

Abstract
Autoimmune diseases have registered an alarming rise worldwide in recent years. Accumulated evidence indicates that the immune system's ability to distinguish self from nonself is negatively impacted by genetic factors and environmental triggers. Genetics is certainly a factor, but since it normally takes a very long time for the human genetic pattern to change enough to register on a worldwide scale, increasingly the attention of studies has been focused on the environmental factors of a rapidly changing and evolving civilization. New technology, new industries, new inventions, new chemicals and drugs, and new foods and diets are constantly and rapidly being introduced in this fast-paced ever-changing world. Toxicants, infections, epitope spreading, dysfunctions of immune homeostasis, and dietary components can all have an impact on the body's delicate immune recognition system. Although the precise etiology and pathogenesis of many autoimmune diseases are still unknown, it would appear from the collated studies that there are common mechanisms in the immunopathogenesis of multiple autoimmune reactivities. Of particular interest is the citrullination of host proteins and their conversion to autoantigens by the aforementioned environmental triggers. The identification of these specific triggers of autoimmune reactivity is essential then for the development of new therapies for autoimmune diseases.
1. Introduction
The immune system walks a fine line to distinguish self from nonself in preserving the integrity of the host [1]. Interference with this fine line can result in overactivity to self-antigens, leading to autoimmunity. During the past 20 years a significant increase has been observed in the incidence of autoimmune disease worldwide. The etiology and pathogenesis of many autoimmune diseases remain unknown. It does appear that a close interplay between environmental triggers and genetic factors is responsible for the loss of immunological tolerance and autoimmunities [2, 3] (Figure 1). Therefore, in relation to the role of heritability in autoimmunity, genome-wide association studies reported that genetics only accounted for a minority of autoimmunity cases, and in many cases disease discordance exists in monozygotic twins [4]. For this reason, research and publications dedicated to environmental factors in autoimmunity have grown by an average of 7% every year since 1997 [2]. This includes toxic chemicals, infections, and dietary components. Indeed, detection of reactive antibodies to various citrullinated peptides and proteins in autoimmune disease is the best indication for gene-environment interactions [5].
Figure 1.
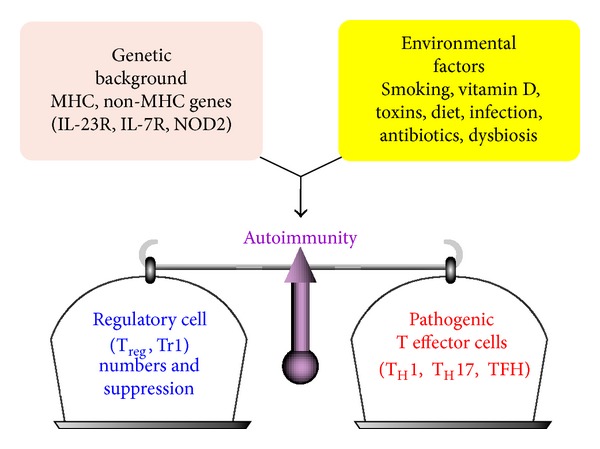
The balance of immunity. A combination of host genetic factors and exposure to environmental triggers promote the development of autoimmune disease. A balance must be maintained between the regulatory T cells and the pathogenic T effector cells.
2. Dysregulation of Immune Homeostasis
The full collaboration of both the innate and adaptive arms of the immune system plays a crucial role in the promotion or inhibition of autoimmune disease. Generally, to clear infections the innate immune cells can upregulate costimulatory molecules and produce a mixture of pro- and anti-inflammatory cytokines such as interleukin-1-beta (IL-1β), IL-12, transforming growth factor-beta (TGF-β), IL-23, tumor necrosis factor-alpha (TNF-α), and IL-6 that regulate the adaptive arm of the immune system. However, a dysregulated immune response to environmental triggers, such as pathogens, microbiota, or toxins, can initiate a chronic inflammatory response through activation of T-helper-1 (Th1), Th17, and TNF-α and the production of IL-17, IL-22, interferon-gamma (IFN-γ), and IL-21, resulting in inflammation, antibody production and tissue injury [6].
Therefore, a dysregulated adaptive immune system is at the core of the pathogenesis of autoimmune and other immune-mediated diseases. Hyperactivation of innate immune response affects the adaptive immune response as well as development effector T and B cells. Paired with defects in the regulatory T cells, this results in the breakdown of immune homeostasis and the development of autoimmunity [7].
To induce an autoimmune response in the lymph nodes, effector T cells first have to acquire a defined cytokine fingerprint and then must migrate to the appropriate target organs where they initiate tissue inflammation. The effector cells that participate in the induction of autoimmunities are IFN-γ-producing Th1 cells, IL-17- and IL-22-producing Th17 cells, and IL-21-producing follicular Th cells or TFH cells. It has been shown that overactivation or expansion of these newly discovered TFH cells causes antibody production and the development of lupus-like disease in an animal model [8]. In fact, high concentrations of circulating T cells that resemble TFH cells have been detected in a subgroup of patients with lupus. This increased frequency of TFH cells correlated with both disease severity and end-organ damage [9]. Unfortunately, a decrease in frequency and function of FOXP3+TREG cell is often seen in autoimmune diseases. This decrease seems to be associated with the inflammatory environment that contributes to the dysregulation of TREG cells [7].
In the environment of immune homeostasis, the actions of autoreactive Th1, Th17, and TFH cells are countered by FOXP3+ regulatory T cells that produce TGF-β and IL-10. But in an inflammatory milieu the deletion of different transcription factors results in the generation of TREG cells that are unable to suppress the autoreactive T cells (Figure 2).
Figure 2.
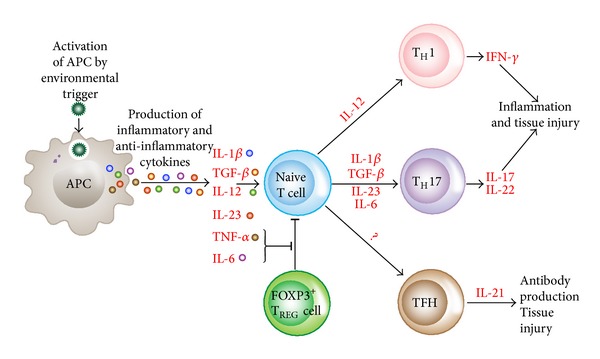
Differentiation of naïve T cells into pathogenic effector T cells. APCs can be activated by numerous factors, resulting in the release of cytokines that promote the differentiation of naïve T cells into various subsets of pathogenic effector T cells that drive inflammation, tissue injury, and autoantibody production. Segmented filamentous bacteria (SFB) can also promote the development of Th17 cells and autoimmune responses in vivo. Proinflammatory cytokines derived from both innate and adaptive immune cells attenuate TREG cell-mediated suppression of effector T cells.
Thus, tight control of autoreactive T cells, in particular TFH cells, by TREG cells is necessary to suppress the development of autoimmune lupus-like disease.
In order to induce long-lasting remission of immune-mediated diseases, two important factors have to be in place: controlling the inflammatory environment and boosting the frequency and function of FOXP3+ regulatory T cells.
3. Toxicants and Autoimmunity
A number of experimental studies and clinical reports have shown that autoimmune reactivity and/or autoimmune diseases are induced in humans and chronic exposure to various chemicals in animal models. These were summarized by Bigazzi in 1997 [10]. Furthermore, very recently, this role of environmental chemicals, in particular, the induction of autoimmunities by toxicants, was summarized by Pollard et al. [11] in his paper, “Toxicology of autoimmune diseases.” The mechanism of toxicant-induced autoimmunity is described by either toxicant induction of aberrant cell death making the hidden cellular material available to anti-gen presenting cells [12, 13] or by immune reactions to xenobiotics through covalent binding of chemicals or haptens to human tissue proteins and formation of neoantigens [14] (Figure 3). This is due to the fact that reactive organic compounds most often bind covalently; that is, their electrophilic properties enable them to react with protein nucleophilic groups such as thiol, amino, and hydroxyl groups. Examples of such reactive, haptenic compounds that frequently lead to sensitization after dermal contact or inhalation are toluene diisocyanate, trimellitic anhydride, phthalic anhydride, benzoquinone, formaldehyde, ethylene oxide, dinitrochlorobenzene, picryl chloride, penicillins, and D-penicillinamine. Sensitizing metal ions react somewhat differently in that they oxidize proteins or form stable protein-metal chelate complexes by undergoing multipoint binding with several amino acid side-chains [12]. For example, in regard to nail polish and its association with primary biliary cirrhosis (PBC), halogenated compounds could bind to mitochondrial proteins, changing their immunogenicity and inducing antimitochondrial antibodies [10, 15, 16].
Figure 3.
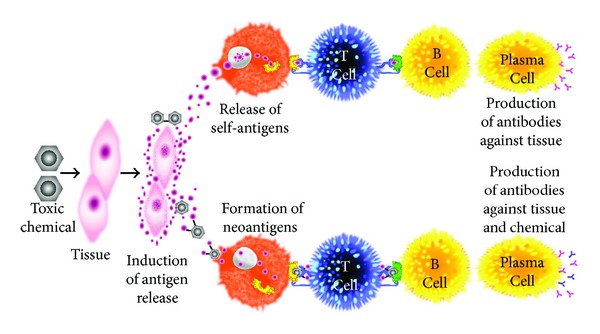
Putative mechanism of chemical-induced autoimmunity.
In contrast to haptenic compounds, most xenobiotics eliciting adverse immune reaction are unable to bind to proteins when entering the body; however, they can do so after conversion to reactive metabolites. These xenobiotics can be considered as prohaptens, which, after metabolization, manage to bind to human tissue proteins and induce antibody production against both the haptenic chemicals as well as tissue proteins.
Another mechanism is the activation of toll-like receptors by xenobiotics. This predisposes individuals to toxicant-induced inflammatory cytokine production, which exacerbates autoimmune diseases [17].
These and other mechanisms of action were explored in relation to exposure to organic solvents as a risk factor for autoimmune disease in a very extensive systemic review of literature and meta-analysis [18]. After reviewing a total of 103 articles and the inclusion of 33 in the meta-analysis, it was concluded that (1) exposure to organic solvents was associated with systemic sclerosis, primary systemic vasculitis, and multiple sclerosis (MS) and (2) individuals who carry genetic factors for autoimmunities should avoid any exposure to organic solvents in order to avoid increasing their risk for autoimmune diseases.
In addition to the above mechanisms of actions (as shown in Figures 3 and 4), these autoimmune responses and diseases can be induced by solvents and other environmental chemicals through a variety of effects at the biochemical and cellular levels.
Chemicals are capable of altering cellular proliferation, Th1, Th2, Th3, Th17, apoptosis, and tissue-specific function.
Chemicals are capable of inducing protein or lipid adducts which activate Th17 cells and induce the production of IL-17 and IL-21.
Chemicals can activate HSP90 and induce production of anti-HSP90 autoantibodies.
Chemicals are capable of inducing DNA-hypermethylation and change in cellular functions.
Chemicals can increase ROS production and the induction of DNA-fragmentation.
Chemicals may compete with thyroid hormones or interfere with iodine transportation and induce oxidative stress that leads to an inflammatory response to the thyroid gland.
Chemicals not only stimulate the release of reactive oxygen species but also stimulate the synthesis of nitric oxide by nitric oxide synthase [18].
Figure 4.
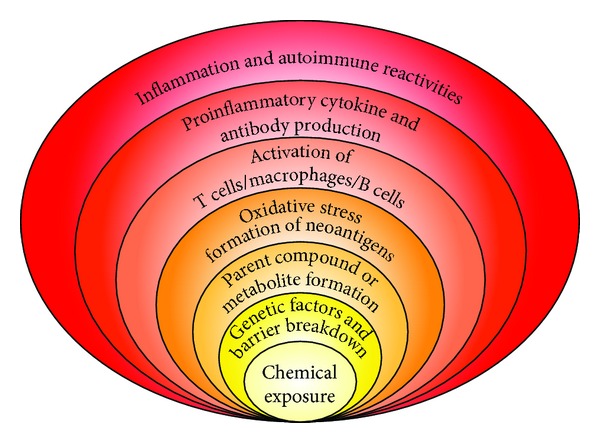
Potential molecular mechanisms implicated in chemical-induced autoimmune reactivities.
Finally, modification of DNA methylation is an additional mechanism by which environmental triggers induce changes in gene expression. For example, environmental pollutants, cigarette smoke and alcohol consumption have been advocated for autoimmunity incidence due to their links with the induction of DNA methylation [10, 19].
Overall, the precise mechanisms responsible for the development of environmentally induced autoimmune disorders are unknown. Additionally, mechanisms involved in the initiation of a disease process might differ from mechanisms responsible for exacerbation of the established illness. Therefore, one or more of these mechanisms either individually or jointly can have strong effects on the development of autoimmune reactivity, which may then be followed by autoimmune disease (Figure 4).
4. Induction of Autoimmunities by Infection
Although some infections can protect individuals from specific autoimmune diseases, infectious agents play a pivotal role in the induction of autoimmune disorders. The question of how infectious agents contribute to autoimmunity has continued to be of interest to clinical and basic researchers and immunologists in general [20].
An autoimmune disease can be induced or triggered by infectious agents, which can also determine its clinical manifestations. Most infectious agents, such as viruses, bacteria, fungi, and parasites, can induce autoimmunity via different mechanisms. In many cases, it is not a single infection but rather the “burden of infections” from childhood that is responsible for the induction of autoimmunity [20].
Almost every autoimmune disease is linked to one or more infectious agent. During the past 50 years molecular techniques have been utilized to explore the interaction between infections and autoimmunities [20–23]. One of the classical examples of this relationship is rheumatic fever, which presents several weeks after infection with beta hemolytic streptococcus. Molecular resemblance between the bacterial M5 protein and human α-myosin results in a breakdown of immunological tolerance and antibody production against α-myosin in genetically susceptible individuals [21, 24]. In the case of antiphospholipid syndrome (APS), anti-cardiolipin and anti-β 2-glycoprotein I pathogenic (β 2GPI) antibodies are detected. Although there is molecular mimicry between β 2GPI and infections, such as cytomegalovirus, Haemophilus influenza, Neisseria gonorrhoeae, rubella, toxoplasma, and tetanus toxoid, and IgM antibodies against them have been detected, the direct connection between these infections and APS has not been established [24]. Another example of associating infection with autoimmune disease is type 1 diabetes. Type 1 diabetes is an autoimmune disease resulting from the destruction of β-islet cells by autoreactive T cells and the concomitant release of various islet cell antigens [21, 25]. The appearance of antibodies against glutamic acid decarboxylase 65 (GAD-65) and tyrosine phosphatase precedes the onset of the disease by 5–10 years [26, 27]. Several lines of evidence link infections with type 1 diabetes.
A search on PubMed using the keywords “association of viruses with type 1 diabetes” produces close to 1,400 manuscripts.
Enteroviruses such as coxsackie B4 virus and rotavirus, the most common cause of childhood gastroenteritis, not only share homology with GAD-65, but can cause the precipitation of type 1 diabetes when introduced. Higher levels of anti-coxsackievirus and rotavirus antibodies are detected in sera from patients with recent onset of type 1 diabetes [28, 29].
Using PCR technology, coxsackie B4 virus was detected in islet cells of 65% of patients versus only 6% of controls [30].
Inoculation of the virus to genetically susceptible strains of mice resulted in insulitis and diabetes, fulfilling Koch's postulates [21, 31, 32].
Both DNA and RNA viruses are capable of initiating antiviral responses that cross-react with insulin, GAD-65, and other islet cell antigens [21, 33, 34].
Altering the balance of gut microbiota toward either a tolerogenic or nontolerogenic state using antibiotics or probiotics may influence the development of type 1 diabetes [35, 36]. Therefore, just as with their viral counterparts, there is sufficient indirect evidence that gut and other microbial agents, for example, Mycobacterium avium, are potential triggers for type 1 diabetes [37].
This multifaceted interaction between genetics, immune dysregulation, various infections, and autoimmune diseases such as rheumatoid arthritis (RA) and thyroid disease reveals many possibilities for pathogenic relationships between different species of infectious agents and autoimmunity [20, 38, 39]. These infectious agents and their association with RA and thyroid autoimmunity are shown in Tables 1 and 2.
Table 1.
Infectious agents associated with rheumatoid arthritis.
| Infection | Reference |
|---|---|
| Porphyromonas gingivalis | Farquharson et al. 2012 [40] |
| Segmented filamentous bacteria | Wu et al. 2010 [41] |
| Yersinia enterocolitica | Gaston and Lillicrap 2003 [42] |
| Salmonella typhi | McColl et al. 2000 [43] |
| Shigella flexneri | Hannu et al. 2005 [44] |
| Proteus mirabilis | Ebringer and Rashid 2006 [45] |
| Campylobacter jejuni | Pope et al. 2007 [46] |
| Klebsiella pneumoniae | Domínguez-López et al. 2000 [47] |
| Clostridium difficile | Cope et al. 1992 [48] |
| Staphylococcus aureus | Liu et al. 2001 [49] |
| Streptococcus pyogenes | Faé et al. 2006 [50] |
| Candida albicans | Hermann et al. 1991 [51] |
| Leptospira pomona | Sutliff et al. 1953 [52] |
| Chlamydia | Carter et al. 2010 [53] |
| Mycoplasma arthritidis | Cole and Ward 1979 [54] |
| Mycobacterium tuberculosis | Kim et al. 2006 [55] |
| Borrelia burgdorferi | Imai et al. 2013 [56] |
| Parvovirus | Kerr et al. 1995 [57] |
| Epstein-Barr virus | Pratesi et al. 2006 [58] |
Table 2.
Infectious agents associated with thyroid autoimmunity.
| Infection | Reference |
|---|---|
| Yersinia enterocolitica | Bech et al. 1978 [59] |
| Epstein-Barr virus | Shimon et al. 2003 [60] |
| Parvovirus | Mori et al. 2007 [61] |
| Hepatitis C | Fernandez-Soto et al. 1998 [62] |
| Mumps | Parmar et al. 2001 [63] |
| Rubella | Ziring et al. 1977 [64] |
| Coxsackievirus | Brouqui et al. 1991 [65] |
| HTLV-1 | Kawai et al. 1992 [66] |
| Human herpes virus types 6 and 7 | Leite et al. 2008 [67] |
4.1. Mechanisms Responsible for the Induction of Autoimmunity by Infection
Autoimmunity can be induced by infectious agents through the following mechanisms: molecular mimicry, epitope spreading, standard activation, viral persistence, polyclonal activation, dysregulation of immune homeostasis, and autoinflammatory activation of innate immunity [20]. In some cases, even if infections are not directly responsible for the induction of autoimmunities, they can often target the site of autoimmune inflammation and amplify the autoimmune disease [68]. In this case, infections can have one of three effects: first, it can exacerbate ongoing disease, leading to greater severity and duration; second, it can induce a relapse; and, third, it can lead to chronic progressive disease.
4.1.1. Molecular Mimicry
In the most likely mechanism by which infection induces autoimmunity, foreign antigens very often may bear sufficient structural similarity to self-antigens. This is called antigenic mimicry or molecular mimicry. Immune response to microbial antigens could result in activation of T cells that are cross-reactive with self-antigens. This is due to the fact that a single T cell can respond to various peptides with similar charge distribution and overall shape [20, 69]. Examples of bacterial or viral antigens, their cross-reactivity with various tissue antigens, and potential ensuing autoimmune diseases are shown in Table 3.
Table 3.
Examples of bacterial and viral antigens that can cross-react with self-antigens with potentially resultant diseases.
| Pathogen antigen | Cross-reactive self-antigen | Autoimmune disease |
|---|---|---|
| Herpes simplex virus | Corneal antigen | Stromal keratitis |
| Campylobacter jejuni | Ganglioside in peripheral nerve | Guillain-Barré syndrome |
| Coxsackievirus | Glutamic acid decarboxylase | Type 1 diabetes |
| Theiler's murine encephalomyelitis virus | Proteolipid protein | Multiple sclerosis |
| Yersinia enterocolitica | Thyrotropin receptor | Thyroid autoimmunity |
| Borrelia burgdorferi | Leukocyte function associated antigen | Lyme arthritis |
| Salmonella typhi and Yersinia enterocolitica | HLA-B27 | Reactive arthritis |
| HHV-6, EBV, Rubeolla, influenza virus, and HPV | Myelin basic protein | Multiple sclerosis |
| Streptococcal M protein | Myosin and other heart valve proteins | Rheumatic fever |
| Trypanosoma cruzi | Cardiac myosis | Chagas heart disease |
Mechanisms of infection-induced autoimmunity through molecular mimicry are shown in Figure 5.
Figure 5.
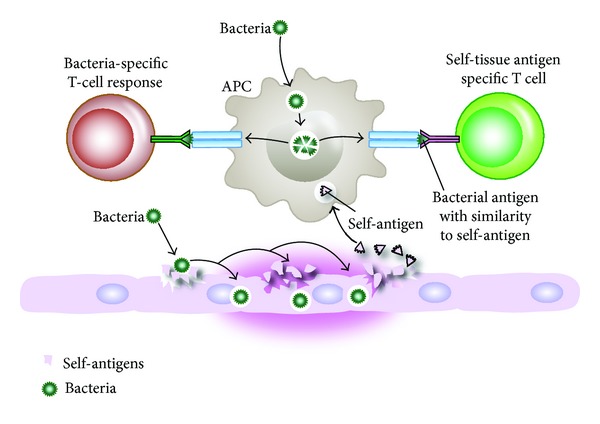
Mechanisms of infection-induced autoimmunity through molecular mimicry. Bacterial induction of self-tissue antigen release and simultaneous presentation of bacterial and self-tissue antigens to T cells; activated T cells can produce antibodies against both bacterial and self-tissue antigens.
Initiation of immune response to the foreign antigens such as coxsackievirus that share identical amino acid residues with self-proteins such as GAD-65 may generate a cross-reactive antibody response that incorrectly recognizes the self-protein as a foreign antigen. When the self-antigen is a cell surface molecule such as GAD-65, the antibody- and cell-mediated immune response can lead to tissue damage [69].
Given the vast numbers of microbial proteins and their cross-reaction with human proteins, immune response against microbial antigens will not always result in autoimmunity. However, such an initial immune response could result in epitope spreading or exposure of other regions of the same self-protein and production of more antibodies [69]. The criteria for the mechanism of autoimmunity induction were reviewed and summarized by Kivity et al. 2009 [20]. In the classical examples of autoimmunities induced by infections summarized in Table 3, all these criteria are present.
4.1.2. Epitope Spreading
Epitope spreading is a phenomenon in which the immune system expands its response beyond the original epitope recognized by T or B cells to induce the release of non-cross-reactive epitopes that are recognized by the immune system later [70]. Epitope spreading can result from a change in protein structure. One such example is protein citrullination, the changing of an amino acid from arginine to citrulline. This can result not only in immune reaction against the original protein or its citrullinated form, but also against other citrullinated proteins.
Epitope spreading is demonstrated in rheumatic fever, in which a chronic autoimmune response against streptococcal M protein and heart valve tissue can result in immune response against collagen or laminin. This immune response against collagen or laminin is no longer specific to the bacterial M protein or its cross-reactive tissue protein. In pemphigus, blistering of the mouth precedes blistering of the skin, and blisters in the mouth are associated with the presence of antibodies against desmoglein-3 protein, which is specific to the mouth epithelial cell antigens. It is only later on when T cells attack skin desmoglein-1 that autoantibodies are produced against skin-specific antigens, and skin blistering develops [71]. In a mouse model of encephalomyelitis, Theiler's murine encephalomyelitis virus T-cell response to myelin develops first against dominant myelin proteolipid (PLP) peptide 139–151. As the disease progresses, response to the different and less dominant epitope PLP peptide 178–191 emerges. This mechanism of infection-induced autoimmunity through epitope spreading is shown in Figure 6.
Figure 6.
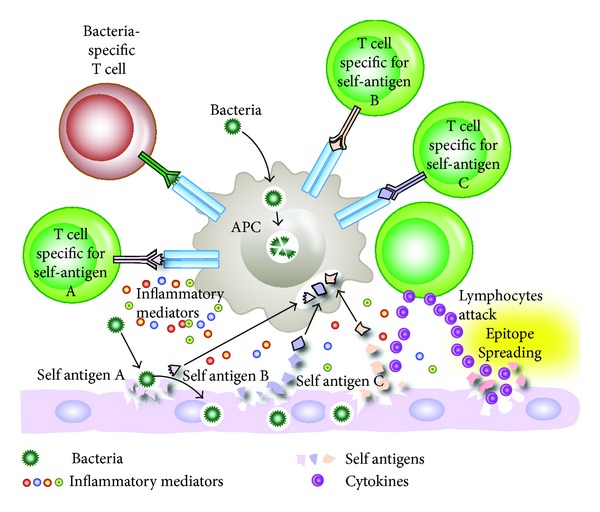
Bacterial infection induces release of tissue antigen and presentation of bacterial and self-tissue antigens resulting in the induction of autoreactive T cells. T cells and inflammatory mediators cause the release of more self-antigens which differ from the original antigens. T-cell responses can then spread to involve T cells specific to other self-antigens. This T-cell response against different epitopes results in antibody production against multiple tissue antigens.
4.1.3. Bystander Activation and Stimulation of Pattern Recognition Receptors
Bystander activation occurs when viral antigens stimulate toll-like receptors and other pattern recognition receptors become activated in the inflammatory environment [72]. This activation of receptors on an antigen-presenting cell (APC) causes the release of proinflammatory cytokines which can induce tissue damage and the release of hidden antigens (Figure 7). The release of tissue antigens can activate autoreactive T cells that initially were not involved in the immune reactivity against the original infection [20]. Additionally, virally infected APCs and the concomitantly released mediators are able to activate autoreactive Th1 or Th17 cells in a bystander manner. Upon recognition of virally infected tissue cells, viral-specific T cells then release cytotoxic granules such as granzymes and cytokines such as TNF-α, IL-17, lymphotoxin, and nitric oxide. This inflammatory environment can lead to the bystander killing of uninfected neighboring cells. Microbial superantigens can induce a broader form of bystander activation by cross-linking MHC class II molecules to TCRs on APCs and T-cell activation (Figure 8). T cells that are stimulated in this manner may contain a subset recognizing specific tissue antigen [73]. Examples of superantigens are staphylococcal antigens, mycoplasma antigens, enteric-microbiota LPS, EBV, retrovirus, and many heat shock proteins. Some of these superantigens do not cause autoimmune disease but are involved in the exacerbation of EAE, arthritis, IBD, and other disorders [1].
Figure 7.
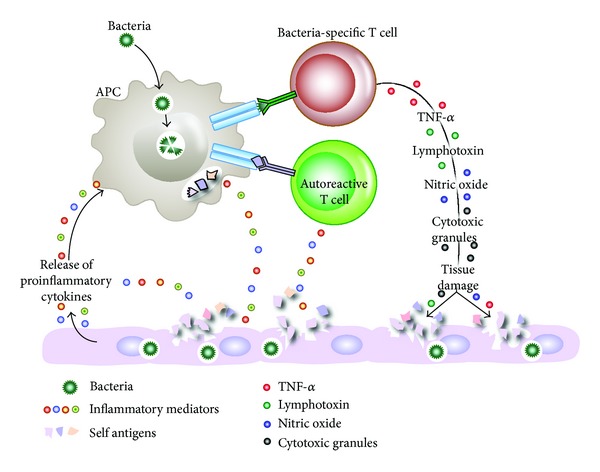
Microbial infection stimulates toll-like receptors (TLRs) and other pattern recognition receptors on antigen-presenting cells (APCs), leading to the production of proinflammatory mediators, which in turn can lead to tissue damage. The release of both tissue antigens and bacterial antigens results in bacterial-specific T cells and autoreactive T cells in the process called bystander activation, which contributes to autoimmunity.
Figure 8.
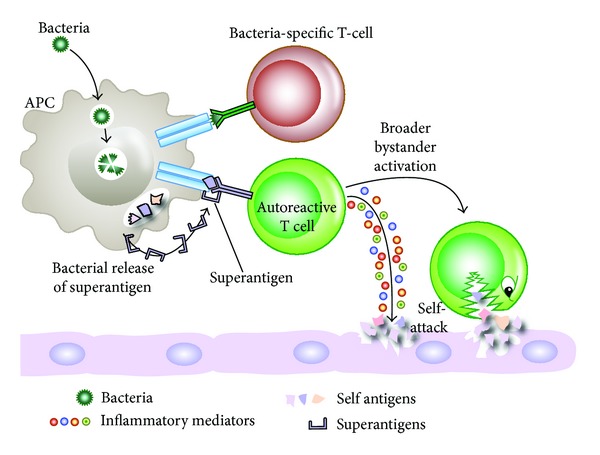
Superantigens and autoimmunity. Infection can lead to the release of superantigens, which can cross-link between MHCII and TCR, causing broader bystander activation, some of which may be specific for self-antigens, leading to attack on self-tissues.
4.1.4. Persistent Infection and Polyclonal Activation of B Cells
In many autoimmune diseases, such as lupus, RA, type 1 diabetes, and MS, B-cell functions are closely correlated with disease activity. Antibodies produced by B-cell-derived plasma cells contribute significantly to disease pathogenesis [69]. In these and other disorders, prolonged infectivity with a virus such as EBV, viral proteins, or viral genomes can lead to autoimmunity by the constant activation and proliferation of B cells.
After a long period of polyclonal B-cell activation, sometimes monospecific clones can emerge, accompanied by very high levels of antibody production and the formation of circulating immune complexes. Finally, this mixture of polyclonal antibodies and immune complexes may cause the autoimmune disease [74], as shown in Figure 9.
Figure 9.
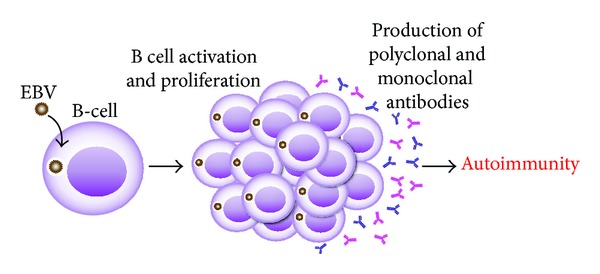
Infections, B cells, and autoimmunity. Prolonged infection with a virus, such as EBV, can lead to constant activation and proliferation of B cells, resulting in the production of monoclonal and polyclonal antibodies as well as immune complexes, causing autoimmune disease.
5. Dietary Components and Autoimmunities
It is undeniable that the diet of the industrialized and urbanized parts of the world today is vastly different from what it was even two or three decades ago, with a whole new range of novel food experiences that come from new food component sources, new breeds of food plants and food animals, genetic modifications, chemical ingredients, flavors, and preservatives. Over recent decades, a significant increase in the incidence of autoimmune diseases such as diabetes and MS in industrialized countries has led to the postulation that diet is a potential environmental risk factor for such disorders. The link between gluten ingestion and gluten sensitive enteropathies is already well established and accepted [3]. High levels of dietary sodium are associated with raised blood pressure and adverse cardiovascular health [75] and have been shown to affect the immune system [76]. Low levels of vitamin D have been linked with MS, systemic lupus erythematosus (SLE), RA, and other autoimmune disorders [3]. Lactose intolerance is no laughing matter for those afflicted with it or other milk-related disorders. The pleasures of a modern diet unfortunately come with caveats and unexpected catches that urgently need investigation.
5.1. Sodium Chloride in Diet and Autoimmune Diseases
For the past five decades various studies have been conducted on the comparative sodium intake levels in different countries [75, 76]. Animal experiments, epidemiological studies, and clinical trials have provided convincing evidence for the detrimental effect of sodium intake on blood pressure (BP), coronary heart disease, and stroke, as well as noncardiovascular diseases [77–81]. These comparative studies have shown that generally the simpler and less modernized a society and culture are, the lower the sodium intake is, with a concomitant lessening of the associated disorders. Understandably, the high salt content of the modern Asian diet is known worldwide, particularly the use of soy sauce as a seasoning [75, 82]. Indeed, in comparison to home-made meals, the salt content of fast foods can be many times higher [75]. The concentration of Na+ in plasma similar to standard culture medium is about 149 mM. The consumption of high-salt processed foods may increase this concentration to a higher level and result in a change in physiological conditions. It has been theorized that the consumption of processed foods containing high amounts of salt may in part be responsible for the increasing incidence of autoimmune diseases. In a recent study it was demonstrated that an excess uptake of salt can affect the innate immune system, in particular, macrophage function [83]. However, until very recently little was known about increased NaCl intake, its direct effect on the T-helper cell populations, and the connection of all this to autoimmune diseases. Upon stimulation of the T-cell receptor and the cytokine environment, the naïve CD4+ T cell can differentiate into functionally distinct effector cell subsets. This differentiation is also driven by key transcriptional regulators such as T-bet for Th1, GATA binding protein-3 for Th2, FOXP3 for Th3, retinoid acid receptor-related orphan receptor gamma t (RORγt) for Th17, and transcriptional regulator B-cell lymphoma 6 (BCL6) for T-follicular-helper (TFH) cells [84].
Among these CD4+ T-cell subsets, the IL-23-dependent IL-17-producing CD4+ helper T cells play a pivotal role in autoimmune disease [85]. Adding salt to the wound of complex autoimmune diseases, it has been shown that sodium chloride can drive autoimmune disease by the activation or induction of pathogenic Th17 cells [86]. These elegant experiments were conducted in a culture medium containing an additional 10–40 mM concentration of salt, mimicking animals fed a high-salt diet. Increased NaCl concentrations markedly induced the conversion of naïve CD4+ T cells to CD4+ T cells expressing IL-17A (Figure 10). This effect was dose dependent, and the optimum IL-17A induction was achieved by increasing the concentration of NaCl by 40 mM. Moreover, the authors demonstrated that a high-salt diet could accelerate neuropathology in a mouse model of multiple sclerosis through cellular signaling pathways involving transcription factor NFAT5, the protein kinase enzyme P38, and salt-sensing kinase SGK1. In comparison with the controls, mice on the high-salt diet not only displayed a much higher number of infiltrating CD3+ and MAC3+ cells but also almost doubled the number of CD4+ T cells expressing IL-17A or pathogenic Th17 cells [86, 87]. This effect of the high-salt diet was specific for Th17 conditions, since the high salt levels did not significantly alter cell death, lymphocyte proliferation, or enhancement of Th1 or Th2 differentiations. The mechanism by which a high-salt diet enhances the differentiation of naïve CD4+ cells to pathogenic Th17 cells is shown in Figure 11.
Figure 10.
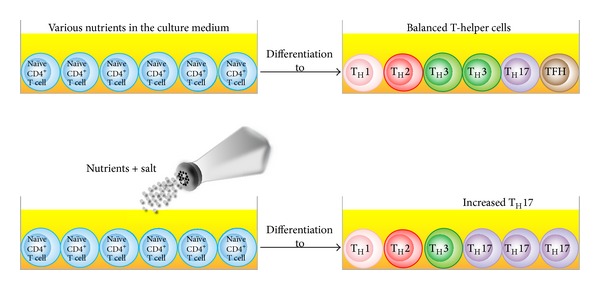
Salt affects the differentiation of naïve CD4+ cells. Increased concentrations of salt resulted in the differentiation of naïve CD4+ T cells into a greater number of TH17 cells.
Figure 11.
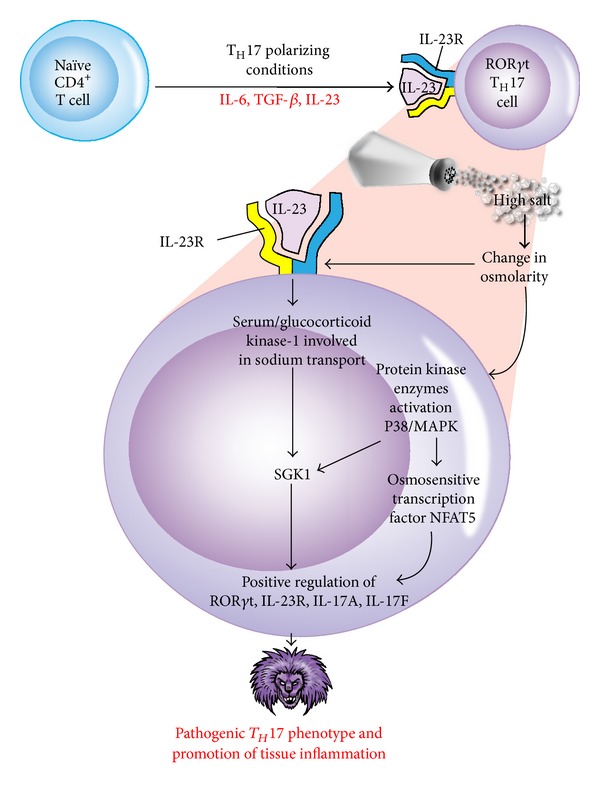
Mechanism by which a high-salt diet enhances the differentiation of naïve CD4+ cells to pathogenic TH17 cells that may exacerbate experimental autoimmune encephalitis. High salt concentration, change in osmolarity, the influence of IL-23 and IL-23 receptor signaling, and the activation of various enzymes drive the expression of TH17-associated cytokines and the formation of pathogenic TH17 phenotype.
Extracellular NaCl concentration through the activation of IL-23 receptor and its binding by IL-23 influences the activity of SGK1 and NFAT5 which drives the expression of transcription factor RORγt, IL-23R, IL-17A, and IL-17F resulting in the phenotype switch from naïve CD4+ T cells to pathogenic Th17 cells in MS, psoriasis, and other autoimmune disorders (Figure 12). The data presented in these manuscripts [86, 87] clearly indicate that high intake of sodium potentiates pathogenic Th17 cell generation in in vitro and in vivo systems in an SGK1-dependent manner and, therefore, has the potential of increasing the risk of promoting autoimmune diseases. Moreover, the elevated in vivo Th17 resulting from a high-salt diet raises the important question of whether or not increased salt in westernized diets and in processed foods contributes to an increased generation of pathogenic Th17 cells and towards an unprecedented increase in autoimmune diseases [87].
Figure 12.
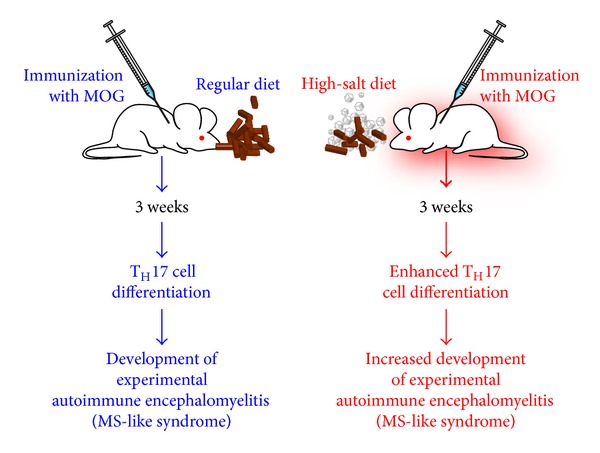
High-salt diet increases risk of autoimmune disease. In two groups of mice, both of which were immunized with MOG to induce EAE, the mice that had been given a high-salt diet (HSD) showed enhanced differentiation of naïve T cells into pathogenic TH17 cells and a subsequent increased, more profligate development of EAE.
Thus, as indicated, dietary salt is just one of many dietary components that can influence T-helper cell differentiation and the development of autoimmune disease. The effect of other dietary nutrients, for example, vitamins, and other diverse environmental factors on metabolism and microbiota should also be investigated [88].
When this information is taken together with the strong and consistent evidence that implicates high salt intake with high BP and other cardiovascular disorders, it is alarming to note as laid out in all these studies that most adult populations have daily salt intakes well over the recommended US daily level of 1.5 g/day for middle-aged and older adults [89]. On the face of it a voluntary decrease in salt consumption seems to be an easy policy to implement, but good sense and good health face the formidable opposing forces of flavor, habit, and culture.
5.2. The Role of Milk and Wheat Components in Autoimmune Diseases
In relation to dietary proteins it has been well established that different proteins and peptides in milk and wheat are involved in autoimmune diseases [90–93]. Milk contains more than 400 different proteins, most of which have over 150 amino acids (AA). AA that mimic collagen may induce RA, while those that mimic neural cell antigens may induce multiple sclerosis or other neuroimmune disorders.
For example, a study reports that, as a consequence of immunological cross-reactivity or molecular mimicry between the extracellular IV-like domain of the milk protein butyrophilin and myelin oligodendrocyte glycoprotein (MOG), butyrophilin can modulate the encephalitogenic T-cell response to MOG in experimental autoimmune encephalitis [92]. Epidemiological and ecological investigations suggest that early infant nutrition, particularly drinking cow's milk, may induce autoimmunity, leading to type 1 diabetes. This autoimmune reactivity is due to cross-reactivity of cow's milk, particularly its albumin component, with islet cell antigen-1 and beta cell surface protein. These studies suggest that dysregulation of oral tolerance triggers a cellular and humoral immune response against various components of milk proteins, and cross-reaction with B-cell molecules may result in autoimmunity [94–97]. In association with various autoimmune disorders, wheat proteins and, more specifically, gluten, have received significant attention [98–100]. Indeed, it has been demonstrated that a wheat-based diet induces not only Th1-type cytokine bias in the gut but also increased T-cell reactivity to gluten, with a higher frequency of diabetes [99–101]. In addition to diabetes, it has been shown that celiac disease (CD) is associated with various extraintestinal autoimmune disorders that involve the thyroid, joints, heart, skin, pancreas, bone, liver, reproductive organs, and the nervous system [102–112].
Although the exact mechanisms for the induction of these autoimmunities are not definitely known, there is a growing body of evidence indicating that these diseases may result from molecular mimicry between gliadin or transglutaminase and various tissue antigens, including nervous system proteins [33–35, 68]. Interestingly, the celiac peptide VVKVGGSSSLGW shares more than 30% homology with the trangslutaminase peptide 476–487 (RIRVGQSMNMGS) [113]. Therefore, antibodies generated against transglutaminase in the intestine can bind to extraintestinal tissues such as those of the liver, pancreas, lymph nodes, muscle, heart, and brain [114–117]. Very recently, we used both affinity-purified and, monoclonal antibodies against α-gliadin 33-mer peptide to examine the cross-reaction between gliadin with different food and tissue antigens [91]. We observed significant immune reactivity when these antibodies were applied to cow's milk, milk chocolate, milk butyrophilin, whey protein, casein, yeast, oats, corn, millet, instant coffee, and rice. With regard to the reaction of α-gliadin antibody with various tested tissue antigens, the most significant binding occurred with asialoganglioside, hepatocyte, glutamic acid decarboxylase 65, adrenal 21-hydroxylase, and various neural antigens [92].
These studies collectively indicate that circulating antibodies present in patients with nonceliac gluten sensitivity (NCGS) and CD interact with different food antigens and transglutaminases in various tissues, which may induce the formation of antigen-antibody aggregates that can trigger the activation of the inflammatory cascade.
While most studies about the implications of cross-reactivity with various autoimmunities are limited to milk and wheat, a thorough investigation and understanding of the immunologic cross-reactivity of other food proteins and peptides are essential for advancing our knowledge about the involvement of these dietary components in the development of many autoimmune disorders. Finally, the identification of triggers of autoimmunity can be used in the development of new therapies for autoimmune diseases.
6. Using Gluten Sensitivity, Celiac Disease, and Oral Pathogens to Understand Autoimmunities
There is a lot that clinicians can learn about autoimmune diseases from looking at gluten sensitivity and celiac disease. Some of the features of CD HLA-DQ2/DQ8 association, target organ (villi) T-cell infiltration, and disease-specific autoantibodies produced against modified antigens such as deamidated gliadin and deamidated gliadin-transglutaminase complex [99, 116] are paralleled in chronic joint disorder [94, 118]. These observations suggest that it might be feasible to use CD to identify disease-relevant epitopes in RA and other autoimmune disorders, such as type 1 diabetes and multiple sclerosis [100]. For example, the key enzymes that catalyze the modification of glutamine to glutamic acid or arginine to citrulline as new epitopes have a central role in CD and RA (Figure 13).
Figure 13.
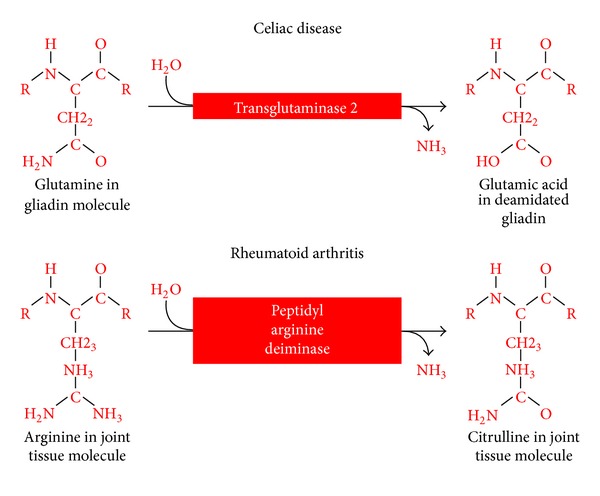
The central role of catalytic enzymes in celiac disease and rheumatoid arthritis. Key enzymes that catalyze the modification of glutamine to glutamic acid or arginine to citrulline as new epitopes have a central role in CD and RA.
It is interesting to note that this process of arginine deamination and the formation of citrullinated proteins and peptides in the joint or other tissues could be potentiated by oral pathogens such as Porphyromonas gingivalis [40, 119–124] (Figure 14). That is why RA can also cause inflammation in other organs, including the skin, lungs, heart, and peripheral nerves, often with serious consequences.
Figure 14.
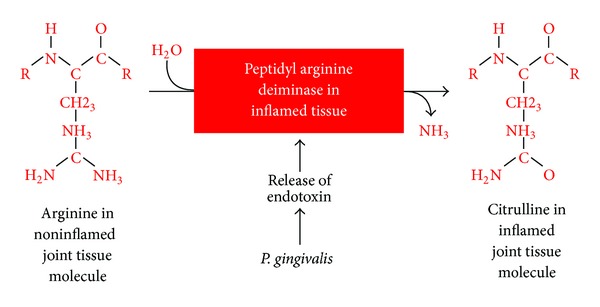
Potentiation by oral pathogens. Oral pathogens such as P. gingivalis can potentiate the deamination of arginine or formation of citrullinated proteins and peptides in joint and other tissues.
This is only one observation suggesting that environmental triggers can change self-tissue antigens to become disease-associated T-cell epitope, resulting in antibody production against the citrulline-containing new epitope. Therefore, if CD as an autoimmune disorder is driven by transglutaminase-2 and deamidated gliadin, then we may state that RA is caused by environmental factors, such as P. gingivalis or EBV. These environmental factors, by causing the formation of various citrullinated self-epitopes such as collagen type II, fibrin, vimentin, keratin, α-enolase, and filaggrin, are involved in the induction of RA [119–125]. Experience with CD has taught scientists that genes, environmental factors, and target tissue antigens are all important issues for consideration in understanding the molecular structure of epitopes recognized by T cells and B cells within the inflamed target organ [100, 124].
6.1. Mechanism Involved in the Induction of Autoimmunity by Oral Pathogen
A similar mechanism applies to the autopathogenic correlation of periodontitis induced by P. gingivalis and RA [40, 119, 124]. It is possible that both diseases share a common aetiopathogenic background [124]. This mechanism includes the posttranslation modification or citrullination of bacterial proteins and self-antigens simultaneously, generating neoepitope structure. This can result in a breakdown in self-tolerance and antibody production against citrullinated bacterial antigens as well as citrullinated host proteins [125]. One such antigen is α-enolase, which features significant homology between human and bacterial α-enolase. Therefore, antibodies produced against citrullinated bacterial α-enolase will react with human α-enolase, and antibodies produced against human citrullinated α-enolase will react strongly against bacterial α-enolase. For this reason, elevated levels of α-enolase antibodies are detected in the synovium of 60% of patients with RA [122]. Indeed, immunological mapping using a library of cyclic citrullinated α-enolase peptides led to the identification of a B-cell-dominant epitope comprising amino acids 5–21 of α-enolase (KIHAREIFDSRGNPTVE) where arginine-9 and arginine-15 are citrullinated, with an 82% sequence similarity with that of P. gingivalis [126, 127]. Immunization with citrullinated human and P. gingivalis α-enolase and citrullinated fibrinogen causes similar pathology in humanized DR4 transgenic mice. This mechanism may be the common denominator between autoimmunity and cardiovascular disease. These findings suggest that, by mimicking the molecular structure of host-citrullinated proteins, P. gingivalis peptidylarginine deiminase-citrullinated bacterial α-enolase could trigger a loss of tolerance to structurally similar host proteins, resulting in expression of anti-citrullinated protein antibodies and the development of RA [128, 129].
These antibodies can be detected up to 10 years before the clinical onset of RA and the production of IgM antibodies against IgG (called rheumatoid factor) in the majority of patients.
In the joint, the specificity of anti-citrullinated peptide is enhanced through epitope spreading to other citrullinated autoantigens such as fibrinogen, collagen, filaggrin, and vimentin (see Figure 15).
Figure 15.
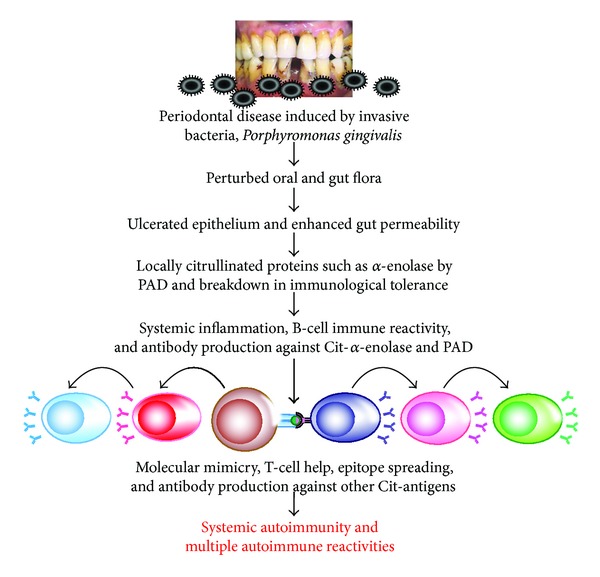
Proposed model for the pathogenesis of multiple autoimmune reactivities by infection. Bacterial generation of autoantigens, local inflammation generating autoantigens by PAD, antibody production against one autoantigen, epitope spreading, antibody production against multiple antigens, systemic inflammation, and multiple autoimmune reactivities.
7. Conclusion
Putting all this information together, it appears that there are common mechanisms in the immunopathogenesis of multiple autoimmune reactivities. In genetically susceptible individuals, environmental triggers such as xenobiotics and P. gingivalis can, respectively, induce the formation of neoantigens, or be capable of inducing the citrullination of host proteins and converting them to autoantigens. These modified proteins can be recognized by the immune system, triggering antibody production and the inflammatory process involved in the clinical manifestations of autoimmune diseases.
To optimize the chances of therapeutic success it is essential to identify the environmental triggers first and then attempt to remove them from the patient's environment (e.g., toxic chemicals and food associated with autoimmunities). In the case of infections, this also helps to guide the clinical use of various medications which are now often used for prophylaxis. Therefore, careful monitoring for the presence of infections in the patient's blood or tissue will be desirable for monitoring the effects of the drug therapy [40, 119–124].
Manipulation of environment triggers and the host immune system during the clinical and in particular preclinical stages of autoimmune disease will offer significant insight and guide early intervention for many autoimmune disorders that, according to the American Autoimmune Related Disease Association, Inc. (http://www.aarda.org/), affects approximately 10% of the world's population [130], while others put it as high as 20%. Finally, identification of triggers of autoimmunities could be used in the development of new therapies for autoimmune diseases.
Conflict of Interests
Aristo Vojdani, PhD, MSc, CLS is the owner, CEO and technical director of Immunosciences Lab., Inc., a clinical reference testing laboratory in Los Angeles, CA, USA.
References
- 1.Munz C, Lunemann JD, Getts MT, et al. Antiviral immune responses: triggers of or triggered by autoimmunity? Nature Reviews Immunology. 2009;9:246–228. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selmi C, Lu Q, Humble MC. Heritability versus the role of environment in autoimmunity. Journal of Autoimmunity. 2012;39(4):249–252. doi: 10.1016/j.jaut.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Miller FW, Pollard KM, Parks CG, et al. Criteria for environmentally associated autoimmune disease. Journal of Autoimmunity. 2012;39(4):253–258. doi: 10.1016/j.jaut.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogdanos DP, Smyk DS, Rigopoulou EI, et al. Twin studies in autoimmune disease: genetics, gender and environment. Journal of Autoimmunity. 2012;38(2-3):J156–J169. doi: 10.1016/j.jaut.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Mahdi H, Fisher BA, Källberg H, et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated α-enolase in the etiology of rheumatoid arthritis. Nature Genetics. 2009;41(12):1319–1324. doi: 10.1038/ng.480. [DOI] [PubMed] [Google Scholar]
- 6.Kuchroo VK, Ohashi PS, Sartor RB, Vinuesa CG. Dysregulation of immune homeostasis in autoimmune diseases. Nature Medicine. 2012;18(1):42–47. doi: 10.1038/nm.2621. [DOI] [PubMed] [Google Scholar]
- 7.Bayry J, Sibéril S, Triebel F, Tough DF, Kaveri SV. Rescuing CD4+CD25+ regulatory T-cell functions in rheumatoid arthritis by cytokine-targeted monoclonal antibody therapy. Drug Discovery Today. 2007;12(13-14):548–552. doi: 10.1016/j.drudis.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Linterman MA, Rigby RJ, Wong RK, et al. Follicular helper T cells are required for systemic autoimmunity. Journal of Experimental Medicine. 2009;206(3):561–576. doi: 10.1084/jem.20081886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simpson N, Gatenby PA, Wilson A, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis and Rheumatism. 2010;62(1):234–244. doi: 10.1002/art.25032. [DOI] [PubMed] [Google Scholar]
- 10.Bigazzi PE. Autoimmunity caused by xenobiotics. Toxicology. 1997;119(1):1–21. doi: 10.1016/s0300-483x(96)03591-3. [DOI] [PubMed] [Google Scholar]
- 11.Pollard KM, Hultman P, Kono DH. Toxicology of autoimmune diseases. Chemical Research in Toxicology. 2010;23(3):455–466. doi: 10.1021/tx9003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pollard KM. Gender differences in autoimmunity associated with exposure to environmental factors. Journal of Autoimmunity. 2012;38(2-3):J177–J186. doi: 10.1016/j.jaut.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Germolec D, Kono DH, Pfau JC, et al. Animal models used to examine the role of the environment in the deveopment of autoimmune disease: findings from an NIEHS expert panel workshop. Journal of Autoimmunity. 2012;39(4):285–293. doi: 10.1016/j.jaut.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griem P, Wulferink M, Sachs B, González JB, Gleichmann E. Allergic and autoimmune reactions to xenobiotics: how do they arise? Immunology Today. 1998;19(3):133–141. doi: 10.1016/s0167-5699(97)01219-x. [DOI] [PubMed] [Google Scholar]
- 15.Gershwin ME, Selmi C, Worman HJ, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42(5):1194–1202. doi: 10.1002/hep.20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rieger R, Gershwin ME. The X and why of xenobiotics in primary biliary cirrhosis. Journal of Autoimmunity. 2007;28(2-3):76–84. doi: 10.1016/j.jaut.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selmi C. Mechanisms of environmental influences on human autoimmunity: a National Institute of Environmental Health Sciences expert panel workshop. Journal of Autoimmunity. 2012;39(4):272–284. doi: 10.1016/j.jaut.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Barragan-Martinez C, Speck-Hernandez CA, Montoya-Ortiz G, et al. Organic solvents as risk factor for autoimmune diseases: a systemic review and meta-analysis. PLOS ONE. 2012;7(12) doi: 10.1371/journal.pone.0051506.e51506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baccarelli A, Wright RO, Bollati V, et al. Rapid DNA methylation changes after exposure to traffic particles. American Journal of Respiratory and Critical Care Medicine. 2009;179(7):572–578. doi: 10.1164/rccm.200807-1097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kivity S, Agmon-Levin N, Blank M, Shoenfeld Y. Infections and autoimmunity—friends or foes? Trends in Immunology. 2009;30(8):409–414. doi: 10.1016/j.it.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 21.Oldstone MBA, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65(2):319–331. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 22.Posnett DN, Yarilin D. Amplification of autoimmune disease by infection. Arthritis Research and Therapy. 2005;7(2):74–84. doi: 10.1186/ar1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faé KC, Da Silva DD, Oshiro SE, et al. Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. Journal of Immunology. 2006;176(9):5662–5670. doi: 10.4049/jimmunol.176.9.5662. [DOI] [PubMed] [Google Scholar]
- 24.Blank M, Krause I, Fridkin M, et al. Bacterial induction of autoantibodies to β2-glycoprotein-I accounts for the infectious etiology of antiphospholipid syndrome. The Journal of Clinical Investigation. 2002;109(6):797–804. doi: 10.1172/JCI12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52(6):1143–1151. doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Robles DT, Abiru N, et al. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(4):1701–1706. doi: 10.1073/pnas.040556697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Notkins AL. New predictors of disease. Scientific American. 2007;296(3):72–79. [PubMed] [Google Scholar]
- 28.Gamble DR, Kinsley ML, FitzGerald MG, Bolton R, Taylor KW. Viral antibodies in diabetes mellitus. British medical journal. 1969;3(671):627–630. doi: 10.1136/bmj.3.5671.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frisk G, Friman G, Tuvemo T, Fohlman J, Diderholm H. Coxsackie B virus IgM in children at onset of Type 1 (insulin-dependent) diabetes mellitus: evidence for IgM induction by a recent or current infection. Diabetologia. 1992;35(3):249–253. doi: 10.1007/BF00400925. [DOI] [PubMed] [Google Scholar]
- 30.Clements GB, Galbraith DN, Taylor KW. Coxsackie B virus infection and onset of childhood diabetes. The Lancet. 1995;346(8969):221–223. doi: 10.1016/s0140-6736(95)91270-3. [DOI] [PubMed] [Google Scholar]
- 31.Yoon JW, Onodera T, Notkins AL. Virus-induced diabetes mellitus. XV. Beta cell damage and insulin-dependent hyperglycemia in mice infected with Coxsackie virus B4. Journal of Experimental Medicine. 1978;148(4):1068–1080. doi: 10.1084/jem.148.4.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon JW, Austin M, Onodera T, Notkins AL. Virus-induced diabetes mellitus. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. The New England Journal of Medicine. 1979;300(21):1173–1179. doi: 10.1056/NEJM197905243002102. [DOI] [PubMed] [Google Scholar]
- 33.Kaufman DL, Erlander MG, Clare-Salzler M, Atkinson MA, Maclaren NK, Tobin AJ. Autoimmunity to two forms of glutamate decarboxylase in insulin-dependent diabetes mellitus. The Journal of Clinical Investigation. 1992;89(1):283–292. doi: 10.1172/JCI115573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadeharju K, Lönnrot M, Kimpimäki T, et al. Enterovirus antibody levels during the first two years of life in prediabetic autoantibody-positive children. Diabetologia. 2001;44(7):818–823. doi: 10.1007/s001250100560. [DOI] [PubMed] [Google Scholar]
- 35.Calcinaro F, Dionisi S, Marinaro M, et al. Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia. 2005;48(8):1565–1575. doi: 10.1007/s00125-005-1831-2. [DOI] [PubMed] [Google Scholar]
- 36.Vaarala O, Atkinson MA, Neu J. The “perfect storm” for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes. 2008;57(10):2555–2562. doi: 10.2337/db08-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sechi LA, Rosu V, Pacifico A, Fadda G, Ahmed N, Zanetti S. Humoral immune responses of type 1 diabetes patients to Mycobacterium avium subsp. paratuberculosis lend support to the infectious trigger hypothesis. Clinical and Vaccine Immunology. 2008;15(2):320–326. doi: 10.1128/CVI.00381-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park H, Bourla AB, Kastner DL, et al. Lighting the fires within: the cell biology of autoinflammatory disease. Nature Reviews Immunology. 2012;12:571–580. doi: 10.1038/nri3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramanian S. Etiopathogenesis of rheumatoid arthritis may be misunderstood of non-infectious—a review on infectious etiology of RA. Asian Journal of Medical Sciences. 2009;1(2):1–9. [Google Scholar]
- 40.Farquharson D, Butcher JP, Culshaw S. Periodontitis, porphyromonas, and the pathogenesis of rheumatoid arthritis. Mucosal Immunology. 2012;5(2):112–120. doi: 10.1038/mi.2011.66. [DOI] [PubMed] [Google Scholar]
- 41.Wu H-J, Ivanov II, Darce J, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaston JSH, Lillicrap MS. Arthritis associated with enteric infection. Best Practice and Research. 2003;17(2):219–239. doi: 10.1016/s1521-6942(02)00104-3. [DOI] [PubMed] [Google Scholar]
- 43.McColl GJ, Diviney MB, Holdsworth RF, et al. HLA-B27 expression and reactive arthritis susceptibility in two patient cohorts infected with Salmonella Typhimurium . Australian and New Zealand Journal of Medicine. 2000;30(1):28–32. doi: 10.1111/j.1445-5994.2000.tb01050.x. [DOI] [PubMed] [Google Scholar]
- 44.Hannu T, Mattila L, Siitonen A, Leirisalo-Repo M. Reactive arthritis attributable to Shigella infection: a clinical and epidemiological nationwide study. Annals of the Rheumatic Diseases. 2005;64(4):594–598. doi: 10.1136/ard.2004.027524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ebringer A, Rashid T. Rheumatoid arthritis is an autoimmune disease triggered by Proteus urinary tract infection. Clinical and Developmental Immunology. 2006;13(1):41–48. doi: 10.1080/17402520600576578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pope JE, Krizova A, Garg AX, Thiessen-Philbrook H, Ouimet JM. Campylobacter reactive arthritis: a systematic review. Seminars in Arthritis and Rheumatism. 2007;37(1):48–55. doi: 10.1016/j.semarthrit.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Domínguez-López ML, Cancino-Díaz ME, Jiménez-Zamudio L, Granados-Arreola J, Burgos-Vargas R, García-Latorre E. Cellular immune response to Klebsiella pneumoniae antigens in patients with HLA-B27+ ankylosing spondylitis. Journal of Rheumatology. 2000;27(6):1453–1460. [PubMed] [Google Scholar]
- 48.Cope A, Anderson J, Wilkins E. Clostridium difficile toxin-induced reactive arthritis in a patient with chronic Reiter’s syndrome. European Journal of Clinical Microbiology and Infectious Diseases. 1992;11(1):40–43. doi: 10.1007/BF01971269. [DOI] [PubMed] [Google Scholar]
- 49.Liu Z-Q, Deng G-M, Foster S, Tarkowski A. Staphylococcal peptidoglycans induce arthritis. Arthritis Research. 2001;3(6):375–380. doi: 10.1186/ar330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faé KC, Da Silva DD, Oshiro SE, et al. Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. Journal of Immunology. 2006;176(9):5662–5670. doi: 10.4049/jimmunol.176.9.5662. [DOI] [PubMed] [Google Scholar]
- 51.Hermann E, Mayet W-J, Klein O, et al. Candida arthritis: cellular immune responses of synovial fluid and peripheral blood lymphocytes to Candida albicans. Annals of the Rheumatic Diseases. 1991;50(10):697–701. doi: 10.1136/ard.50.10.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sutliff WD, Shephard R, Dunham WB. Acute Leptospira pomona arthritis and myocarditis. Annals of internal medicine. 1953;39(1):134–140. doi: 10.7326/0003-4819-39-1-134. [DOI] [PubMed] [Google Scholar]
- 53.Carter JD, Espinoza LR, Inman RD, et al. Combination antibiotics as a treatment for chronic Chlamydia-induced reactive arthritis: a double-blind, placebo-controlled, prospective trial. Arthritis and Rheumatism. 2010;62(5):1298–1307. doi: 10.1002/art.27394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cole BC, Ward JR. Mycoplasmas as arthritogenic agents. In: Tully JG, Whitcomb RF, editors. The Mycoplasmas. New York, NY, USA: Academic Press; 1979. pp. 367–398. [Google Scholar]
- 55.Kim HR, Kim EY, Cerny J, Moudgil KD. Antibody responses to mycobacterial and self heat shock protein 65 in autoimmune arthritis: epitope specificity and implication in pathogenesis. Journal of Immunology. 2006;177(10):6634–6641. doi: 10.4049/jimmunol.177.10.6634. [DOI] [PubMed] [Google Scholar]
- 56.Imai D, Holden K, Velasquez EM, et al. Influence of arthritis-related protein (BBF01) on infectivity of Borrelia burgdorferi B31. BMC Microbiology. 2013;13(100) doi: 10.1186/1471-2180-13-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kerr JR, Cartron JP, Curran MD, Moore JE, Elliott JRM, Mollan RAB. A study of the role of parvovirus B19 in rheumatoid arthritis. British Journal of Rheumatology. 1995;34(9):809–813. doi: 10.1093/rheumatology/34.9.809. [DOI] [PubMed] [Google Scholar]
- 58.Pratesi F, Tommasi C, Anzilotti C, Chimenti D, Migliorini P. Deiminated Epstein-Barr virus nuclear antigen 1 is a target of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis and Rheumatism. 2006;54(3):733–741. doi: 10.1002/art.21629. [DOI] [PubMed] [Google Scholar]
- 59.Bech K, Clemmensen O, Larsen JH. Cell-mediated immunity to Yersinia enterocolitica serotype 3 in patients with thyroid diseases. Allergy. 1978;33(2):82–88. doi: 10.1111/j.1398-9995.1978.tb01513.x. [DOI] [PubMed] [Google Scholar]
- 60.Shimon I, Pariente C, Shlomo-David J, Grossman Z, Sack J. Transient elevation of triiodothyronine caused by triiodothyronine autoantibody associated with acute Epstein-Barr-virus infection. Thyroid. 2003;13(2):211–215. doi: 10.1089/105072503321319530. [DOI] [PubMed] [Google Scholar]
- 61.Mori K, Munakata Y, Saito T, et al. Intrathyroidal persistence of human parvovirus B19 DNA in a patient with Hashimoto’s thyroiditis. Journal of Infection. 2007;55(2):e29–e31. doi: 10.1016/j.jinf.2007.05.173. [DOI] [PubMed] [Google Scholar]
- 62.Fernandez-Soto L, Gonzalez A, Escobar-Jimenez F, et al. Increased risk of autoimmune thyroid disease in hepatitis C vs hepatitis B before, during, and after discontinuing interferon therapy. Archives of Internal Medicine. 1998;158(13):1445–1448. doi: 10.1001/archinte.158.13.1445. [DOI] [PubMed] [Google Scholar]
- 63.Parmar RC, Bavdekar SB, Sahu DR, Warke S, Kamat JR. Thyroiditis as a presenting feature of mumps. Pediatric Infectious Disease Journal. 2001;20(6):637–638. doi: 10.1097/00006454-200106000-00023. [DOI] [PubMed] [Google Scholar]
- 64.Ziring PR, Gallo G, Finegold M. Chronic lymphocytic thyroiditis: identification of rubella virus antigen in the thyroid of a child with congenital rubella. Journal of Pediatrics. 1977;90(3):419–420. doi: 10.1016/s0022-3476(77)80705-1. [DOI] [PubMed] [Google Scholar]
- 65.Brouqui P, Raoult D, Conte-Devolx B. Coxsackie thyroiditis. Annals of Internal Medicine. 1991;114(12):1063–1064. doi: 10.7326/0003-4819-114-12-1063_2. [DOI] [PubMed] [Google Scholar]
- 66.Kawai H, Inui T, Kashiwagi S, et al. HTLV-I infection in patients with autoimmune thyroiditis (Hashimoto’s thyroiditis) Journal of Medical Virology. 1992;38(2):138–141. doi: 10.1002/jmv.1890380212. [DOI] [PubMed] [Google Scholar]
- 67.Leite JL, Bufalo NE, Santos RB, et al. Human herpes virus type 6 (HHV6) and human herpes virus type 7 (HHV7) may increase the susceptibility to Graves disease in individuals with inherited diminished TP53 apoptotic function. Proceedings of the 33rd annual meeting of the European Thyroid Association; 2008; Thessalonique; [Google Scholar]
- 68.Davies TF. Infection and autoimmune thyroid disease. Journal of Clinical Endocrinology and Metabolism. 2008;93(3):674–676. doi: 10.1210/jc.2008-0095. [DOI] [PubMed] [Google Scholar]
- 69.Ray S, Sonthalia N, Kundu S, et al. Autoimmune disorders: an overview of molecular and cellular basis in today's perspective. Journal of Clinical & Cellular Immunology. 2012 [Google Scholar]
- 70.Gershwin ME. The mosaic of autoimmunity. Autoimmunity Reviews. 2008;7(3):161–163. doi: 10.1016/j.autrev.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 71.Tuohy VK, Yu M, Weinstock-Guttman B, Kinkel RP. Diversity and plasticity of self recognition during the development of multiple sclerosis. The Journal of Clinical Investigation. 1997;99(7):1682–1690. doi: 10.1172/JCI119331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duke RC. Self recognition by T cells. I. Bystander killing of target cells bearing syngeneic MHC antigens. Journal of Experimental Medicine. 1989;170(1):59–71. doi: 10.1084/jem.170.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. The Journal of Clinical Investigation. 2001;108(8):1097–1104. doi: 10.1172/JCI14235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Agmon-Levin N, Ram M, Barzilai O, et al. Prevalence of hepatitis C serum antibody in autoimmune diseases. Journal of Autoimmunity. 2009;32(3-4):261–266. doi: 10.1016/j.jaut.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 75.Brown IJ, Tzoulaki I, Candeias V, Elliott P. Salt intakes around the world: implications for public health. International Journal of Epidemiology. 2009;38(3):791–813. doi: 10.1093/ije/dyp139. [DOI] [PubMed] [Google Scholar]
- 76.Rose G, Stamler J, Stamler R, et al. Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. British Medical Journal. 1988;297(6644):319–328. doi: 10.1136/bmj.297.6644.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Elliott E. Change in salt intake affects blood pressure of chimpanzees: implications for human populations. Circulation. 2007;116(14):1563–1568. doi: 10.1161/CIRCULATIONAHA.106.675579. [DOI] [PubMed] [Google Scholar]
- 78.Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the dietary approaches to stop hypertension (dash) diet. The New England Journal of Medicine. 2001;344(1):3–10. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- 79.Graudal NA, Galløe AM, Garred P. Effects of sodium restriction on blood pressure, renin, aldosterone, catecholamines, cholesterols, and triglyceride: a meta-analysis. Journal of the American Medical Association. 1998;279(17):1383–1391. doi: 10.1001/jama.279.17.1383. [DOI] [PubMed] [Google Scholar]
- 80.Cook NR, Cutler JA, Obarzanek E, et al. Long term effects of dietary sodium reduction on cardiovascular disease outcomes: observational follow-up of the trials of hypertension prevention (TOHP) British Medical Journal. 2007;334(7599):885–888. doi: 10.1136/bmj.39147.604896.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xie JX, Sasaki S, Joossens JV, Kesteloot H. The relationship between urinary cations obtained from the INTERSALT study and cerebrovascular mortality. Journal of Human Hypertension. 1992;6(1):17–21. [PubMed] [Google Scholar]
- 82.Sasaki N. High blood pressure and the salt intake of the Japanese. International Heart Journal. 1962;3:313–324. doi: 10.1536/ihj.3.313. [DOI] [PubMed] [Google Scholar]
- 83.MacHnik A, Neuhofer W, Jantsch J, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nature Medicine. 2009;15(5):545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 84.Cretney E, Kallies A, Nutt SL. Differentiation and function of FOXP3+ effector regulatory T cells. Trends in Immunology. 2013;34(2):74–80. doi: 10.1016/j.it.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 85.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annual Review of Immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 86.Kleinewietfeld M, Manzel A, Titze J, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu C, Yosef N, Thalhamer T, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.O' Shea JJ, Jones RG. Autoimmunity: rubbing salt in the wound. Nature. 2013;496:437–439. doi: 10.1038/nature11959. [DOI] [PubMed] [Google Scholar]
- 89.Institute of Medicine. Dietary Reference Intakes For Water, Potassium, Sodium, Chloride, and Sulphate. Washington, DC, USA: National Academies Press; 2004. [Google Scholar]
- 90.Vojdani A, Campbell AW, Anyanwu E, Kashanian A, Bock K, Vojdani E. Antibodies to neuron-specific antigens in children with autism: possible cross-reaction with encephalitogenic proteins from milk, Chlamydia pneumoniae and Streptococcus group A. Journal of Neuroimmunology. 2002;129(1-2):168–177. doi: 10.1016/s0165-5728(02)00180-7. [DOI] [PubMed] [Google Scholar]
- 91.Vojdani A, Tarash I. Cross-reaction between gliadin and different food and tissue antigens. Food and Nutrition Sciences. 2013;4:20–32. [Google Scholar]
- 92.Stefferl A, Schubart A, Storch M, et al. Butyrophilin, a milk protein, modulates the encephalitogenic T cell response to myelin oligodendrocyte glycoprotein in experimental autoimmune Encephalomyelitis. Journal of Immunology. 2000;165(5):2859–2865. doi: 10.4049/jimmunol.165.5.2859. [DOI] [PubMed] [Google Scholar]
- 93.Molberg Ø, Sollid LM. A gut feeling for joint inflammation—using coeliac disease to understand rheumatoid arthritis. Trends in Immunology. 2006;27(4):188–194. doi: 10.1016/j.it.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 94.Karjalainen J, Martin JM, Knip M, et al. A bovine albumin peptide as a possible trigger of insulin-dependent diabetes mellitus. The New England Journal of Medicine. 1992;327(5):302–307. doi: 10.1056/NEJM199207303270502. [DOI] [PubMed] [Google Scholar]
- 95.Van Belle TL, Coppieters KT, Von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiological Reviews. 2011;91(1):79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 96.Cavallo MG, Fava D, Monetini L, Barone F, Pozzilli P. Cell-mediated immune response to β casein in recent-onset insulin-dependent diabetes: implications for disease pathogenesis. The Lancet. 1996;348(9032):926–928. doi: 10.1016/S0140-6736(95)12065-3. [DOI] [PubMed] [Google Scholar]
- 97.Virtanen SM, Laara E, Hypponen E, et al. Cow's milk consumption, HLA-DQB1 genotype, and type 1 diabetes: a nested case-control study of sibling of children with diabetes. Diabetes. 2000;49(9):p. 1617. doi: 10.2337/diabetes.49.6.912. [DOI] [PubMed] [Google Scholar]
- 98.Volta U, Bonazzi C, Pisi E, Salardi S, Cacciari E. Antigliadin and antireticulin antibodies in coeliac disease and at onset of diabetes in children. The Lancet. 1987;2(8566):1034–1035. doi: 10.1016/s0140-6736(87)92608-0. [DOI] [PubMed] [Google Scholar]
- 99.MacFarlane AJ, Burghardt KM, Kelly J, et al. A type 1 diabetes-related protein from wheat (Triticum aestivum): cDNA clone of a wheat storage globulin, Glb1, linked to islet damage. The Journal of Biological Chemistry. 2003;278(1):54–63. doi: 10.1074/jbc.M210636200. [DOI] [PubMed] [Google Scholar]
- 100.Vojdani A. The characterization of the repertoire of wheat antigens and peptides involved in the humoral immune responses in patients with gluten sensitivity and Crohn's disease. ISRN Allergy. 2011;2011:12 pages. doi: 10.5402/2011/950104.950104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sumnik Z, Kolouskova S, Malcova H, et al. High prevalence of coeliac disease in siblings of children with type 1 diabetes. European Journal of Pediatrics. 2005;164(1):9–12. doi: 10.1007/s00431-004-1547-1. [DOI] [PubMed] [Google Scholar]
- 102.Hvatum M, Kanerud L, Hällgren R, Brandtzaeg P. The gut-joint axis: cross reactive food antibodies in rheumatoid arthritis. Gut. 2006;55(9):1240–1247. doi: 10.1136/gut.2005.076901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sugai E, Cherñavsky A, Pedreira S, et al. Bone-specific antibodies in Sera from patients with celiac disease: characterization and implications in osteoporosis. Journal of Clinical Immunology. 2002;22(6):353–362. doi: 10.1023/a:1020786315956. [DOI] [PubMed] [Google Scholar]
- 104.Frustaci A, Cuoco L, Chimenti C, et al. Celiac disease associated with autoimmune myocarditis. Circulation. 2002;105(22):2611–2618. doi: 10.1161/01.cir.0000017880.86166.87. [DOI] [PubMed] [Google Scholar]
- 105.Hadjivassiliou M, Mäki M, Sanders DS, et al. Autoantibody targeting of brain and intestinal transglutaminase in gluten ataxia. Neurology. 2006;66(3):373–377. doi: 10.1212/01.wnl.0000196480.55601.3a. [DOI] [PubMed] [Google Scholar]
- 106.Vojdani A, O’Bryan T, Green JA, et al. Immune response to dietary proteins, gliadin and cerebellar peptides in children with autism. Nutritional Neuroscience. 2004;7(3):151–161. doi: 10.1080/10284150400004155. [DOI] [PubMed] [Google Scholar]
- 107.Alaedini A, Okamoto H, Briani C, et al. Immune cross-reactivity in celiac disease: anti-gliadin antibodies bind to neuronal synapsin I. Journal of Immunology. 2007;178(10):6590–6595. doi: 10.4049/jimmunol.178.10.6590. [DOI] [PubMed] [Google Scholar]
- 108.Chin LC, Jones MK, Kingham JGC. Celiac disease and autoimmune thyroid disease. Clinical Medicine and Research. 2007;5(3):184–192. doi: 10.3121/cmr.2007.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bodvarsson S, Jonsdottir I, Freysdottir J, Leonard JN, Fry L, Valdimarsson H. Dermatitis herpetiformis—an autoimmune disease due to cross-reaction between dietary glutenin and dermal elastin? Scandinavian Journal of Immunology. 1993;38(6):546–550. doi: 10.1111/j.1365-3083.1993.tb03239.x. [DOI] [PubMed] [Google Scholar]
- 110.Shor DB-A, Barzilai O, Ram M, et al. Gluten sensitivity in multiple sclerosis: experimental myth or clinical truth. Annals of the New York Academy of Sciences. 2009;1173:343–349. doi: 10.1111/j.1749-6632.2009.04620.x. [DOI] [PubMed] [Google Scholar]
- 111.O' Leary C, Walsj CH, Wieneke P, et al. Coeliac disease and autoimmune Addison's disease: a clinical pitfall. Quarterly Journal of Medicine. 2002;95(2):79–82. doi: 10.1093/qjmed/95.2.79. [DOI] [PubMed] [Google Scholar]
- 112.Naiyer AJ, Shah J, Hernandez L, et al. Tissue transglutaminase antibodies in individuals with celiac disease bind to thyroid follicles and extracellular matrix and may contribute to thyroid dysfunction. Thyroid. 2008;18(11):1171–1178. doi: 10.1089/thy.2008.0110. [DOI] [PubMed] [Google Scholar]
- 113.Zanoni G, Navone R, Lunardi C, et al. In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes. PLoS Medicine. 2006;3(9):1637–1653. doi: 10.1371/journal.pmed.0030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sategna-Guidetti C, Franco E, Martini S, Bobbio M. Binding by serum IgA antibodies from patients with coeliac disease to monkey heart tissue. Scandinavian Journal of Gastroenterology. 2004;39(6):540–543. doi: 10.1080/00365520410008764. [DOI] [PubMed] [Google Scholar]
- 115.Marietta EV, Camilleri MJ, Castro LA, Krause PK, Pittelkow MR, Murray JA. Transglutaminase autoantibodies in dermatitis herpetiformis and celiac sprue. Journal of Investigative Dermatology. 2008;128(2):332–335. doi: 10.1038/sj.jid.5701041. [DOI] [PubMed] [Google Scholar]
- 116.Toscano V, Conti FG, Anastasi E, et al. Importance of gluten in the induction of endocrine autoantibodies and organ dysfunction in adolescent celiac patients. American Journal of Gastroenterology. 2000;95(7):1742–1748. doi: 10.1111/j.1572-0241.2000.02187.x. [DOI] [PubMed] [Google Scholar]
- 117.Rozenberg O, Lerner A, Pacht A, et al. A new algorithm for the diagnosis of celiac disease. Cellular and Molecular Immunology. 2011;8(2):146–149. doi: 10.1038/cmi.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meyer O. Is the celiac disease model relevant to rheumatoid arthritis? Joint Bone Spine. 2004;71(1):4–6. doi: 10.1016/j.jbspin.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 119.Burkhardt H, Sehnert B, Bockermann R, Engström Å, Kalden JR, Holmdahl R. Humoral immune response to citrullinated collagen type II determinants in early rheumatoid arthritis. European Journal of Immunology. 2005;35(5):1643–1652. doi: 10.1002/eji.200526000. [DOI] [PubMed] [Google Scholar]
- 120.Masson-Bessière C, Sebbag M, Girbal-Neuhauser E, et al. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the α-and β-chains of fibrin. Journal of Immunology. 2001;166(6):4177–4184. doi: 10.4049/jimmunol.166.6.4177. [DOI] [PubMed] [Google Scholar]
- 121.Vossenaar ER, Després N, Lapointe E, et al. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis research & therapy. 2004;6(2):R142–150. doi: 10.1186/ar1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kinloch A, Tatzer V, Wait R, et al. Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis research & therapy. 2005;7(6):R1421–1429. doi: 10.1186/ar1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weinberg JB, Pippen AMM, Greenberg CS. Extravascular fibrin formation and dissolution in synovial tissue of patients with osteoarthritis and rheumatoid arthritis. Arthritis and Rheumatism. 1991;34(8):996–1005. doi: 10.1002/art.1780340809. [DOI] [PubMed] [Google Scholar]
- 124.Routsias JG, Goules JD, Goules A, Charalampakis G, Pikazis D. Autopathogenic correlation of periodontitis and rheumatoid arthritis. Rheumatology. 2011;50(7):1189–1193. doi: 10.1093/rheumatology/ker090.ker090 [DOI] [PubMed] [Google Scholar]
- 125.Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1∗0401 MHC class II molecule. Journal of Immunology. 2003;171(2):538–541. doi: 10.4049/jimmunol.171.2.538. [DOI] [PubMed] [Google Scholar]
- 126.Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. Periodontitis in RA-the citrullinated enolase connection. Nature Reviews Rheumatology. 2010;6(12):727–730. doi: 10.1038/nrrheum.2010.139. [DOI] [PubMed] [Google Scholar]
- 127.Lundberg K, Kinloch A, Fisher BA, et al. Antibodies to citrullinated α-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis and Rheumatism. 2008;58(10):3009–3019. doi: 10.1002/art.23936. [DOI] [PubMed] [Google Scholar]
- 128.Kinloch AJ, Alzabin S, Brintnell W, et al. Immunization with porphyromonas gingivalis enolase induces autoimmunity to mammalian α-enolase and arthritis in DR4-IE-transgenic mice. Arthritis and Rheumatism. 2011;63(12):3818–3823. doi: 10.1002/art.30639. [DOI] [PubMed] [Google Scholar]
- 129.Hill JA, Bell DA, Brintnell W, et al. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. Journal of Experimental Medicine. 2008;205(4):967–979. doi: 10.1084/jem.20072051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. American Autoimmune Related Disease Association, http://www.aarda.org.