]
- 10.Sun IO, Hong YA, Park HS, Choi SR, Kang SH, Chung BH, Choi BS, Choi YJ, Yang CW, Kim YS, Park CW: Type III membranoproliferative glomerulonephritis in a patient with primary Sjögren’s syndrome. Clin Nephrol 79: 171–174, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Sethi S, Zand L, Leung N, Smith RJ, Jevremonic D, Herrmann SS, Fervenza FC: Membranoproliferative glomerulonephritis secondary to monoclonal gammopathy. Clin J Am Soc Nephrol 5: 770–782, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cortez MS, Sturgill BC, Bolton WK: Membranoproliferative glomerulonephritis with primary Sjögren’s syndrome. Am J Kidney Dis 25: 632–636, 1995 [DOI] [PubMed] [Google Scholar]
- 13.Zand L, Fervenza FC, Nasr SH, Sethi S: MPGN secondary to autoimmune diseases. Am J Kidney Dis 61: A1–A100, 2013
- 14.KDIGO: KDIGO clinical practice guidelines for the prevention, diagnosis, evaluation, and treatment of hepatitis c in chronic kidney disease. Kidney Int 73[Suppl 109]:S69–S77, 2008 [DOI] [PubMed]
- 15.Leung N, Bridoux F, Hutchison CA, Nasr SH, Cockwell P, Fermand JP, Dispenzieri A, Song KW, Kyle RA; International Kidney and Monoclonal Gammopathy Research Group: Monoclonal gammopathy of renal significance: When MGUS is no longer undetermined or insignificant. Blood 120: 4292–4295, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Martínez-Barricarte R, Heurich M, Valdes-Cañedo F, Vazquez-Martul E, Torreira E, Montes T, Tortajada A, Pinto S, Lopez-Trascasa M, Morgan BP, Llorca O, Harris CL, Rodríguez de Córdoba S: Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest 120: 3702–3712, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbour TD, Pickering MC, Cook HT: Recent insights into C3 glomerulopathy. Nephrol Dial Transplant 28: 1685–1693, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walport MJ: Complement. First of two parts. N Engl J Med 344: 1058–1066, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Walport MJ: Complement. Second of two parts. N Engl J Med 344: 1140–1144, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ, Botto M: Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet 31: 424–428, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Lesher AM, Zhou L, Kimura Y, Sato S, Gullipalli D, Herbert AP, Barlow PN, Eberhardt HU, Skerka C, Zipfel PF, Hamano T, Miwa T, Tung KS, Song WC: Combination of factor H mutation and properdin deficiency causes severe C3 glomerulonephritis. J Am Soc Nephrol 24: 53–65, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Meyer NC, Wang K, Nishimura C, Frees K, Jones M, Katz LM, Sethi S, Smith RJ: Causes of alternative pathway dysregulation in dense deposit disease. Clin J Am Soc Nephrol 7: 265–274, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zand L, Kattah A, Fervenza FC, Smith RJ, Nasr SH, Zhang Y, Vrana JA, Leung N, Cornell LD, Sethi S: C3 glomerulonephritis associated with monoclonal gammopathy: A case series. Am J Kidney Dis 62: 506–514, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Servais A, Frémeaux-Bacchi V, Lequintrec M, Salomon R, Blouin J, Knebelmann B, Grünfeld JP, Lesavre P, Noël LH, Fakhouri F: Primary glomerulonephritis with isolated C3 deposits: A new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet 44: 193–199, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, McLean AG, Pusey CD, Pierides A, Kyriacou K, Athanasiou Y, Voskarides K, Deltas C, Palmer A, Frémeaux-Bacchi V, de Cordoba SR, Maxwell PH, Pickering MC: Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 376: 794–801, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Athanasiou Y, Voskarides K, Gale DP, Damianou L, Patsias C, Zavros M, Maxwell PH, Cook HT, Demosthenous P, Hadjisavvas A, Kyriacou K, Zouvani I, Pierides A, Deltas C: Familial C3 glomerulopathy associated with CFHR5 mutations: Clinical characteristics of 91 patients in 16 pedigrees. Clin J Am Soc Nephrol 6: 1436–1446, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, Theis JD, Dogan A, Smith RJ: C3 glomerulonephritis: Clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int 82: 465–473, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tortajada A, Yébenes H, Abarrategui-Garrido C, Anter J, García-Fernández JM, Martínez-Barricarte R, Alba-Domínguez M, Malik TH, Bedoya R, Cabrera Pérez R, López Trascasa M, Pickering MC, Harris CL, Sánchez-Corral P, Llorca O, Rodríguez de Córdoba S: C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation. J Clin Invest 123: 2434–2446, 2013 [DOI] [PMC free article] [PubMed]
- 29.Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, Moulin B, Grünfeld JP, Niaudet P, Lesavre P, Frémeaux-Bacchi V: Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int 82: 454–464, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Sandhu G, Bansal A, Ranade A, Jones J, Cortell S, Markowitz GS: C3 glomerulopathy masquerading as acute postinfectious glomerulonephritis. Am J Kidney Dis 60: 1039–1043, 2012 [DOI] [PubMed] [Google Scholar]
- 31.Frémeaux-Bacchi V, Weiss L, Demouchy C, May A, Palomera S, Kazatchkine MD: Hypocomplementaemia of poststreptococcal acute glomerulonephritis is associated with C3 nephritic factor (C3NeF) IgG autoantibody activity. Nephrol Dial Transplant 9: 1747–1750, 1994 [PubMed] [Google Scholar]
- 32.Sethi S, Fervenza FC, Zhang Y, Zand L, Meyer NC, Borsa N, Nasr SH, Smith RJ: Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int 83: 293–299, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andresdottir MB, Assmann KJ, Koene RA, Wetzels JF: Immunohistological and ultrastructural differences between recurrent type I membranoproliferative glomerulonephritis and chronic transplant glomerulopathy. Am J Kidney Dis 32: 582–588, 1998 [DOI] [PubMed] [Google Scholar]
- 34.Braun MC, Stablein DM, Hamiwka LA, Bell L, Bartosh SM, Strife CF: Recurrence of membranoproliferative glomerulonephritis type II in renal allografts: The North American Pediatric Renal Transplant Cooperative Study experience. J Am Soc Nephrol 16: 2225–2233, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Briganti EM, Russ GR, McNeil JJ, Atkins RC, Chadban SJ: Risk of renal allograft loss from recurrent glomerulonephritis. N Engl J Med 347: 103–109, 2002 [DOI] [PubMed] [Google Scholar]
- 36.Angelo JR, Bell CS, Braun MC: Allograft failure in kidney transplant recipients with membranoproliferative glomerulonephritis. Am J Kidney Dis 57: 291–299, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Hamburger J, Crosnier J, Noël LH: Recurrent glomerulonephritis after renal transplantation. Annu Rev Med 29: 67–72, 1978 [DOI] [PubMed] [Google Scholar]
- 38.Lorenz EC, Sethi S, Leung N, Dispenzieri A, Fervenza FC, Cosio FG: Recurrent membranoproliferative glomerulonephritis after kidney transplantation. Kidney Int 77: 721–728, 2010 [DOI] [PubMed] [Google Scholar]
- 39.Vernon KA, Gale DP, de Jorge EG, McLean AG, Galliford J, Pierides A, Maxwell PH, Taube D, Pickering MC, Cook HT: Recurrence of complement factor H-related protein 5 nephropathy in a renal transplant. Am J Transplant 11: 152–155, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bomback AS, Appel GB: Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol 8: 634–642, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Faedda R, Satta A, Tanda F, Pirisi M, Bartoli E: Immunosuppressive treatment of membranoproliferative glomerulonephritis. Nephron 67: 59–65, 1994 [DOI] [PubMed] [Google Scholar]
- 42.Habbig S, Mihatsch MJ, Heinen S, Beck B, Emmel M, Skerka C, Kirschfink M, Hoppe B, Zipfel PF, Licht C: C3 deposition glomerulopathy due to a functional factor H defect. Kidney Int 75: 1230–1234, 2009 [DOI] [PubMed] [Google Scholar]
- 43.Radhakrishnan S, Lunn A, Kirschfink M, Thorner P, Hebert D, Langlois V, Pluthero F, Licht C: Eculizumab and refractory membranoproliferative glomerulonephritis. N Engl J Med 366: 1165–1166, 2012 [DOI] [PubMed] [Google Scholar]
- 44.Vivarelli M, Pasini A, Emma F: Eculizumab for the treatment of dense-deposit disease. N Engl J Med 366: 1163–1165, 2012 [DOI] [PubMed] [Google Scholar]
- 45.Daina E, Noris M, Remuzzi G: Eculizumab in a patient with dense-deposit disease. N Engl J Med 366: 1161–1163, 2012 [DOI] [PubMed] [Google Scholar]
- 46.McCaughan JA, O’Rourke DM, Courtney AE: Recurrent dense deposit disease after renal transplantation: An emerging role for complementary therapies. Am J Transplant 12: 1046–1051, 2012 [DOI] [PubMed] [Google Scholar]
- 47.Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, Stokes MB, Markowitz GS, D’Agati VD, Canetta PA, Radhakrishnan J, Appel GB: Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol 7: 748–756, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herlitz LC, Bomback AS, Markowitz GS, Stokes MB, Smith RN, Colvin RB, Appel GB, D’Agati VD: Pathology after eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol 23: 1229–1237, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]