

ABSTRACT
Andes virus (ANDV) is the only hantavirus known to spread from person to person and shown to cause highly lethal hantavirus pulmonary syndrome (HPS) in patients and Syrian hamsters. Hantaviruses replicate in human endothelial cells and accomplish this by restricting the early induction of beta interferon (IFN-β)- and IFN-stimulated genes (ISGs). Our studies reveal that the ANDV nucleocapsid (N) protein uniquely inhibits IFN signaling responses directed by cytoplasmic double-stranded RNA (dsRNA) sensors RIG-I and MDA5. In contrast, N proteins from Sin Nombre, New York-1, and Prospect Hill hantaviruses had no effect on RIG-I/MDA5-directed transcriptional responses from IFN-β-, IFN-stimulated response element (ISRE)-, or κB-containing promoters. Ablating a potential S-segment nonstructural open reading frame (ORF) (NSs) within the ANDV plasmid expressing N protein failed to alter IFN regulation by ANDV N protein. Further analysis demonstrated that expressing the ANDV N protein inhibited downstream IFN pathway activation directed by MAVS, TBK1, and IκB kinase ε (IKKε) but failed to inhibit transcriptional responses directed by constitutive expression of active interferon regulatory factor IRF3-5D or after stimulation by alpha interferon (IFN-α) or tumor necrosis factor alpha (TNF-α). Consistent with IFN pathway-specific regulation, the ANDV N protein inhibited TBK1-directed IRF3 phosphorylation (phosphorylation of serine 396 [pS396]) and TBK1 autophosphorylation (pS172). Collectively, these findings indicate that the ANDV N inhibits IFN signaling responses by interfering with TBK1 activation, upstream of IRF3 phosphorylation and NF-κB activation. Moreover, our findings reveal that ANDV uniquely carries a gene encoding a virulence determinant within its N protein that is capable of restricting ISG and IFN-β induction and provide a rationale for the novel pathogenesis and spread of ANDV.
IMPORTANCE
Andes virus (ANDV) is distinguished from other hantaviruses by its unique ability to spread from person to person and cause lethal hantavirus pulmonary syndrome (HPS)-like disease in Syrian hamsters. However, virulence determinants that distinguish ANDV from other pathogenic hantaviruses have yet to be defined. Here we reveal that ANDV uniquely contains a virulence determinant within its nucleocapsid (N) protein that potently inhibits innate cellular signaling pathways. This novel function of the N protein provides a new mechanism for hantaviruses to regulate interferon (IFN) and IFN-stimulated gene (ISG) induction that is likely to contribute to the enhanced ability of ANDV to replicate, spread, and cause disease. These findings differentiate ANDV from other HPS-causing hantaviruses and provide a potential target for viral attenuation that needs to be considered in vaccine development.
INTRODUCTION
Hantaviruses are carried by specific small mammal hosts and spread to humans by the inhalation of aerosolized excreted virus (1). Hantaviruses predominantly infect the endothelial cell lining of the vasculature and nonlytically cause 2 diseases associated with capillary leakage (1–4). Hantaan virus (HTNV), Puumala virus (PUUV), and other Old World hantaviruses cause hemorrhagic fever with renal syndrome (HFRS), while Andes virus (ANDV), Sin Nombre virus (SNV), and New York-1 virus (NY-1V) are New World North and South American hantaviruses that cause a highly lethal hantavirus pulmonary syndrome (HPS) (1, 5, 6). Viruses that cause both HPS and HFRS infect vast capillary beds of pulmonary and renal endothelial cells, and pulmonary and renal disease manifestations can be found in either syndrome (7, 8). As a result, hantavirus pathogenesis is not purely a function of organ-specific endothelial cell targeting by HPS- or HFRS-causing hantaviruses. Consistent with this, Tula virus (TULV) and Prospect Hill virus (PHV) hantaviruses infect human endothelial cells but are not associated with any human disease (2, 9). Collectively, these findings suggest that pathogenicity is conferred by virulence determinants encoded within genes of discrete hantaviruses.
Hantaviruses are enveloped viruses with three negative-sense RNA segments (L, M, and S) that encode a polymerase, heterodimeric surface glycoproteins (Gn and Gc) and a highly expressed cytoplasmic nucleocapsid (N) protein (1, 10, 11). Gn and Gc are integral membrane proteins trafficked to the endoplasmic reticulum (ER)/cis-Golgi and acquired on hantavirus surfaces by viral budding into the lumen of the Golgi bodies (1). Pathogenic hantaviruses engage inactive conformations of αvβ3 integrins for cell entry and later virion accumulation on the surfaces of human endothelial cells (12–16). In contrast, nonpathogenic TULV and PHV interact with β1 integrins, but not β3 integrins, and fail to alter β3 integrin-regulated barrier functions of endothelial cells (12, 14, 16–18). Although hantavirus surface glycoprotein (GnGc) interactions with specific integrins have not been resolved, the prominent roles of endothelial cell and platelet β3 integrins in regulating vascular permeability (16, 17, 19) suggest that hantavirus GnGc proteins are pathogenic determinants present on the virion’s surface.
In order for hantaviruses to be human pathogens, it is essential that they productively replicate in human endothelial cells (2, 20). Hantavirus replication is blocked by the addition of type I interferon (alpha/beta interferon [IFN-α/β]), and hantaviruses are grown in IFN locus-deficient cells (2, 20–22). In fact, PHV lacks the ability to be a pathogen in part because it rapidly induces IFN-β and IFN-stimulated genes (ISGs) that restrict PHV replication in human endothelial cells (20–22). This suggests that the permissive replication of hantaviruses within human endothelial cells requires the expression of virulence determinants that inhibit the early induction of IFN-β and ISG responses (20–25).
Cytosolic retinoic acid-inducible gene 1 (RIG-I)/melanoma differentiation-associated protein 5 (MDA5) sensors detect small amounts of double-stranded RNA (dsRNA) generated by RNA viruses and direct signaling responses that induce IFN-β in endothelial cells (26, 27). Signals emanating from cellular sensors converge on the activation of TANK binding kinase 1 (TBK1), and its homologue IκB kinase (IKKε), which are critical inducers of IFN signaling (26–29). Activation of TBK1 causes its autophosphorylation, and in turn, TBK1 phosphorylates IFN regulatory factors (IRF)3/5/7 and activates NF-κB in order to transcriptionally induce IFN-β (27, 28, 30–33). With the exception of PHV, expression of hantavirus Gn proteins or their cytoplasmic tails (GnTs) has been shown to regulate RIG-I-directed transcriptional responses from IFN-stimulated response element (ISRE)- or IFN-β enhanceosome-containing promoters (20–25, 34). These reports suggest that the Gn protein cytoplasmic tail contains virulence determinants that contribute to the early regulation of innate immune responses.
ANDV is the only hantavirus spread person to person and the only hantavirus that causes lethal HPS-like disease in Syrian hamsters (35–38). However, mechanisms that account for the enhanced viral spread and pathogenesis of ANDV in humans or animal models have yet to be defined (39). N proteins are highly expressed in hantavirus-infected cells (1, 11), and our surprising new findings reveal that the ANDV N protein uniquely inhibits RIG-I-directed ISRE, κB, and IFN-β promoter induction. In contrast, N proteins from other hantaviruses had no effect on IFN signaling responses. This suggests that ANDV carries a gene encoding a novel IFN-regulating virulence determinant within its N protein that may promote ANDV pathogenesis and spread.
Here we examined N proteins from ANDV, NY-1V, SNV, and PHV for their ability to regulate IFN signaling pathway-directed phosphorylation and transcriptional responses. Our findings demonstrate that in contrast to other hantavirus N proteins, the ANDV N protein regulates RIG-I, MDA5, MAVS (mitochondrial antiviral signaling protein), TBK1, and IKKε induced transcriptional responses from ISRE, κB, and IFN-β promoters. However, the ANDV N protein failed to inhibit transcriptional responses directed by expressing the constitutively active IRF3-5D protein (48). Consistent with this, the ANDV N protein uniquely inhibited TBK1 autophosphorylation as well as the phosphorylation of IRF3. In contrast, the ANDV N protein failed to regulate transcriptional responses induced by the addition of tumor necrosis factor alpha (TNF-α) or IFN-α. Thus, the ANDV N protein selectively regulates cellular IFN signaling pathways by inhibiting the activation of TBK1/IKKε. Our results indicate that only the ANDV N protein regulates innate cell signaling pathway responses and suggest that the ANDV N protein is a unique hantavirus virulence determinant. These findings suggest a potential role for the N protein in the enhanced pathogenesis and spread of ANDV.
RESULTS
Previous studies suggested that hantavirus N proteins fail to inhibit IFN signaling responses directed by RIG-I or TBK1 (21–24, 34, 40). However, when we initially evaluated the ANDV N protein, we were surprised to find that it potently inhibited RIG-I-directed transcriptional responses from ISRE-, IFN-β-, and κB-directed luciferase reporters (Fig. 1A). In contrast, comparable expression of the SNV N protein had no effect on RIG-I-directed responses. Quantitative reverse transcription-PCR (qRT-PCR) analysis of cellular mRNAs in response to RIG-I activation similarly demonstrated that only expression of the ANDV N protein potently inhibited IFN-β mRNA induction (Fig. 1B). These findings suggested that the ANDV N protein has the unique ability to regulate IFN signaling responses and prompted an in-depth analysis of ANDV N protein functions.
FIG 1 .
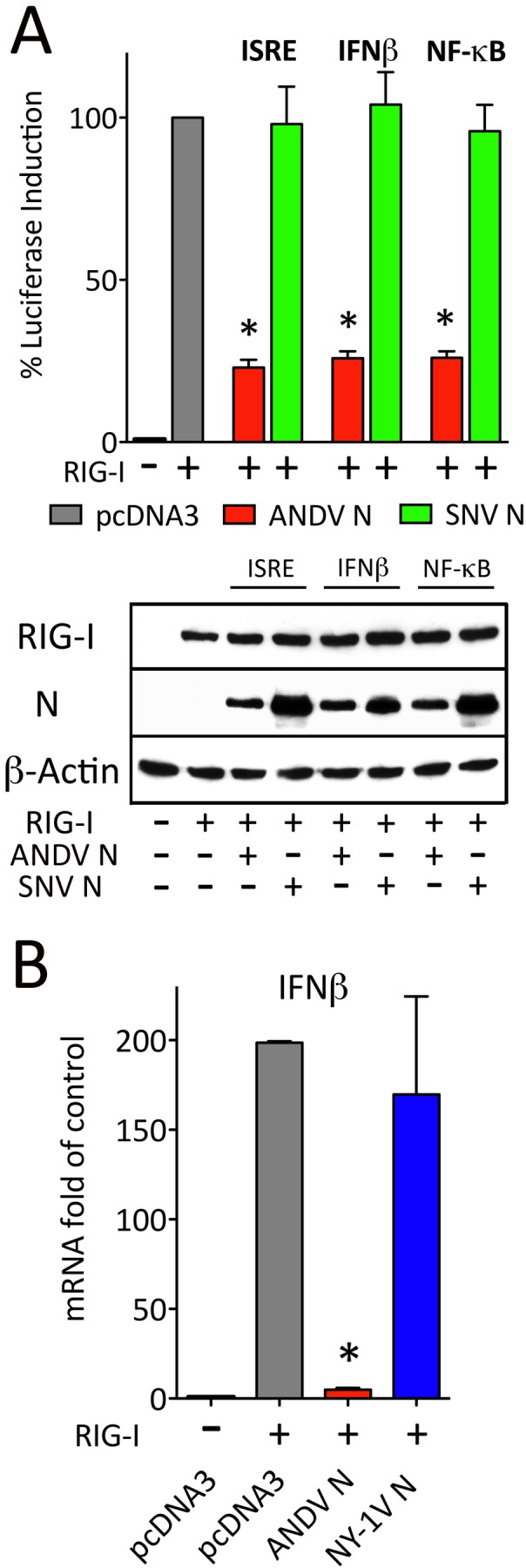
ANDV N protein regulates ISRE, NF-κβ, and IFN-β promoter transcription. (A) HEK293T cells were transfected with ISRE, κβ, or IFN-β promoter-directed luciferase reporter and Renilla luciferase-expressing plasmids (34). Cells were cotransfected with a TBK1-expressing plasmid and plasmids expressing either ANDV N protein or SNV N protein. Cells were harvested 1 day posttransfection and assayed for firefly luciferase activity. Results are presented as the percent induction compared to pcDNA3 induction control (100%) after standardization to Renilla luciferase levels as previously described (34). (B) HEK293T cells were transfected with plasmids expressing ANDV or NY-1V N protein (+) or a pcDNA control, and 24 h later, cells were activated by transfection with RIG-I CARD-expressing plasmid (+) as indicated. IFN-β mRNA levels were analyzed 12 h later by qRT-PCR and standardized to GAPDH mRNA levels by the 2−ΔCT method as previously described (61). Values that are significantly different (P < 0.05) by Student’s t test are indicated by an asterisk.
ANDV N protein uniquely inhibits the RIG-I/MDA5 interferon signaling pathway.
RNA viruses generate small amounts of dsRNA products that are sensed by RIG-I and MDA5 helicases in the cytoplasm of infected cells (26). RIG-I and MDA5 direct IFN induction by engaging downstream MAVS and TBK1/IKKε signaling effectors that activate IRF3/5/7 and NF-κB transcription factors required for the induction of type I IFN (26–28, 30, 31, 41). Here we determined the point at which the ANDV N protein regulates this IFN signaling pathway. Plasmids expressing N proteins from ANDV, NY-1V, PHV, and SNV or the ANDV GnGc were cotransfected into HEK293T cells along with plasmids expressing RIG-I, MDA5, or MAVS and ISRE-luciferase reporters. We found that the ANDV N and GnGc proteins inhibited ISRE transcriptional responses induced by RIG-I, MDA5, or MAVS by ~70% to 90% (Fig. 2A to C) (34). In contrast, comparable expression of N proteins from SNV, NY-1V, or PHV had no effect on IFN signaling responses (Fig. 2A to C). These findings are consistent with ANDV N protein regulation of cytoplasmic helicase-directed IFN signaling pathways (26, 27).
FIG 2 .

ANDV N protein blocks RIG-I-, MDA5-, and MAVS-directed ISRE induction. HEK293T cells were transfected with an ISRE-driven firefly luciferase reporter and a plasmid constitutively expressing Renilla luciferase in the presence or absence of vectors expressing RIG-I CARD (A), MDA5 (B), or MAVS (C). The cells were cotransfected with plasmids expressing ANDV, NY-1V, SNV, or PHV N protein or ANDV GnGc protein. Empty vector (pcDNA3) was used to maintain constant DNA levels and as a negative control. Luciferase (Luc) activity was determined 24 h posttransfection, normalized to Renilla luciferase activity, and reported as the fold increase compared to that of controls lacking inducer (34). Assays were performed in triplicate with similar results in at least 3 separate experiments. Asterisks indicate statistical significance (P < 0.05) as determined by Student’s t test. Viral proteins, inducers, and β-actin (loading control) were detected by Western blot analysis as described previously (34) and are shown in the blots below the bar graphs. neg, negative.
ANDV N protein blocks TBK1/IKKε transcriptional responses.
RIG-I/MDA5/MAVS signaling responses activate homologous downstream kinases TBK1 and IKKε which phosphorylate IRF3 and activate NF-κB (26–28, 31). Expression of TBK1 or IKKε induces transcriptional responses from κB, ISRE, and IFN-β promoters. In a comparative analysis of N-protein functions, we found that the ANDV N protein inhibited TBK1-directed ISRE, κB, or IFN-β transcriptional responses >70% to 80% (Fig. 3A to C), while comparably expressed N proteins from NY-1V, PHV, and SNV failed to regulate TBK1-directed responses from any promoter.
FIG 3 .
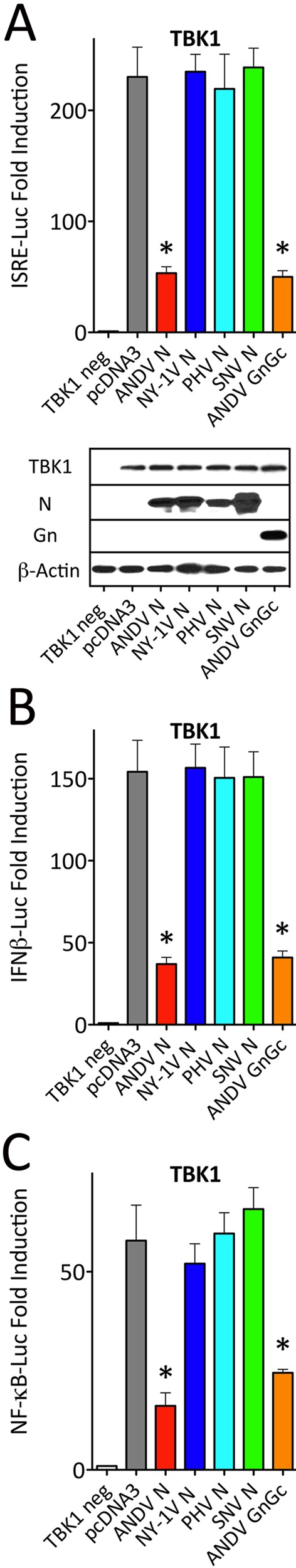
ANDV N protein blocks TBK1-induced transcriptional responses. (A to C) HEK293T cells were transfected with an ISRE (A), IFN-β (B), or κB (C) promoter-driven firefly luciferase reporter and a plasmid constitutively expressing Renilla luciferase in the presence or absence of TBK1-expressing plasmid (TBK1 neg). Cells were cotransfected with plasmids expressing ANDV, NY-1V, SNV, or PHV N protein or ANDV GnGc protein. Luciferase activity within lysates was determined and reported with statistical significance as described in the legend to Fig. 2. Western blot analysis was performed as described in the legend to Fig. 2 to detect N protein, Gn, actin, and TBK1.
Further analysis demonstrated that ANDV N protein regulation is dose dependent and inhibited transcriptional responses to a similar extent as ANDV GnGc or NY-1V GnT proteins (Fig. 4A) (34). A potential truncated nonstructural protein open reading frame (ORF) (NSs; 63 residues) is present within the ANDV S segment N protein-coding region (42). NSs is associated with IFN regulation by the Puumala hantavirus and La Crosse and Rift Valley fever viruses (Bunyaviridae family), but it is not essential in Bunyamwera virus (43–46). In order to determine whether the NSs ORF is responsible for observed TBK1 regulation, we generated 2 termination codons within NSs (residues 13 and 28) by mutagenesis without changing ANDV N protein-coding sequences (ΔNSs). Although there are no reagents to analyze NSs expression, wild-type (WT) ANDV N and N ΔNSs were comparably expressed (Fig. 4B). However, we observed no difference in the ability of WT N or the N ΔNSs mutant to regulate TBK1-directed ISRE transcription (Fig. 4B).
FIG 4 .

ANDV N protein, not NSs, regulates IFN induction. (A) HEK293T cells were transfected with an ISRE promoter-driven firefly luciferase reporter and a plasmid constitutively expressing Renilla luciferase in the presence or absence of TBK1-expressing plasmid. The cells were cotransfected with increasing or constant amounts of plasmids expressing ANDV N protein, ANDV GnGc protein, NY-1V N protein, or NY-1V Gn tail (GnT) as indicated. Luciferase activity and protein expression within lysates was determined as described in the legend to Fig. 2. The lines between the sections of the Western blots indicate splicing of discrete expression responses. (B) HEK293T cells were transfected with ISRE-driven reporter plasmid and Renilla control luciferase plasmid in the presence or absence of TBK1 expression vector. The cells were cotransfected with pcDNA3 (control) or with vectors expressing ANDV N ΔNSs, ANDV N protein, or NY-1V N protein. The cells were harvested 24 h posttransfection and assayed as described in the legend to Fig. 2 by Western blot analysis to detect cellular proteins.
IKKε shares 49% identity and 65% similarity with TBK1, and both IKKε and TBK1 are functionally homologous kinases that phosphorylate IRF3/7 and activate NF-κB (28, 32, 47). Similar to TBK1-directed responses, we found that the ANDV N protein also inhibited IKKε-directed ISRE transcriptional responses, while N proteins from NY-1V, PHV, and SNV had no effect on IKKε signaling (Fig. 5A). These findings demonstrate that only the ANDV N protein inhibits TBK1/IKKε-directed signaling responses required for IRF3 and NF-κB activation.
FIG 5 .

ANDV N protein blocks IKKε signaling response, but not IRF3-5D, IFN-α, and TNF-α signaling responses. (A and B) HEK293T cells were transfected with an ISRE-driven firefly luciferase reporter and a plasmid constitutively expressing Renilla luciferase in the presence or absence of vectors expressing IKKε (A) or IRF3-5D (B) (34). The cells were cotransfected with plasmids expressing hantavirus N proteins (ANDV, NY-1V, SNV, and PHV) or ANDV GnGc protein and assayed as described in the legend to Fig. 2 for luciferase induction and protein expression. (C and D) HEK293T cells were transfected with an ISRE (C) or κB (D) promoter-driven firefly luciferase reporter and a plasmid constitutively expressing Renilla luciferase. The cells were cotransfected with plasmids expressing hantavirus N proteins (ANDV, NY-1V, SNV, and PHV) or ANDV GnGc and either SOCS1 (C) or IκBα-SR (D) positive-control expression vectors. (C) IFN-α (500 U/well) was added 6 h prior to cell lysis. (D) TNF-α (50 ng/ml) was added 12 h prior to cell lysis. The cells were assayed as described in the legend to Fig. 2 for luciferase induction 24 h posttransfection.
Mutating 5 serine residues to aspartic acid within IRF3 results in a constitutively active IRF3-5D protein that does not require TBK1 or IKKε to direct ISRE transcriptional responses (48). In contrast to TBK1/IKKε regulation, we found that the ANDV N protein had no effect on IRF3-5D-directed transcription from ISRE promoters (Fig. 5B). Similarly, expressing N proteins from NY-1V, PHV, and SNV or the ANDV GnGc protein had no effect on IRF3-5D-directed ISRE transcription. These findings indicate that the ANDV N protein regulates the IFN signaling pathway at a point upstream of phosphorylated IRF3 and at the level of TBK1/IKKε complexes.
The addition of IFN-α or TNF-α to cells activates the cognate IFN or TNF receptor signaling pathway and induces ISRE or NF-κB transcriptional response, respectively, independent of TBK1 or IKKε (27, 28, 47). In order to determine whether the ANDV N protein is a broadly active signaling inhibitor, we assayed the ability of N to inhibit ISRE and κB transcriptional responses induced by IFN-α or TNF-α addition. We found that expressing N proteins from ANDV, NY-1V, PHV, or SNV had no effect on IFN-α- or TNF-α-induced ISRE or κB transcriptional responses (Fig. 5C and D). In contrast, transfection of cells with pathway-specific inhibitors SOCS1 (suppressor of cytokine signaling 1) and IκB superrepressor (IκB-SR), respectively, blocked ISRE and κB transcriptional responses (Fig. 5C and D). As a result, the ANDV N protein selectively inhibits TBK1/IKKε-directed κB, ISRE, and IFN-β transcriptional responses, but it is not a ubiquitous inhibitor of NF-κB or type I IFN receptor signaling pathways.
ANDV N protein inhibits TBK1 autophosphorylation.
The results of the studies discussed above indicate that the ANDV N protein inhibits TBK1/IKKε-directed transcriptional responses. However, TBK1 requires autophosphorylation at serine 172 prior to its activation and subsequent phosphorylation of IRF3 (28, 30, 47). In addition, following RNA virus infection, phosphorylation of serine 396 (pS396) on IRF3 plays a pivotal role in IRF3 activation (30, 31, 47). Collectively, these findings suggest that the ANDV N protein could act at several points to inhibit TBK1-directed signaling responses. Here we evaluated TBK1 and IRF3 phosphorylation in the presence of N proteins from ANDV, SNV, PHV, and NY-1V. We observed that only the ANDV N protein inhibited TBK1-directed phosphorylation of IRF3 (pS396) (Fig. 6) and that comparably expressed N proteins from SNV, NY-1V, and PHV had no effect on IRF3 phosphorylation. Interestingly, there was also a dramatic decrease in the phosphorylation of serine 172 (pS172) of TBK1 in the presence of the ANDV N protein that was not observed following coexpression of other N proteins (Fig. 6). These findings indicate that the ANDV N protein inhibits TBK1-directed IRF3 phosphorylation and NF-κB activation by preventing the autophosphorylation and activation of TBK1 (Fig. 7).
FIG 6 .

ANDV N protein inhibits TBK1 (pS172) and IRF3 (pS396) phosphorylation. HEK293T cells were cotransfected with empty vector (pcDNA3) or with plasmid expressing ANDV N protein, NY-1V N protein, PHV N protein, or SNV N protein along with IRF3 expression vector in the presence or absence of TBK1-Flag expression vector as indicated. After 24 h, the cells were harvested and analyzed by Western blotting for N protein, phosphorylated TBK1 (pTBK1) (pSer172), phosphorylated IRF3 (pIRF3) (pS396), total IRF3, or total TBK1 as indicated and compared to β-actin (loading control) (34).
FIG 7 .
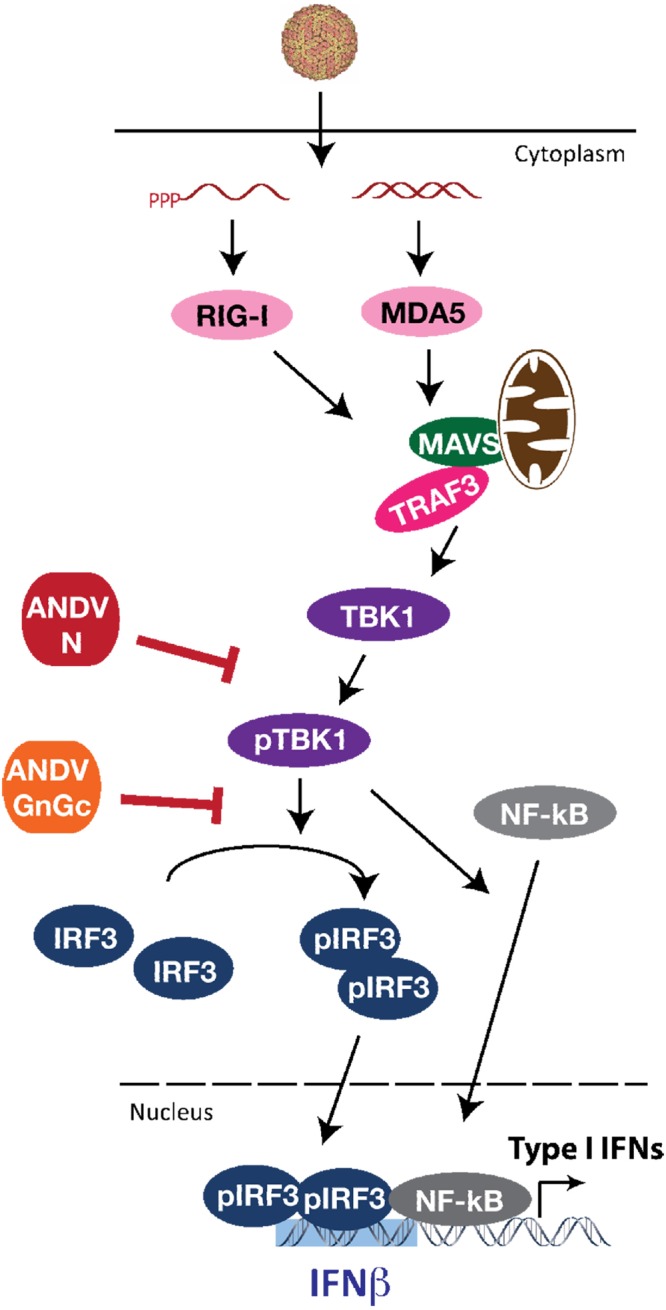
Schematic of ANDV N and GnGc protein regulation of TBK1 signaling. TBK1 autophosphorylation and its activation of IRF3 by phosphorylation are vital downstream steps in the type I IFN signaling pathway leading to IFN induction following hantavirus infection (26–31). The results of our collective studies revealed novel roles for the ANDV N and GnGc proteins in regulation of RIG-I/MDA5 signaling pathway responses that induce ISGs and IFN-β. ANDV N protein blocks TBK1 activation by inhibiting its autophosphorylation through a yet unknown mechanism that may involve regulation of TBK1 ubiquitination, dimerization, dephosphorylation, or recruitment to scaffolding protein complexes. ANDV GnGc does not inhibit TBK1 autophosphorylation but blocks TBK1-directed IRF3 phosphorylation. Thus, ANDV carries genes encoding two virulence determinants, the N and GnGc proteins, which restrict IFN induction by impeding discrete steps in TBK1-directed signaling responses that mediate RIG-I/MDA5-directed type I IFN induction.
DISCUSSION
ANDV is a South American hantavirus that causes highly lethal HPS (5, 38). However, unlike other hantaviruses that cause HPS or HFRS, ANDV is reportedly transmitted from person to person (5, 38) and ANDV uniquely causes fatal HPS-like disease in Syrian hamsters (36). Although several factors may combine to differentiate ANDV from SNV (39), at least one contributing factor likely stems from ANDV’s ability to bypass normal innate immune responses and cause viremia in Syrian hamsters (35, 37). Moreover, patient and animal findings suggest that ANDV contains unique virulence determinants that permit its enhanced replication and spread (5, 35, 37).
Hantavirus replication is highly sensitive to prior or early addition of type I IFN (20–22). However, at late times postinfection, hantaviruses paradoxically induce high-level ISG responses and appear resistant to the effects of IFN-α (20, 21, 49). Except for PHV, expressing hantavirus GnGc or cytoplasmic GnT domains inhibits IRF3 and NF-κB activation and reduces IRF3 phosphorylation (21–25, 34). However, since both ANDV and SNV GnTs regulate IRF3 phosphorylation (34), these findings do not distinguish ANDV- from SNV-regulated responses.
In contrast to Gn, N proteins from NY-1V, SNV, and PHV were observed to have no effect on RIG-I-directed signaling (21–24, 34), and as a result, we were surprised to find that the ANDV N protein potently inhibited RIG-I-directed transcriptional responses. Systematic analysis of this cytoplasmic signaling pathway determined that the ANDV N protein uniquely inhibits RIG-I-, MDA5-, MAVS-, and TBK1/IKKε-directed ISRE, κB, and IFN-β transcriptional induction, but fails to block responses induced by constitutively active IRF3-5D. Consistent with this, ANDV N protein inhibited TBK1 autophosphorylation which is required for helicase-directed IRF3 phosphorylation, NF-κB activation, and IFN-β induction (Fig. 7) (34).
Some hantaviruses carry a gene encoding a short ~90-residue NSs protein within an alternate S segment ORF within the N protein-coding region. In ANDV and other hantaviruses causing HPS, this ORF is truncated further to ~63 residues, although there is little understanding of the functions of NSs in HPS-causing viruses (42). In the Bunyaviridae family, the NSs ORF has been shown to regulate IFN responses in Rift Valley fever virus, Puumala hantavirus, and La Crosse virus, while the NSs of Bunyamwera virus is not essential (43–46). In order to determine whether the encoded ANDV NSs protein is responsible for IFN regulation, we ablated the NSs ORF by the insertion of 2 termination codons without altering the N protein ORF. We found that there was no effect of the NSs ORF on IFN regulation by the ANDV N protein, and these data indicate that the ANDV N protein is responsible for IFN pathway regulation.
The successful replication of hantaviruses within human endothelial cells is at least partly due to the ability to regulate the early induction of IFN-β (2, 20, 21, 25). Gn protein cytoplasmic tails inhibit RIG-I signaling (34) but also serve a matrix protein function coordinating virion assembly and budding into intraluminal ER/cis-Golgi compartments for exocytosis (1, 11). Although the mechanism of viral resistance to IFN addition late in infection remains to be resolved, Gn’s availability for IFN regulation prior to virion assembly could explain the transience of early IFN regulation during infection. The added ability of ANDV N protein to regulate IFN signaling could potentially contribute to the more robust early replication by ANDV observed during infection.
Unlike GnGc, the hantavirus N protein is highly expressed during infection, and early hantavirus studies showed that the N protein forms extensive lattice-like arrays within infected cells (1, 10). Structurally, the N protein encapsidates RNA and engages the polymerase and GnT to nucleate virion assembly (1, 11). However, this does not explain why high levels of N protein are found in infected cells or why N protein levels, following infection or plasmid expression, are not cytotoxic. In fact, hantaviruses persistently infect cells, and one possibility is that N protein expression may foster cell survival and viral persistence (1, 49). These findings and the function of ANDV N in IFN regulation suggest that the roles for N protein in regulating cellular responses are only beginning to be revealed.
Reports indicate that the HTNV N protein, but not ANDV and SNV N proteins, reduces TNF-α-directed nuclear translocation of NF-κB and suggest that this occurs through HTNV N protein interactions with nuclear importins or by binding to NF-κB (50–52). Consistent with this, our findings indicate that the N proteins from ANDV, SNV, NY-1V, and PHV have no effect on TNF-α-induced NF-κB activation (Fig. 6). This indicates that the ANDV N protein selectively inhibits helicase-directed IFN signaling pathway responses mediated by the activation of TBK1 or IKKε and that the ANDV N protein is not a ubiquitous inhibitor of κB transcription.
One report suggested that neither the ANDV N protein nor the GnGc protein inhibits IFN-β induction resulting from the infection of cells with Sendai virus (SeV) and instead found only a ~40% reduction in SeV-induced responses following cotransfection of N and GnGc plasmids (40). In contrast, our findings indicate that each individually expressed ANDV N or GnGc protein is fully capable of regulating pathway-specific RIG-I/MDA5-directed IFN signaling responses (34). These differing results are likely explained by our use of pathway-specific RIG-I/MDA5 activators rather than induction following infection of cells with Sendai virus (23, 24, 34, 40). ANDV proteins may not compensate for the rapid replication of SeV or expressed SeV proteins may interact with or interfere with ANDV N and GnGc proteins or their cellular regulatory targets. Cotransfection synergy of N and GnGc in the prior study also suggests the possibility that individual protein expression levels were inadequate to regulate SeV-induced responses (40). In comparison to SeV, hantaviruses replicate very slowly, and an analysis of ANDV, HTNV, or NY-1V infection versus PHV infection finds that IFN regulation occurs within the initial 24-h viral replication cycle (20, 23–25, 34). The pathway-specific inducers used here demonstrate the unique function of the ANDV N protein in regulating IFN-β transcriptional responses and define its ability to interfere with IRF3 and NF-κB activation by inhibiting the pathway-specific activation of TBK1 and IKKε.
The mechanism by which the ANDV N protein uniquely inhibits TBK1 and IKKε activation remains to be revealed. We were surprised that N proteins of closely related HPS-causing SNV or NY-1V N proteins are unable to inhibit IFN induction, since these proteins are 87% identical and 93% similar to the N protein of ANDV (EMBL accession no. AAB69170.1). In fact, N- and C-terminal domains (residues 1 to 241 and 290 to 428) of ANDV and SNV share 93% identity (99% similarity) but are dissimilar between residues 242 and 289 (only 52% identical). These differences suggest divergent N protein domains that may determine IFN regulation, but further investigation is required to define the regulatory elements uniquely encoded within the ANDV N protein gene.
On the basis of prior reports and current findings, ANDV is unique in having both GnGc and N proteins that regulate TBK1-directed responses (21–24, 34, 40), although regulatory mechanisms occur at discrete points for each protein (Fig. 7). Recent findings indicate that expressing the ANDV GnGc or GnT protein inhibits RIG-I-directed IRF3 activation by reducing IRF3 phosphorylation, yet GnGc had no effect on the phosphorylation of TBK1 (34). In contrast, the ANDV N protein inhibits TBK1 autophosphorylation required for its activation and the subsequent phosphorylation of IRF3. These findings suggest that N and GnGc proteins impact discrete steps in the type I IFN signaling pathway (Fig. 7) (34). This provides ANDV with an additional IFN-regulating determinant within the N protein that acts upstream and independent of GnGc regulation.
TBK1 and its homologue IKKε (26, 28, 47) are downstream mediators of RIG-I, MDA5, MAVS, and other cytoplasmic sensors that direct the phosphorylation of IRF3/5/7 and activation of NF-κB (26, 27, 29, 31, 41). TBK1 and IKKε contain a kinase domain, a ubiquitin-like domain, and a scaffold/dimerization domain. TBK1 dimers, K63-linked ubiquitination, and autophosphorylation of S172 are required for TBK1 activation and to mediate viral/RIG-I pathway activation of IRF3 (28, 47). TBK1 also binds the polyubiquitin binding protein optineurin, and K63-linked ubiquitination is a prerequisite for its kinase domain to phosphorylate S172 (28, 47, 53). These facets provide many points for TBK1 interactions with cellular targets and for regulation by deubiquitinases (CYLD, A20, and DUBA), phosphatases (SHIP-1) and scaffold-specific complexes (TRAF3, TRAF2, TANK, MAVS, NEMO, optineurin) (53–56).
Mechanisms by which the ANDV N protein regulates TBK1 are currently elusive, as our studies failed to reveal the coprecipitation of the ANDV N protein with TBK1 or TBK1 complex components TANK or TRAF3. TBK1 was not degraded by ANDV N protein expression (Fig. 6), suggesting that degradative ubiquitination is not a regulatory mechanism, although K63-linked ubiquitination required for TBK1 activation could be uniquely inhibited by ANDV N protein (53, 56). Additionally, ANDV N protein may interfere with the phosphorylation or dimerization of TBK1 by recruiting phosphatases or deubiquitinases to TBK1 or through aberrant interactions with cellular scaffolding proteins or regulators (TAX1BP, RNF11, ABIN, OTUB1/2, NAP1, NDRP1, TRIMs, MIB1, and SINTBAD) (28, 31, 32, 47, 53–57). Further studies of ANDV N protein interactions with cellular TBK1 regulatory proteins and complexes are needed to resolve this mechanism and may provide a means to block ANDV’s ability to bypass innate immune responses and prevent enhanced ANDV viremia and spread.
Currently, ANDV infection of Syrian hamsters is the only lethal HPS disease model, and this distinguishes ANDV from SNV and other hantaviruses that cause asymptomatic Syrian hamster infections (35–37). Findings of experiments with Syrian hamsters demonstrate that ANDV uniquely causes viremia, and this was suggested as a means for ANDV to circumvent innate immune responses of Syrian hamsters in order to cause disease (37). Enhanced viremia in ANDV patients may similarly be mediated by enhanced regulation of innate immune responses that permit ANDV to be spread from patient to patient (38). Although the role of N protein IFN regulation in ANDV-induced viremia is unknown, our finding of a novel innate immune-regulating determinant in the highly expressed ANDV N protein provides a potential mechanism for enhanced ANDV virulence that distinguishes it from other HPS-causing hantaviruses.
Conclusions.
Virulence factors that distinguish ANDV from other pathogenic hantaviruses have yet to be defined. Here we reveal that ANDV uniquely carries a gene encoding a virulence determinant within its N protein that potently inhibits innate cellular signaling pathways. The findings presented here suggest potential cellular targets and mechanisms of N protein regulation that specifically target IRF kinases TBK1 and IKKε. This ANDV-specific N protein function provides a new mechanism for hantaviruses to regulate early IFN and ISG induction during infection. Data presented support a role for the ANDV N protein as a virulence factor that may contribute to enhanced ANDV replication and spread. These findings distinguish ANDV from other hantaviruses that cause HPS and suggest targets for ANDV attenuation that need to be considered in vaccine development.
MATERIALS AND METHODS
Cells and antibodies.
HEK293T cells (ATCC) were grown in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS) as previously described (14, 21–23, 34). The following antibodies were purchased: anti-β-actin monoclonal antibody (MAb) (A5441; Sigma); MAb and polyclonal anti-Gal4 (sc-510 and sc-577; Santa Cruz); ANDV Gn MAb (H1808-50; US Biologicals). Antibodies to TBK1 (3504), phospho-TBK1 (Ser172) (5483), IRF3 (4302), phospho-IRF3 (pSer396) (4947), IKKε (2905), IRF3 (4302), and Flag (2368) were all purchased from Cell Signaling. Anti-N polyclonal serum directed at the New York-1 virus nucleocapsid protein was previously described (20, 21). Horseradish peroxidase (HRP)-conjugated sheep anti-mouse and goat anti-rabbit antibodies were purchased from GE Healthcare.
Plasmids.
Plasmids expressing N proteins from New York-1 virus (U36802.1), Andes virus (ANDV) (CHI-7913; AY228237.1), Sin Nombre virus (SNV) (JQ690276.1), and Prospect Hill virus (PHV) (M34011.1) as well as ANDV GnGc (CHI-7913; AY228238.1) were generated in pcDNA3 vectors as previously described (21–23, 34). An ANDV N protein expression construct deficient in the nonstructural protein ORF (ANDV N ΔNSs) was generated by site-directed mutagenesis that created stop codons at residues 13 and 28 within the NSs ORF. Constitutively active RIG-I-Flag (RIG-I CARD, residues 1 to 284) was obtained from Michael Gale. Human IKKε-Flag plasmid was obtained from Chris Basler (58). pCMV-IRF3-T7 and IRF3-5D plasmids were obtained from John Hiscott (48), and Iκβα mutant was provided by Ken Marcu. Murine SOCS1-Flag plasmid was obtained from Robyn Starr (59). The following constructs were purchased from Addgene: human TBK-1-Flag, human MAVS-Flag, and human MDA5-Flag (60). Firefly luciferase ISRE, κΒ, and IFN-β reporter plasmids were purchased from Clontech, and the pRL-null Renilla reporter was purchased from Promega.
Transcriptional reporter assays.
HEK293T cells were seeded (~100,000 cells/20 mm well) one day before transfection with 1 to 2 µg of plasmid DNA using polyethyleneimine (PEI) at a 3:1 µg PEI/DNA ratio. The cells were transfected with a constant amount of total DNA using the indicated plasmids expressing N, GnGc proteins, pcDNA3.1, firefly luciferase reporter, Renilla reporter, and plasmids expressing pathway activators (50 to 100 ng RIG-I, MDA5, MAVS, TBK1, IKKε, or IRF3-5D). The indicated samples were treated 18 h posttransfection with 50 ng/ml TNF-α or 12 h posttransfection with 500 units of IFN-α (R&D Systems). Cells were lysed 24 h posttransfection with 1× passive lysis buffer (Promega). Luciferase assays were performed using a dual-luciferase reporter assay (Promega) (22, 23, 34). Firefly luciferase activity was normalized to Renilla luciferase activity, and promoter induction was reported as fold increase of controls lacking inducer or as indicated as a percentage of inducer plus empty vector levels. Representative results are presented with assays performed in triplicate and replicated at least 3 times with similar results.
qRT-PCR analysis.
HEK293T cells were transfected using PEI with plasmids expressing ANDV N protein or NY-1V N protein or with pcDNA control plasmid. One day later, the cells were transfected with RIG-I CARD or a pcDNA control plasmid. Total RNA was extracted 12 h later with RNeasy (Qiagen), digested with DNaseI (NEB M0303S), and then cDNA was generated with the Transcriptor First Strand cDNA synthesis kit (Roche). mRNA levels were analyzed by Sybr green qRT-PCR on an ABI 7300 using IFN-β primers from Operon and GAPDH primers as previously described (61). Responses were normalized to GAPDH mRNA levels, and the change in induction (fold induction) was calculated using the 2−ΔCT method (61).
Western blot analysis.
Protein expression was analyzed by Western blotting of HEK293T cells transfected with PEI using the same plasmids and conditions described above and as described previously (34). Cells were harvested 1 day posttransfection in NP-40 lysis buffer (0.5% NP-40, 140 mM NaCl, 50 mM Tris [pH 8.2], 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], Sigma cocktail inhibitor, 1 mM NA3VO4, 10 mM beta-glycerophosphate, 10 mM NaF). Protein lysates were analyzed by SDS-PAGE and immunoblotting techniques as previously described (23, 34). Proteins were detected by using primary and secondary antibodies and the Luminata Forte system (Millipore).
Statistical analysis
Representative results are presented with assays performed in triplicate and replicated ≥3 times with similar results. Statistical analysis was performed using paired Student’s t test in GraphPad Prism. Values were statistically significantly different if P < 0.05.
ACKNOWLEDGMENTS
This work was supported by grants AI75022, AI1092191, AI097951, and AI093792 from the National Institutes of Health.
We thank I. Gavrilovskaya, E. Gorbunova, K. Marcu, and N. Reich for insightful discussions.
Footnotes
Citation Cimica V, Dalrymple NA, Roth E, Nasonov A, Mackow ER. 2014. An innate immunity-regulating virulence determinant is uniquely encoded encoded within the Andes virus nucleocapsid protein. mBio 5(1):e01088-13. doi:10.1128/mBio.01088-13.
REFERENCES
- 1. Schmaljohn C. 2001. Bunyaviridae and their replication, p 1581–1602 In Fields virology, vol 1 Lippincott Raven, Philadelphia, PA. [Google Scholar]
- 2. Yanagihara R, Silverman DJ. 1990. Experimental infection of human vascular endothelial cells by pathogenic and nonpathogenic hantaviruses. Arch. Virol. 111:281–286. 10.1007/BF01311063 [DOI] [PubMed] [Google Scholar]
- 3. Zaki SR, Greer PW, Coffield LM, Goldsmith CS, Nolte KB, Foucar K, Feddersen RM, Zumwalt RE, Miller GL, Khan AS, Rollin P, Ksiazek T, Nichol S, Peters C. 1995. Hantavirus pulmonary syndrome: pathogenesis of an emerging infectious disease. Am. J. Pathol. 146:552–579 [PMC free article] [PubMed] [Google Scholar]
- 4. Macneil A, Nichol ST, Spiropoulou CF. 2011. Hantavirus pulmonary syndrome. Virus Res. 162:138–147. 10.1016/j.virusres.2011.09.017 [DOI] [PubMed] [Google Scholar]
- 5. Enría D, Padula P, Segura EL, Pini N, Edelstein A, Posse CR, Weissenbacher MC. 1996. Hantavirus pulmonary syndrome in Argentina. Possibility of person to person transmission. Medicina 56:709–711 [PubMed] [Google Scholar]
- 6. López N, Padula P, Rossi C, Lázaro ME, Franze-Fernández MT. 1996. Genetic identification of a new hantavirus causing severe pulmonary syndrome in Argentina. Virology 220:223–226. 10.1006/viro.1996.0305 [DOI] [PubMed] [Google Scholar]
- 7. Vaheri A, Strandin T, Hepojoki J, Sironen T, Henttonen H, Mäkelä S, Mustonen J. 2013. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 11:539–550. 10.1038/nrmicro3066 [DOI] [PubMed] [Google Scholar]
- 8. Koster F, Mackow E. 2012. Pathogenesis of the hantavirus pulmonary syndrome. Future Virol. 7:41–51. 10.2217/fvl.11.138 [DOI] [Google Scholar]
- 9. Plyusnin A, Vapalahti O, Lankinen H, Lehväslaiho H, Apekina N, Myasnikov Y, Kallio-Kokko H, Henttonen H, Lundkvist A, Brummer-Korvenkontio M, Gavrilovskaya I, Vaheri A. 1994. Tula virus: a newly detected hantavirus carried by European common voles. J. Virol. 68:7833–7839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tao H, Xia SM, Chan ZY, Song G, Yanagihara R. 1987. Morphology and morphogenesis of viruses of hemorrhagic fever with renal syndrome. II. Inclusion bodies—ultrastructural markers of hantavirus-infected cells. Intervirology 27:45–52. 10.1159/000149714 [DOI] [PubMed] [Google Scholar]
- 11. Hepojoki J, Strandin T, Lankinen H, Vaheri A. 2012. Hantavirus structure—molecular interactions behind the scene. J. Gen. Virol. 93:1631–1644. 10.1099/vir.0.042218-0 [DOI] [PubMed] [Google Scholar]
- 12. Gavrilovskaya IN, Brown EJ, Ginsberg MH, Mackow ER. 1999. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J. Virol. 73:3951–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gavrilovskaya IN, Gorbunova EE, Mackow ER. 2010. Pathogenic hantaviruses direct the adherence of quiescent platelets to infected endothelial cells. J. Virol. 84:4832–4839. 10.1128/JVI.02405-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER. 1998. β3 integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. U. S. A. 95:7074–7079. 10.1073/pnas.95.12.7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gorbunova E, Gavrilovskaya IN, Mackow ER. 2010. Pathogenic hantaviruses Andes virus and Hantaan virus induce adherens junction disassembly by directing vascular endothelial cadherin internalization in human endothelial cells. J. Virol. 84:7405–7411. 10.1128/JVI.00576-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raymond T, Gorbunova E, Gavrilovskaya IN, Mackow ER. 2005. Pathogenic hantaviruses bind plexin-semaphorin-integrin domains present at the apex of inactive, bent alphavbeta3 integrin conformers. Proc. Natl. Acad. Sci. U. S. A. 102:1163–1168. 10.1073/pnas.0406743102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gavrilovskaya IN, Gorbunova EE, Mackow NA, Mackow ER. 2008. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 82:5797–5806. 10.1128/JVI.02397-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matthys VS, Gorbunova EE, Gavrilovskaya IN, Mackow ER. 2010. Andes virus recognition of human and Syrian hamster beta3 integrins is determined by an L33P substitution in the PSI domain. J. Virol. 84:352–360. 10.1128/JVI.01013-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robinson SD, Reynolds LE, Wyder L, Hicklin DJ, Hodivala-Dilke KM. 2004. Beta3-integrin regulates vascular endothelial growth factor-A-dependent permeability. Arterioscler. Thromb. Vasc. Biol. 24:2108–2114. 10.1161/01.ATV.0000143857.27408.de [DOI] [PubMed] [Google Scholar]
- 20. Geimonen E, Neff S, Raymond T, Kocer SS, Gavrilovskaya IN, Mackow ER. 2002. Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proc. Natl. Acad. Sci. U. S. A. 99:13837–13842. 10.1073/pnas.192298899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alff PJ, Gavrilovskaya IN, Gorbunova E, Endriss K, Chong Y, Geimonen E, Sen N, Reich NC, Mackow ER. 2006. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 80:9676–9686. 10.1128/JVI.00508-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alff PJ, Sen N, Gorbunova E, Gavrilovskaya IN, Mackow ER. 2008. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. J. Virol. 82:9115–9122. 10.1128/JVI.00290-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matthys V, Gorbunova EE, Gavrilovskaya IN, Pepini T, Mackow ER. 2011. The C-terminal 42 residues of the Tula virus Gn protein regulate interferon induction. J. Virol. 85:4752–4760. 10.1128/JVI.01945-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matthys V, Mackow ER. 2012. Hantavirus regulation of type I interferon responses. Adv. Virol. 2012:524024. 10.1155/2012/524024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spiropoulou CF, Albariño CG, Ksiazek TG, Rollin PE. 2007. Andes and Prospect Hill hantaviruses differ in early induction of interferon although both can downregulate interferon signaling. J. Virol. 81:2769–2776. 10.1128/JVI.02402-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoneyama M, Fujita T. 2007. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 18:545–551 [DOI] [PubMed] [Google Scholar]
- 27. Hiscott J. 2007. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 282:15325–15329. 10.1074/jbc.R700002200 [DOI] [PubMed] [Google Scholar]
- 28. Tu D, Zhu Z, Zhou AY, Yun CH, Lee KE, Toms AV, Li Y, Dunn GP, Chan E, Thai T, Yang S, Ficarro SB, Marto JA, Jeon H, Hahn WC, Barbie DA, Eck MJ. 2013. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 3:747–758. 10.1016/j.celrep.2013.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496. 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- 30. Honda K, Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6:644–658. 10.1038/nri1900 [DOI] [PubMed] [Google Scholar]
- 31. Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW, Nikolich-Zugich J, Moses AV, Gale M, Jr, Früh K, Diamond MS. 2013. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 9:e1003118. 10.1371/journal.ppat.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shen RR, Hahn WC. 2011. Emerging roles for the non-canonical IKKs in cancer. Oncogene 30:631–641. 10.1038/onc.2010.493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Panne D. 2008. The enhanceosome. Curr. Opin. Struct. Biol. 18:236–242. 10.1016/j.sbi.2007.12.002 [DOI] [PubMed] [Google Scholar]
- 34. Matthys VS, Cimica V, Dalrymple N, Glennon NB, Bianco C, Mackow ER. 3 January 2014. Hantavirus GnT elements mediate TRAF3 binding and inhibit RIG-I/TBK1 directed IFNβ transcription by blocking IRF3 phosphorylation. J. Virol. 10.1128/JVI.02647-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hammerbeck CD, Hooper JW. 2011. T cells are not required for pathogenesis in the Syrian hamster model of hantavirus pulmonary syndrome. J. Virol. 85:9929–9944. 10.1128/JVI.05356-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hooper JW, Larsen T, Custer DM, Schmaljohn CS. 2001. A lethal disease model for hantavirus pulmonary syndrome. Virology 289:6–14. 10.1006/viro.2001.1133 [DOI] [PubMed] [Google Scholar]
- 37. Wahl-Jensen V, Chapman J, Asher L, Fisher R, Zimmerman M, Larsen T, Hooper JW. 2007. Temporal analysis of Andes virus and Sin Nombre virus infections of Syrian hamsters. J. Virol. 81:7449–7462. 10.1128/JVI.00238-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Padula PJ, Edelstein A, Miguel SD, López NM, Rossi CM, Rabinovich RD. 1998. Hantavirus pulmonary syndrome outbreak in Argentina: molecular evidence for person-to-person transmission of Andes virus. Virology 241:323–330. 10.1006/viro.1997.8976 [DOI] [PubMed] [Google Scholar]
- 39. McElroy AK, Smith JM, Hooper JW, Schmaljohn CS. 2004. Andes virus M genome segment is not sufficient to confer the virulence associated with Andes virus in Syrian hamsters. Virology 326:130–139. 10.1016/j.virol.2004.05.018 [DOI] [PubMed] [Google Scholar]
- 40. Levine JR, Prescott J, Brown KS, Best SM, Ebihara H, Feldmann H. 2010. Antagonism of type I interferon responses by New World hantaviruses. J. Virol. 84:10790–10801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pomerantz JL, Baltimore D. 1999. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18:6694–6704. 10.1093/emboj/18.23.6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vera-Otarola J, Solis L, Soto-Rifo R, Ricci EP, Pino K, Tischler ND, Ohlmann T, Darlix JL, López-Lastra M. 2012. The Andes hantavirus NSs protein is expressed from the viral small mRNA by a leaky scanning mechanism. J. Virol. 86:2176–2187. 10.1128/JVI.06223-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bridgen A, Weber F, Fazakerley JK, Elliott RM. 2001. Bunyamwera bunyavirus nonstructural protein NSs is a nonessential gene product that contributes to viral pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 98:664–669. 10.1073/pnas.98.2.664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jääskeläinen KM, Kaukinen P, Minskaya ES, Plyusnina A, Vapalahti O, Elliott RM, Weber F, Vaheri A, Plyusnin A. 2007. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 79:1527–1536. 10.1002/jmv.20948 [DOI] [PubMed] [Google Scholar]
- 45. Billecocq A, Spiegel M, Vialat P, Kohl A, Weber F, Bouloy M, Haller O. 2004. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J. Virol. 78:9798–9806. 10.1128/JVI.78.18.9798-9806.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blakqori G, Delhaye S, Habjan M, Blair CD, Sánchez-Vargas I, Olson KE, Attarzadeh-Yazdi G, Fragkoudis R, Kohl A, Kalinke U, Weiss S, Michiels T, Staeheli P, Weber F. 2007. La Crosse bunyavirus nonstructural protein NSs serves to suppress the type I interferon system of mammalian hosts. J. Virol. 81:4991–4999. 10.1128/JVI.01933-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC. 2012. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. U. S. A. 109:9378–9383. 10.1073/pnas.1121552109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Odorizzi PM, Wherry EJ. 2013. Immunology. An interferon paradox. Science 340:155–156. 10.1126/science.1237568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taylor SL, Krempel RL, Schmaljohn CS. 2009. Inhibition of TNF-alpha-induced activation of NF-kappaB by hantavirus nucleocapsid proteins. Ann. N. Y. Acad. Sci. 1171(Suppl 1):E86–E93. 10.1111/j.1749-6632.2009.05049.x [DOI] [PubMed] [Google Scholar]
- 51. Ontiveros SJ, Li Q, Jonsson CB. 2010. Modulation of apoptosis and immune signaling pathways by the Hantaan virus nucleocapsid protein. Virology 401:165–178. 10.1016/j.virol.2010.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taylor SL, Frias-Staheli N, García-Sastre A, Schmaljohn CS. 2009. Hantaan virus nucleocapsid protein binds to importin alpha proteins and inhibits tumor necrosis factor alpha-induced activation of nuclear factor kappa B. J. Virol. 83:1271–1279. 10.1128/JVI.00986-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maelfait J, Beyaert R. 2012. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol. Mol. Biol. Rev. 76:33–45. 10.1128/MMBR.05012-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Belgnaoui SM, Paz S, Samuel S, Goulet ML, Sun Q, Kikkert M, Iwai K, Dikic I, Hiscott J, Lin R. 2012. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe 12:211–222. 10.1016/j.chom.2012.06.009 [DOI] [PubMed] [Google Scholar]
- 55. Gleason CE, Ordureau A, Gourlay R, Arthur JS, Cohen P. 2011. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J. Biol. Chem. 286:35663–35674. 10.1074/jbc.M111.267567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rajsbaum R, García-Sastre A. 2013. Viral evasion mechanisms of early antiviral responses involving regulation of ubiquitin pathways. Trends Microbiol. 21:421–429. 10.1016/j.tim.2013.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Charoenthongtrakul S, Gao L, Parvatiyar K, Lee D, Harhaj EW. 2013. RING finger protein 11 targets TBK1/IKKi kinases to inhibit antiviral signaling. PLoS One 8:e53717. 10.1371/journal.pone.0053717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cárdenas WB, Loo YM, Gale M, Jr, Hartman AL, Kimberlin CR, Martínez-Sobrido L, Saphire EO, Basler CF. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 80:5168–5178. 10.1128/JVI.02199-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. 1997. A family of cytokine-inducible inhibitors of signalling. Nature 387:917–921. 10.1038/43206 [DOI] [PubMed] [Google Scholar]
- 60. Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. 2005. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 175:5260–5268 [DOI] [PubMed] [Google Scholar]
- 61. Pepini T, Gorbunova EE, Gavrilovskaya IN, Mackow JE, Mackow ER. 2010. Andes virus regulation of cellular microRNAs contributes to hantavirus induced endothelial cell permeability. J. Virol. 84:11929–11936. 10.1128/JVI.01658-10 [DOI] [PMC free article] [PubMed] [Google Scholar]