

Abstract
We have previously shown that FKBP12 associates with RyR2 in cardiac muscle and that it modulates RyR2 function differently to FKBP12.6. We now investigate how these proteins affect the single-channel behavior of RyR1 derived from rabbit skeletal muscle. Our results show that FKBP12.6 activates and FKBP12 inhibits RyR1. It is likely that both proteins compete for the same binding sites on RyR1 because channels that are preactivated by FKBP12.6 cannot be subsequently inhibited by FKBP12. We produced a mutant FKBP12 molecule (FKBP12E31Q/D32N/W59F) where the residues Glu31, Asp32, and Trp59 were converted to the corresponding residues in FKBP12.6. With respect to the functional regulation of RyR1 and RyR2, the FKBP12E31Q/D32N/W59F mutant lost all ability to behave like FKBP12 and instead behaved like FKBP12.6. FKBP12E31Q/D32N/W59F activated RyR1 but was not capable of activating RyR2. In conclusion, FKBP12.6 activates RyR1, whereas FKBP12 activates RyR2 and this selective activator phenotype is determined within the amino acid residues Glu31, Asp32, and Trp59 in FKBP12 and Gln31, Asn32, and Phe59 in FKBP12.6. The opposing but different effects of FKBP12 and FKBP12.6 on RyR1 and RyR2 channel gating provide scope for diversity of regulation in different tissues.
Introduction
FK506-binding proteins (FKBPs) bind tightly to ryanodine receptor (RyR) channels in cardiac and skeletal muscle, and evidence suggests that they play an important regulatory role that is impaired in disease, for example, in heart failure (1–4) and certain skeletal muscle disorders (5). It is reported that FKBPs provide a stabilizing effect on RyR channel function by lowering open probability (Po) and preventing subconductance state gating and that, at the cellular level, this leads to fewer leaky RyR channels and fewer aberrant Ca2+-release events (6–8). It is generally thought that FKBP12 stabilizes RyR1 and FKBP12.6 stabilizes RyR2 (1,9–11). This is a controversial area, however, because not all investigators agree that low Po or substate gating in RyR channels is dependent on the binding of FKBPs (12–15). Moreover, we have recently shown that FKBP12 binds with high affinity to RyR2, causing channel activation and stimulating Ca2+-induced Ca2+-release in isolated cardiac myocytes (15). We demonstrated that FKBP12.6 cannot itself lower RyR2 Po, but because it is a partial agonist of very low efficacy, it can act as an antagonist of FKBP12 and indirectly reduce RyR2 Po and sarcoplasmic reticulum (SR) Ca2+-release in cardiac cells. Thus, the gating of RyR2 in cardiac muscle is under the dual modulation of FKBP12 and FKBP12.6, rather than the exclusive influence of FKBP12.6.
Other experiments also suggest that FKBP12 and FKBP12.6 do not selectively regulate RyR1 and RyR2, respectively. In an FKBP12 knockout mouse model, skeletal muscle function appeared normal, whereas cardiac hypertrophy and disturbances in cardiac excitation-contraction coupling were very severe (16). It has also been shown that FKBP12.6 can affect skeletal muscle function (17–19) indicating that, similar to RyR2 in cardiac muscle, RyR1 may also be subjected to dual regulation by FKBP12 and FKBP12.6.
We have therefore investigated how FKBP12 and FKBP12.6 affect the single-channel behavior of RyR1 and, to understand the mechanisms underlying their functional effects, we have made direct comparisons of their effects on RyR2 gating. FKBP12 and FKBP12.6 share the same number of amino acids (108) and with 82% sequence identity have similar molecular masses (11.8 kDa, FKBP12.6; 11.9 kDa, FKBP12 (15)). X-ray crystallography highlights the close structural similarity of the two proteins, providing few clues about the residues that may be important for binding to RyR1 and RyR2 or for transducing distinct functional effects. Previous work has indicated that the residues Gln3, Arg18, and Met49 are necessary for FKBP12 binding to RyR1 (20). Likewise, mutation of Asp37 to Val or Ser is reported to enhance the affinity of FKBP12.6 for RyR2 (21,22). Although affinity is important, the particular amino acid residues important for efficacy (the ability of FKBP12/12.6 to act as agonist or antagonist of RyR1/RyR2) have not been investigated previously. By locating the steric and electrostatic differences between FKBP12 and FKBP12.6 we identified three amino acid residues that could be relevant in this respect. We generated a mutant FKBP12 molecule (FKBP12E31Q/D32N/W59F), in which these three residues were mutated to the corresponding residues found in FKBP12.6 and investigated if the mutant possessed increased or decreased ability to act as an activator (agonist) of RyR1 and/or RyR2.
We find that FKBP12 is an inhibitor of RyR1 but an activator of RyR2. Conversely, FKBP12.6 is an activator of RyR1 but has barely detectable ability to activate RyR2. The triple mutant, FKBP12E31Q/D32N/W59F, lost all ability to activate RyR2 and inhibit RyR1 but instead, caused significant activation of RyR1. Thus, the ability for conferring selectivity of RyR agonist behavior is contained within a few key amino acids.
Materials and Methods
Isolation of membrane fractions and bilayer techniques
Heavy sarcoplasmic reticulum (HSR) vesicles were obtained from sheep hearts (collected from an abattoir) or rabbit skeletal muscle, as described previously (23). SR vesicles were fused with planar phosphatidylethanolamine lipid bilayers as described (23). Incorporation of RyR always occurred in the same fixed orientation with the cis side corresponding to the cytosolic channel side and the trans side to the luminal face. The trans-chamber was held at ground and the cis-chamber clamped at potentials relative to ground. After fusion, the cis-chamber was perfused with 250 mM HEPES, 80 mM Tris, 10 μM free Ca2+, pH 7.2. The trans-chamber was perfused with 250 mM glutamic acid and 10 mM HEPES, pH to 7.2 with Ca(OH)2 (free [Ca2+], ∼50 mM). Experiments were performed at 22 ± 2°C. Free [Ca2+] and pH were maintained constant during experiments and were determined using a Ca2+ electrode (Orion 93–20, Thermo Fisher Scientific Inc., Waltham, MA) and Ross-type pH electrode (Orion 81–55, Thermo Fisher Scientific Inc., Waltham, MA) as previously described (23). FKBP12 and FKBP12.6 were added to the cis-chamber. Both proteins were stored in a buffer containing 10 mM HEPES, 50 mM NaCl, 0.5 mM DTT and volumes added to the cis-chamber were <2% of the total volume. Control experiments demonstrated that the buffer alone caused no effects on RyR function. Rapamycin treatment of the skeletal HSR membrane fraction was obtained by incubating vesicles with 20 μM rapamycin for 15 min at 36°C. Following incubation, the membrane fraction was sedimented at 180,000 × g for 15 min at room temperature.
Data acquisition and analysis
Single-channel currents were monitored under voltage-clamp conditions using a BC-525C amplifier (Warner Instruments, Hamden, CT). Channel recordings were low-pass filtered at 10 kHz with a 4-pole Bessel filter, digitized at 100 kHz using an ITC-18 data acquisition interface (HEKA Elektronik, Lambrecht/Pfalz, Germany) and recorded on a computer hard drive using WinEDR 3.05 software (John Dempster, University of Strathclyde, UK). The recordings were subsequently filtered at 800 Hz (−3 dB) using a low-pass digital filter implemented in WinEDR 3.05. Channel events were detected by the 50% threshold method (24) using TAC 4.2.0 software (Bruxton, Seattle, WA). Po and lifetime distributions were calculated from 3 min of continuous recording using TACfit 4.2.0 software (Bruxton). Po diary plots were obtained using Clampfit 10.2 (Molecular Devices, Sunnyvale, CA).
Preparation of wild-type and mutant FKBPs
Human FKBP12 and rabbit FKBP12.6 were cloned, expressed, and purified as previously described (15). The preparation of FKBP12E31Q/D32N/W59F triple mutant is described in the Supporting Material. FKBP12 was also purchased from Sigma-Aldrich (Dorset, UK).
Statistics
Data are expressed as mean ± SE where n ≥4. For n = 3, SD is given. Where appropriate, Student’s t-test was used to assess the difference between treatments. Where multiple treatments were compared, analysis of variance followed by a modified t-test was used to assess the difference between treatments. A p value of <0.05 was taken as significant.
Materials
All chemicals were obtained from VWR (Poole, UK) or Sigma-Aldrich. All solutions were prepared in MilliQ deionized water (Millipore, Harrow, UK) and those for use in bilayer experiments were filtered through a Millipore membrane filter with 0.45 μm pore diameter.
Results
We have previously demonstrated that both FKBP12 and FKBP12.6 behave as very high affinity, partial agonists of RyR2 (15). FKBP12 activates RyR2 at low picomolar concentrations. In contrast, the stimulatory effects of FKBP12.6 on RyR2 are usually imperceptible because although FKBP12.6 can bind to RyR2, it has extremely low efficacy. The Supporting Material (Fig. S1) shows typical examples of how FKBP12 and FKBP12.6 affect RyR2 gating. To make direct comparisons, identical recording conditions were used to investigate the effects of FKBP12 and FKBP12.6 on rabbit skeletal RyR1 gating. Unexpectedly, FKBP12.6 caused an increase in RyR1 Po. This effect was irreversible on the timescale of a single-channel experiment as washout of protein from the cytosolic chamber did not lower Po (Fig. 1 A). The irreversibility of the FKBP12.6 effect suggests a high affinity interaction between FKBP12.6 and RyR1 and this is confirmed by the fact that concentrations as low as 10 pM FKBP12.6 can significantly increase Po. Fig. 1 B shows the mean data for different concentrations of FKBP12.6. The mean open and closed times derived from experiments where only single channels were present in the bilayer were 1.68 ± 0.35 ms and 91.2 ± 34.9 ms, respectively, before and 2.26 ± 0.27 ms and 58.4 ± 37.3 ms (SD; n = 3), respectively, after addition of 200 nM FKBP12.6 indicating that FKBP12.6 primarily increases channel opening frequency with little effect on open lifetime duration. Lifetime analysis (see Fig. S2) confirms this mechanism of action; the open lifetime distribution is not altered even at high concentrations of FKBP12.6.
Figure 1.
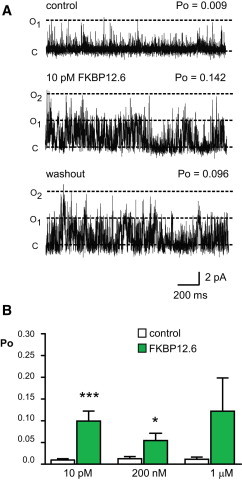
FKBP12.6 activates rabbit skeletal RyR1. (A) A typical single-channel experiment showing marked activation of RyR1 by 10 pM FKBP12.6. The bottom trace shows washout of FKBP12.6 from the cytosolic chamber. Po values are indicated. Dashed lines indicate open (O1, O2) and closed (C) channel levels, respectively. (B) Mean Po data obtained before and after addition of 10 pM, 200 nM, and 1 μM FKBP12.6 (SE; n = 5–11; ∗∗∗p < 0.001; ∗p < 0.05). Each concentration represents a set of independent experiments. To see this figure in color, go online.
In contrast, FKBP12 always reduced the Po of RyR1. A typical experiment is shown in Fig. 2 A with mean data shown in Fig. 2 B. Washout of cytosolic solutions to remove unbound FKBP12 did not reverse the inhibition as shown in the bottom trace. In four experiments where FKBP was washed out and Po was recorded for a further 3 min, Po was 0.007 ± 0.005 before and 0.002 ± 0.001 (SE; n = 4) after washout. The diary plot of channel activity during a typical experiment is shown in Fig. 2 C. This illustrates the modal gating that is characteristic of RyR channels (25,26) and the irreversibility of the effect of FKBP12. Detailed lifetime analysis was not possible because of the low number of events that occurred after addition of FKBP12. However, mean open and closed times were 2.41 ± 0.16 ms and 22.9 ± 3.94 ms (SE; n = 4), respectively, before and 2.04 ± 0.04 ms and 332 ± 142 ms (SE; n = 4), respectively, after the addition of 500 nM FKBP12 showing that the main effect of FKBP12 is to reduce channel opening frequency.
Figure 2.
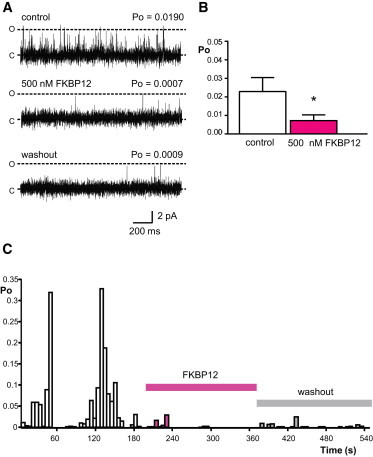
Inhibitory effects of FKBP12 on rabbit skeletal RyR1. (A) A representative experiment showing that RyR1 Po is reduced by 500 nM FKBP12. The bottom trace shows that perfusion of the cis chamber back to control solutions did not reverse the reduction in Po. The dashed lines indicate open (O) and closed (C) channel levels, respectively. Po values are indicated above each trace. (B) Mean data showing that 500 nM FKBP12 inhibits RyR1 activity (SE; n = 8; ∗p < 0.05). (C) Diary plot showing RyR1 Po in the presence of 10 μM cytosolic Ca2+ (control), after addition of 500 nM FKBP12 and after cis chamber perfusion back to control solutions. Single-channel traces were subdivided into 10 s sections and Po was measured for each section and plotted against time. Note that even during control periods, RyR1 channels display marked variability of gating over time with random switching between low and high Po modes. The bars indicate the times of incubation with FKBP12 and washout of FKBP12. To see this figure in color, go online.
It is generally assumed that FKBP12.6 is not important for RyR1 function in skeletal muscle because FKBP12.6 is present at very low levels, whereas FKBP12 is present at micromolar levels (1–3 μM) (12,27,28). The data shown in Fig. 1, however, indicates that activation of RyR1 results from a high affinity interaction with FKBP12.6 and so an important question to answer is whether FKBP12 and FKBP12.6 could compete for the same binding sites. If so, then preaddition of FKBP12.6 should prevent FKBP12 from causing the immediate inhibition of RyR1 that is shown in Fig. 2 C. Fig. 3 A and B confirm that this is the case. Preaddition of FKBP12.6 (200 nM) prevents FKBP12 (500 nM) from causing channel inhibition (on the timescale of a single-channel experiment). There are two further issues to consider. 1), If the affinities of FKBP12/FKBP12.6 for RyR1 are as high as Figs. 1 and 2 suggest and the dissociation of FKBPs from RyR1 is very slow, then even picomolar quantities of either isoform should prevent the binding of the other isoform. 2), Physiological levels of FKBP12 are likely to be at least 5–10 times the levels of FKBP12.6, therefore, what happens in the competition studies if FKBP12 is present at 10 times the levels of FKBP12.6? To address these questions, we have performed experiments at 10 pM FKBP12.6 and 100 pM FKBP12 (pretreating first with 10 pM FKBP12.6 (Fig. 3 C) or 100 pM FKBP12 (Fig. 3 D). The experiments demonstrate pharmacological consistency; the high affinity binding and slow dissociation rate of either isoform of FKBP blocks the binding of the subsequently added isoform of FKBP.
Figure 3.
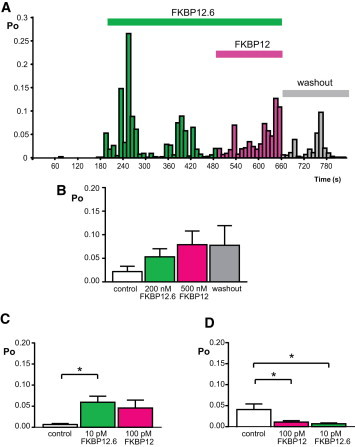
Preaddition of FKBP12.6 prevents the inhibition of rabbit skeletal RyR1 by FKBP12. (A) A representative diary plot of RyR1 Po changes following preincubation with 200 nM FKBP12.6 and subsequent addition of 500 nM FKBP12 (incubation periods are shown by the bars). Control solutions contained 10 μM cytosolic Ca2+ as the sole channel activator. Subsequent addition of 500 nM FKBP12 did not override the activating effects of FKBP12.6. After washout of the cis chamber, Po was not completely reversed to control values. (B) Bar chart showing the mean Po data for control, preaddition of 200 nM FKBP12.6, subsequent addition of 500 nM FKBP12, and washout to control solutions (SE; n = 4). Pretreatment with picomolar levels of FKBP12.6 (C) or FKBP12 (D) at the 10:1 ratio of FKBP12:FKBP12.6 that is expected in situ, was able to block the subsequent addition of the other FKBP isoform (SE; n = 10–11; ∗p < 0.05). To see this figure in color, go online.
To identify the amino acid residues in FKBP12 that are important for the selective ability of FKBP12 to reduce the Po of RyR1 and increase the Po of RyR2, we compared the specific amino acids of FKBP12 and FKBP12.6 that differ. Fig. 4 compares the primary sequences of FKBP12 and FKBP12.6 and their three-dimensional structures (FKBP12 purple; Protein Data Bank (PDB) 2DG3; FKBP12.6 green; PDB 1C9H) and illustrates their high structural similarity. Indeed, the root mean-squared deviation between the superimposed backbone atoms is only 0.44 Å. On the basis of their steric and electrostatic properties, we identified three residues that could have particular relevance. Glu31 and Asp32 are negatively charged residues of FKBP12, whereas the corresponding residues in FKBP12.6 are neutral (Gln31 and Asn32). Trp59 in FKBP12 is located within the hydrophobic binding pocket for rapamycin and has a larger side chain (an indole) compared with Phe59 (a phenyl group) in FKBP12.6. The locations of these three amino acid residues in FKBP12 and the corresponding residues in FKBP12.6 are highlighted. We produced a triple mutant of FKBP12 where these three highlighted amino acids were mutated to the corresponding residues in FKBP12.6 thus converting Glu31 to Gln, Asp32 to Asn, and Trp59 to Phe. The affinity of various mutants of FKBPs for RyR proteins have previously been investigated, (20,29) although the ability to influence RyR single-channel function (efficacy) has not been studied. The FKBP12E31Q/D32N/W59F triple mutant retains affinity for RyR channels (20,29) and therefore is a molecule of choice for studying efficacy (because we must use mutant molecules that bind to RyR channels to study efficacy).
Figure 4.
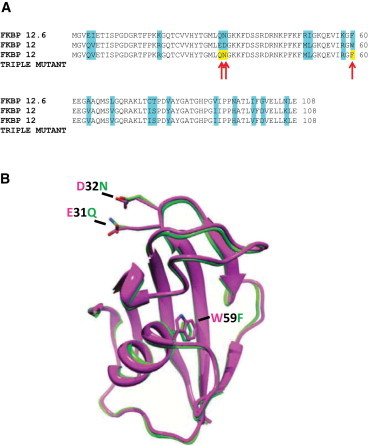
Comparison of FKBP12 and FKBP12.6 proteins to highlight the amino acid substitutions of the FKBP12E31Q/D32N/W59F mutant. (A) Amino acid sequence alignments of human FKBP12, human FKBP12.6, and FKBP12E31Q/D32N/W59F. The residues of FKBP12 and FKBP12.6 that differ are highlighted in blue. The red arrows indicate the three amino acid residues that were substituted into the FKBP12E31Q/D32N/W59F mutant. (B) Overlaid ribbon diagrams of human FKBP12 and human FKBP12.6 based on their crystal structures (RCSB PDB accession codes 2DG3 and 1C9H, respectively) showing the three residues highlighted as most likely relevant amino acid substitutions between the two proteins. The dashes indicate these residues: Glu31 in FKBP12 is replaced by Gln31 from FKBP12.6 (E31Q); Asp32 in FKBP12 is replaced by Asn32 from FKBP12.6 (D32N); Trp59 in FKBP12 is replaced by Phe59 from FKBP12.6 (W59F). Mammalian FKBP12 and FKBP12.6 are highly conserved and Fig. S3 shows sequence alignment of several species demonstrating that the three residues that we chose to mutate were absolutely conserved in FKBP12 and were different but, again, absolutely conserved in FKBP12.6.
We then investigated the ability of FKBP12E31Q/D32N/W59F to influence the gating of RyR1 and RyR2 under identical experimental conditions to those used with the wild-type proteins. Representative examples of the effects of FKBP12E31Q/D32N/W59F on RyR2 gating are shown in Fig. 5. FKBP12E31Q/D32N/W59F produced no observable changes in RyR2 gating even at nanomolar or micromolar concentrations. Clearly, the triple mutant protein, FKBP12E31Q/D32N/W59F, has lost the normal ability of FKBP12 to activate RyR2 and behaves instead like FKBP12.6; efficacy has been lost.
Figure 5.
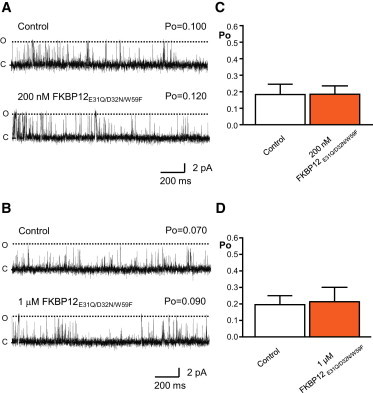
Effects of FKBP12E31Q/D32N/W59F on sheep cardiac RyR2 gating. A and B are typical examples of single-channel experiments demonstrating that neither 200 nM (A) or 1 μM (B) FKBP12E31Q/D32N/W59F cause any observable effects on RyR2 activity. Dashed lines indicate open (O) and closed (C) channel levels, respectively. Po values are indicated. C and D illustrate mean Po before and after addition of 200 nM (C) and 1 μM (D) FKBP12E31Q/D32N/W59F, respectively (SE; n = 5). To see this figure in color, go online.
When FKBP12E31Q/D32N/W59F was added to RyR1 channels, an increase in Po was observed (Fig. 6), very similar to that observed with FKBP12.6 (see Fig. 1). The representative experiment shown in Fig. 6 A illustrates the increase in the frequency of channel openings that was induced by FKBP12E31Q/D32N/W59F and demonstrates that washout of the mutant protein from the cytosolic chamber could not reverse the effects of the mutant. To observe the variations in Po that occur with time, diary plots of Po against time were recorded (see Fig. 6 B). The effects of a range of concentrations of FKBP12E31Q/D32N/W59F were investigated and in all cases, FKBP12E31Q/D32N/W59F activated RyR1 (Fig. 6 C).
Figure 6.
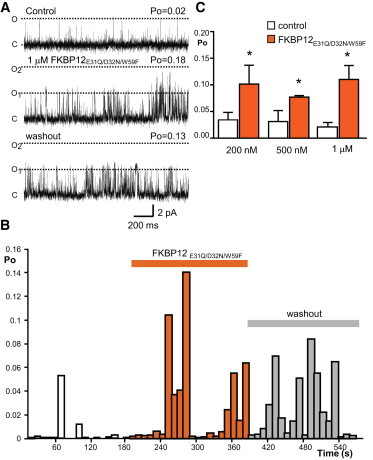
FKBP12E31Q/D32N/W59F activates rabbit skeletal RyR1. (A) A typical experiment showing that 1 μM FKBP12E31Q/D32N/W59F activates RyR1. The effects were not reversed after washout of the cis chamber (bottom trace). Open (O1, O2) and closed (C) channel levels are marked with dashed lines. (B) Diary plot of a typical RyR1 single-channel experiment. After washout of the cis chamber, Po did not reverse to control values. The bars indicate the incubation time with the mutant protein and subsequent washout of the protein from the cis chamber. (C) Mean Po values before and after addition of 200 nM, 500 nM, and 1 μM FKBP12E31Q/D32N/W59F (SE; n = 7–11; ∗p < 0.05). Each concentration represents a set of independent experiments. To see this figure in color, go online.
It is possible that some endogenous FKBP could still be associated with the channels in the bilayer before we add exogenous FKBPs. However, because we observe reproducible effects with both FKBP12 and FKBP12.6 and at both RyR1 and RyR2, it is obvious that there are always vacant FKBP binding sites on both channels that are functionally relevant. We do not know how many molecules of FKBP12 or FKBP12.6 must bind to RyR channels to produce their effect. The irreversible nature of the binding makes this difficult to examine at the single-channel level. Many previous reports have used drugs such as rapamycin or FK-506 to strip FKBPs from RyRs (9,11,30–33). However, even in these studies, it was not possible to be certain that all FKBP molecules were removed because Western blot was the only proof of FKBP dissociation and this is not a technique able to detect low levels of proteins. In fact, later work shows that these treatments do not achieve complete displacement of bound FKBPs (34,35). We have therefore treated SR vesicles with rapamycin (20 μM) using previously published methods (30) and performed mass spectrometry to detect the FKBPs because this is a more sensitive method of protein detection than Western blot analysis. We find that FKBP12 is still detected with high confidence (false discovery rate < 1%; Table S1) demonstrating that rapamycin treatment does not remove all FKBP proteins from rabbit skeletal SR. Western blot analysis shown in Fig. 7, A and B, shows that rapamycin is very effective at dissociating FKBPs from the SR but that there is a residual amount left which, depending on the sensitivity of the antibody, may not always be immunodetectable. In the literature, there is heavy reliance on the ability of rapamycin to dissociate FKBPs from RyR channels to infer mechanistic insight into FKBP effects on RyR function. However, such reasoning is questionable because there are reports that rapamycin may affect RyR channel behavior directly (35,36). We have therefore examined if rapamycin affects RyR1 gating by incorporating the rapamycin pretreated SR vesicles into bilayers. In line with the literature, we find that the Po of RyR1 channels pretreated with rapamycin is significantly higher than that of control channels (Fig. 7, C and D). Of importance, however, we find that addition of FKBP12 does not reduce Po values back to control levels (see second trace and Fig. 7 D) suggesting that the rapamycin-induced elevation of Po was not related to dissociation of FKBP12. The rapamycin-induced increase in Po was not caused by nonspecific damage to the channels because lowering cytosolic [Ca2+] to subactivating levels (third trace) completely closed the channels showing they were still sensitive to Ca2+.
Figure 7.
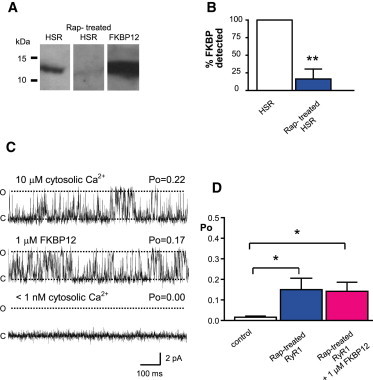
Rapamycin irreversibly activates rabbit skeletal RyR1. (A) Immunochemical detection of FKBP associated with skeletal HSR before (left lane) and after (middle lane) rapamycin treatment (as described in Methods). HSR was loaded at a protein concentration of 25 μg. Recombinant purified FKBP12 was loaded at 500 ng as a positive control (right lane). (B) The percentage of FKBP detected in HSR after rapamycin treatment (Rap-treated HSR) is compared to control levels (HSR) (SD, n = 3, ∗∗p < 0.01). (C) Shows a representative example of a rapamycin pretreated RyR1 channel reconstituted into a bilayer. (O) and (C) indicate the open and the closed channel levels, respectively. Addition of 1 μM FKBP12 to the cytosolic side of the channel (second trace) did not lower Po. Lowering the cytosolic [Ca2+] to <1 nM (by addition of 10 mM EGTA) shut the channel (bottom trace). (D) Mean Po values for control channels (untreated), rapamycin-treated channels (Rap-treated RyR1), and Rap-treated channels after addition of 1 μM FKBP12 (Rap-treated RyR1 + 1 μM FKBP12) are illustrated (SE; n = 3–7; ∗p < 0.05). To see this figure in color, go online.
A rapamycin-induced increase in cardiac RyR2 Po has also been used to infer that dissociation of FKBP12.6 increases RyR2 Po (11) so we performed similar experiments with RyR2 (see Fig. S4). As with RyR1, we found that rapamycin increased RyR2 activity, although the effect was more marked. Washout of rapamycin did not lower Po, nor did the addition of FKBP12.6, although the channels retained sensitivity to cytosolic Ca2+ and were closed by reducing [Ca2+]. Because rapamycin does not fully dissociate FKBP12 from RyR1 and because it irreversibly alters RyR1 and RyR2 channel gating, these experiments highlight the need to use native RyR channels that have not been chemically treated to probe the functional effects of FKBP12/FKBP12.6.
Discussion
To our knowledge, a novel and unexpected finding of this study is that FKBP12.6 is a potent activator of the rabbit skeletal RyR1, whereas FKBP12 is an inhibitor. The results contrast with the situation in cardiac muscle where FKBP12 is an activator of RyR2 and FKBP12.6 is an antagonist of FKBP12 (15) but does not, itself, lower Po. We performed site-directed mutagenesis of the FKBP12 molecule and found that the selective ability of FKBP12 to activate RyR2 and inhibit RyR1 is contained within the three amino acid residues; Glu31, Asp32, and Trp59. Mutation of these key residues to those found in FKBP12.6, caused a complete switch from FKBP12-like action to FKBP12.6-like action.
Experiments where sequential addition of FKBP12 and FKBP12.6 are made to the cytosolic face of RyR channels indicate that these proteins would compete for the same binding sites on RyR1 and RyR2. This is not surprising in view of the structural similarity of FKBP12 and FKBP12.6 but also in the light of cryo-electron microscopy and single-particle three-dimensional reconstruction studies indicating that FKBPs bind within similar binding pockets on RyR1 and RyR2 (37–39). Use of fluorescence resonance energy transfer also suggests that FKBP12 and FKBP12.6 would bind in the same locations on RyR1 and RyR2 and with similar orientation (40).
We show that FKBP12 and FKBP12.6 exert different effects on the gating of both RyR1 and RyR2. This indicates that the binding sites on the RyR1 and RyR2 channel proteins could be composed of different amino acid residues. Confirmation of this idea comes from binding studies (12,28) using radiolabeled FKBP12/12.6, which indicate that canine cardiac and rabbit skeletal RyR have different affinity for FKBP12/12.6 (although it should be remembered that subsequent work demonstrated that canine RyR2 has unusually high affinity for FKBP12.6 that is not shared by other mammalian RyR2 isoforms (41)). Another explanation for the different functional effects of FKBP12/12.6 could be that the binding interactions between FKBPs/RyR1 and FKBPs/RyR2 produce different changes in channel gating simply because of subtle differences in RyR1 and RyR2 channel gating mechanisms. For example, RyR1 and RyR2 are regulated slightly differently by cytosolic Ca2+, especially at inactivating [Ca2+] (42–44), and there are subtle differences in the mechanisms by which other ligands such as ATP, caffeine, or suramin activate RyR1 and RyR2 (45–48). We show that activation of RyR1 by FKBP12.6 involves an increase in the frequency of channel opening. This is similar to the mechanism by which cytosolic Ca2+ activates RyR1 (49) and therefore FKBP12.6 may be sensitizing the channel to cytosolic Ca2+. We have previously demonstrated that FKBP12 sensitizes RyR2 to cytosolic Ca2+ (15) and therefore this may be a common mechanism by which FKBPs regulate RyR channels. We cannot be so confident about the mechanism by which FKBP12 inhibits RyR1; it may reduce RyR1 sensitivity to cytosolic Ca2+ but because Po is lowered so much, we cannot collect enough opening events for lifetime analysis. The important, take-home message, though, is that FKBP12 and FKBP12.6 drive Po in opposite directions for both RyR1 and RyR2. This has important consequences for skeletal muscle function if the relative ability of FKBP12 and FKBP12.6 to bind to RyR1 is affected in stress, exercise, aging, or disease.
Although we and others have previously reported that FKBP12.6 does not, itself, reduce the Po of RyR2 (12,14,28), opinion to the contrary still persists. Our experiments with rapamycin now indicate why this could be. Rapamycin is a frequently used tool for dissociating FKBPs from RyR channels and irreversible increases in RyR Po after rapamycin treatment have led to the conclusion that RyRs must be bound by FKBPs to ensure stable low Po channel gating (11,31,35,50). However, FKBP12 or FKBP12.6 was not added back in the bilayer experiments to examine if these proteins could reverse a rapamycin-induced increase in Po. We have now demonstrated that use of rapamycin may produce misleading results because it can irreversibly increase RyR Po in a manner that is independent of the presence or absence of FKBPs. FK506 is another drug, used extensively to dissociate FKBPs from RyRs. However, studies conducted on intact skeletal fibers have also shown effects of both rapamycin and FK506 on excitation-contraction coupling that are unrelated to the removal of FKBPs (51,52). Again, incorrect conclusions could be drawn from using this compound if it was not completely removed during a subsequent experiment.
Another problem is that [3H]ryanodine binding has often been used to indicate RyR channel activity following rapamycin treatment rather than examining the detailed single-channel behavior of RyR1/RyR2 channels directly following incorporation into bilayers. [3H]ryanodine binding can provide an incorrect approximation of the effect of an intervention on RyR activity if that intervention also directly affects the binding of ryanodine. For example, calmodulin increases RyR2 Po but decreases [3H]ryanodine binding because calmodulin also reduces the rate of association of ryanodine to RyR2 (53). There is no study that has investigated whether FKBPs could directly alter the rate of association or dissociation of ryanodine to RyRs or, particularly pertinent to this study, whether rapamycin itself influences ryanodine binding independently of Po. It is very relevant that a single-channel report by Ahern et al. (1997) (33) suggests that ryanodine modification of RyR1 may be reversible after treatment with FK506. Any such increase in the rate of dissociation of ryanodine from RyR channels would, naturally, affect the binding of [3H]ryanodine to cardiac or skeletal SR. We (and others (34,35)) have also shown that rapamycin-treated skeletal SR vesicles will still have residual FKBP12 molecules associated (even if below immunodetection levels) and this will be a significant confounding factor given the very high affinity of FKBPs for RyR channels. Endogenous FKBPs will be present within the incubation medium for the [3H]ryanodine binding assay and will already be bound to an unquantified number of RyR channels. Using [3H]ryanodine binding to skeletal SR vesicles to assay the functional actions of FKBPs will therefore provide results that are difficult to interpret.
Our mutant experiments have begun to distinguish those regions of the FKBP proteins that are important for efficacy. The FKBP12 mutant, FKBP12E31Q/D32N/W59F, clearly has efficacy as an activator of RyR1 but appears to have no or low efficacy as a regulator of RyR2. Our experiments do not provide accurate measurement of the affinity of FKBP12E31Q/D32N/W59F for RyR1 or RyR2, however, they suggest that FKBP12E31Q/D32N/W59F retains high affinity for both RyR1 and RyR2 because the effects of adding FKBP12E31Q/D32N/W59F to the cytosolic chamber are irreversible after washout (see Fig. 6, A and C), and preaddition of FKBP12E31Q/D32N/W59F prevents FKBP12 from activating RyR2 (data not shown). Indeed, this same triple FKBP12 mutant has previously been shown to be capable of displacing 35S-labeled FKBPs bound to skeletal and cardiac SR vesicles indicating that high affinity binding to RyR1 and RyR2 is retained (29). The FKBP12 single point mutants E31Q, D32N, and W59F also bind tightly to rabbit skeletal RyR1 as evidenced by coimmunoprecipitation of GST-mutant FKBP12 and RyR1 (20). Crystallographic data show that Trp59 in FKBP12 and Phe59 in FKBP12.6 are located within the rapamycin hydrophobic binding pocket. This region is also suggested to provide the hydrophobic cavity involved in binding RyR channels (37,38,54,55). Mutations at position 59 in FKBP12/FKBP12.6 clearly do not abolish the binding of FKBPs to RyRs (20,29), although some change in affinity is indicated (29). The larger amino acid in FKBP12 does not alter the fold of the protein but it could influence the particular interactions between FKBPs and RyR2 that lead to changes in channel gating.
The residues in position 31 and 32 point away from the hydrophobic core of the proteins, and there are no structural differences between FKBP12 and FKBP12.6 in this region, however these residues could be relevant for electrostatic reasons, as FKBP12 carries two negative charges very close to each other, which are not present in FKBP12.6. A recent publication (39) suggests involvement of electrostatic charges in the interactions between FKBP12 and RyR1 and the formation of a salt bridge between Asp32 on FKBP12 and Arg976 on RyR1, which stabilizes the binding. If this hypothesis is correct, the mutations E31Q and D32N (where two negative charges in FKBP12 are substituted by the corresponding neutral residues of FKBP12.6), could lead to a critical change in protein function as these residues may be crucial for producing the precise binding interactions that enable FKBP12 to inhibit RyR1 and activate RyR2.
Physiological and pathophysiological perspectives
Our study calls for a fresh evaluation of the roles of FKBPs in cardiac and skeletal muscle and, in fact, in all tissues where RyR1 or RyR2 are present. It has generally been assumed that FKBP12 is the only relevant physiological regulator of RyR1, whereas FKBP12.6 is the only important isoform that influences the function of cardiac muscle and other tissues where RyR2 is expressed. Because our experiments clearly demonstrate that both isoforms of FKBP can modulate both isoforms of RyR at extremely low concentrations, and since both FKBP12 and FKBP12.6 are present in skeletal and cardiac cells (12,27,28), SR Ca2+-release in cardiac and skeletal muscle may depend on the competitive, dual regulation by both FKBP12 and FKBP12.6. The literature reports levels of 1–3 μM FKBP12 in cardiac and skeletal muscle but much lower (100–200 nM (56)) or undetectable (12,27,28) levels of FKBP12.6. Regarding RyR2, it now appears that canine RyR2 is somewhat of an outlier in having particularly high affinity for FKBP12.6 (41). For all other species of RyR2 investigated, RyR2 appears to also have high affinity for FKBP12 (15,41). In sheep, mass spectrometry demonstrated that FKBP12 can be detected in membrane fractions containing RyR2 with high confidence, whereas FKBP12.6 can only be detected with low confidence (15). Recent immunoblot analysis also showed that FKBP12 is present in cardiac cells (and in membrane fractions containing RyR2) of three mammalian species (pig, rabbit, mouse) at much higher levels than FKBP12.6 (34). In skeletal muscle, there is evidence that FKBP12 is more likely to be associated with RyR1 than FKBP12.6 (34,57). However, because FKBP12.6 is present in skeletal muscle and because we show a functional effect of FKBP12.6 at very low concentrations, we conclude that FKBP12.6 may also play a role in regulating RyR1 in skeletal muscle.
It is clear that the relative level of expression of FKBP12 and FKBP12.6 is important. In this regard, it is notable that FKBP binding to RyRs has been reported to be reduced in certain skeletal muscle pathologies (2,3,5). This may reflect an altered ratio in the expression levels of FKBP12 and FKBP12.6 or could be due to changes to RyR1 that affect its relative affinity for FKBP12/FKBP12.6. For example, a reduction in RyR1/FKBP12 binding as reported for dystrophic muscle (5) and sarcopenia (58,59), would be expected to lead to increased RyR1 Po due to dissociation of FKBP12 (which reduces RyR1 Po) and the possible increased binding by FKBP12.6 (which increases RyR1 Po). Fig. 8 suggests how the dual regulation of SR Ca2+-release by FKBP12 and FKBP12.6 may operate in skeletal muscle under physiological and pathophysiological conditions.
Figure 8.
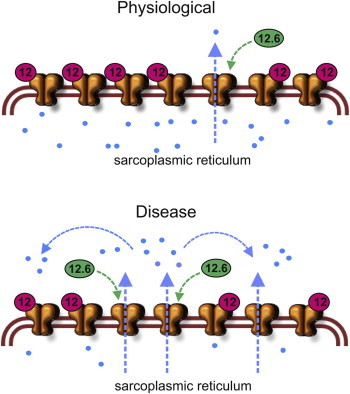
Proposed model of FKBP regulation of SR Ca2+ release in skeletal muscle. Under normal conditions, evidence suggests that RyR1 is predominantly bound by FKBP12, which maintains RyR1 in a low Po mode. The occasional binding of FKBP12.6 to RyR1 would lead to a minority of channels with increased Po. In disease or aging, it is reported that less FKBP12 is associated with RyR1. We speculate that a greater fraction of RyR1 channels may be bound by FKBP12.6, which would increase the numbers of leaky RyR1 channels. To see this figure in color, go online.
Our study has clear implications for other tissues where both RyRs and FKBPs are expressed. We should not assume that FKBP12 will exclusively bind to RyR1 or that FKBP12.6 will only bind to RyR2, rather, that both proteins can interact with either RyR isoform and that the absolute binding stoichiometry may vary from tissue to tissue. There are many reports indicating that FKBPs play an important role in intracellular Ca2+ release in a wide range of different tissues and cell types. These include bladder smooth muscle (60), pancreatic β-cells (61), vascular smooth muscle (62), endothelial cells (63), and various neuronal cells (64). Changes to the relative FKBP12/FKBP12.6 expression level in these cells may have important pathological implications.
Conclusion
We have shown that RyR1 is activated by FKBP12.6 but inhibited by FKBP12 and use of the FKBP12E31Q/D32N/W59F mutant demonstrates that efficacy is controlled by very small changes in FKBP structure. The experiments highlight the need to understand the functional consequences of FKBP12/FKBP12.6 binding to RyR channels at the single-channel level to recognize the putative physiological roles of FKBP/RyR interactions in different cell types and the changes that occur in disease and aging.
Acknowledgments
We thank Dr Kate Heesom for help and advice with the identification of FKBP proteins.
This work was funded by the British Heart Foundation.
Footnotes
Elisa Venturi and Elena Galfré contributed equally to this work.
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Marx S.O., Reiken S., Marks A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 2.Wehrens X.H., Lehnart S.E., Marks A.R. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc. Natl. Acad. Sci. USA. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reiken S., Lacampagne A., Marks A.R. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J. Cell Biol. 2003;160:919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellinger A.M., Mongillo M., Marks A.R. Stressed out: the skeletal muscle ryanodine receptor as a target of stress. J. Clin. Invest. 2008;118:445–453. doi: 10.1172/JCI34006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellinger A.M., Reiken S., Marks A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prestle J., Janssen P.M., Hasenfuss G. Overexpression of FK506-binding protein FKBP12.6 in cardiomyocytes reduces ryanodine receptor-mediated Ca(2+) leak from the sarcoplasmic reticulum and increases contractility. Circ. Res. 2001;88:188–194. doi: 10.1161/01.res.88.2.188. [DOI] [PubMed] [Google Scholar]
- 7.Yano M., Kobayashi S., Matsuzaki M. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107:477–484. doi: 10.1161/01.cir.0000044917.74408.be. [DOI] [PubMed] [Google Scholar]
- 8.Gellen B., Fernández-Velasco M., Mercadier J.-J. Conditional FKBP12.6 overexpression in mouse cardiac myocytes prevents triggered ventricular tachycardia through specific alterations in excitation-contraction coupling. Circulation. 2008;117:1778–1786. doi: 10.1161/CIRCULATIONAHA.107.731893. [DOI] [PubMed] [Google Scholar]
- 9.Brillantes A.B., Ondrias K., Marks A.R. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 10.Marx S.O., Ondrias K., Marks A.R. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors) Science. 1998;281:818–821. doi: 10.1126/science.281.5378.818. [DOI] [PubMed] [Google Scholar]
- 11.Kaftan E., Marks A.R., Ehrlich B.E. Effects of rapamycin on ryanodine receptor/Ca(2+)-release channels from cardiac muscle. Circ. Res. 1996;78:990–997. doi: 10.1161/01.res.78.6.990. [DOI] [PubMed] [Google Scholar]
- 12.Barg S., Copello J.A., Fleischer S. Different interactions of cardiac and skeletal muscle ryanodine receptors with FK-506 binding protein isoforms. Am. J. Physiol. 1997;272:C1726–C1733. doi: 10.1152/ajpcell.1997.272.5.C1726. [DOI] [PubMed] [Google Scholar]
- 13.Stewart R., Song L., Sitsapesan R. Single-channel characterization of the rabbit recombinant RyR2 reveals a novel inactivation property of physiological concentrations of ATP. J. Membr. Biol. 2008;222:65–77. doi: 10.1007/s00232-008-9102-z. [DOI] [PubMed] [Google Scholar]
- 14.Xiao J.M., Tian X.X., Chen S.R.W. Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J. Biol. Chem. 2007;282:34828–34838. doi: 10.1074/jbc.M707423200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galfré E., Pitt S.J., Sitsapesan R. FKBP12 activates the cardiac ryanodine receptor Ca2+-release channel and is antagonized by FKBP12.6. PLoS ONE. 2012;7:e31956. doi: 10.1371/journal.pone.0031956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shou W., Aghdasi B., Matzuk M.M. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 17.Avila G., Lee E.H., Dirksen R.T. FKBP12 binding to RyR1 modulates excitation-contraction coupling in mouse skeletal myotubes. J. Biol. Chem. 2003;278:22600–22608. doi: 10.1074/jbc.M205866200. [DOI] [PubMed] [Google Scholar]
- 18.Eltit J.M., Feng W., Perez C.F. Ablation of skeletal muscle triadin impairs FKBP12/RyR1 channel interactions essential for maintaining resting cytoplasmic Ca2+ J. Biol. Chem. 2010;285:38453–38462. doi: 10.1074/jbc.M110.164525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eltit J.M., Szpyt J., Perez C.F. Reduced gain of excitation-contraction coupling in triadin-null myotubes is mediated by the disruption of FKBP12/RyR1 interaction. Cell Calcium. 2011;49:128–135. doi: 10.1016/j.ceca.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee E.H., Rho S.-H., Kim H. N-terminal region of FKBP12 is essential for binding to the skeletal ryanodine receptor. J. Biol. Chem. 2004;279:26481–26488. doi: 10.1074/jbc.M309574200. [DOI] [PubMed] [Google Scholar]
- 21.Seidler T., Teucher N., Maier L.S. Limitations of FKBP12.6-directed treatment strategies for maladaptive cardiac remodeling and heart failure. J. Mol. Cell. Cardiol. 2011;50:33–42. doi: 10.1016/j.yjmcc.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Huang F., Shan J., Marks A.R. Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: rescue of heart failure by calstabin2 in mice. Proc. Natl. Acad. Sci. USA. 2006;103:3456–3461. doi: 10.1073/pnas.0511282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sitsapesan R., Montgomery R.A.P., Williams A.J. Sheep cardiac sarcoplasmic reticulum calcium-release channels: modification of conductance and gating by temperature. J. Physiol. 1991;434:469–488. doi: 10.1113/jphysiol.1991.sp018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colquhoun D., Sigworth F.J. Fitting and statistical analysis of single-channel recording. In: Sakmann B., Neher E., editors. Single-Channel Recording. Plenum; New York & London: 1983. pp. 191–263. [Google Scholar]
- 25.Saftenku E., Williams A.J., Sitsapesan R. Markovian models of low and high activity levels of cardiac ryanodine receptors. Biophys. J. 2001;80:2727–2741. doi: 10.1016/S0006-3495(01)76241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Armisen R., Sierralta J., Suarez-Isla B.A. Modal gating in neuronal and skeletal muscle ryanodine-sensitive Ca2+ release channels. Am. J. Physiol. 1996;271:C144–C153. doi: 10.1152/ajpcell.1996.271.1.C144. [DOI] [PubMed] [Google Scholar]
- 27.Lam E., Martin M.M., Wiederrecht G.J. A novel FK506 binding protein can mediate the immunosuppressive effects of FK506 and is associated with the cardiac ryanodine receptor. J. Biol. Chem. 1995;270:26511–26522. doi: 10.1074/jbc.270.44.26511. [DOI] [PubMed] [Google Scholar]
- 28.Timerman A.P., Onoue H., Fleischer S. Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J. Biol. Chem. 1996;271:20385–20391. doi: 10.1074/jbc.271.34.20385. [DOI] [PubMed] [Google Scholar]
- 29.Xin H.B., Rogers K., Fleischer S. Three amino acid residues determine selective binding of FK506-binding protein 12.6 to the cardiac ryanodine receptor. J. Biol. Chem. 1999;274:15315–15319. doi: 10.1074/jbc.274.22.15315. [DOI] [PubMed] [Google Scholar]
- 30.Timerman A.P., Ogunbumni E., Fleischer S. The calcium release channel of sarcoplasmic reticulum is modulated by FK-506-binding protein. Dissociation and reconstitution of FKBP-12 to the calcium release channel of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1993;268:22992–22999. [PubMed] [Google Scholar]
- 31.Timerman A.P., Wiederrecht G., Fleischer S. Characterization of an exchange reaction between soluble FKBP-12 and the FKBP.ryanodine receptor complex. Modulation by FKBP mutants deficient in peptidyl-prolyl isomerase activity. J. Biol. Chem. 1995;270:2451–2459. doi: 10.1074/jbc.270.6.2451. [DOI] [PubMed] [Google Scholar]
- 32.Ahern G.P., Junankar P.R., Dulhunty A.F. Single channel activity of the ryanodine receptor calcium release channel is modulated by FK-506. FEBS Lett. 1994;352:369–374. doi: 10.1016/0014-5793(94)01001-3. [DOI] [PubMed] [Google Scholar]
- 33.Ahern G.P., Junankar P.R., Dulhunty A.F. Subconductance states in single-channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys. J. 1997;72:146–162. doi: 10.1016/S0006-3495(97)78654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zissimopoulos S., Seifan S., Lai F.A. Disparities in the association of the ryanodine receptor and the FK506-binding proteins in mammalian heart. J. Cell Sci. 2012;125:1759–1769. doi: 10.1242/jcs.098012. [DOI] [PubMed] [Google Scholar]
- 35.Ahern G.P., Junankar P.R., Dulhunty A.F. Ryanodine receptors from rabbit skeletal muscle are reversibly activated by rapamycin. Neurosci. Lett. 1997;225:81–84. doi: 10.1016/s0304-3940(97)00193-6. [DOI] [PubMed] [Google Scholar]
- 36.Ahern G.P., Junankar P.R., Dulhunty A.F. Effects of ivermectin and midecamycin on ryanodine receptors and the Ca2+-ATPase in sarcoplasmic reticulum of rabbit and rat skeletal muscle. J. Physiol. 1999;514:313–326. doi: 10.1111/j.1469-7793.1999.313ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagenknecht T., Radermacher M., Fleischer S. Locations of calmodulin and FK506-binding protein on the three-dimensional architecture of the skeletal muscle ryanodine receptor. J. Biol. Chem. 1997;272:32463–32471. doi: 10.1074/jbc.272.51.32463. [DOI] [PubMed] [Google Scholar]
- 38.Sharma M.R., Jeyakumar L.H., Wagenknecht T. Three-dimensional visualization of FKBP12.6 binding to an open conformation of cardiac ryanodine receptor. Biophys. J. 2006;90:164–172. doi: 10.1529/biophysj.105.063503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu L., Zhong X., Liu Z. Modeling a ryanodine receptor N-terminal domain connecting the central vestibule and the corner clamp region. J. Biol. Chem. 2013;288:903–914. doi: 10.1074/jbc.M112.429670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cornea R.L., Nitu F.R., Fruen B.R. Mapping the ryanodine receptor FK506-binding protein subunit using fluorescence resonance energy transfer. J. Biol. Chem. 2010;285:19219–19226. doi: 10.1074/jbc.M109.066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeyakumar L.H., Ballester L., Fleischer S. FKBP binding characteristics of cardiac microsomes from diverse vertebrates. Biochem. Biophys. Res. Commun. 2001;281:979–986. doi: 10.1006/bbrc.2001.4444. [DOI] [PubMed] [Google Scholar]
- 42.Chu A., Fill M., Entman M.L. Cytoplasmic Ca2+ does not inhibit the cardiac muscle sarcoplasmic reticulum ryanodine receptor Ca2+ channel, although Ca2+-induced Ca2+ inactivation of Ca2+ release is observed in native vesicles. J. Membr. Biol. 1993;135:49–59. doi: 10.1007/BF00234651. [DOI] [PubMed] [Google Scholar]
- 43.Laver D.R., Roden L.D., Dulhunty A.F. Cytoplasmic Ca2+ inhibits the ryanodine receptor from cardiac muscle. J. Membr. Biol. 1995;147:7–22. doi: 10.1007/BF00235394. [DOI] [PubMed] [Google Scholar]
- 44.Copello J.A., Barg S., Fleischer S. Heterogeneity of Ca2+ gating of skeletal muscle and cardiac ryanodine receptors. Biophys. J. 1997;73:141–156. doi: 10.1016/S0006-3495(97)78055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sitsapesan R., Williams A.J. Mechanisms of caffeine activation of single calcium-release channels of sheep cardiac sarcoplasmic reticulum. J. Physiol. 1990;423:425–439. doi: 10.1113/jphysiol.1990.sp018031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reference deleted in proof.
- 47.Sitsapesan R., Williams A.J. Modification of the conductance and gating properties of ryanodine receptors by suramin. J. Membr. Biol. 1996;153:93–103. doi: 10.1007/s002329900113. [DOI] [PubMed] [Google Scholar]
- 48.Kermode H., Williams A.J., Sitsapesan R. The interactions of ATP, ADP, and inorganic phosphate with the sheep cardiac ryanodine receptor. Biophys. J. 1998;74:1296–1304. doi: 10.1016/S0006-3495(98)77843-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith J.S., Coronado R., Meissner G. Single channel measurements of the calcium release channel from skeletal muscle sarcoplasmic reticulum. Activation by Ca2+ and ATP and modulation by Mg2+ J. Gen. Physiol. 1986;88:573–588. doi: 10.1085/jgp.88.5.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao R.P., Valdivia H.H., Cheng H. The immunophilin FK506-binding protein modulates Ca2+ release channel closure in rat heart. J. Physiol. 1997;500:343–354. doi: 10.1113/jphysiol.1997.sp022025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lamb G.D., Stephenson D.G. Effects of FK506 and rapamycin on excitation-contraction coupling in skeletal muscle fibres of the rat. J. Physiol. 1996;494:569–576. doi: 10.1113/jphysiol.1996.sp021514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.duBell W.H., Wright P.A., Rogers T.B. Effect of the immunosuppressant FK506 on excitation-contraction coupling and outward K+ currents in rat ventricular myocytes. J. Physiol. 1997;501:509–516. doi: 10.1111/j.1469-7793.1997.509bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sigalas C., Mayo-Martin M.B., Sitsapesan R. Ca2+-calmodulin increases RyR2 open probability yet reduces ryanoid association with RyR2. Biophys. J. 2009;97:1907–1916. doi: 10.1016/j.bpj.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samsó M., Shen X.H., Allen P.D. Structural characterization of the RyR1-FKBP12 interaction. J. Mol. Biol. 2006;356:917–927. doi: 10.1016/j.jmb.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 55.Fulton K.F., Jackson S.E., Buckle A.M. Energetic and structural analysis of the role of tryptophan 59 in FKBP12. Biochemistry. 2003;42:2364–2372. doi: 10.1021/bi020564a. [DOI] [PubMed] [Google Scholar]
- 56.Guo T., Cornea R.L., Bers D.M. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ. Res. 2010;106:1743–1752. doi: 10.1161/CIRCRESAHA.110.219816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carmody M., Mackrill J.J., O’Neill C. FKBP12 associates tightly with the skeletal muscle type 1 ryanodine receptor, but not with other intracellular calcium release channels. FEBS Lett. 2001;505:97–102. doi: 10.1016/s0014-5793(01)02787-9. [DOI] [PubMed] [Google Scholar]
- 58.Andersson D.C., Betzenhauser M.J., Marks A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14:196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Russ D.W., Grandy J.S., Ward C.W. Ageing, but not yet senescent, rats exhibit reduced muscle quality and sarcoplasmic reticulum function. Acta Physiol. (Oxf.) 2011;201:391–403. doi: 10.1111/j.1748-1716.2010.02191.x. [DOI] [PubMed] [Google Scholar]
- 60.Zheng J., Wenzhi B., Ji G. Ca(2+) release induced by cADP-ribose is mediated by FKBP12.6 proteins in mouse bladder smooth muscle. Cell Calcium. 2010;47:449–457. doi: 10.1016/j.ceca.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 61.Noguchi N., Yoshikawa T., Sugawara A. FKBP12.6 disruption impairs glucose-induced insulin secretion. Biochem. Biophys. Res. Commun. 2008;371:735–740. doi: 10.1016/j.bbrc.2008.04.142. [DOI] [PubMed] [Google Scholar]
- 62.Georgeon-Chartier C., Menguy C., Morel J.-L. Effect of aging on calcium signaling in C57Bl6J mouse cerebral arteries. Pflugers Arch. - Eur. J. Physiol. 2013;465:829–838. doi: 10.1007/s00424-012-1195-7. [DOI] [PubMed] [Google Scholar]
- 63.Long C., Cook L.G., Mitchell B.M. Removal of FKBP12/12.6 from endothelial ryanodine receptors leads to an intracellular calcium leak and endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2007;27:1580–1586. doi: 10.1161/ATVBAHA.107.144808. [DOI] [PubMed] [Google Scholar]
- 64.Suzuki M., Nagai Y., Koike T. Calcium leak through ryanodine receptor is involved in neuronal death induced by mutant huntingtin. Biochem. Biophys. Res. Commun. 2012;429:18–23. doi: 10.1016/j.bbrc.2012.10.107. [DOI] [PubMed] [Google Scholar]
- 65.Sigworth F.J., Sine S.M. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys. J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.