

Abstract
Late kidney transplant dysfunction may be a harbinger of graft failure. For many years, calcineurin inhibitor toxicity was felt to be the main cause for graft dysfunction with fibrosis and transplant loss. Recently this idea has come into question. With the observation that peritubular capillary C4d staining in kidney allografts may indicate antibody-mediated injury in conjunction with biopsy study findings, an appreciation for antibody-mediated rejection as a major cause of late graft dysfunction and loss has emerged. Twenty percent to 30% of patients develop de novo donor-specific antibodies after kidney transplantation. There are no US Food and Drug Administration–approved treatments for antibody-mediated rejection, nor have any randomized controlled trials assessed efficacy. Off-label treatment strategies include some combination of plasma exchange, intravenous immunoglobulin, and rituximab. Other approaches, including splenectomy, bortezomib, and eculizumab, have also been tried.
Introduction
A 63-year-old man who received a deceased-donor kidney transplant 26 years ago was found to have an increase in his serum creatinine from its usual range of 1.5–1.9 mg/dl to 2.2 mg/dl on routine laboratory evaluation. He was advised to increase his fluid intake, and 1 week later, when repeated, his creatinine was 2.8 mg/dl and a spot urine protein-to-creatinine ratio was 0.09. Medications included allopurinol, atorvastatin, clopidogrel, cyclosporine, diltiazem, ezetimibe, furosemide, losartan, mycophenolate sodium, methylprednisolone, and omega-3 acid-ethyl esters.
The patient’s medical history was notable for hypertension, gout, dyslipidemia, obesity, a transient ischemic attack from basilar artery stenosis, and ESRD secondary to mesangioproliferative GN. He received a deceased-donor kidney transplant in November 1986, 18 months after initiating peritoneal dialysis. His post-transplant course was complicated by three episodes of presumed rejection in November 1986, April 1987, and October 1988. All were treated with pulse steroids. An allograft biopsy performed in September 1996 for an increase in his serum creatinine revealed arteriolar hyaline sclerosis, mild mesangial sclerosis, and mild interstitial fibrosis. He moved and transferred his care to our institution in July 2000.
Case Discussion
Differential Diagnosis
Consideration of which anatomic compartment might be responsible for AKI in a transplanted organ is the same as that for native kidneys. The approach to AKI begins by categorizing the potential lesions into clinical processes with a prerenal, intrarenal, or postrenal physiologic basis. The list of potential specific causes may differ between a kidney transplant recipient and someone without a transplant, because unique issues affect the former. Examples focused on AKI causes specific to late kidney allograft dysfunction are outlined in Table 1.
Table 1.
Transplant-specific causes of late kidney dysfunction
| Physiologic Basis and Cause |
| Prerenal |
| Calcineurin inhibitor–induced vasoconstriction |
| Renal artery stenosis due to progressive atherosclerotic disease in anastomosed artery |
| Intrarenal |
| Immunologically mediated occurrences: cellular and antibody-mediated rejection |
| BK nephropathy |
| Post-transplant lymphoproliferative disorder |
| Recurrences of original diseases |
| Diabetic nephropathy, recurrent or de novo if due to new-onset diabetes after transplant |
| Calcineurin inhibitor nephrotoxicity |
| Pyelonephritis in the transplanted kidney |
| Postrenal |
| Obstruction from ureteral stricture due to BK or surgical technique or rejection |
With this framework in mind, an evaluation was initiated when our patient was found to have a creatinine level of 2.8 mg/dl. To this end, any factors that might be contributing to a prerenal state were considered. An immediate assessment of volume status was hampered by the patient’s being out of town (he underwent laboratory tests in the afternoon just before heading to the airport). This made physical evaluation of the patient at the time impossible. Because of the timing of his laboratory check, a cyclosporine level had not been measured. To minimize any potential contribution of a prerenal state, the patient’s diuretic and angiotensin-receptor blocker were held. Three days later, his serum creatinine came down to 2.3 mg/dl. His serum creatinine levels were then monitored closely for the next 6 weeks; they fluctuated between 2.2 and 2.4 mg/dl. They never returned to his pre-AKI levels. Ultrasonography to evaluate for hydronephrosis and biopsy were scheduled.
Biopsy
On the day of the biopsy, the patient was obese (body mass index, 37 kg/m2) and with a blood pressure of 144/76 mmHg and a heart rate of 75 beats/min. The remainder of his examination was normal. There was no tenderness over the kidney. The ultrasonography findings were normal, without hydronephrosis.
The kidney biopsy specimen shown in Figures 1–3 was interpreted by Dr. Anthony Chang, associate professor of pathology, University of Chicago, as demonstrating diffuse C4d peritubular capillary deposition and capillaritis with focal features of chronic transplant glomerulopathy, consistent with acute and chronic antibody-mediated rejection (AMR).
Figure 1.
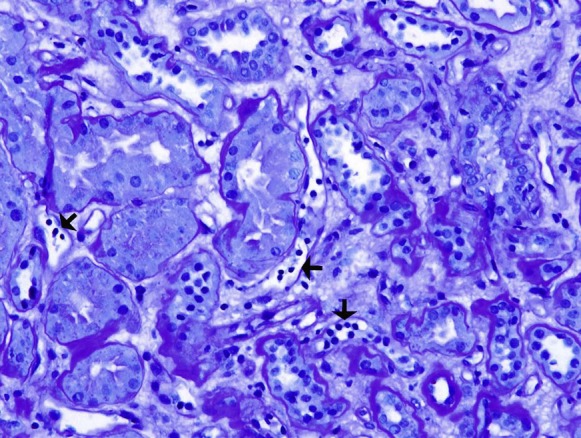
Several peritubular capillaries (arrows) contain increased numbers of leukocytes, which is a histologic feature suggestive of antibody-mediated rejection. Stain: periodic acid–Schiff; original magnification, ×200.
Figure 3.
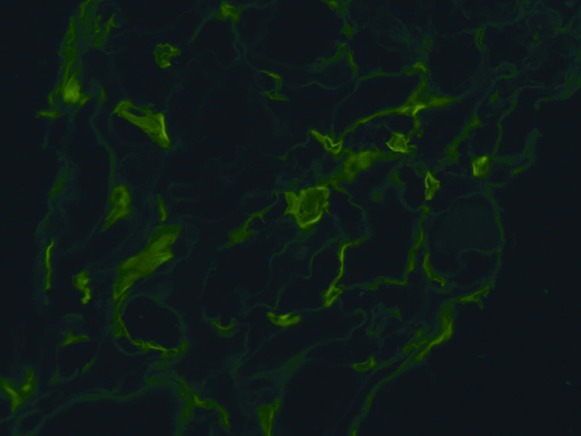
Strong C4d deposition of the peritubular capillaries is a characteristic feature of antibody-mediated rejection in the allograft kidney. Indirect immunofluorescence microscopy; original magnification, ×200.
Dr. Anthony Chang
The biopsy specimen was composed of one tissue core that consisted of both renal cortex and medulla. There was prominent congestion of the peritubular capillaries by leukocytes (Figure 1), which is a characteristic feature of AMR. There was also occasional duplication of the glomerular basement membranes (Figure 2), which is also termed chronic transplant glomerulopathy and represents a chronic feature of AMR. Finally, the presence of strong C4d peritubular capillary deposition (Figure 3), as detected by indirect immunofluorescence microscopy, further supported the diagnosis of AMR. The identification of donor-specific antibodies (DSAs) remains important in establishing the diagnosis of AMR, but other non-HLA antigens may be emerging as potential contributors to this injury. The histologic finding of peritubular capillaritis in a kidney allograft should raise the diagnostic consideration of AMR. The presence of duplication of the glomerular basement membranes can be observed in chronic thrombotic microangiopathy and recurrent or de novo membranoproliferative GN, but in the kidney allograft chronic AMR is definitely an important consideration. Finally, the presence of C4d peritubular capillary deposition indicates that an active component of AMR is present, but this is a common finding in ABO-incompatible kidney allografts and does not necessarily indicate AMR in this special circumstance.
Figure 2.
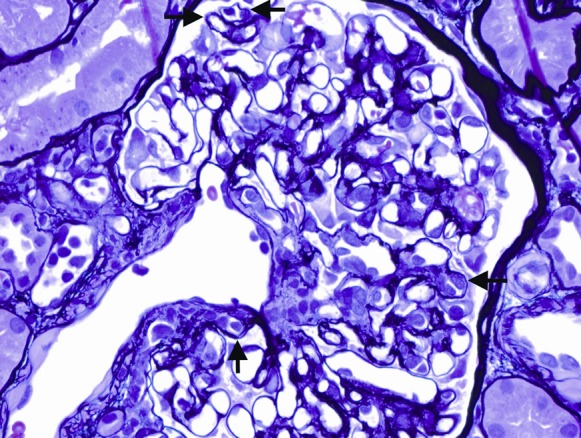
This glomerulus demonstrates focal duplication of the glomerular basement membranes (arrows), a pathologic finding consistent with chronic antibody-mediated rejection. Stain: Jones methenamine silver; original magnification, ×400.
In the early 1990s, Feucht and colleagues were the first to show peritubular capillary C4d deposition in biopsy specimens obtained from kidney transplant recipients with graft dysfunction (1,2). C4d is a degradation product of the classic complement pathway. After an antigen–antibody complex fixes complement, a cascade of events ensues, with activation of several complement proteins. The complement protein C4 is split into C4a and C4b. C4b is then converted to C4d. C4d covalently binds to the endothelial and collagen basement membranes (Figure 4A). Because of the binding, C4d eludes removal and, thus, becomes a valuable clue or “footprint” to the pathologist that complement activation and antibody activity is present. C4d is detected by immunohistochemical or immunofluorescence staining (Figure 4B). The latter technique is more sensitive.
Figure 4.

C4d deposition. (A) Schematic depiction. (B) Detection by indirect immunofluorescence.
Not all C4d indicates pathologic antibody activity. Location of the C4d deposition is important in determining whether the presence of C4d portends a pathologic immunologic process. For example, C4d is detectable in the mesangium and at the vascular pole in normal kidneys because some turnover of immune complexes normally occurs. It is the finding of C4d deposited in peritubular capillaries that indicates abnormal antibody activity. Why it occurs in this location is unknown because DSAs target HLA antigens that are found in the glomerulus, as well as in the peritubular capillaries. Although C4d can be detected, antibody cannot. This observation may be due to C4d covalently binding to the endothelial cells and collagen basement membrane, leading to it being anchored in place, while the immunoglobulins are removed and, thus, only transiently present. C4d’s binding properties make it a useful biomarker (3,4).
The finding of C4d deposition alone is not sufficient to diagnose AMR, although it makes one highly suspicious. In the original classification of AMR (5), criteria that had to be met, aside from immunologic evidence of antibody activity against the graft (i.e., C4d), included morphologic evidence of acute tissue injury (in this case manifested by capillaritis) and serologic evidence of antibodies against the donor. This latter could be manifested as antibody to donor HLA or other donor antigens (in other words, DSAs).
Our understanding of AMR is a work in progress, and, consequently, criteria are constantly evolving. To that end, attendees at the Banff 2007 meeting examined the issue of C4d deposition without morphologic evidence of rejection in patients with DSAs. This entity was added to the Banff classification, although the prognostic implications were felt to be unclear (6). AMR can be diagnosed in the absence of C4d deposition as well (7).
Diagnosis and Treatment
Biopsy findings help us decide whether AMR is acute or chronic. As noted by Dr. Chang, the presence of transplant glomerulopathy indicates chronicity. Transplant glomerulopathy is a consequence of repeated episodes of antibody-mediated injury to the endothelium, with ensuing endothelial repair and formation of a new basement membrane (8). Transplant glomerulopathy is not specific to chronic humoral rejection; it is a pattern of glomerular injury that can also be found in other settings: hepatitis C virus infection and thrombotic microangiopathy. Transplant glomerulopathy is not accompanied by DSAs or C4d in these scenarios (9).
In our patient, the biopsy findings led us to ask whether he had AMR. Making the diagnosis initially proved a bit of a challenge, because he received his kidney transplant at another institution and the donor HLA results were not immediately available, although HLA antibodies were tested for and were found to be present. However, we could not be sure if these HLA antibodies were part of a donor-specific process or not. Once the donor HLA information was obtained, we identified some of the HLA antibodies as directed against the donor’s HLA. Several other HLA antibodies were present as well. These had not been tested for in the donor (probably because of the limitations of available HLA identification assays in 1986), making it unclear whether they were donor specific. With identification of the DSAs, the diagnosis of AMR became clear. Our patient had anti-HLA antibodies to HLA DR4 and DR11, both class II donor specific. He is one of the 20%–30% of transplant patients who develop DSAs after transplant. In a study of 189 consecutive nonsensitized patients receiving their first kidney transplant (non-HLA identical), protocol testing for DSAs indicated that 25% of patients developed de novo DSA (dnDSA) within 10 years: 11.2% within the first year and then at a rate of between 1% and 5% annually thereafter (10). De novo antibodies formed after transplant are most commonly directed against class II antigens (11,12).
The biopsy findings were reviewed with the patient. Treatment options and their risks were discussed, as well as the uncertainty of efficacy. After careful consideration, the patient decided to proceed with therapy. He was given three pulses of intravenous, methylprednisolone (500 mg each), and four plasma exchanges replacing 1 plasma volume with 5% albumin every other day. Each of the first 3 exchanges was followed by 100 mg/kg sucrose-free iso-osmolar intravenous immunoglobulin (IVIG). The fourth exchange was followed by 500 mg/kg sucrose-free iso-osmolar IVIG. Rituximab, 375 mg/m2, was then given, after which another course of three plasma exchanges (with replacement of 1.3 plasma volumes using 5% albumin and saline) and IVIG treatments was given.
Late Allograft Dysfunction
Despite improvements in short-term outcomes, long-term kidney allograft survival has not markedly improved (13). Late graft dysfunction is concerning, because it can be a harbinger of graft failure, although the exact cause of graft failure is debated. The pendulum of consensus opinion on the likely culprit of late graft dysfunction and failure has swung between different theories over time.
Understanding the cause of late graft dysfunction is important so that it can be effectively reversed. To that end, El-Zoghby et al. analyzed 1317 kidney transplant recipients who had received allografts between 1996 and 2006. Surveillance biopsies were performed as part of standard clinical follow-up at the time of transplant and at 4 months and 1, 2, and 5 years after transplant. Death with function was the most common cause of graft loss in that series (43.4%). The following pathologic abnormalities were most commonly found in the recipients who lost their kidneys after 5 years (death censored): 45% had interstitial fibrosis and tubular atrophy (IF/TA), 37.5% had glomerular pathology (recurrent disease, transplant glomerulopathy, and other de novo glomerular diseases, including FSGS and membranoproliferative GN), and 5% had acute rejection (14). IF/TA has replaced the term “chronic allograft nephropathy” (15) because the latter did not convey the underlying cause or disease process, only the pattern of graft destruction. Ironically, IF/TA is not a specific disease either, but a pattern of injury that can have many underlying causes. Nevertheless, El-Zoghby and colleagues were able to identify the underlying cause of IF/TA in most cases, indicating that with enough clinical and histologic information, the cause of most kidney allograft failure can be understood.
For many years, the main cause for graft fibrosis and loss was attributed to chronic calcineurin inhibitor toxicity. Recently, this belief has come into question on the basis of many factors, including the observation that histologic features of calcineurin inhibitor toxicity are not specific and may be a consequence of other causes (16). Supporting this idea are the findings of Snanoudj et al. (17) from their retrospective study of 141 kidney transplant recipients, some of whom had received cyclosporine treatment for 10 years and some of whom had never received it. Biopsies from these individuals revealed arteriolar hyalinosis in 92% of patients who received cyclosporine and in 65% of those who had not. When the investigators looked at lesions deemed “more specific” to calcineurin inhibitor toxicity (muscular arteriopathy), the “more specific” lesions were found in 68% of those who received cyclosporine and in 28% of those who did not. In other words, so-called cyclosporine nephrotoxicity is not always a consequence of cyclosporine (17).
Putting this issue into historical perspective, the concern over the toxicity of calcineurin inhibitors is understandable. Starting with the report by Myers et al. documenting histologic evidence of tubulointerstitial injury and focal glomerulosclerosis in cyclosporine-treated cardiac transplant recipients, it was clear that calcineurin inhibitors were nephrotoxic (18). At the same time, it was observed that cyclosporine provided greater immunologic potency than the alternative nonsteroid immunosuppressive medication of the time, azathioprine. Recognition of this adverse effect led to strategies for cyclosporine conversion or dose minimization (19). These approaches were attempts to tip the precarious balance between the immunologic benefit of calcineurin inhibitors and the long-term nephrotoxicity to the patient’s advantage. Nankivell and colleagues’ 10-year prospective biopsy study documenting the natural history of chronic allograft nephropathy in 120 kidney transplant recipients with type 1 diabetes mellitus reinforced the idea that calcineurin inhibitors were a major contributor to graft failure (20). However, despite the negative publicity surrounding calcineurin inhibitors and the finding by some investigators of benefit to calcineurin inhibitor withdrawal (21), others observed an unacceptably high rate of rejection following calcineurin inhibitor reduction or withdrawal (22,23). Thus, calcineurin inhibitor withdrawal or minimization has not turned out to be the answer to preventing graft dysfunction and eventual loss.
As early acute cellular rejection rates plunged and 1-year graft survival rates improved, the transplant community turned its attention to the issue of long-term graft survival. To uncover the common causes of late graft dysfunction and failure and more effectively prevent or treat dysfunction, a multicenter study called the Long-Term Deterioration of Kidney Allograft Function (DeKAF) study was initiated. This study focused on the “troubled transplant” phenotype. The DeKAF study examined two cohorts of patients: those at least 4 months post-transplant with new-onset late graft dysfunction (a cross-sectional group) and a group longitudinally followed from the time of transplant. All 173 patients in the cross-sectional group underwent kidney biopsy; 48% were diagnosed with “chronic allograft nephropathy,” 30% with calcineurin inhibitor toxicity, and 26% with acute rejection (24). These groups were further subdivided into clusters based on the similarity of biopsy findings. The clusters had different outcomes, suggesting that these groupings were useful. For example, kidneys with both fibrosis and inflammation had a poor outcome, while for kidneys with biopsy specimens that showed fibrosis but no inflammation the outcome was excellent. Inflammation in previously stable biopsy specimens was strongly associated with risk of graft failure (25). In the cross-sectional part of the study, 99 of the 173 patients had DSAs in the serum and/or C4d staining on the biopsy specimen, which was associated with a poor outcome. By contrast, graft loss within 3 years of follow-up was rare for the group that had neither C4d nor DSAs (26). Although there is no doubt that calcineurin inhibitors are nephrotoxic and there is still traction for the argument that long-term calcineurin inhibitor toxicity is damaging and contributes to graft failure (27), the idea that calcineurin inhibitor toxicity is the main or even a major cause of late kidney failure is under scrutiny (28). As well, as illustrated by Snanoudj and colleagues’ study, the specificity of so-called calcineurin inhibitor lesions is up for debate (29). Thus, the field of transplantation is in the midst of a paradigm shift.
Ideological debates and paradigm shifts are nothing new in the transplant field and date back to the early days of transplantation, as illustrated by the controversy between Peter Gorer’s view of the centrality of the antibody in the immune response and Peter Medawar’s focus on the cellular response (30). Although for decades the latter was our focus in immunosuppression development and strategy, based on the notion that rejection is overwhelmingly a T cell–mediated response, attention to the former is increasing. The transplant community has begun to appreciate that, even with excellent control of the cellular arm of the immune response achieved with current immunosuppression, inadequate control of humoral immunity leads to allograft dysfunction and failure. This is a relatively recent turn of events as the entity of chronic AMR was only first recognized in 2001 (31). Over the last few years, results from studies, including the DeKAF study and other biopsy studies, support the central role of antibody-mediated damage in late kidney failure (26,32,33). This focus on the damage produced by antibodies in long-term allografts is a consequence of what has been learned with C4d staining, as well as a growing appreciation that antibodies can play a destructive role even when C4d staining is negative (34).
Wiebe et al. demonstrated the natural history of dnDSAs in a longitudinal study of 315 consecutive kidney transplant recipients without pretransplant DSAs by prospectively testing for HLA antibody development. Fifteen percent of these individuals developed dnDSAs after 6 months or later (mean, 4.6±3.0 years after transplant). The 10-year graft survival for this group was significantly lower than for patients who did not develop dnDSAs. As found in our patient, the antibodies were mostly directed at class II antigens. Risk factors for development of dnDSAs included nonadherence and cellular rejection within the first 6 months of transplant (35). This latter finding is consistent with observations by El Ters et al., who studied 797 patients without pretransplant DSAs undergoing a 1-year protocol biopsy. Those with acute rejections within the first year had significantly increased development of new class II antibodies and transplant glomerulopathy. In this study, acute rejection with subsequent histologic abnormalities was associated with decreased graft survival. Transplant glomerulopathy was identified as the single most common cause of graft loss (36). These studies indicate that dnDSA development and transplant glomerulopathy are an important cause of graft loss, but they also indicate that we should not turn our attention away from T cell–mediated rejections. They both can play a role and may often be intertwined. T-cell rejections may lead to inflammation and eventual interstitial fibrosis, along with class II DSA development and transplant glomerulopathy (37).
Treatment Strategies and Options
There are no US Food and Drug Administration–approved treatments of AMR; consequently, the ensuing discussion is one of off-label use of these drugs. Treatment strategies for antibody-mediated rejection include, but are not limited to, the removal of deleterious anti-HLA antibodies with plasmapheresis; blocking anti-HLA antibody activity with IVIG; and prevention of further antibody production through depletion of B cells, which are antigen-presenting cells and precursors of mature plasma cells with rituximab, an anti-CD20 chimeric antibody. Plasmapheresis rapidly reduces antibody titers, although the effect is short lived, with anti-HLA levels rebounding within days (38). By contrast, the effect of IVIG, as measured by panel reactive antibody reduction, persists on the order of 6 months (39). Rituximab also has a longer-lasting effect. B-cell depletion from rituximab can be seen within a day of treatment and persists for 6 months to a year. Subsequent reconstitution of B cells is characterized by young or naive B cells returning more quickly and abundantly than memory B cells (40). Use of treatment combinations is based on clinical experience with few prospective controlled trials (41–46). Optimal dosing and duration, as well as efficacy, are important issues that are not well defined. Whether patients should be treated differently if they develop dnDSAs after transplant compared with whether they are sensitized and have preexisting antibodies at the time of transplant is also not known. Other reported approaches to the treatment of antibody-mediated rejection include splenectomy, bortezomib (a proteasomal inhibitor approved for use in multiple myeloma), and eculizumab (a complement protein C5 antibody that inhibits the formation of the membrane attack complex). Despite the increasing AMR treatment options, the optimal treatment approach has yet to be defined (47–51).
Back to Our Patient
The patient’s anti-HLA antibody levels came down with treatment. Before receiving steroids, plasma exchange, IVIG, and rituximab, his DSA levels, based on single-antigen bead testing, had mean fluorescence intensity values in a range high enough to cause a positive flow cytometric cross-match. After treatment, the DSAs were still present, but at a level below that associated with a positive cross-match, indicating a clinically significant reduction. After treatment, his creatinine continued to fluctuate, with a nadir of 2.1 mg/dl. In the months to come, the patient’s proteinuria increased, and by 1 year after treatment, the protein-to-creatinine ratio was 2.5. Looking back at the patient’s history, we find clues that speak to his risk for developing dnDSAs: first he had three “presumed” acute rejections within the first year after transplant (two within the first 6 months), and second, he mentioned that in the early years after his transplant, his cyclosporine level had been kept low in order to avoid calcineurin inhibitor toxicity, perhaps unintentionally leaving him with subtherapeutic immunosuppression levels.
Questions
Dr. Abhijit Naik, transplant nephrology fellow, University of Chicago, Chicago, Illinois: What is the difference between class I and class II antibodies, and does the presence of one versus the other portend a difference in prognosis or management?
Dr. Josephson: Studies by Everly et al. (10) and Weibe et al. (35) both show that kidney transplant recipients develop predominately class II dnDSAs after transplant. Everly found that the likelihood of developing class I antibodies decreased after 6 months post-transplant. He did not find any significant difference in graft survival based on the class of DSA that developed after transplant (class I, class II, or class I and II). By comparison, Bentall et al. studied the 5-year outcome of patients who received a living donor transplant after desensitization for a positive cross-match and who achieved a negative CDC cross-match on the day of transplant. Five-year graft survival was higher in recipients who had antibodies to class I donor antigens detected before transplant compared with individuals who had pretransplant antibodies to class II (alone or in combination with class I) (52). Despite these findings, and given that an effective approach to managing AMR has not been well defined, there are not enough available outcome data to establish that AMR from class I, class II, or mixed DSAs should be managed differently.
Dr. Divya Jain Arwindekar, transplant nephrologist, Advocate Christ Hospital, Oak Lawn, Illinois: How do his other medications affect his cyclosporine levels?
Dr. Josephson: Diltiazem can alter his cyclosporine level. Like cyclosporine, diltiazem is metabolized by the CYP3A4 enzyme system in the liver. When cyclosporine and diltiazem are taken together, diltiazem is a competitive inhibitor for CYP3A4 metabolism of cyclosporine. Consequently, the concentration of cyclosporine and its metabolites increase in the blood. This is an important issue, because if diltiazem is stopped or the dose is reduced, the cyclosporine level would decrease, putting the patient at risk for rejection. Conversely, if the diltiazem dose was increased, his cyclosporine level could rise. Atorvastatin levels are increased when taken with cyclosporine, and, thus, side effects, such as muscle aches and rhabdomyolysis, can be much more pronounced and occur at lower doses of atorvastatin in patients taking cyclosporine. Cyclosporine levels can be mildly reduced by atorvastatin.
The bottom line to take away is that there is the potential for a number of drug–drug interactions when patients are on calcineurin inhibitors and drug levels are important to monitor.
Dr. Ling-Xin Chen, first-year nephrology fellow, University of Chicago, Chicago, Illinois: Is there any role for prophylactic treatment (e.g., periodic plasmapheresis, IVIG, or rituximab) in patients who are found to have positivity for DSA, but who have yet to develop graft dysfunction? What do you do in clinical practice with such patients?
Dr. Josephson: I am going to answer this question by focusing on patients like the patient described in Attending Rounds who develops dnDSAs. Your question brings up the question of whether or not we should prospectively monitor these patients for dnDSAs routinely if they do not have graft dysfunction, and, if we find dnDSAs, what we should do about them? Akin to other areas of transplant, the answers are not so straightforward. Cooper et al. (53) studied prospective DSA screening. They found that patients in whom DSAs were detected in the setting of an indication (decreased kidney function, acute rejection) had decreased kidney survival, while those in whom dnDSAs were found on basis of a protocol and not for indication did not have a worse outcome than those who did not develop dnDSAs. In this study, screening for DSAs without a clinical indication did not identify people at risk for acute rejection or poor short-term outcomes. This study suggests that screening for DSAs is not worthwhile. Although it did not address the issue of whether prophylactic treatment improves outcome, it implies that prophylactic treatment is not needed as outcomes do not differ.
By contrast, Everly et al. (10) treated 26 patients with bortezomib-based regimens to pre-emptively remove dnDSA before the serum creatinine increased. Those who responded with complete DSA removal were found to have significantly better kidney function than those who relapsed with return of DSAs. These findings suggest that monitoring and pre-emptively treating may be beneficial. However, there are a couple of important considerations. The patients in this trial received donor-specific transfusions prior to transplant. Although they were said to develop dnDSAs, it is possible that through these transfusions they received some degree of sensitization prior to transplant. Also, it is important to consider that although kidney function was better in those who responded to the bortezomib-based treatment regimens, this is a less rigorous endpoint than graft survival.
In considering our patient with kidney dysfunction, AMR, and dnDSAs, there are data to indicate that reducing or removing DSAs during acute rejections improves kidney allograft survival (54). Regarding your specific question, at this point I think it is fair to say that the jury is still out on whether monitoring for dnDSAs in the absence of clinical signs is useful. Currently, our program does not monitor for development of dnDSAs in asymptomatic patients, though I am sure there are programs that do. The field of DSAs and AMR in kidney transplantation is a rapidly developing area and as the transplant community learns more about dnDSAs, as we are certain to, I will be better able to answer your question as to whether one should or should not monitor and treat in the absence of clinical findings.
Disclosures
None.
Acknowledgments
Thank you to Dr. Divya Jain Arwindekar for assistance with the patient’s history, Maya Daiter for drafting Figure 4, A and B, and Dr. Anthony Chang for providing pathology slides and interpretation.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Feucht HE, Felber E, Gokel MJ, Hillebrand G, Nattermann U, Brockmeyer C, Held E, Riethmüller G, Land W, Albert E: Vascular deposition of complement-split products in kidney allografts with cell-mediated rejection. Clin Exp Immunol 86: 464–470, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feucht HE, Schneeberger H, Hillebrand G, Burkhardt K, Weiss M, Riethmüller G, Land W, Albert E: Capillary deposition of C4d complement fragment and early renal graft loss. Kidney Int 43: 1333–1338, 1993 [DOI] [PubMed] [Google Scholar]
- 3.Cornell LD: Renal allograft pathology in the sensitized patient. Curr Opin Organ Transplant 18: 327–336, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Racusen LC, Haas M: Antibody-mediated rejection in renal allografts: Lessons from pathology. Clin J Am Soc Nephrol 1: 415–420, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Racusen LC, Colvin RB, Solez K, Mihatsch MJ, Halloran PF, Campbell PM, Cecka MJ, Cosyns JP, Demetris AJ, Fishbein MC, Fogo A, Furness P, Gibson IW, Glotz D, Hayry P, Hunsickern L, Kashgarian M, Kerman R, Magil AJ, Montgomery R, Morozumi K, Nickeleit V, Randhawa P, Regele H, Seron D, Seshan S, Sund S, Trpkov K: Antibody-mediated rejection criteria — an addition to the Banff 97 classification of renal allograft rejection. Am J Transplant 3: 708–714, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Solez K, Colvin RB, Racusen LC, Haas M, Sis B, Mengel M, Halloran PF, Baldwin W, Banfi G, Collins AB, Cosio F, David DS, Drachenberg C, Einecke G, Fogo AB, Gibson IW, Glotz D, Iskandar SS, Kraus E, Lerut E, Mannon RB, Mihatsch M, Nankivell BJ, Nickeleit V, Papadimitriou JC, Randhawa P, Regele H, Renaudin K, Roberts I, Seron D, Smith RN, Valente M: Banff 07 classification of renal allograft pathology: Updates and future directions. Am J Transplant 8: 753–760, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Haas M: C4d-negative antibody-mediated rejection in renal allografts: Evidence for its existence and effect on graft survival. Clin Nephrol 75: 271–278, 2011 [DOI] [PubMed] [Google Scholar]
- 8.Sis B, Campbell PM, Mueller T, Hunter C, Cockfield SM, Cruz J, Meng C, Wishart D, Solez K, Halloran PF: Transplant glomerulopathy, late antibody-mediated rejection and the ABCD tetrad in kidney allograft biopsies for cause. Am J Transplant 7: 1743–1752, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Baid-Agrawal S, Farris AB, 3rd, Pascual M, Mauiyyedi S, Farrell ML, Tolkoff-Rubin N, Collins AB, Frei U, Colvin RB: Overlapping pathways to transplant glomerulopathy: Chronic humoral rejection, hepatitis C infection, and thrombotic microangiopathy. Kidney Int 80: 879–885, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Everly MJ, Rebellato LM, Haisch CE, Ozawa M, Parker K, Briley KP, Catrou PG, Bolin P, Kendrick WT, Kendrick SA, Harland RC, Terasaki PI: Incidence and impact of de novo donor-specific alloantibody in primary renal allografts. Transplantation 95: 410–417, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Hidalgo LG, Campbell PM, Sis B, Einecke G, Mengel M, Chang J, Sellares J, Reeve J, Halloran PF: De novo donor-specific antibody at the time of kidney transplant biopsy associates with microvascular pathology and late graft failure. Am J Transplant 9: 2532–2541, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Loupy A, Hill GS, Jordan SC: The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol 8: 348–357, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Lodhi SA, Lamb KE, Meier-Kriesche HU: Solid organ allograft survival improvement in the United States: The long-term does not mirror the dramatic short-term success. Am J Transplant 11: 1226–1235, 2011 [DOI] [PubMed] [Google Scholar]
- 14.El-Zoghby ZM, Stegall MD, Lager DJ, Kremers WK, Amer H, Gloor JM, Cosio FG: Identifying specific causes of kidney allograft loss. Am J Transplant 9: 527–535, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Solez K, Colvin RB, Racusen LC, Sis B, Halloran PF, Birk PE, Campbell PM, Cascalho M, Collins AB, Demetris AJ, Drachenberg CB, Gibson IW, Grimm PC, Haas M, Lerut E, Liapis H, Mannon RB, Marcus PB, Mengel M, Mihatsch MJ, Nankivell BJ, Nickeleit V, Papadimitriou JC, Platt JL, Randhawa P, Roberts I, Salinas-Madriga L, Salomon DR, Seron D, Sheaff M, Weening JJ: Banff ’05 Meeting Report: Differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN’). Am J Transplant 7: 518–526, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Naesens M, Kuypers DR, Sarwal M: Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 4: 481–508, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Snanoudj R, Royal V, Elie C, Rabant M, Girardin C, Morelon E, Kreis H, Fournet JC, Noël LH, Legendre C: Specificity of histological markers of long-term CNI nephrotoxicity in kidney-transplant recipients under low-dose cyclosporine therapy. Am J Transplant 11: 2635–2646, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Myers BD, Ross J, Newton L, Luetscher J, Perlroth M: Cyclosporine-associated chronic nephropathy. N Engl J Med 311: 699–705, 1984 [DOI] [PubMed] [Google Scholar]
- 19.Morris PJ, Chapman JR, Allen RD, Ting A, Thompson JF, Dunnill MS, Wood RF: Cyclosporin conversion versus conventional immunosuppression: Long-term follow-up and histological evaluation. Lancet 1: 586–591, 1987 [DOI] [PubMed] [Google Scholar]
- 20.Nankivell BJ, Borrows RJ, Fung CL, O’Connell PJ, Allen RD, Chapman JR: The natural history of chronic allograft nephropathy. N Engl J Med 349: 2326–2333, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Mourer JS, Hartigh J, van Zwet EW, Mallat MJ, Dubbeld J, de Fijter JW: Randomized trial comparing late concentration-controlled calcineurin inhibitor or mycophenolate mofetil withdrawal. Transplantation 93: 887–894, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Åsberg A, Apeland T, Reisaeter AV, Foss A, Leivestad T, Heldal K, Thorud LO, Eriksen BO, Hartmann A; NILS Study Group: Long-term outcomes after cyclosporine or mycophenolate withdrawal in kidney transplantation — results from an aborted trial. Clin Transplant 27: E151–E156, 2013 [DOI] [PubMed] [Google Scholar]
- 23.Opelz G, Döhler B: Effect on kidney graft survival of reducing or discontinuing maintenance immunosuppression after the first year posttransplant. Transplantation 86: 371–376, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Gourishankar S, Leduc R, Connett J, Cecka JM, Cosio F, Fieberg A, Gaston R, Halloran P, Hunsicker L, Kasiske B, Rush D, Grande J, Mannon R, Matas A: Pathological and clinical characterization of the ‘troubled transplant’: Data from the DeKAF study. Am J Transplant 10: 324–330, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mannon RB, Matas AJ, Grande J, Leduc R, Connett J, Kasiske B, Cecka JM, Gaston RS, Cosio F, Gourishankar S, Halloran PF, Hunsicker L, Rush D; DeKAF Investigators: Inflammation in areas of tubular atrophy in kidney allograft biopsies: A potent predictor of allograft failure. Am J Transplant 10: 2066–2073, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaston RS, Cecka JM, Kasiske BL, Fieberg AM, Leduc R, Cosio FC, Gourishankar S, Grande J, Halloran P, Hunsicker L, Mannon R, Rush D, Matas AJ: Evidence for antibody-mediated injury as a major determinant of late kidney allograft failure. Transplantation 90: 68–74, 2010 [DOI] [PubMed] [Google Scholar]
- 27.Chapman JR: Chronic calcineurin inhibitor nephrotoxicity—lest we forget. Am J Transplant 11: 693–697, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Matas AJ: Chronic progressive calcineurin nephrotoxicity: An overstated concept. Am J Transplant 11: 687–692, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mengel M, Mihatsch M, Halloran PF: Histological characteristics of calcineurin inhibitor toxicity—there is no such thing as specificity! Am J Transplant 11: 2549–2550, 2011 [DOI] [PubMed] [Google Scholar]
- 30.Arias M, Serón D, Moreso F, Bestard O, Praga M: Chronic renal allograft damage: Existing challenges. Transplantation 91[Suppl]: S4–S25, 2011 [DOI] [PubMed] [Google Scholar]
- 31.Mauiyyedi S, Pelle PD, Saidman S, Collins AB, Pascual M, Tolkoff-Rubin NE, Williams WW, Cosimi AA, Schneeberger EE, Colvin RB: Chronic humoral rejection: identification of antibody-mediated chronic renal allograft rejection by C4d deposits in peritubular capillaries. J Am Soc Nephrol 12: 574–582, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Sellarés J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, Hidalgo LG, Famulski K, Matas A, Halloran PF: Understanding the causes of kidney transplant failure: The dominant role of antibody-mediated rejection and nonadherence. Am J Transplant 12: 388–399, 2012 [DOI] [PubMed] [Google Scholar]
- 33.Einecke G, Sis B, Reeve J, Mengel M, Campbell PM, Hidalgo LG, Kaplan B, Halloran PF: Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant 9: 2520–2531, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Sis B, Jhangri GS, Bunnag S, Allanach K, Kaplan B, Halloran PF: Endothelial gene expression in kidney transplants with alloantibody indicates antibody-mediated damage despite lack of C4d staining. Am J Transplant 9: 2312–2323, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Wiebe C, Gibson IW, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, Goldberg A, Birk PE, Rush DN, Nickerson PW: Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant 12: 1157–1167, 2012 [DOI] [PubMed] [Google Scholar]
- 36.El TM, Grande JP, Keddis MT, Rodrigo E, Chopra B, Dean PG, Stegall MD, Cosio FG: Kidney allograft survival after acute rejection, the value of follow-up biopsies. Am J Transplant 13: 2334–2341 [DOI] [PubMed] [Google Scholar]
- 37.Nickerson PW, Rush DN: Rejection: An integrated response. Am J Transplant 13: 2239–2240, 2013 [DOI] [PubMed] [Google Scholar]
- 38.Jordan SC, Pescovitz MD: Presensitization: The problem and its management. Clin J Am Soc Nephrol 1: 421–432, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Jordan SC, Tyan D, Stablein D, McIntosh M, Rose S, Vo A, Toyoda M, Davis C, Shapiro R, Adey D, Milliner D, Graff R, Steiner R, Ciancio G, Sahney S, Light J: Evaluation of intravenous immunoglobulin as an agent to lower allosensitization and improve transplantation in highly sensitized adult patients with end-stage renal disease: Report of the NIH IG02 trial. J Am Soc Nephrol 15: 3256–3262, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Sidner RA, Book BK, Agarwal A, Bearden CM, Vieira CA, Pescovitz MD: In vivo human B-cell subset recovery after in vivo depletion with rituximab, anti-human CD20 monoclonal antibody. Hum Antibodies 13: 55–62, 2004 [PubMed] [Google Scholar]
- 41.Siskind E, Bhaskaran M, Boctor F, Shah K, Molmenti E: Treatment of late class II antibody-mediated rejection status postkidney transplantation: Two case reports. Int J Angiol 21: 107–110, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rocha PN, Butterly DW, Greenberg A, Reddan DN, Tuttle-Newhall J, Collins BH, Kuo PC, Reinsmoen N, Fields T, Howell DN, Smith SR: Beneficial effect of plasmapheresis and intravenous immunoglobulin on renal allograft survival of patients with acute humoral rejection. Transplantation 75: 1490–1495, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Jordan SC, Vo AA, Tyan D, Nast CC, Toyoda M: Current approaches to treatment of antibody-mediated rejection. Pediatr Transplant 9: 408–415, 2005 [DOI] [PubMed] [Google Scholar]
- 44.Slatinska J, Honsova E, Burgelova M, Slavcev A, Viklicky O: Plasmapheresis and intravenous immunoglobulin in early antibody-mediated rejection of the renal allograft: A single-center experience. Ther Apher Dial 13: 108–112, 2009 [DOI] [PubMed] [Google Scholar]
- 45.Becker YT, Becker BN, Pirsch JD, Sollinger HW: Rituximab as treatment for refractory kidney transplant rejection. Am J Transplant 4: 996–1001, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Lefaucheur C, Nochy D, Andrade J, Verine J, Gautreau C, Charron D, Hill GS, Glotz D, Suberbielle-Boissel C: Comparison of combination Plasmapheresis/IVIg/anti-CD20 versus high-dose IVIg in the treatment of antibody-mediated rejection. Am J Transplant 9: 1099–1107, 2009 [DOI] [PubMed] [Google Scholar]
- 47.Locke JE, Zachary AA, Haas M, Melancon JK, Warren DS, Simpkins CE, Segev DL, Montgomery RA: The utility of splenectomy as rescue treatment for severe acute antibody mediated rejection. Am J Transplant 7: 842–846, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Roberts DM, Jiang SH, Chadban SJ: The treatment of acute antibody-mediated rejection in kidney transplant recipients—a systematic review. Transplantation 94: 775–783, 2012 [DOI] [PubMed] [Google Scholar]
- 49.González-Roncero F, Suñer M, Bernal G, Cabello V, Toro M, Pereira P, Angel Gentil M: Eculizumab treatment of acute antibody-mediated rejection in renal transplantation: Case reports. Transplant Proc 44: 2690–2694, 2012 [DOI] [PubMed] [Google Scholar]
- 50.Kocak B, Arpali E, Demiralp E, Yelken B, Karatas C, Gorcin S, Gorgulu N, Uzunalan M, Turkmen A, Kalayoglu M: Eculizumab for salvage treatment of refractory antibody-mediated rejection in kidney transplant patients: Case reports. Transplant Proc 45: 1022–1025, 2013 [DOI] [PubMed] [Google Scholar]
- 51.Tzvetanov I, Spaggiari M, Joseph J, Jeon H, Thielke J, Oberholzer J, Benedetti E: The use of bortezomib as a rescue treatment for acute antibody-mediated rejection: Report of three cases and review of literature. Transplant Proc 44: 2971–2975, 2012 [DOI] [PubMed] [Google Scholar]
- 52.Bentall A, Cornell LD, Gloor JM, Park WD, Gandhi MJ, Winters JL, Chedid MF, Dean PG, Stegall MD: Five-year outcomes in living donor kidney transplants with a positive crossmatch. Am J Transplant 13: 76–85, 2013 [DOI] [PubMed] [Google Scholar]
- 53.Cooper JE, Gralla J, Cagle L, Goldberg R, Chan L, Wiseman AC: Inferior kidney allograft outcomes in patients with de novo donor-specific antibodies are due to acute rejection episodes. Transplantation 91: 1103–1109, 2011 [DOI] [PubMed] [Google Scholar]
- 54.Everly MJ, Everly JJ, Arend LJ, Brailey P, Susskind B, Govil A, Rike A, Roy-Chaudhury P, Mogilishetty G, Alloway RR, Tevar A, Woodle ES: Reducing de novo donor-specific antibody levels during acute rejection diminishes renal allograft loss. Am J Transplant 9: 1063–1071, 2009 [DOI] [PubMed] [Google Scholar]