

Abstract
Next generation sequencing (RNA-seq) technology was used to evaluate the effects of the Ligon lintless-2 (Li2) short fiber mutation on transcriptomes of both subgenomes of allotetraploid cotton (Gossypium hirsutum L.) as compared to its near-isogenic wild type. Sequencing was performed on 4 libraries from developing fibers of Li2 mutant and wild type near-isogenic lines at the peak of elongation followed by mapping and PolyCat categorization of RNA-seq data to the reference D5 genome (G. raimondii) for homeologous gene expression analysis. The majority of homeologous genes, 83.6% according to the reference genome, were expressed during fiber elongation. Our results revealed: 1) approximately two times more genes were induced in the AT subgenome comparing to the DT subgenome in wild type and mutant fiber; 2) the subgenome expression bias was significantly reduced in the Li2 fiber transcriptome; 3) Li2 had a significantly greater effect on the DT than on the AT subgenome. Transcriptional regulators and cell wall homeologous genes significantly affected by the Li2 mutation were reviewed in detail. This is the first report to explore the effects of a single mutation on homeologous gene expression in allotetraploid cotton. These results provide deeper insights into the evolution of allotetraploid cotton gene expression and cotton fiber development.
Introduction
Cotton is the major source of natural fibers used in the textile industry. There are four cultivated species: AA genome diploids, Gossypium arboretum L. and G. herbaceum L.; and AADD genome allotetraploids, G. hirsutum L. and G. barbadense L. Upland cotton (G. hirsutum) represents about 95% of world cotton production [1]. Allotetraploid species originated around 1–2 million years ago from inter-specific hybridization between an AA-genome diploid native to Africa and Mexican DD-genome diploid [1], [2].
Cotton fibers are single-celled trichomes that emerge from the ovule epidermal cells. About 25–30% of the seed epidermal cells differentiate into spinnable fibers [3]. Fiber length ranges from short (fuzz <6 mm) to long (lint). Lint fibers of economically important G. hirsutum generally grow up to about 30–40 mm in length. Cotton fiber development undergoes four distinctive but overlapping stages: initiation, elongation, secondary cell wall biosynthesis, and maturation [4]. The rate and duration of each developmental stage is important to the quality attributes of the mature fiber. Cell elongation is crucial for fiber length, whereas secondary cell wall thickening is important for fiber fineness and strength.
Cotton fiber mutants are useful tools to elucidate biological processes of cotton fiber development. A cotton plant with abnormally short lint fibers was discovered in a breeding nursery of the Texas Agricultural Experiment Station in 1984. This mutant had short lint fibers (<6 mm) visually similar to those produced by Ligon lintless-1 (Li1); however, unlike the stunted and deformed vegetative morphology caused by the Li1 mutation, this fiber mutant had normal vegetative growth. The trait was controlled by one dominant gene named Ligon lintless-2 (Li2) [5]. This gene was mapped to chromosome 18 (DT subgenome of G. hirsutum) using several approaches [6]–[8]. In a fiber developmental study, Kohel et al. [9] observed that elongation is restricted in Li2 fibers, however secondary wall development proceeds normally in proportion to fiber length. Two near-isogenic lines (NILs) of Li2 with the Upland cotton variety DP5690 were developed in a backcross program at Stoneville, MS [6]. Morphological evaluation of developing fibers did not reveal apparent differences between WT and Li2 NILs during initiation or early elongation up to 5 days post-anthesis (DPA). Transcript and metabolite evaluations revealed significant changes in biological processes associated with cell expansion in the Li2 mutant line at peak of fiber elongation, including reactive oxygen species, hormone homeostasis, nitrogen metabolism, carbohydrate biosynthesis, cell wall biogenesis, and cytoskeleton [6], [10]. Therefore, the Li2 mutation can be considered as a factor affecting cotton fiber elongation process, making it an excellent model system to study cotton fiber elongation.
In previous reports, we used microarray techniques to investigate global gene expression in Li2 NILs [6], [10]. However, by using the genome sequence of G. raimondii [11], RNA-seq can provide a more comprehensive and accurate transcriptome analysis based on the reference DNA sequences [12]. RNA-seq offers a larger dynamic range of quantification, reduced technical variability, and higher accuracy for distinguishing and quantifying expression levels of homeologous copies than DNA microarrays [12]. Because of the limited sequence divergence between the AT and DT subgenomes in cotton [13], a pipeline was developed to map and categorize RNA-seq reads as originating from the AT or DT subgenomes [14].
In the present study we compared quantitative gene expression levels of RNA-seq data between developing fibers of Li2 and its WT NILs. We investigated the Li2 mutation’s effect on global transcriptional changes in subgenomes and on the functional distribution of homeologous genes during fiber elongation. These results provide deeper insights into the evolution of allotetraploid cotton gene expression.
Results
RNA-seq of Wild Type and Li2 Developing Fibers at Peak of Elongation
Considering the cost of deep sequencing, only one time point, at the peak of elongation, was selected for RNA-seq analysis, including two biological replicates for wild type and mutant NILs. The time points 8 and 12 days post-anthesis (DPA) represent peak rates of fiber elongation. The time point 8 DPA was selected because: 1) our earlier research revealed significant transcript and metabolite changes between the Li2 and wild type NILs during this time of fiber development [6], [10]; 2) the transcript level of the elongation stage-related gene GhExp1 significantly decreased in Li2 mutant fiber at 8 DPA [6].
A total of 639 million reads (each 101 bp in length) from 4 libraries were obtained by paired-end Illumina sequencing. Approximately 2.3% more reads were obtained from Li2 than wild type fiber transcriptomes. From 84.4% to 90.2% of the reads were mapped to the D5-genome reference sequence of G. raimondii (Table 1). Not all the reads mapped to the reference genome sequence, probably since some of the genes were not included in the 13 large pseudo-molecules and transcripts mapped to genomic regions were outside of annotated genes. Of the mapped reads, between 29.3%–31.4% were mapped to the AT subgenome and between 23.4%–25.1% were mapped to the DT subgenomes of G. hirsutum. If the mapped reads overlapped a homeologous SNP position (SNPs between the AT and DT subgenomes), they were categorized as belonging to one of the two subgenomes or as a chimeric read (A-reads, D-reads, and X-reads, respectively; [14]). If a read did not overlap a homeologous SNP position, the read was unable to be categorized as originating from either the AT or DT subgenome (N reads; Table 1). Notably, more reads from each library were aligned to the AT subgenome than to the DT subgenome. Among the 37,223 genes on the 13 chromosomes of the G. raimondii genome, 34,692 genes (93%) had at least one mapped read from developing fibers at peak of elongation (Table 2).
Table 1. Results of mapping reads.
| Library | Li2 BR1 | Li2 BR2 | WT BR1 | WT BR2 | ||||
| count | % | count | % | count | % | count | % | |
| Number reads | 155,057,542 | 100.0 | 168,114,870 | 100.0 | 149,831,738 | 100.0 | 165,935,842 | 100.0 |
| A reads | 45,382,923 | 29.3 | 51,937,609 | 30.9 | 47,115,502 | 31.4 | 52,072,768 | 31.4 |
| D reads | 36,234,551 | 23.4 | 41,617,562 | 24.8 | 37,675,249 | 25.1 | 41,512,224 | 25.0 |
| X reads | 7,972,844 | 5.1 | 9,130,944 | 5.4 | 8,068,891 | 5.4 | 8,896,820 | 5.4 |
| N reads | 41,252,612 | 26.6 | 46,157,152 | 27.5 | 42,291,972 | 28.2 | 46,818,360 | 28.2 |
| Mapped total | 130,842,930 | 84.4 | 148,843,267 | 88.5 | 135,151,614 | 90.2 | 149,300,172 | 90.0 |
Table 2. Count of expressed genes.
| Sample | Li2 BR 1 | Li2 BR 2 | Li2 Total | WT BR1 | WT BR2 | WT Total | Total |
| Expressed (≥1) | 32,413 | 31,845 | 33,553 | 32,170 | 32,481 | 33,460 | 34,692 |
| % of annotated genes of G. raimondii | 87.1 | 85.6 | 90.1 | 86.4 | 87.3 | 89.9 | 93.2 |
| Expressed (≥10) | 29,919 | 29,820 | 30,842 | 28,796 | 29,041 | 29,603 | 31,114 |
| % of annotated genes of G. raimondii | 80.4 | 80.1 | 82.9 | 77.4 | 78.0 | 79.5 | 83.6 |
Differential Gene Expression in Developing Fibers
Counts of mapped reads were evaluated in wild type and mutant fiber transcriptomes. Genes were considered to be expressed if they had ≥10 reads mapped in one sample. Genes that were not considered to be expressed were not included in further analyses. Approximately 3% more expressed genes were detected in Li2 than in wild type. Of the 37,223 genes on the 13 chromosomes of the G. raimondii D5 reference genome 29,603 (79.5%) genes were expressed in wild type and 30,842 (82.9%) genes were expressed in Li2 fiber (Table 2).
Many genes had altered expression levels as a result of the Li2 mutation (Table S1in File S1). Some genes were expressed in one treatment (such as Li2) but not the other treatment. For example, expressions of genes annotated as SAUR-like auxin-responsive protein (Gorai.005G257000), bHLH (Gorai.003G034700) and NAC domain transcription factor (Gorai.009G170700) were only detected in wild-type fiber. Cytokinin response factor 6 (Gorai.007G105600), UGT73C14 (Gorai.002G107900), cystein proteinase (Gorai.007G329600), MYB-like 102 (Gorai.012G132200) and WRKY transcription factor (Gorai.009G157300) were only detected in Li2 fiber. The majority of these genes have not yet been functionally characterized in cotton, except of glycosyltransferase UGT73C14, which has been shown recently to be involved in ABA homeostasis [15].
The quantitative levels of mapped reads were evaluated for differential expression between elongating fibers of Li2 and wild type NILs. A gene-by-gene ANOVA determined that 7,163 of 31,114 expressed genes were differentially expressed (FDR corrected p-value <0.05) and had ≥2-fold difference in at least one of the following comparisons: fiber type (Li2 versus wild type), AT/DT subgenomes, and combinations of these factors (statistical data for significantly regulated genes are provided in Data S1). The highest numbers of significantly differentially expressed genes were identified between the AT and DT subgenomes in wild type and Li2; whereas approximately 3 times fewer differentially expressed genes were detected between fiber type comparisons (Figure 1A). Of the 29,603 expressed homeologous pairs in wild type, 4,578 (wtA/wtD, 15.5%) showed significantly different expression level between subgenomes; whereas in mutant fiber of the 30,842 expressed genes, 3,967 (LiA/LiD, 12.9%) were differentially expressed between subgenomes. Therefore, the homeolog expression bias was significantly (p-value <0.0001; Chi square) reduced in Li2 fiber transcriptome.
Figure 1. Overview of significantly regulated genes in developing fiber of Li2 NILs across all comparisons.
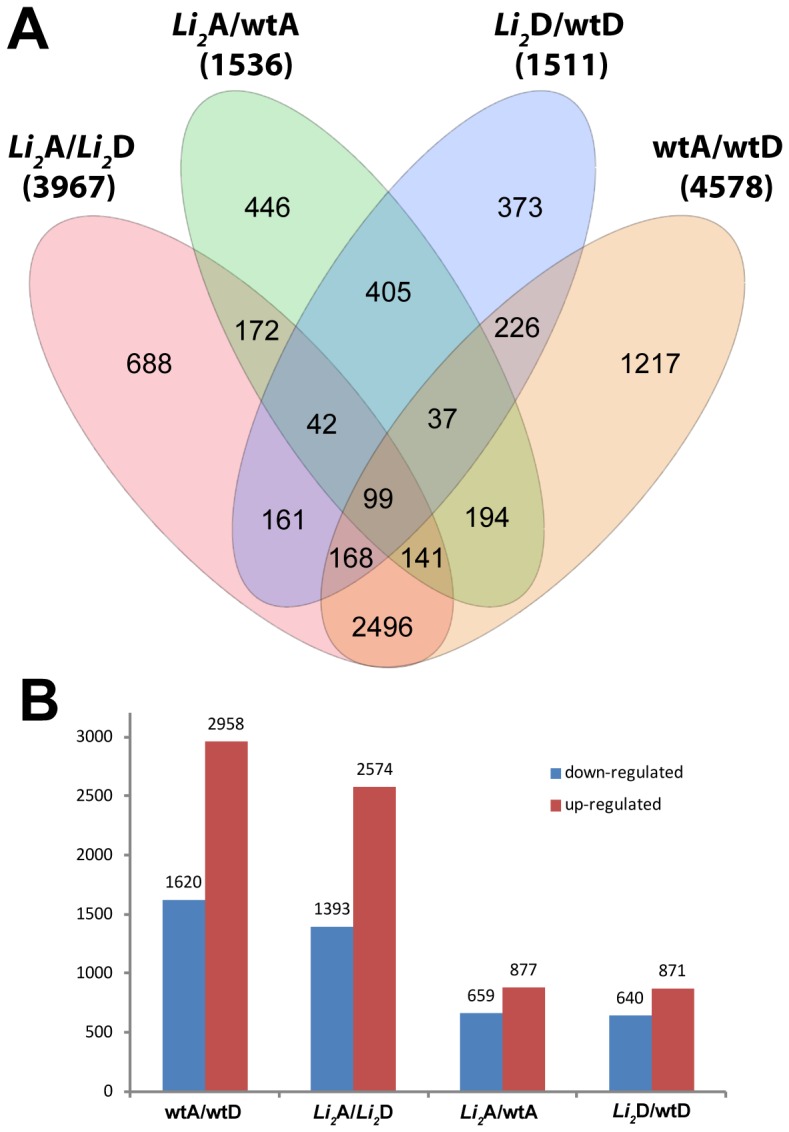
(A)Venn diagram of regulated genes in Li2 versus wild type in AT/DT subgenomes. Total number of significantly regulated genes in each comparison is indicated in parentheses. (B) The chart represents up- and down- regulated genes between subgenomes and fiber type comparisons.
In general, the AT subgenome contributed more differentially expressed genes to fiber transcriptome than did the DT subgenome. Approximately two times more genes were differentially expressed in the AT subgenome compared to the DT subgenome in wild type (AT - 2958 vs. DT - 1620; Figure 1B) and mutant fiber (AT - 2574 vs. DT - 1393). Comparison between fiber types showed more genes were upregulated in Li2 versus wild type in both subgenomes (Figure 1B). It should be noted that only about 38% (583 genes out of 1,536 in AT and 1,511 in DT subgenome) of significantly regulated genes between mutant and wild type overlapped between subgenomes (Figure 1A).
Mutation Effects on Transcriptome of AT and DT Subgenomes of Allotetraploid G. hirsutum
The effect of Li2 mutation on the transcriptome of each subgenome was evaluated. The genes significantly (FDR corrected p-value <0.05) up-regulated (≥2-fold) in one subgenome of wild type were considered to have biased expression. Of the 2,958 AT biased genes, 26.5% (784) had significantly changed the expression levels in both subgenomes of Li2 as a result of mutation. However, of the 1,620 DT biased genes, 35.9% (582) had significantly changed the expression levels in both subgenomes of the Li2 mutant (Figure 2). Therefore Li2 has a significantly greater effect (p-value <0.0001; Chi square) on DT biased genes than AT biased genes. Importantly, the majority of biased genes had significantly reduced expression levels (8.6% and 12.4%), whereas only a small portion of genes increased expression levels (1.6% and 2.7%) in the mutant. However, more genes, which were down-regulated in homeologous subgenome in wild type, had increased expression levels in the mutant fiber: 11.9% and 14% were up-regulated, whereas 4.4% and 6.8% were down-regulated (Figure 2).
Figure 2. Mutation effects on AT and DT subgenomes of allotetraploid G. hirsutum.

The bar chart represents percent of AT and DT biased genes regulated in subgenomes of Li2 mutant fiber.
Furthermore, a few homeolog pairs had reciprocal expression biases between two subgenomes as a result of mutation. Expression levels for three of these genes were tested by RT-qPCR across eight developmental time points from DOA to 20 DPA, representing initiation, elongation, and beginning of secondary cell wall biosynthesis stages (Figure 3). Interestingly, the direction of expression bias changed between developmental stages in these three genes. For example, expression of homeolog pair was biased in favor of the DT subgenome for Gorai.002G223800 at initiation (1 DPA), but switched to favor the AT subgenome at elongation (5–16 DPA) in wild type developing fibers, whereas expression was biased in favor of the DT subgenome across all evaluated time points in mutant fibers. These results demonstrate that the Li2 mutation had a greater effect on the DT subgenome and also influenced direction of expression bias for some genes across developmental stages.
Figure 3. RNA-seq and rt-qPCR analysis detected reciprocal expression biases as a result of mutation.

Original RNA-seq data are shown on left. Asterisks indicate significant (p-value <0.05) difference in gene expression level between AT vs. DT subgenomes in wild type (black) and mutant (blue) developing fibers. Error bars represent standard deviation from two biological replicates for RNA-seq data and three biological replicates for RT-qPCR.
Mutation Effects on Functional Distribution of Homeologous Genes during Fiber Elongation
The greater effect of Li2 on DT biased genes was observed in overall transcript data. In general, the subgenomes contributed unequally to different biological processes [16]; therefore diverse mutation effects could be expected on different functional categories of genes. To determine which biological processes were affected by the mutation, MapMan ontology was used (Data S2).
The distribution of genes from the AT and DT subgenomes with significantly changed expression levels in the mutant were categorized into MapMan functional categories (Figure 4; Table S2 in File S1). Relative gene frequencies in functional categories were represented in percents of biased genes in each subgenome (2,958 AT biased genes and 1,620 DT biased genes). Most functional categories were biased in favor of the DT subgenome with the exception of photosynthesis and redox, which only contained AT homeologs. Two functional categories were significantly (p-value <0.05; Fisher’s exact test) biased in enrichment among DT biased genes: secondary metabolism and stress (Table S2 in File S1). These results demonstrate that different biological processes were unequally affected by Li2 mutation.
Figure 4. Mutation effects on functional distribution of homeolog genes during fiber elongation.

Relative gene frequencies in functional categories were represented in percents from amount of biased genes in each subgenome. Asterisks indicate significant (p-value <0.05; Fisher’s exact test) enrichment of functional category between subgenomes with genes that changed expression in result of mutation. Table S2 in File S1 provides Fisher’s exact test results. MapMan BIN structure was used for functional categorization of genes regulated by Li2 mutation. Only functional categories with more than 0.06% gene representation are shown here. Carbohydrates combine 6 BIN classes, including major and minor carbohydrates, glycolysis, fermentation, gluconeogenesis and oxidative pentose phosphate pathway.
Transcriptional regulators
Transcriptional regulators (TRs) were identified in the G. raimondii genome based on similarity to Arabidopsis TRs and categorized into 76 families. Among them, 229 homeolog pairs were AT biased and 111 were DT biased in elongating cotton fibers. Of the 229 AT biased TRs, 21 (9.2%) of them changed transcription level, whereas of the 111 DT biased TRs 14 (12.6%) of them changed transcription level (Table 3), but this difference was statistically insignificant. Expression levels for the majority of subgenome biased homeologs decreased as the result of Li2 mutation. However, six TRs (including both homeologs) had increased expression levels in mutant fibers. Three classes of TRs were the most abundant, including Aux/IAA (6 members), bHLH (5 members) and MYB (3 members). Interestingly, two of the three members of MYB TRs had increased expression due to mutation.
Table 3. Subgenome biased transcriptional regulators affected by mutation.
| Class | Identifier | TAIR10 best hit | TAIR 10 symbol | AT biased (wtA/wtD) | Li2A/wtA | DT biased (wtD/wtA) | Li2D/wtD |
| AP2/EREBP | Gorai.006G222600 | AT4G13620 | 3.78 | 0.35 | 0.26 | 1.47 | |
| AP2/EREBP | Gorai.007G010200 | AT5G52020 | 2.22 | 0.28 | 0.45 | 0.68 | |
| bHLH | Gorai.007G005700 | AT1G73830 | BEE3 | 0.14 | 1.39 | 7.36 | 0.37 |
| bHLH | Gorai.006G115500 | AT2G27230 | LHW | 2.71 | 2.45 | 0.37 | 2.87 |
| bHLH | Gorai.007G157700 | AT3G07340 | 5.10 | 0.50 | 0.20 | 1.77 | |
| bHLH | Gorai.001G275600 | AT3G26744 | ICE1,SCRM | 5.82 | 0.22 | 0.17 | 0.64 |
| bHLH | Gorai.009G219100 | AT4G33880 | RSL2 | 0.37 | 1.53 | 2.68 | 0.31 |
| C2C2(Zn) CO-like | Gorai.009G065600 | AT5G24930 | COL4 | 2.38 | 2.17 | 0.42 | 2.23 |
| C2H2(Zn) | Gorai.009G092800 | AT2G29660 | 3.56 | 0.40 | 0.28 | 0.77 | |
| C3H(Zn) | Gorai.011G126400 | AT5G51980 | 31.56 | 0.44 | 0.03 | 1.88 | |
| CPP(Zn) | Gorai.013G121300 | AT3G22780 | TSO1 | 0.33 | 1.27 | 2.99 | 0.38 |
| G2-like | Gorai.008G224400 | AT1G32240 | KAN2 | 0.31 | 0.93 | 3.20 | 0.41 |
| Homeobox | Gorai.003G094500 | AT3G01470 | HAT5,HB-1 | 0.27 | 0.76 | 3.76 | 0.47 |
| MYB | Gorai.006G195700 | AT4G22680 | MYB85 | 12.21 | 0.31 | 0.08 | 0.56 |
| MYB | Gorai.001G015200 | AT5G35550 | MYB123,TT2 | 3.05 | 2.13 | 0.33 | 2.08 |
| MYB | Gorai.007G037100 | AT5G62470 | MYB96 | 4.72 | 2.30 | 0.21 | 5.78 |
| WRKY | Gorai.009G066900 | AT4G31550 | WRKY11 | 6.87 | 0.44 | 0.15 | 0.77 |
| bZIP | Gorai.013G258300 | AT3G44460 | DPBF2 | 0.27 | 0.78 | 3.68 | 0.37 |
| AS2, LOB | Gorai.013G016900 | AT3G11090 | LBD21 | 0.29 | 1.67 | 3.48 | 0.18 |
| Aux/IAA | Gorai.009G132300 | AT3G15540 | IAA19,MSG2 | 0.33 | 0.97 | 3.07 | 0.41 |
| Aux/IAA | Gorai.010G227800 | AT3G15540 | IAA19,MSG2 | 0.10 | 0.20 | 9.65 | 0.21 |
| Aux/IAA | Gorai.001G242900 | AT3G23050 | AXR2,IAA7 | 0.24 | 0.15 | 4.23 | 0.06 |
| Aux/IAA | Gorai.006G246000 | AT4G14550 | IAA14,SLR | 4.08 | 0.28 | 0.24 | 0.43 |
| Aux/IAA | Gorai.001G043900 | AT4G32280 | IAA29 | 2.39 | 2.20 | 0.42 | 2.60 |
| Aux/IAA | Gorai.007G277000 | AT5G43700 | IAA4 | 0.19 | 0.32 | 5.28 | 0.25 |
| General | Gorai.011G240900 | AT3G52270 | 3.94 | 0.40 | 0.25 | 0.40 | |
| GATA type (Zn) | Gorai.005G230900 | AT1G10200 | WLIM1 | 3.86 | 0.30 | 0.26 | 0.62 |
| TAZ domain | Gorai.007G056400 | AT5G67480 | BT4 | 0.09 | 0.99 | 11.08 | 0.19 |
| GRP | Gorai.008G029100 | AT4G39260 | GRP8 | 3.23 | 0.47 | 0.31 | 0.74 |
| unclassified | Gorai.013G029900 | AT2G01818 | 3.23 | 0.26 | 0.31 | 0.39 | |
| unclassified | Gorai.013G258400 | AT2G27580 | 2.85 | 0.38 | 0.35 | 0.82 | |
| unclassified | Gorai.012G146800 | AT3G18870 | 0.47 | 1.31 | 2.14 | 0.45 | |
| unclassified | Gorai.008G221500 | AT4G19050 | 2.08 | 2.23 | 0.48 | 2.01 | |
| unclassified | Gorai.010G120300 | AT5G10770 | 0.06 | 1.17 | 17.63 | 0.36 | |
| unclassified | Gorai.004G118800 | AT5G23750 | 2.79 | 0.48 | 0.36 | 0.47 |
The genes significantly (FDR corrected p-value <0.05) down-regulated or up- regulated more than 2-fold are shown in boldface and underlined.
Cell wall
In the cell wall functional category, 60 homeologs were AT biased and 40 were DT biased in fiber transcriptome. Ten (16.7%) of the AT biased homeologs changed expression levels; whereas 12 (30%) of the DT biased homeologs changed expression levels (Table 4), indication a higher, but statistically insignificant effect on DT biased homeologs. Interestingly, more DT homeologs (11) than AT homeologs (4) increased transcript levels as a result of the Li2 mutation. Genes encoding enzymes involved in polysaccharide degradation (14 genes) and cell wall proteins (9 genes) were the most abundant classes.
Table 4. Subgenome biased genes encoding enzyme involved in cell wall biosynthesis changed expression level as a result of Li2 mutation.
| Description | Identifier | TAIR10 best hit | AT biased (wtA/wtD) | Li2A/wtA | DT biased wtD/wtA | Li2D/wtD |
| Hemicellulose synthesis | ||||||
| Exostosin family protein | Gorai.011G272700 | AT3G45400 | 4.82 | 1.16 | 0.21 | 2.03 |
| Xyloglucan β-galactosyltransferase; MUR3 | Gorai.007G150100 | AT2G20370 | 3.36 | 0.41 | 0.30 | 0.97 |
| Xylosyltransferase; IRX9 | Gorai.006G168500 | AT2G37090 | 0.28 | 0.64 | 3.63 | 0.42 |
| Cell wall proteins | ||||||
| Fasciclin-like arabinogalactan 7 | Gorai.001G219000 | AT2G04780 | 9.65 | 0.41 | 0.10 | 1.72 |
| Arabinogalactan protein 16 | Gorai.007G025300 | AT2G46330 | 3.07 | 0.42 | 0.33 | 0.44 |
| Fasciclin-like arabinogalactan | Gorai.003G132100 | AT3G46550 | 0.05 | 0.25 | 20.68 | 0.43 |
| Fasciclin-like arabinogalactan 10 | Gorai.007G092300 | AT3G60900 | 0.09 | 2.30 | 11.31 | 2.30 |
| Arabinogalactan protein 18 | Gorai.013G048900 | AT4G37450 | 3.01 | 0.91 | 0.33 | 2.51 |
| Fasciclin-like arabinogalactan 17 | Gorai.006G147500 | AT5G06390 | 2.08 | 0.42 | 0.48 | 0.59 |
| Fasciclin-like arabinogalactan 1 | Gorai.013G152900 | AT5G55730 | 3.71 | 2.62 | 0.27 | 1.65 |
| Proline-rich protein 2 | Gorai.002G193500 | AT2G21140 | 0.23 | 0.89 | 4.35 | 0.37 |
| Reversibly glycosylated polypeptide 1 | Gorai.001G090200 | AT3G02230 | 0.33 | 2.04 | 3.07 | 1.99 |
| Degradation | ||||||
| Peptidoglycan-binding LysM domain-containing protein | Gorai.008G281400 | AT5G62150 | 19.29 | 0.35 | 0.05 | 1.68 |
| Glycosyl hydrolase 9B8 | Gorai.007G170600 | AT2G32990 | 0.10 | 1.57 | 10.48 | 0.37 |
| Glycosyl hydrolase | Gorai.006G118100 | AT5G20950 | 0.17 | 1.33 | 6.02 | 0.49 |
| α-L-fucosidase 1 | Gorai.008G282700 | AT2G28100 | 4.69 | 0.86 | 0.21 | 2.36 |
| β-Xylosidase 1 | Gorai.011G198200 | AT5G49360 | 2.01 | 1.88 | 0.50 | 2.25 |
| β-Xylosidase 2 | Gorai.008G140600 | AT1G02640 | 0.31 | 0.71 | 3.18 | 0.22 |
| Rhamnogalacturonate lyase | Gorai.002G138200 | AT1G09890 | 3.63 | 1.60 | 0.28 | 3.51 |
| Rhamnogalacturonate lyase | Gorai.007G231600 | AT2G22620 | 0.40 | 2.06 | 2.53 | 2.33 |
| Rhamnogalacturonate lyase | Gorai.008G089300 | AT2G22620 | 11.16 | 0.20 | 0.09 | 6.32 |
| Polygalacturonase 2 | Gorai.002G161400 | AT1G70370 | 0.35 | 0.46 | 2.85 | 0.38 |
| Polygalacturonase 2 | Gorai.005G136300 | AT1G70370 | 5.03 | 0.39 | 0.20 | 1.55 |
| Pectin lyase-like | Gorai.004G138900 | AT4G23500 | 0.36 | 1.48 | 2.81 | 2.50 |
| Pectin methylesterase 1 | Gorai.009G147800 | AT1G53840 | 11.39 | 2.27 | 0.09 | 3.18 |
| Pectinacetylesterase | Gorai.004G176200 | AT4G19410 | 0.38 | 2.11 | 2.66 | 1.89 |
| Modification | ||||||
| Expansin A4 | Gorai.003G131000 | AT2G39700 | 2.97 | 0.51 | 0.34 | 0.48 |
| Expansin A4 | Gorai.007G376300 | AT2G39700 | 2.07 | 1.66 | 0.48 | 2.27 |
| Expansin A4 | Gorai.012G104400 | AT2G39700 | 2.10 | 1.56 | 0.48 | 2.50 |
| Expansin A8 | Gorai.012G014400 | AT2G40610 | 0.24 | 0.32 | 4.11 | 0.23 |
| Xyloglucan endotransglucosylase/hydrolase | Gorai.004G030500 | AT4G25810 | 4.32 | 0.40 | 0.23 | 0.43 |
| Xyloglucan endotransglucosylase/hydrolase, GhXTH1 | Gorai.007G057400 | AT4G37800 | 0.16 | 0.97 | 6.06 | 0.41 |
The genes significantly (FDR corrected p-value <0.05) down-regulated or up- regulated more than 2-fold are shown in boldface and underlined.
Validation of Illumina RNA-seq Expression and Subgenome Specific Categorization of Reads by RT-qPCR Analysis
To test the reliability of Illumina sequencing and SNP-based categorization of reads to the AT or DT subgenome of allotetraploid G. hirsutum, RT-qPCR analysis was performed for a subset of 8 genes (selected from Table S1 in File S1) expressed only in WT or Li2 NILs, and for a subset of 11 genes (selected from Tables 3 and 4) that showed subgenome biased expression. Overall, the results of RT-qPCR analysis were consistent with results of RNA-seq analysis for 19 selected genes (Figures S1 and S2). RT-qPCR analysis confirmed silencing or activation of the expression by the Li2 mutation for the subset of 8 genes (Figure S1). Correlation analysis of the expression patterns revealed strong correlations between RNA-seq and RT-qPCR data. In the subset of 11subgenome biased genes, 7 genes showed 100% correlation (p-value <0.05) and 4 genes showed 99% correlation (p-value >0.05; Figure S2).
Discussion
Our results demonstrate that the AT subgenome in general contributed approximately two times more significantly induced genes to the fiber transcriptome than the DT subgenome; however, the Li2 mutation had greater effects on the DT subgenome than the AT subgenome.
Global Transcript Changes in Subgenomes of G. hirsutum Following Li2 Mutation
The role of the AT and DT subgenomes in determination of fiber quality in allotetraploid cotton has been extensively discussed in the literature. Allopolyploidization resulted in significant improvements in the desirable agronomic fiber traits in the allotetraploid species in comparison with the diploid progenitors [17], [18]. The first evidences showing that QTLs for fiber quality (including length, strength and fineness) were associated with DNA markers mapped to the DT subgenome rather than the AT subgenome was published by Paterson’s group [18]. Review of numerous QTLs published from 1998 to 2007 confirmed the observation that the DT subgenome plays a larger role in genetic control of fiber traits [19]. A microarray study published by Wendel’s group found that the homeolog expression in G. hirsutum was biased in favor of the DT subgenome in fiber cells [16]. Similar results were reported by Lacape and coauthors utilizing deep sequencing approach to analyze the fiber transcriptome of two allotetraploid species G. hirsutum and G. barbadense [20]. From an evolutionary point of view, these observations are surprising since the genes responsible for improved fiber properties evolved in the diploid AA genome before polyploidization [21]. None of the DD genome species produce spinnable fibers [22].
There are discrepancies in the literature regarding homeolog bias in contribution to fiber traits. Using a core set of 111 RFLP markers, Ulloa and coauthors revealed that the AT subgenome exhibited 68% of QTLs from the five chromosomes, whereas the DT subgenome exhibited only 32% of QTLs from the three chromosomes [23]. Another study utilizing combinations of markers found more fiber trait QTLs in the AT subgenome than in the DT subgenome [24]. The expression analysis of ESTs derived from immature ovules of G. hirsutum TM-1 revealed significant enrichment in all functional categories for AT subgenome ESTs [25].
These inconsistencies could be explained by technical limitations. The QTL studies reported in the current literature are detecting only a small subset of the genes related to fiber traits that may not cover the whole genome and could be insufficient to conclude which subgenome more significantly contributes to fiber properties [19]. The microarray studies evaluated a limited number of homeologous gene pairs, resulting in limited statistical power [16], [26]. Lacape and coauthors used next generation DNA sequencing technology for fiber transcriptome analysis; however, they evaluated only 617,000 good quality reads from four libraries without biological replication [20]. Unlike previous studies we obtained ∼160 million reads per sample for each of two biological replicates (Table 1), providing ∼5.6 times coverage of the G. hirsutum genome (∼2.83 Gb per haploid [27]), which is more than enough to deliver statistically powerful transcriptional analysis. Our observation of higher expression of AT than DT genes in the fiber transcriptome is consistent with the results of cotton ovules ESTs analysis [25] and reflects the evolutionary role of the AA diploid progenitor in fiber quality traits of allotetraploid cotton.
It is interesting to note that the Li2 mutation coincides with an increase in the number of expressed genes, but the homeolog expression bias was significantly decreased in Li2 fiber. How expression of homeologous genes is regulated in polyploids is still unclear, although it could involve altered regulatory interactions and rapid genetic and epigenetic changes in subgenomes [28]. The evolution homeolog-specific expression after polyploidization has been extensively studied in allotetraploid cotton. Higher rates of homeolog expression bias in natural allotetraploids than in hybrid and synthetic polyploid cottons suggested that the extent of homeolog expression bias increases over time from hybridization through evolution [26], [29], [30]. The Li2 mutation is negative for desirable fiber quality traits, resulting in extremely short lint fiber. Significant reduction of homeolog expression bias in short fiber suggests that the extent of homeolog expression bias is also important for fiber quality characteristics.
We observed a reciprocal switch for some genes in expression bias between homeologs during fiber developmental stages in the mutant. A high degree of expression differences between homeologous genes that are developmentally and stress regulated was reported in cotton [17], [31], [32]. A high-resolution genome-specific study of expression profiling for 63 gene pairs in 24 tissues in allopolyploid and their diploid progenitor cotton species demonstrated that the majority of expression differences between homeologs are caused by cis-regulatory divergence between the diploid progenitors; however, some degree of transcriptional neofunctionalization was detected as well [32].
The Li2 mutation was mapped to the DT subgenome [6]–[8]; however, the mutated gene and the nature of mutation are currently unknown. The greater mutation effect on the DT than on the AT subgenome observed here suggests two possible mechanisms. The network of regulatory interactions may have been interrupted by a mutation in the DT subgenome resulting in transactivation or repression of individual gene expression levels and expression cascades. Alternatively an epigenetic modulation may preferentially target the DT subgenome. It has been shown that small RNAs can control gene expression and epigenetic regulation in response to hybridization [33]–[36]. For example, miRNAs in allopolyploid Arabidopsis triggered unequal degradation of parental target genes [33]. Similarly, in rice hybrids small RNA populations inherited from parents were responsible for biased expression [36]. Additional investigations of epigenetic and chromatin level modifications will provide insights into causes of gene expression variation between subgenomes.
TRs and Cell Wall Functional Categories of Genes Regulated by Li2 Mutation
Previous transcriptomics and metabolomics studies have shown that the Li2 mutation terminated the cotton fiber elongation process [6], [10]; therefore, genes with changed expression level in the mutant could be involved in elongation. In the present work, we described in detail TRs and cell wall functional categories, which are critical for fiber developmental processes. Many genes in this list (Table 3 and Table 4) were not functionally characterized in cotton; although, based on sequence similarity to genes characterized in Arabidopsis, they could be involved with fiber elongation and represent candidates for further functional analysis in cotton.
Transcriptional regulators
Many TRs regulated by Li2 mutation are involved in hormonal signaling and development. Particularly, two AP2/EREBPs and six Aux/IAAs were in the pool of TRs affected by Li2 mutation (Table 3). Plant hormones are important for fiber development. It is well documented that exogenous applications of auxins and gibberellic acid stimulate the differentiation of fibers and promote elongation, while abscisic acid and cytokinins inhibit fiber growth in an in vitro cotton ovule culture system [37], [38]. Among the auxin responsive genes, Gorai.009G132300 and Gorai.010G227800, whose transcript abundances were significantly reduced in the DT genome of Li2, showed sequence similarity to grapevine VvIAA19 regulator [39]. Transgenic Arabidopsis plants over expressing VvIAA19 exhibited faster growth, including root elongation and floral transition, than the control, suggesting that grape Aux/IAA19 protein is likely to play a crucial role as a plant growth regulator. In the group of bHLH family of TRs, Gorai.007G005700 transcript abundance was significantly reduced in the DT genome of Li2 and showed sequence similarity to Arabidopsis BEE3, one of several redundant positive regulators of brassinosteroids signaling required for normal growth and development [40].
The actin cytoskeleton plays an important role in cell morphogenesis; down-regulation of GhACT1 disrupted the actin cytoskeleton network in fibers that resulted in inhibition of fiber elongation [41]. A GATA type TR Gorai.005G230900, a homolog of Arabidopsis WLIM1, was down-regulated in the DT subgenome of Li2; a recent study revealed that plant LIM-domain containing proteins (LIMs) define a highly specialized actin binding protein family, which contributes to the regulation of actin bundling in virtually all plant cells [42].
Cell wall
The plant cell wall has a dual role during elongation: to sustain the large mechanical forces caused by cell turgor and to permit controlled polymer extension generating more space for protoplast enlargement [43]. The active biosynthesis of matrix polysaccharides along with increased activity of cell wall loosening enzymes has been considered to be associated with cell wall extension [44]–[48]. Expression levels of genes encoding enzymes involved in xyloglucan and glucuronoxylan biosynthesis were decreased as a result of Li2 mutation. Particularly, xyloglucan β-galactosyltransferase (Arabidopsis homolog, MUR3 [49]) and xylosyltransferase (IRX9 [50]) were down-regulated in the AT or DT subgenomes of mutant fibers (Table 4).
Among cell wall proteins arabinogalactans were the most abundant members. Arabinogalactan-proteins have been implicated in many processes involved in plant growth and development, including cell expansion [51], [52].
Primary cell wall expansibility and strength is in part mediated by a group of enzymes that comprise a large family of cell wall modifying proteins, the xyloglucan endotransglycosylase/hydrolases (XTHs). XTHs are apoplast-localized enzymes that cleave and reattach xyloglucan polymers [53], [54]. The role of XTHs in cotton fiber elongation has been demonstrated: transgenic over-expression of GhXTH1 in cotton increased fiber length up to 20% [55]. DT biased Gorai.007G057400 corresponding to GhXTH1 was down-regulated in mutant fiber.
Conclusion
Repeated polyploidization over evolutionary time has played a significant role in adding genetic variation to the genomes of plant species. The evolution of the homeolog expression after polyploidization has been extensively studied in cotton comparing expression profiling between parental diploids and natural and synthetic allopolyploid species. This is the first report that explored the effects of a single mutation on the homeolog expression of allotetraploid cotton. Our results showed that significant reduction of the homeolog expression bias in mutant fiber correlates with negative fiber traits, indicating that the extent of homeolog expression bias is important for fiber quality characteristics. In addition, we observed significantly greater mutation effects on the DT than on the AT subgenome that might be explained by localization of the mutated gene. Additional studies using numerous naturally occurring cotton fiber mutations are needed to confirm these observations. This work will lead to an understanding of how gene regulation between AT and DT homeologs contributes to enhanced fiber morphology in cultivated cotton allopolyploids.
Materials and Methods
Plant Material and RNA Isolation
The cotton short fiber mutant Li2 was developed as a near-isogenic line (NIL) with the WT upland cotton line DP5690 as described before [6]. Growth conditions and fiber sampling were previously described [6]. Cotton bolls were harvested at the following time-points during development: day of anthesis (DOA), 1, 3, 5, 8, 12, 16, and 20 days post-anthesis (DPA). Cotton fibers were isolated from developing ovules using a glass bead shearing technique to separate fibers from the ovules [56]. Total RNA was isolated from detached fibers using the Sigma Spectrum Plant Total RNA Kit (Sigma-Aldrich, St. Louis, MO) with the optional on column DNase1 digestion according to the manufacturer’s protocol. The concentration of each RNA sample was determined using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE). The RNA quality for each sample was determined by RNA integrity number (RIN) using an Agilent Bioanalyzer 2100 and the RNA 6000 Nano Kit Chip (Agilent Technologies Inc., Santa Clara, CA) with 250 ng of total RNA per sample.
RT-qPCR Analysis
The experimental procedures and data analysis related to RT-qPCR were performed according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines [57]. Eight fiber developmental time-points mentioned above were used for RT-qPCR analyses of homeolog pairs which showed reciprocal expression biases. Only one time point, 8 DPA, was used for RT-qPCR confirmation of RNA-seq data of selected genes. The detailed description of reverse transcription, qPCR and calculation were previously reported [6]. Single nucleotide polymorphisms that distinguish the AT and DT subgenome copies of the selected genes were identified by aligning reads from the RNA-seq data to the G. raimondii reference mRNA sequences [11]. These homeologous SNPs were used to design subgenome specific primers by the SNAPER approach, whereby an additional mismatch is included near the end of the SNP-specific primers to increase stringency [58]. Primer sequences are provided in Table S3 and Table S4 in File S1. Correlations of biased expression patterns between RNA-seq and RT-qPCR data were calculated using GraphPad Prism 5 software (Pearson test).
Library Preparation and Sequencing
RNA samples from Li2 and wild type cotton fiber at 8 DPA (in two biological replicates) were subjected to paired-end Illumina mRNA sequencing (RNA-seq). Library preparation and sequencing were conducted by Data2Bio LLC (2079 Roy J. Carver Co-Laboratory, Ames, Iowa). Indexed libraries were prepared using the Illumina protocol outlined in the TruSeq RNA Sample Prep Guide (Part# 15008136 Rev. A, November 2010). The library size and concentration were determined using an Agilent Bioanalyzer. The indexed libraries were combined and seeded onto one lane of the flowcell. The libraries were sequenced using 101cycles of chemistry and imaging, resulting in paired end (PE) sequencing reads with length of 2×101 bp. The raw reads were submitted to the Sequence Read Archive (accession number SRP026301).
Processing of Illumina RNA-Seq Reads and Mapping to AT and DT Subgenomes of Gossypium hirsutum
The reads were trimmed with SICKLE (https://github.com/najoshi/sickle) using a quality score cutoff of 20. Mapping the reads (in pairs where both reads of a pair passed trimming) to the 13 chromosomes of the G. raimondii genome D5 v2 reference sequence was performed using GSNAP [59]. Default parameters were used, but with the flags “-n 1 -Q” which means that only a single mapping was reported for each read, and reads with multiple equally good hits were thrown away rather than randomly mapped. We used a cotton SNP index generated between DD genome G. raimondii and the AA genome G. arboreum to categorize reads of the allotetraploid G. hirsutum as belonging to the AT or DT subgenomes according to the method reported previously [14].
Digital Gene Expression Analysis
The comparison of the number of reads mapped to the genes of G. raimondii reference genome was used as an indicator of the relative digital gene expression (DGE). The JMP/Genomics 6.0 (SAS, Cary, NC, USA) was used for data normalization and statistical analysis. The data was normalized using TMM (Trimmed Mean of M component) method [60]. Genes with less than 10 reads in one sample were removed before normalization; from 37,223 genes assigned to chromosomes, 31,114 genes passed filtering conditions and were processed for normalization. The ANOVA process was fit to the normalized data, with the data following a Poisson distribution. This was accomplished with a generalized linear mixed model for each gene: Yij = Ti + Gj + TGij + Eijk, where T is the treatment effect for the ith biological treatment (Li2 or wild-type fiber), G is the specific subgenome type effect for the jth subgenome type (AT, DT, X and N categorized reads), their interaction (TG), and the error term (E). The linear model was used to test the null hypothesis that expression of a given gene was not different. Specifically, multiple comparisons were made between fiber type (Li2 versus wild type) and AT/DT subgenomes as well as combinations of these factors, such as fiber type in AT and DT subgenomes. We identified genes for which the difference in expression levels within these a priori questions were significantly different (false discovery rate≤0.05) [61].
Functional Categorization of Genes
Functional categorization of genes was performed using MapMan ontology [62]; the MapMan mapping for G. raimondii is available at http://mapman.gabipd.org/. Fisher’s exact test was used to estimate enrichment or depletion relative to background of functional categories with differentially regulated genes.
Supporting Information
RT-qPCR confirmation of silencing or activation of genes as a result of mutation. Bar charts represent RNA-seq and RT-qPCR data (side by side) at 8 DPA of fiber development for 8 randomly selected genes from Table S1 in File S1. Error bars indicate standard deviation from two biological replicates for RNA-seq data and three biological replicates for RT-qPCR.
(TIF)
RT-qPCR confirmation of biased expression of homeolog pairs. Bar charts represent RNA-seq and RT-qPCR data (side by side) at 8 DPA of fiber development for 11 randomly selected genes from Table 3 and Table 4. Pearson correlation (GraphPad Prism 5 software) of expression patterns for selected genes between RNA-seq and RT-qPCR data is provided in the table; correlation coefficients with p-value less than 0.05 are shown in boldface and underlined. Error bars indicate standard deviation from two biological replicates for RNA-seq data and three biological replicates for RT-qPCR.
(TIF)
Supporting tables. Table S1. Silencing or activation of genes as a result of mutation. Table S2. Mutation effects on functional distribution of homeolog genes. Fisher’s exact test results. Table S3. Primer’s sequences for detection expression of homeolog pairs. Table S4. Primer’s sequences.
(DOCX)
Statistical data for significantly regulated genes.
(TXT)
AT/DT biased genes annotated by MapMan ontology.
(TXT)
Acknowledgments
We thank Data2Bio for sequencing service. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture that is an equal opportunity provider and employer.
Funding Statement
This research was funded by United States Department of Agriculture-Agricultural Research Service project number 6435-21000-017-00D and Cotton Incorporated project number 58-6435-2-663. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wendel J, Cronn RC (2002) Polyploidy and the evolutionary history of cotton. Advances in Agronomy 78: 139–186. [Google Scholar]
- 2. Wendel JF (1989) New World tetraploid cottons contain Old World cytoplasm. Proceedings of the National Academy of Sciences of the United States of America 86: 4132–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Basra AS, Malik AC (1984) Development of the cotton fiber. International Review of Cytology 89: 65–113. [Google Scholar]
- 4. Kim HJ, Triplett BA (2001) Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant physiology 127: 1361–1366. [PMC free article] [PubMed] [Google Scholar]
- 5. Narbuth EV, Kohel RJ (1990) Inheritance and Linkage Analysis of a New Fiber Mutant in Cotton. Journal of Heredity 81: 131–133. [Google Scholar]
- 6. Hinchliffe DJ, Turley RB, Naoumkina M, Kim HJ, Tang Y, et al. (2011) A combined functional and structural genomics approach identified an EST-SSR marker with complete linkage to the Ligon lintless-2 genetic locus in cotton (Gossypium hirsutum L.). BMC genomics 12: 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kohel RJ, Stelly DM, Yu J (2002) Tests of six cotton (Gossypium hirsutum L.) mutants for association with aneuploids. The Journal of Heredity 93: 130–132. [DOI] [PubMed] [Google Scholar]
- 8. Rong J, Pierce GJ, Waghmare VN, Rogers CJ, Desai A, et al. (2005) Genetic mapping and comparative analysis of seven mutants related to seed fiber development in cotton. Theoretical and applied genetics 111: 1137–1146. [DOI] [PubMed] [Google Scholar]
- 9. Kohel RJ, Narbuth EV, Benedict CR (1992) Fiber development of Ligon lintless-2 mutant of cotton. Crop Science 32: 733–735. [Google Scholar]
- 10. Naoumkina M, Hinchliffe D, Turley R, Bland J, Fang D (2013) Integrated metabolomics and genomics analysis provides new insights into the fiber elongation process in ligon lintless-2 mutant cotton (Gossypium hirsutum L.). BMC genomics 14: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, et al. (2012) Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492: 423–427. [DOI] [PubMed] [Google Scholar]
- 12. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews Genetics 10: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doyle JJ, Flagel LE, Paterson AH, Rapp RA, Soltis DE, et al. (2008) Evolutionary genetics of genome merger and doubling in plants. Annual review of genetics 42: 443–461. [DOI] [PubMed] [Google Scholar]
- 14. Page JT, Gingle AR, Udall JA (2013) PolyCat: a resource for genome categorization of sequencing reads from allopolyploid organisms. G3: Genes|Genomes|Genetics 3: 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gilbert MK, Bland JM, Shockey JM, Cao H, Hinchliffe DJ, et al. (2013) A transcript profiling approach reveals an abscisic acid specific glycosyltransferase (UGT73C14) induced in developing fiber of Ligon lintless-2 mutant of cotton (Gossypium hirsutum L.). PloS one 8: e75268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hovav R, Udall JA, Chaudhary B, Rapp R, Flagel L, et al. (2008) Partitioned expression of duplicated genes during development and evolution of a single cell in a polyploid plant. Proceedings of the National Academy of Sciences of the United States of America 105: 6191–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adams KL, Cronn R, Percifield R, Wendel JF (2003) Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proceedings of the National Academy of Sciences of the United States of America 100: 4649–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang C, Wright RJ, El-Zik KM, Paterson AH (1998) Polyploid formation created unique avenues for response to selection in Gossypium (cotton). Proceedings of the National Academy of Sciences of the United States of America 95: 4419–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chee PW, Campbell BT (2009) Bridging classical and molecular genetics of cotton fiber quality and development. In: Paterson AH, editor. Genetics and genomics of cotton. New York: Springer. 283–311.
- 20. Lacape JM, Claverie M, Vidal RO, Carazzolle MF, Guimaraes Pereira GA, et al. (2012) Deep sequencing reveals differences in the transcriptional landscapes of fibers from two cultivated species of cotton. PloS one 7: e48855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brubaker CL, Paterson AH, Wendel JF (1999) Comparative genetic mapping of allotetraploid cotton and its diploid progenitors. Genome 42: 184–203. [Google Scholar]
- 22. Kohel R, Yu J, Park Y-H, Lazo G (2001) Molecular mapping and characterization of traits controlling fiber quality in cotton. Euphytica 121: 163–172. [Google Scholar]
- 23. Ulloa M, Saha S, Jenkins JN, Meredith WR, McCarty JC, et al. (2005) Chromosomal Assignment of RFLP Linkage Groups Harboring Important QTLs on an Intraspecific Cotton (Gossypium hirsutum L.) Joinmap. Journal of Heredity 96: 132–144. [DOI] [PubMed] [Google Scholar]
- 24. Mei M, Syed NH, Gao W, Thaxton PM, Smith CW, et al. (2004) Genetic mapping and QTL analysis of fiber-related traits in cotton (Gossypium). Theoretical and applied genetics 108: 280–291. [DOI] [PubMed] [Google Scholar]
- 25. Yang SS, Cheung F, Lee JJ, Ha M, Wei NE, et al. (2006) Accumulation of genome-specific transcripts, transcription factors and phytohormonal regulators during early stages of fiber cell development in allotetraploid cotton. The Plant journal 47: 761–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flagel L, Udall J, Nettleton D, Wendel J (2008) Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC biology 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grover CE, Kim H, Wing RA, Paterson AH, Wendel JF (2004) Incongruent patterns of local and global genome size evolution in cotton. Genome research 14: 1474–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Osborn TC, Pires JC, Birchler JA, Auger DL, Chen ZJ, et al. (2003) Understanding mechanisms of novel gene expression in polyploids. Trends in genetics : TIG 19: 141–147. [DOI] [PubMed] [Google Scholar]
- 29. Flagel LE, Wendel JF (2010) Evolutionary rate variation, genomic dominance and duplicate gene expression evolution during allotetraploid cotton speciation. The New phytologist 186: 184–193. [DOI] [PubMed] [Google Scholar]
- 30. Yoo MJ, Szadkowski E, Wendel JF (2013) Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110: 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dong S, Adams KL (2011) Differential contributions to the transcriptome of duplicated genes in response to abiotic stresses in natural and synthetic polyploids. The New phytologist 190: 1045–1057. [DOI] [PubMed] [Google Scholar]
- 32. Chaudhary B, Flagel L, Stupar RM, Udall JA, Verma N, et al. (2009) Reciprocal silencing, transcriptional bias and functional divergence of homeologs in polyploid cotton (Gossypium). Genetics 182: 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ha M, Lu J, Tian L, Ramachandran V, Kasschau KD, et al. (2009) Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proceedings of the National Academy of Sciences of the United States of America 106: 17835–17840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Madlung A, Tyagi AP, Watson B, Jiang H, Kagochi T, et al. (2005) Genomic changes in synthetic Arabidopsis polyploids. The Plant journal 41: 221–230. [DOI] [PubMed] [Google Scholar]
- 35. Salmon A, Ainouche ML, Wendel JF (2005) Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Molecular Ecology 14: 1163–1175. [DOI] [PubMed] [Google Scholar]
- 36. He G, Zhu X, Elling AA, Chen L, Wang X, et al. (2010) Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. The Plant cell 22: 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beasley CA, Ting IP (1973) The effects of plant growth substances on in-vitro fiber development from fertilized cotton ovules. American Journal of Botany 60: 130–139. [Google Scholar]
- 38. Beasley CA, Ting IP (1974) Effects of plant growth substances on in-vitro fiber development from unfertilized cotton ovules. American Journal of Botany 61: 188–194. [Google Scholar]
- 39. Kohno M, Takato H, Horiuchi H, Fujita K, Suzuki S (2012) Auxin-nonresponsive grape Aux/IAA19 is a positive regulator of plant growth. Molecular biology reports 39: 911–917. [DOI] [PubMed] [Google Scholar]
- 40. Friedrichsen DM, Nemhauser J, Muramitsu T, Maloof JN, Alonso J, et al. (2002) Three redundant brassinosteroid early response genes encode putative bHLH transcription factors required for normal growth. Genetics 162: 1445–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li XB, Fan XP, Wang XL, Cai L, Yang WC (2005) The cotton ACTIN1 gene is functionally expressed in fibers and participates in fiber elongation. The Plant cell 17: 859–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Papuga J, Hoffmann C, Dieterle M, Moes D, Moreau F, et al. (2010) Arabidopsis LIM proteins: a family of actin bundlers with distinct expression patterns and modes of regulation. The Plant cell 22: 3034–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cosgrove DJ (2001) Wall structure and wall loosening. A look backwards and forwards. Plant physiology 125: 131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takeda T, Furuta Y, Awano T, Mizuno K, Mitsuishi Y, et al. (2002) Suppression and acceleration of cell elongation by integration of xyloglucans in pea stem segments. Proceedings of the National Academy of Sciences of the United States of America 99: 9055–9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park YW, Baba K, Furuta Y, Iida I, Sameshima K, et al. (2004) Enhancement of growth and cellulose accumulation by overexpression of xyloglucanase in poplar. FEBS letters 564: 183–187. [DOI] [PubMed] [Google Scholar]
- 46. Hayashi T, Delmer DP (1988) Xyloglucan in the cell walls of cotton fiber. Carbohydrate research 181: 273–277. [Google Scholar]
- 47. Huwyler HR, Franz G, Meier H (1979) Changes in the composition of cotton fiber cell walls during development. Planta 146: 635–642. [DOI] [PubMed] [Google Scholar]
- 48. Shimizu Y, Aotsuka S, Hasegawa O, Kawada T, Sakuno T, et al. (1997) Changes in levels of mRNAs for cell wall-related enzymes in growing cotton fiber cells. Plant & cell physiology 38: 375–378. [DOI] [PubMed] [Google Scholar]
- 49. Madson M, Dunand C, Li X, Verma R, Vanzin GF, et al. (2003) The MUR3 gene of Arabidopsis encodes a xyloglucan galactosyltransferase that is evolutionarily related to animal exostosins. The Plant cell 15: 1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee C, Zhong R, Ye ZH (2012) Arabidopsis family GT43 members are xylan xylosyltransferases required for the elongation of the xylan backbone. Plant & cell physiology 53: 135–143. [DOI] [PubMed] [Google Scholar]
- 51. Lee KJ, Sakata Y, Mau SL, Pettolino F, Bacic A, et al. (2005) Arabinogalactan proteins are required for apical cell extension in the moss Physcomitrella patens. The Plant cell 17: 3051–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang J, Showalter AM (2007) Expression and localization of AtAGP18, a lysine-rich arabinogalactan-protein in Arabidopsis. Planta 226: 169–179. [DOI] [PubMed] [Google Scholar]
- 53. Nishitani K, Tominaga R (1992) Endo-xyloglucan transferase, a novel class of glycosyltransferase that catalyzes transfer of a segment of xyloglucan molecule to another xyloglucan molecule. The Journal of biological chemistry 267: 21058–21064. [PubMed] [Google Scholar]
- 54. Fry SC, Smith RC, Renwick KF, Martin DJ, Hodge SK, et al. (1992) Xyloglucan endotransglycosylase, a new wall-loosening enzyme activity from plants. The Biochemical journal 282 (Pt 3): 821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee J, Burns TH, Light G, Sun Y, Fokar M, et al. (2010) Xyloglucan endotransglycosylase/hydrolase genes in cotton and their role in fiber elongation. Planta 232: 1191–1205. [DOI] [PubMed] [Google Scholar]
- 56. Taliercio EW, Boykin D (2007) Analysis of gene expression in cotton fiber initials. BMC plant biology 7: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, et al. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry 55: 611–622. [DOI] [PubMed] [Google Scholar]
- 58. Drenkard E, Richter BG, Rozen S, Stutius LM, Angell NA, et al. (2000) A simple procedure for the analysis of single nucleotide polymorphisms facilitates map-based cloning in Arabidopsis. Plant physiology 124: 1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu TD, Nacu S (2010) Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological) 57: 289–300. [Google Scholar]
- 62. Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, et al. (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. The Plant journal 37: 914–939. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RT-qPCR confirmation of silencing or activation of genes as a result of mutation. Bar charts represent RNA-seq and RT-qPCR data (side by side) at 8 DPA of fiber development for 8 randomly selected genes from Table S1 in File S1. Error bars indicate standard deviation from two biological replicates for RNA-seq data and three biological replicates for RT-qPCR.
(TIF)
RT-qPCR confirmation of biased expression of homeolog pairs. Bar charts represent RNA-seq and RT-qPCR data (side by side) at 8 DPA of fiber development for 11 randomly selected genes from Table 3 and Table 4. Pearson correlation (GraphPad Prism 5 software) of expression patterns for selected genes between RNA-seq and RT-qPCR data is provided in the table; correlation coefficients with p-value less than 0.05 are shown in boldface and underlined. Error bars indicate standard deviation from two biological replicates for RNA-seq data and three biological replicates for RT-qPCR.
(TIF)
Supporting tables. Table S1. Silencing or activation of genes as a result of mutation. Table S2. Mutation effects on functional distribution of homeolog genes. Fisher’s exact test results. Table S3. Primer’s sequences for detection expression of homeolog pairs. Table S4. Primer’s sequences.
(DOCX)
Statistical data for significantly regulated genes.
(TXT)
AT/DT biased genes annotated by MapMan ontology.
(TXT)