

Abstract
Despite the importance of G-protein coupled receptors (GPCRs) their biogenesis is poorly understood. Like vertebrates, C. elegans uses a large family of GPCRs as chemoreceptors. A subset of these receptors, such as ODR-10, requires the odr-4 and odr-8 genes to be appropriately localized to sensory cilia. The odr-4 gene encodes a conserved tail-anchored transmembrane protein; the molecular identity of odr-8 is unknown. Here, we show that odr-8 encodes the C. elegans ortholog of Ufm1-specific protease 2 (UfSP2). UfSPs are cysteine proteases identified biochemically by their ability to liberate the ubiquitin-like modifier Ufm1 from its pro-form and protein conjugates. ODR-8/UfSP2 and ODR-4 are expressed in the same set of twelve chemosensory neurons, and physically interact at the ER membrane. ODR-4 also binds ODR-10, suggesting that an ODR-4/ODR-8 complex promotes GPCR folding, maturation, or export from the ER. The physical interaction between human ODR4 and UfSP2 suggests that this complex's role in GPCR biogenesis may be evolutionarily conserved. Unexpectedly, mutant versions of ODR-8/UfSP2 lacking catalytic residues required for protease activity can rescue all odr-8 mutant phenotypes tested. Moreover, deleting C. elegans ufm-1 does not alter chemoreceptor traffic to cilia, either in wild type or in odr-8 mutants. Thus, UfSP2 proteins have protease- and Ufm1-independent functions in GPCR biogenesis.
Author Summary
Despite the importance of G-protein coupled receptors (GPCRs), we know little about their biogenesis. Olfactory receptors form a large and divergent group of GPCRs. We investigate their biogenesis in C. elegans. We show that maturation of a subset of these GPCRs, including the diacetyl receptor ODR-10, requires Ufm1 specific protease 2 (UfSP2), which corresponds to odr-8. Biochemical studies suggest mouse UfSP2 activates the Ubiquitin-like molecule Ufm1 and cleaves it from protein conjugates. However, neither the protease active site nor ufm-1 is required for UfSP2/ODR-8 to promote ODR-10 maturation. C. elegans UfSP2 is expressed in the same chemosensory neurons as ODR-4, a tail-anchored transmembrane protein also required for ODR-10 maturation. ODR-4 resides in the endoplasmic reticulum (ER); UfSP2 is cytosolic but associates with ER membranes. In odr-4 and odr-8 mutants ODR-10-GFP is retained in the ER, suggesting these genes are required to fold GPCRs or traffic them out of the ER. ODR-4 interacts biochemically with ODR-8 and ODR-10 to form an ER complex. ODR-4 and UfSP2 are conserved from plants to man, and human ODR4 can bind human UfSP2 and recruit it to ER membranes. Both proteins are expressed widely in mammals, suggesting a broader role in GPCR biogenesis.
Introduction
Molecular chaperones ensure the correct folding, assembly, quality control, traffic, and sub-cellular targeting of newly made proteins. Failure of these processes results in protein aggregation, with potential pathological consequences [1]. In the nervous system a growing number of chaperones have been identified that are specialized to facilitate the biogenesis of specific molecules [2]–[3]. Together with general chaperones, these molecules prevent accumulation of protein aggregates in neurons and provide protection against neurodegeneration [4].
GPCRs form a large family of polytopic transmembrane proteins that share a common fold [5]. Despite their importance, the biogenesis of GPCRs is poorly understood. GPCRs are co-translationally targeted to and inserted into the endoplasmic reticulum (ER) membrane using the canonical translocon machinery [6]. In the ER, GPCRs are often N-glycosylated and fold with the help of chaperones [7]–[9]. General chaperones implicated in GPCR folding include calnexin and calreticulin. More specific chaperones have also been identified for a few receptors, including RTP1 and RTP2 for some mammalian olfactory receptors [10], XPORT and NinaA for fly rhodopsin [11], and DRiP78 for the D1 dopamine receptor [12]. GPCR assembly in the ER is monitored by poorly defined quality control systems, and improperly folded receptors are targeted for refolding or degradation by the ubiquitin-proteasome system [9]. Molecules passing ER quality control are exported to downstream compartments of the secretory pathway. Motifs involved in GPCR ER export have been defined for some receptors, [12], although some of these may be involved in correct GPCR folding, a pre-requisite for export [13]. In the Golgi and trans-Golgi network GPCRs are further glycosylated, and assembled into vesicles appropriate for traffic to their appropriate sub-cellular localization. In neurons these destinations can include dendrites, axons or synapses. While the molecular machineries mediating many steps in GPCR biogenesis are unknown, it is clear that ER retention of GPCRs due to misfolding leads to disease, including retinitis pigmentosa [14], nephrogenic diabetes insipidus [15], and hypogonadotropic hypogonadism [16].
In the nematode C. elegans, like in vertebrates, GPCRs play central roles in chemoreception [17]–[19]. The C. elegans genome encodes more than 1300 predicted GPCR chemoreceptors of widely divergent sequence [18]. Many of these GPCRs appear to be expressed in one or more of 12 gustatory and olfactory neuron types [17]. The most intensively studied of these receptors, ODR-10, mediates C. elegans chemotaxis to the volatile odor diacetyl. ODR-10 is specifically expressed in the AWA olfactory neuron, and localizes to sensory cilia [19]. Screens for odortaxis defective mutants have identified two loci that disrupt ODR-10 localization to cilia, odr-4 and odr-8 [20]. In these mutants ODR-10-GFP is retained in endomembranes in the cell body. odr-4 and odr-8 are only required for localization of a subset of GPCR chemoreceptors, and do not affect localization of other signal transduction components, such as G-proteins and ion channels, to cilia [20]. These genetic data suggest that odr-4 and odr-8 encode accessory proteins involved in the maturation, traffic or localization of GPCRs.
As well as being required for olfactory responses, odr-4 and odr-8 are required for npr-1 animals to aggregate efficiently [21] and to respond to variation in ambient oxygen [22]. Our interest in the molecular basis of aggregation behaviour motivated us to understand ODR-4 and ODR-8. Earlier work had established that odr-4 encodes a tail-anchored transmembrane protein selectively expressed in 12 C. elegans chemosensory neurons and localized to unidentified intracellular membrane compartments [20]. The molecular identity of odr-8 has remained unknown. Here, we identify the gene encoded by odr-8 and provide a characterization of its relationship to odr-4 and its role in GPCR biogenesis.
Results
Mutants defective in UFM1-specific protease 2 exhibit behavioral defects
We mutagenized npr-1 animals, isolated mutants that failed to aggregate, and sequenced their genomes. At the same time we sequenced two strains bearing alleles of odr-8, ky173 and ky31, which we previously showed disrupt aggregation behavior [21]. Genome sequence analysis identified only one open reading frame that had lesions in ky31 and ky173 alleles and mapped to the interval where odr-8 maps genetically [20]. Three other aggregation-defective strains in our sequenced collection also had mutations in this gene, which is called F38A5.1. Of the five mutations in F38A5.1, three introduced stop codons (Figure 1A).
Figure 1. odr-8 mutants are defective in Ufm1 specific protease 2.

(A) The UfSP2 gene corresponds to ORF F38A5.1. Shown are the exon/intron structure of the gene and the location of the Cysteine (blue), Histidine (green) and Aspartate (pink) boxes containing catalytic residues in the papain fold. The location of stop codons associated with the db866, ky173 and ky31 mutations is also indicated. (B–D). A transgene carrying F38A5.1a genomic fragment rescues odr-8 defects in olfaction (B), aggregation (C), and responses to changes in O2 (D). **, p<0.01 compared to non-transgenic controls (ANOVA, Bonferroni's multiple comparisons test).
To establish if defects in F38A5.1 caused odr-8 phenotypes we carried out transgenic rescue experiments. Genomic DNA bearing the F38A5.1 open reading frame rescued odr-8 defects in odortaxis, aggregation behavior, and responses to oxygen (Figure 1B–D), confirming the molecular identity of odr-8.
F38A5.1 encodes the C. elegans ortholog of Ufm1-specific protease 2 (UfSP2). Ufm1 (Ubiquitin fold modifier) is a ubiquitin-like post-translational modifier [23]. Its tertiary structure is very similar to that of ubiquitin (Ub) although its primary sequence shows no obvious sequence similarity to Ub [24]. Like Ub, Ufm1 is synthesized as an inactive precursor that is proteolytically cleaved to expose a glycine C-terminal residue. This glycine can then be conjugated to target proteins at lysine residues, although only one Ufm1 target has been identified to date, the ER protein C20orf116 [25]. In vitro studies show that mouse UfSP2 can both activate pro-Ufm1 and cleave it from native protein conjugates, a process termed de-ufmylation (Figure S1) [25]–[27]. Ufm1 and UfSP2 are highly conserved across eukaryotes (Figure S1B and Figure S2), but their in vivo roles are unknown.
ODR-8/UfSP2 is co-expressed with ODR-4 in chemosensory neurons
To determine where ODR-8/UfSP2 is expressed, we fused DNA encoding the fluorescent protein mCherry upstream of the odr-8 open reading frame and expressed this transgene from the odr-8 promoter. The construct rescued all the defects observed in odr-8 mutants, and revealed mCherry-ODR-8 expression in a small number of head neurons and two tail neurons.
The phenotypes of odr-8 mutants closely resemble those of odr-4 defective animals [20]. ODR-4 is a conserved tail-anchored transmembrane protein (Figure S3). To examine if odr-4 or odr-8 were expressed in the same cells, we made animals that carried both podr-4::odr-4::gfp and podr-8::mCherry::odr-8 transgenes. Green and red fluorescence in these animals overlapped, consistent with ODR-4 and ODR-8 functioning in the same neurons (Figure 2A). We explicitly identified odr-8 expressing neurons using the stereotyped position of C. elegans neurons and DiO dye-filling and a podr-3::gfp transgene as fiduciary markers. We observed mcherry-ODR-8 expression in 10 head neurons, the amphid neurons ADL, ASI, ASH, ASJ, ASG, ADF, ASK, AWA, AWB, AWC, and in two tail neurons, the phasmid neurons PHA and PHB. These are all the cells previously reported to express odr-4 [20]. We did not detect expression in any additional neurons.
Figure 2. ODR-8 is co-expressed with ODR-4 in chemosensory neurons.
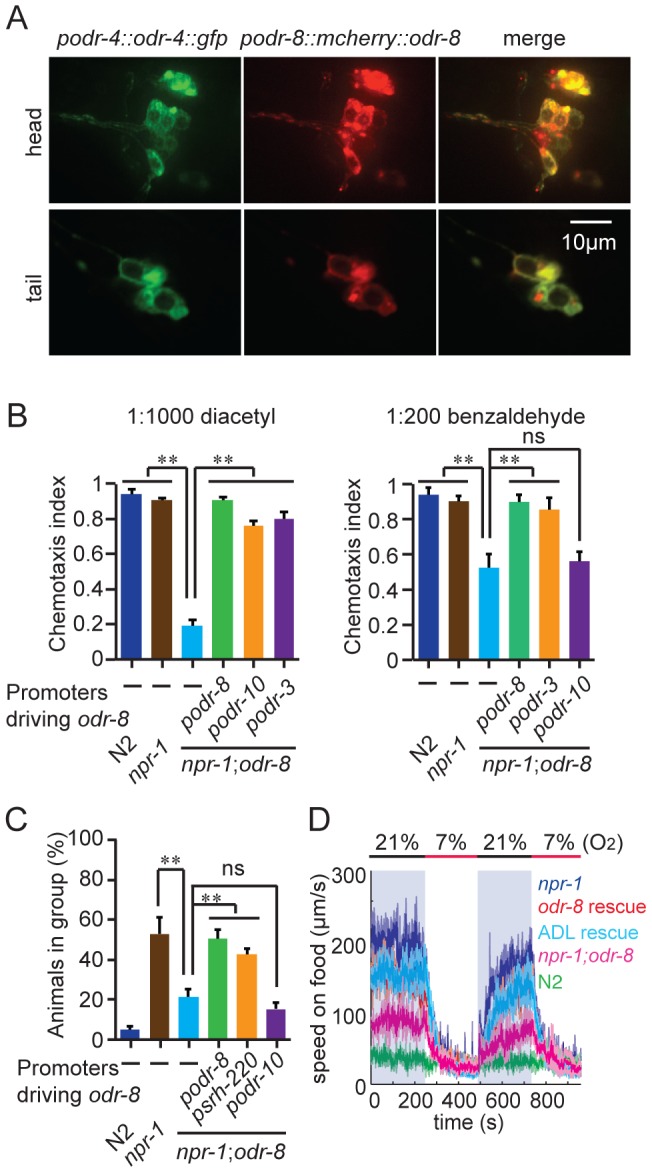
(A) ODR-4 and ODR-8 are co-expressed in the same head and tail neurons. (B–D) Cell-specific rescue of odr-8(ky31) phenotypes. Like ODR-4, ODR-8 is required in AWA neurons for odortaxis to diacetyl (B), and in ADL neurons to sustain aggregation behavior (C) and high locomotory activity in 21% oxygen (D). The podr-10 promoter drives expression only in the AWA neurons; psrh-220 drives expression in ADL neurons; podr-3 drives expression in AWA, AWB, AWC, ASH and ADF. ** p<0.01 (ANOVA, Bonferroni's multiple comparisons test) compared to non-transgenic controls.
ODR-4 functions in the AWA neurons to promote chemotaxis to the odor diacetyl [20], and in the ADL neurons to promote aggregation [21]. Expressing odr-8 specifically in AWA neurons, using the odr-10 promoter, rescued odr-8 odortaxis to diacetyl but not to benzaldehyde, which is sensed by the AWC neurons (Figure 2B). Similarly, expressing odr-8 in ADL, using the srh-220 promoter, restored aggregation behavior (Figure 2C) and oxygen responses (Figure 2D). Expressing odr-8 from the odr-3 promoter, which drives expression in the AWC olfactory neuron and a small number of other neurons including AWA, rescued odr-8 chemotaxis to both benzaldehyde and diacetyl (Figure 2B). These data indicate that ODR-8 and ODR-4 function in the same neurons to promote specific behavioral responses.
Olfactory receptors are retained in the ER in odr-4 and odr-8
The behavioral defects of odr-8 and odr-4 mutants appear to reflect failure in the localization of a subset of G-protein coupled receptors (GPCR) to sensory cilia [20]. ODR-10 is an olfactory GPCR that mediates attraction to diacetyl [19]. In wild type animals ODR-10-GFP is localized to the sensory cilia of AWA olfactory neurons. In odr-4 or odr-8 mutants, ODR-10-GFP is predominantly found in unidentified endomembrane compartments [20]. We observed similar defects when we compared localization of two other GPCRs of the STR olfactory receptor family, STR-112 and STR-113, in the AWA neurons of wild type and odr-8 mutants (Figure 3A–B and Figure S4).
Figure 3. ODR-8 is required for the GPCR trafficking to the AWA cilia.

(A) Schematic drawing of the C. elegans head with the arrow indicating the AWA cell body and the arrowhead pointing to the AWA cilia. (B) ODR-8 is required to localize GFP-tagged STR-112 and STR-113 odorant receptors to AWA cilia. Cilia localization in wild type and odr-8 mutants is quantified to the right of the panel. (C) ODR-8 acts cell-autonomously to permit localization of ODR-10-GFP to the cilia of the AWA neurons. **, p<0.01 (ANOVA, Bonferroni's multiple comparisons test).
To confirm that defects in C. elegans UfSP2 caused the ODR-10-GFP trafficking phenotype, we performed transgenic rescue experiments. ODR-10-GFP was correctly localized to AWA cilia in odr-8 mutants bearing the F38A5.1 transgene, whereas in non-transgenic siblings ODR-10-GFP was localized to the cell body (Figure 3C). Expressing the F38A5.1a open reading frame specifically in the AWA neurons restored cilia localization of ODR-10-GFP, whereas expression in the ADL neurons did not, suggesting that ODR-8 functions cell-autonomously (Figure 3C and data not shown).
The small size of C. elegans neurons makes sub-cellular localization of fluorescently-tagged proteins challenging. Nevertheless, to investigate the fate of ODR-10-GFP in odr-4 and odr-8 mutants, we co-localized it with mCherry-tagged markers for different membrane compartments that we expressed specifically in AWA (Figure 4A). We included in our studies the rough ER marker TRAM-1, the Golgi marker alpha-mannosidase II [28], the ER exit site markers SEC-16 and SEC-23 [29], the early endosome marker RAB-5 [30], the lysosomal marker LMP-1 [31] [32], and RAB-8 which in sensory neurons marks vesicles destined for traffic to the cilia [33]. As expected, in wild type animals, ODR-10-GFP was predominantly localized to cilia, with some fluorescence in the cell body (Figure 4A and Figure S5). The cell body fluorescence co-localized most extensively with mCherry-RAB-8, in foci that were closely apposed to Golgi (Figure 4A and Figure S5). We also observed some co-localization with the lysosomal marker LMP-1 (Figure 4A and Figure S5). Co-localization of ODR-10-GFP with mCherry-RAB-8 has been reported previously when the receptor was expressed heterologously in the AWB and phasmid neurons [33]. In odr-8 mutants co-localization of ODR-10-GFP and mCherry-RAB-8 was lost; instead ODR-10-GFP co-localized most extensively with markers for the ER (Figure 4A and Figure S5). These data suggest that in odr-8 mutants the ODR-10-GFP receptor either does not exit the ER, or undergoes retrograde traffic back to the ER, or is degraded following ER exit.
Figure 4. ODR-10-GFP is retained in the ER in odr-8 mutants.

(A) ODR-10-GFP co-localizes predominantly with RAB-8 and LMP-1 markers in wild type animals, but with ER markers in odr-8(ky31) mutants. (B–C) ODR-4 co-localizes with the ER marker TRAM-1 (B), and ODR-4 and ODR-8 partially co-localize with each other (C).
ODR-4 and ODR-8 proteins localize to the ER
We sought to establish the sub-cellular compartments to which ODR-4 and ODR-8 proteins predominantly localized to in AWA neurons. We made transgenes expressing ODR-4-GFP or mCherry-ODR-8 in AWA, and crossed them into animals in which different compartments were highlighted in AWA neurons using mCherry-tagged markers. ODR-4-GFP co-localized with the ER marker TRAM-1 (Figure 4B), suggesting ODR-4 functions in this compartment. mCherry-ODR-8 showed some co-localization with ODR-4, although we also observed diffuse staining consistent with cytoplasmic localization (Figure 4C).
Disrupting AP-1 does not alter ER retention of ODR-10-GFP in odr-8 mutants
We sought to design genetic experiments that shed light on where ODR-4 and ODR-8 function in the secretory pathway. Previous work indicates that post-Golgi sorting of ODR-10-GFP involves formation of clathrin-coated vesicles via adapter complex 1 (AP-1) [33]–[34]. Mutants lacking unc-101, which encodes the mu1 subunit of AP1, fail to traffic ODR-10-GFP to sensory cilia and instead traffic the receptor to the cell membrane. We reasoned that if odr-8 mutations disrupt ODR-10-GFP receptor maturation prior to its reaching the Golgi, then disrupting unc-101 should not alter ODR-10-GFP localization in odr-8 mutants. Consistent with this, we observed no difference in ODR-10-GFP localization between odr-8 and odr-8; unc-101 animals (Figure 5A–B).
Figure 5. ODR-8 acts in an early step in ODR-10 trafficking to the AWA cilia.

(A) Localization of ODR-10-GFP in AWA neurons of odr-8(ky31), unc-101(m1) and odr-8(ky31); unc-101(m1) mutants. Thresholded images were obtained as described in the methods. (B) Quantitative analysis of ODR-10 and TRAM-1 co-localization in AWA neurons of odr-8(ky31), unc-101(m1) and odr-8(ky31); unc-101(m1) mutants. (C). Localization of ODR-10-GFP in AWA neurons in rab-2 mutants. Arrowheads indicate the AWA cilia, white arrows point to the AWA cell body and red arrows show the AWA processes in the nerve ring. In rab-2(n777) mutants, 23 out of 29 animals displayed normal ODR-10-GFP localization in AWA cilia, and 6 out of 29 show the ODR-10-GFP localization defects in AWA cilia. (D–E). Quantification of ODR-10-GFP signals in the cilia (D) and axons (including cell body) (E) of AWA neurons. **, p<0.01 (ANOVA, Bonferroni's multiple comparisons test); for E, rab-2(n777) shows p<0.01 compared to all other genotypes plotted.
To extend our studies we imaged ODR-10-GFP localization in animals defective in or expressing dominant negative or dominant active version of the RAB GTPases RAB-1, RAB-2, RAB-6.1 and RAB-6.2. These RABs play roles at different steps in the secretory pathway [35], and some have previously been implicated in GPCR traffic [36]. In yeast, Rab1 is thought to be important for ER exit and traffic to the Golgi, Rab2 is implicated in retrograde traffic to the ER from the Golgi, and Rab6 plays a role in intra-Golgi traffic. For rab-1, rab-6.1 and rab-6.2 we expressed dominant negative and dominant active transgenes in AWA using the odr-10 promoter (see methods). For rab-2 we used the deletion allele nu415, which is predicted to be null, and the n777 allele, which is associated with a S149F mutation that constitutively activates RAB-2 [37]. None of the perturbations of RAB protein function completely blocked traffic of ODR-10-GFP to AWA cilia in wild type animals, although there were clear and significant reductions of ODR-10 cilia localization in all cases except when we expressed rab-6.1(Q70L) (Figure S6). In rab-2(n777) mutants, ODR-10 was not correctly localized to AWA cilia in 6 out of 29 animals (Figure 5C–E), and more strikingly, a significant amount of ODR-10 was mis-sorted to axons (Figure 5C–E). Previous work in C. elegans has implicated RAB-2 in biogenesis of dense core vesicles [37], [38] and in post-endocytic trafficking [39], suggesting a more complex role for this RAB protein. Axonal mislocalization of ODR-10 was not observed in rab-2(n777); odr-8 double mutants, consistent with odr-8 being required early in the secretory pathway (Figure 5C–E). For the remaining rab gene perturbations, odr-8; rab double mutants, and odr-8 animals expressing mutant rab transgenes resulted in an ODR-10-GFP localization phenotype that was indistinguishable from that of odr-8 mutants. Together, these observations and the unc-101 data suggest that ODR-8 functions at an early step in GPCR trafficking.
Disrupting ufm-1 does not alter ODR-10-GFP localization in AWA neurons
Biochemical studies indicate that mouse UfSP2 de-ufmylase can activate pro-Ufm1 and cleave it from native protein conjugates [26] [25]. We therefore examined whether disrupting the C. elegans orthologs of Uba5 or Ufc1, the only known E1 and E2 ligases implicated in ufmylation [23] (Figure S1), or deleting C. elegans ufm-1 itself, altered ODR-10 localization. C. elegans UBA-5 is encoded by T03F1.1 [40] whereas C. elegans UFC-1 is encoded by C40H1.6. UFM-1 is encoded by tag-277. Deletion mutations that are likely to be complete loss-of-function alleles are available for all three genes [40] (Figure S7A and Figure 6).
Figure 6. Deleting ufm-1 does not alter ODR-10 localization.

(A) Molecular nature of ufm-1(gk379) deletion allele. The gk379 deletion disrupts two adjacent operons – one comprising ufm-1 and tomm-7 and one including coq-5 and tag-307. gk379 homozygous animals die as embryos or young larvae. To score the ODR-10-GFP localization in adult animals lacking ufm-1 we rescued the gk379 lethal phenotype using a 9 kb genomic fragment in which the ufm-1 open reading frame is interrupted by a stop codon or by a combination of a stop codon and a frameshift mutation, as indicated. (B) Disrupting ufm-1 does not change the ODR-10-GFP phenotype either in wild type animals or odr-8(ky31) mutants. (C) Mutations generated by CRISPR-Cas in the ufm-1 gene. PAM refers to the protospacer adjacent motif. (D–E) Mutations in ufm-1 do not block ODR-10 trafficking to AWA cilia in wild type animals, or rescue the ODR-10 localization defect in an odr-8 mutant background. The ODR-10 cilia localization in (D) was quantified in (E).
If the mutant phenotypes of odr-8/ufsp2 reflected a failure to activate pro-ufm1, then disrupting ufmylation should recapitulate odr-8 phenotypes. Conversely, if the primary defect of odr-8/ufsp2 is failure to cleave UFM-1 from native protein conjugates, then disrupting ufm-1 or inhibiting ufmylation might suppress odr-8 phenotypes. Deleting Ce-uba-5 or Ce-ufc-1 did not affect viability, and had no visible effect on diacetyl chemotaxis or on ODR-10-GFP localization at AWA cilia in an otherwise wild type background (Figure S7B–C). No change in the phenotype of an odr-8 mutant was observed upon further deletion of Ce-uba-5 or Ce-ufc-1 (Figure S7B–C). The only available allele that deletes Ce-ufm-1, gk379, also removes part of an operon that includes the ubiquinone biosynthetic enzyme coq-5, and results in embryonic lethality (Figure 6A). We suspected that lethality reflected disruption of the coq-5 operon, rather than loss of ufm-1. Consistent with this, we could rescue the gk379 embryonic lethality with a 9.1 kb genomic fragment of the region in which we truncated the ufm-1 open reading frame by a premature stop codon or a frame shift mutation (Figure 6A). This allowed us to examine ODR-10-GFP localization at AWA cilia in adult animals lacking UFM-1. We did not observe any significant change in ODR-10-GFP localization, either in odr-8(+) or in odr-8(−) animals that also lacked ufm-1 (Figure 6B). Furthermore, we could not detect UFM-1 expression in AWA neurons (Figure S7D–E).
To confirm our observations, we generated three independent ufm-1 loss-of-function mutations using CRISPR-Cas gene-editing (Figure 6C) [41]. Two mutations caused frameshifts of the ufm-1 open reading frame, and presumably are null alleles (Figure 6C). None of the mutations disrupted ODR-10-GFP cilia localization in odr-8(+) animals, or rescued the ODR-10-GFP cilia localization defect in an odr-8 mutant background (Figure 6D–E). These data suggest that the primary role of ODR-8 UfSP2 in biogenesis of ODR-10-GFP is independent of ufmylation.
ODR-8 UfSP2 functions through a non-catalytic mechanism
Biochemistry and crystallography have highlighted the active sites of the UfSP1 and UfSP2 proteases [25]–[27]. Like in several de-ubiquitinases, the active sites of these enzymes include highly conserved cysteine and histidine boxes (Figure 7A). The cysteine box contains the catalytic cysteine residue that is thought to undergo de-protonation, enabling nucleophilic attack of the carbonyl carbon of incoming substrates. The histidine in the histidine box assists this de-protonation. In vitro biochemical studies show that mutating the active site cysteine residue to a serine, or the histidine to an alanine, abolishes UfSP1 and UfSP2 proteolytic activity [25], [27].
Figure 7. Non-catalytic action of ODR-8 Ufm1-specific protease 2.

(A) Conservation of Cysteine and Histidine boxes in UfSP2 from worm, mouse and man. The catalytic cysteine and histidine residues are marked by an arrowhead. (B–D) odr-8 transgenes in which the catalytic cysteine or histidine residues are mutated retain the ability to rescue odr-8(ky31) mutant phenotypes for odorant responses to diacetyl and benzaldehyde (B), oxygen responses (C) and biogenesis and localization of ODR-10 GPCR in AWA neurons (D). In D, ODR-10 localization in AWA cilia was scored using a scale from 0 to 4, where 0 reflects no observable localization and 4 reflects strong ODR-10 accumulation. Representative images for each genotype are shown. The odr-8 allele rescued in these assays was odr-8(ky31). ** indicates p<0.01 (ANOVA, Bonferroni's multiple comparisons test).
To examine if an intact active site was required for ODR-8/UfSP2 in vivo functions, we made a transgene encoding an ODR-8 in which the active site cysteine was mutated to a serine. Unexpectedly, this transgene rescued all the behavioral defects of odr-8 mutants we tested, and restored ODR-10-GFP localization to AWA cilia (Figure 7B–D). Since serine residues can engage in nucleophilic attack, we also created a transgene encoding an ODR-8 in which the active site cysteine was mutated to an alanine. This Cys-to-Ala active site mutant also rescued the behavioral and cell biological defects of odr-8 mutants (Figure 7D and data not shown). We next mutated the active site histidine residue to an alanine, and tested the biological function of the protein in transgenic animals. The His-to-Ala odr-8 transgene also substantially although not completely rescued the behavioral and cell biological phenotypes of odr-8 mutants. Incomplete rescue could be due to effects of this mutation on protein structure (Figure 7B–D). Together, these data suggest that the protease activity of ODR-8 UfSP2 is not essential for its in vivo function in GPCR maturation.
ODR-4 and ODR-8 form an ER complex that promotes GPCR trafficking
The expression patterns of ODR-8 and ODR-4, the subcellular localizations of the two proteins, and the phenotypes displayed by odr-8 and odr-4 mutants all suggested that these two proteins function together to promote ODR-10 trafficking to the cilia. However, the small size of AWA neurons and the limits of light microscopy prevented high-resolution co-localization of ODR-4 and ODR-8. In addition, biochemical analyses of ODR-4/ODR-8/ODR-10 protein interactions in worm extracts were precluded by the restricted expression patterns of these proteins in vivo. To circumvent these obstacles, we heterologously expressed ODR-4, ODR-8, and ODR-10 in cultured mammalian cells and analyzed their biochemical interactions and sub-cellular localization. Epitope-tagged ODR-8 and ODR-4 (two isoforms, ODR-4a and ODR-4b) were expressed either individually or in combination in HEK293 cells and analyzed by immunoprecipitation (IP). HA-ODR-8 co-precipitated with either ODR-4a-FLAG or ODR-4b-FLAG, regardless of which component was subjected to IP (Figure 8A). Immunoprecipitation of ODR-4 could also co-precipitate ODR-10-GFP (Figure 8B), while ODR-8 did not seem to directly interact with ODR-10 (Data not shown). Since ODR-4 and ODR-8 are conserved, we asked if their human orthologs also interacted biochemically. Immunoprecipitation of hODR4-FLAG brought with it HA-UfSP2 (Figure 8C), suggesting that like the C. elegans proteins, the human orthologs can form a complex in human cells.
Figure 8. ODR-4, ODR-8 and ODR-10 form a complex at the ER.

(A) HEK293 cells were co-transfected with ODR-4a-Flag, ODR-4b-FLAG and HA-ODR-8, and cell lysates were subjected to immunoprecipitation using indicated antibodies. The resulting precipitates were examined by immunoblot analysis with mouse anti-FLAG and mouse anti-HA antibodies. The asterisk indicates a non-specific band. (B) HEK293 cells were co-transfected with ODR-10-GFP with or without ODR-4b-FLAG, and cells lysates were subjected to immunoprecipitation using mouse anti-HA and rabbit anti-GFP antibodies. (C) HEK293 cells were co-transfected with human ODR4-FLAG and human HA-UfSP2, and cell lysates were subjected to immunoprecipitation using a FLAG antibody. The resulting precipitates were examined by immunoblot analysis with mouse anti-FLAG and mouse anti-HA antibodies. (D) HeLa cells were transiently transfected with ODR-4b-FLAG for 3 days. After fixation, the cells were subjected to immunocytochemistry using mouse anti-Flag and rabbit anti-TRAPα antibodies. (E) HeLa cells co-transfected with HA-ODR-8 and ODR-4b-FLAG as in panel D. fixed, and stained using rabbit anti-Flag and mouse anti-HA antibodies. Bar, 5 µm. (F) HeLa cells were transiently transfected with HA-ODR-8 alone or with HA-ODR-8 and ODR-4b-FLAG and cultivated for 3 days. In the panel marked ‘semi-permeabilized’ cells were treated with digitonin for 5 min prior to fixation, whereas in the panel labeled ‘intact’ the digitonin in step was omitted. Immunocytochemistry was performed using rabbit anti-Flag and mouse anti-HA antibodies.
Immunofluorescence studies localized the site of these interactions to the ER. Here, we found that ODR-4 co-localized with the ER resident protein TRAPα (Figure 8D). By contrast, ODR-8 was diffusely cytosolic when expressed alone, but somewhat reticular when co-expressed with ODR-4 (Figure 8E). This suggested that ODR-8 was recruited to the ER via its physical interaction with ODR-4. This was confirmed by demonstrating that ODR-8 was efficiently released from cells by permeabilization of the plasma membrane, but partially retained when co-expressed with ODR-4 (Figure 8F). The retained ODR-4 and ODR-8 were co-localized, consistent with their physical interaction (Figure 8E). Thus, ODR-4 and ODR-8 form a physical complex at the ER membrane.
ODR-10-GFP was also ER-localized in HeLa cells (Figure S8A). This localization remained qualitatively similar in cells co-expressing ODR-4 and/or ODR-8 (Figure S8B). This suggests that while ODR-4 and ODR-8 are clearly necessary for proper ODR-10 surface trafficking in AWA neurons, they may not be sufficient in the heterologous HeLa cell system. This remains to be investigated further. Nevertheless, the analysis in cells, together with the genetic and (lower resolution) localization data in AWA neurons, indicates that ODR-4 and ODR-8 form an ER-localized complex that facilitates ODR-10 surface expression.
Discussion
ODR-8, the C. elegans ortholog of Ufm1 specific protease 2, is involved in the biogenesis of GPCRs via a non-catalytic mechanism that does not require Ufm1. odr-8/UfSP2 mutants, like odr-4 mutants, fail to localize a subset of olfactory receptors to sensory cilia. This defect occurs at an early step in secretory pathway, most likely prior to exit from the endoplasmic reticulum. Consistent with this, ODR-4, ODR-8 and ODR-10 form a complex in the ER of HEK cells.
UfSP2 and its relative UfSP1 were first identified biochemically in mouse by virtue of their ability to process the ubiquitin-like protein Ufm1 [26]. UfSP2 is found throughout eukaryotes, from plants to man. UfSP1 is found in mouse, man, and flies but not C. elegans. Crystallography shows that UfSP1 and UfSP2 have a papain-like domain with a Cysteine-Aspartate-Histidine catalytic triad [25], [27]. The fold structure is most similar to that of the autophagy gene Atg4, which de-conjugates Atg8 ubiquitin-like proteins from phosphatidylethanolamine and promotes autophagosome formation. Mutagenesis of active site residues confirms the importance of these residues for in vitro UfSP protease activity: UfSP1 and UfSP2 are catalytically dead if the catalytic cysteine is mutated to a serine. The residues comprising the papain fold, including the catalytic residues, are highly conserved across UfSP orthologs, with 40% identity from grape vine to man.
Unexpectedly, we find that active site residues of ODR-8 UfSP2 are not required for any of its known in vivo functions. Mutating the catalytic cysteine or histidine residues of C. elegans UfSP2 does not abolish these functions. Thus, at least in C. elegans, and perhaps more generally, the major functions of UfSP2 do not require proteolytic activity. In addition, deleting Ce-ufm1 does not perturb localization of the ODR-10-GFP GPCR in AWA neurons, either in wild-type or in odr-8 mutants. This suggests that ODR-8 UfSP2 and UFM-1 do not necessarily have to function together in vivo.
ODR-4 localizes predominantly to the ER in vivo, and the soluble ODR-8 UfSP2 protein also shows some ER localization. Previous work showed that mouse UfSP2 is recruited to the ER when expressed with its client protein C20orf116 [25]. We show that ODR-4 can also recruit ODR-8 UfSP2 to the ER in HeLa cells. Disrupting either ODR-4 or ODR-8 results in some olfactory GPCRs, including ODR-10, being retained in the ER rather than trafficking in RAB-8-containing vesicles to sensory cilia. Neither ODR-4 nor ODR-8 accumulates preferentially at ER exit sites, and ODR-10-GFP does not appear to accumulate at ER exit sites in odr-4 or odr-8 mutants, suggesting these proteins are not involved in ER exit per se. ODR-4 and ODR-8, and ODR-4 and ODR-10 interact biochemically in HEK cells, A simple interpretation of all our data is that ODR-4 and ODR-8/UfSP2 form a complex in the ER with some olfactory receptors, including ODR-10, that allows these GPCRs to mature and become competent to exit the ER. In the absence of maturation, these GPCRs are retained in the ER. Quality control of folding in the ER is thought to involve cycles of ubiquitination and de-ubiquitination [42]–[44]. One highly speculative model is ODR-8/UfSP2 binds to ubiquitin or a ubiquitin-like molecule to regulate maturation of GPCRs.
In C. elegans ODR-4 and ODR-8/UfSP2 are expressed in the same small set of chemosensory neurons, and their function appears limited to maturation of a subset of GPCR chemoreceptors. Orthologues of ODR-8/UfSP2 and ODR-4 are found in organisms that do not have a nervous system, such as plants. Moreover, the mouse orthologs of ODR-4 and ODR-8 are expressed in many non-neuronal tissues. A simple explanation is that outside C. elegans these proteins participate in maturation of non-olfactory GPCRs. Interestingly, human ODR-4 and human UfSP2 form a complex in HEK cells, suggesting they also function together in humans.
Methods
Strains
Animals were grown under standard conditions [45]. Strains used are listed in Table S1.
Behavioral assays
Aggregation behavior, chemotaxis, and responses to changes in oxygen levels were measured as described previously [46]–[48].
Molecular biology
Whole genome sequencing was carried out on the Illumina HiSeq 2000 platform.
Plasmid construction: Plasmids were constructed using the multi-site Gateway system (Invitrogen). Promoters used include podr-10 (1.2 kb), podr-8 (3.1 kb), psrh-220 (2.1 kb), pstr-1(4 kb), podr-3 (2.7 kb), and pufm-1(4.1 kb); numbers refer to DNA upstream of the initiation codon. Promoters were cloned into the first position of the Gateway system, genes of interest were cloned at the second position, and the unc-54 3′UTR or the SL2::mCherry sequence at the third position. To rescue the odr-8 mutant phenotypes, we used a genomic fragment encoding F38A5.1a (Figure S9). To generate in frame C-terminal GFP or mCherry fusions, genes of interest were cloned in the second Gateway position without a stop codon, and the third position was occupied by GFP or mCherry DNA. For constructs encoding N-terminus GFP or mCherry fusion proteins, GFP or mCherry sequences without stop codons were placed at the second position and genes of interest at the third position. When the third position was occupied by sequences other than unc-54 3′UTR, we used a destination vector that placed unc-54 3′UTR sequences downstream of the 3rd Gateway insert. We rescued the lethality associated with the gk379 deletion using a ∼9.1 kb genomic fragment centered on the ufm-1 gene. This fragment was PCR amplified using primers flanked with attB1 and attB2 sites and cloned into pDonr221 using the BP reaction. To target the ufm-1 gene by CRISPR, a ufm-1 gene-specific sequence was cloned into the sgRNA vector bearing rpr-1 promoter using the Gibson assembly kit, as described [41].
Site-specific mutagenesis: Mutagenesis was performed using the Quikchange II XL kit from Agilent. Changes made include: odr-8(C421S: TGT to TCT); odr-8(C421A: TGT to GCT); odr-8(H547A: CAT to GCT); ufm-1(S2stop: TCG to TAA); ufm-1(S2stop:TCG to TAA; nucleotide A at position 12 deleted); rab-1(GDP bound S25N: TCG to AAT); rab-1(GTP bound Q70L: CAG to CTG); rab-6.1(GDP bound T25N: ACT to AAT); rab-6.2(GTP bound Q70L: CAG to CTG); rab-6.2(GDP bound T24N: ACC to AAC) and rab-6.2(GTP bound Q69L: CAG to CTG).
Transgenic strains: the ufm-1 rescue plasmid and fusion genes that express fluorescent markers of sub-cellular compartments were injected at 2–4 ng/ul together with 50 ng/ul of coelomocyte marker and 1 kb ladder. Other constructs were microinjected at 50 ng/ul.
Microscopy
Animals were mounted on 2% agarose pads containing 50 mM sodium azide. Confocal images were taken on a Nikon Eclipse Ti inverted microscope coupled to the Andor Ixon EMCCD camera and spinning disk confocal unit. GFP signals were captured with a 100 ms exposure time at an EMCCD Gain of 300, except for the ODR-4-4 images, which were captured with 5 ms exposure times. The mCherry signals of subcellular markers were imaged with different exposure times, according to their signal intensities. The subcellular localization images were generated by averaging 32 images.
Co-localization of ODR-8 with various markers was quantified as described [39] with slight modifications. We used the co-localization analysis package in Huygens software (Figure S5). A region of interest was cropped around the AWA cell body. The pixel intensity representing the brightest 4% of pixels in each image was extracted for both green and red channels. The pixel intensity values corresponding to the cut-off of the brightest 4% of pixels were used to threshold each image. The thresholded images were used to calculate the percentage of co-localization between ODR-8 and different markers.
To quantify the amount of ODR-10::GFP in the cilia and cell body, z-stack images were taken on a spinning disk confocal microscope using a 100× lens and 100 ms exposure time. The 3-D images were reconstituted with the aid of IMARIS software (Figure S4). GFP pixel intensities brighter than 1200 were cropped by creating a surface with 0.25 µm details. The total pixel intensities inside the surface were calculated.
CRISPR-Cas9 induced mutations
Frameshift mutations in ufm-1 were obtained using the CRISPR-Cas9 system, as described [41]. Briefly, Cas9 was expressed from the eft-3 promoter, while the sgRNA targeting the ufm-1 gene was expressed from the rpr-1 promoter. The Cas9 and sgRNA constructs were injected into the worm gonad with co-injection marker (cc::GFP). 38 ccGFP-positive F1 animals were singled, and genotyped after the eggs were laid.
Immunofluorescence for mammalian cells
HeLa cells grown on coverslips were washed with PBS and fixed in 3.7% formaldehyde in PBS for 15 min. For semi-permeabilization, cells were treated with 50 µg/ml digitonin in KHM buffer (110 mM KAc, 20 mM HEPES (pH 7.4) and 2 mM MgCl2) for 5 min at 4°C before fixation. Fixed cells were permeabilized with 0.1% Triton ×100 in PBS for 5 min, blocked with 10% fetal bovine serum in PBS for 30 min, and incubated with primary antibodies for 1 h. After washing, cells were incubated with AlexaFluor 488-conjugated goat anti-mouse IgG and/or AlexaFluor 564-conjugated goat anti-rabbit IgG secondary antibodies (Invitrogen) for 60 min. Plasma membrane was stained using 0.01% PKH26, according to the manufacturer's instructions (Sigma). Images were acquired on a confocal laser microscope (LSM 780, Zeiss) using a 63× oil-immersion objective lens with a numerical aperture (NA) of 1.42.
Immunoprecipitation and immunoblotting
Cell lysates were prepared in a lysis buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM phenylmethanesulfonyl fluoride (PMSF) and protease inhibitor cocktail (complete EDTA-free protease inhibitor, Roche)). The lysates were clarified by centrifugation at 15,000 rpm and subjected to immunoprecipitation using anti-Flag M2 affinity gel and anti-HA agarose (Sigma). Precipitated immunocomplexes were washed five times in a washing buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) and boiled in sample buffer. Samples were subsequently separated by SDS-PAGE and transferred to Nitrocellulose membranes (Biorad). Immunoblot analysis was performed with anti-GFP, anti-Flag M2 antibody and anti-HA (clone 16B12), and visualized with Super Signal West Pico Chemiluminescent substrate (Pierce).
Supporting Information
The Ufm1 conjugation pathway. (A) The Ufm1 conjugation pathway highlighted to date by biochemistry [49]. Pro-Ufm1 is activated by UfSP1 or UfSP2 cysteine proteases. Mature Ufm1 is activated by the Uba5 E1 like enzyme, and then transferred to the E2 like enzyme Ufc1. The E3 like enzyme Ufl1 conjugates Ufm1 onto target substrate. Ufmylated substrates can be de-ufmylated by UfSP1 or UfSP2. (B) Alignment of Ufm1 sequences from different species, indicating where we generated frame shift mutations using CRISPR. Colors refer to amino acid types.
(TIF)
ODR-8 UfSP2 across phylogeny. (A) Alignment of UfSP2 from mouse, human, C. elegans and A. thaliana. Colors refer to amino acid types. (B) Phylogenetic tree of UFSP2/ODR-8 orthologues, showing that this protein is found in many eukaryotes, including sponges, plants and most metazoan phyla.
(TIF)
ODR-4 across phylogeny. (A) Alignment of ODR-4 from mouse, human, C. elegans and fish. Colors refer to amino acid types. (B) Phylogenetic tree of ODR-4 orthologues, showing that this protein, like UfSP2, is found in many eukaryotes.
(TIF)
Quantification of ODR-10-GFP localization in the cilia of AWA neurons. A region of interest (ROI) was cropped and thresholded with 1200 signal intensity. Pixels with intensities over 1200 were computed and summed.
(TIF)
Co-localization of ODR-10-GFP with various markers in AWA neurons. (A–D) Examples of co-localization analysis of ODR-10-GFP with different markers in wild type (A–B) and odr-8 mutants (C–D). See Methods for details. (E–F). Quantification of ODR-10-GFP co-localization with different markers in wild type (E), and odr-8 mutants (F). ** indicates p<0.01 by ANOVA, Bonferroni's multiple comparisons test.
(TIF)
ODR-10-GFP localization in mutants expressing defective rab proteins. (A) Localization of ODR-10-GFP in animals expressing defective rab proteins in AWA neurons of npr-1 and odr-8(ky31); npr-1 animals. For rab-1, rab-6.1 and rab-6.2, the indicated dominant active and dominant negative versions of the RAB proteins were transgenically expressed in AWA from the odr-10 promoter. For rab-2, we used the null allele nu415 and the dominant active allele n777 [37] [39]. (B) Quantitation of ODR-10-GFP accumulation in cilia in different rab backgrounds. **, p<0.01 (ANOVA, Bonferroni's multiple comparisons test).
(TIF)
Deleting ufc-1 or uba-5 does not alter ODR-10 function. (A) Molecular nature of mutations in ufc-1 and uba-5 deletion alleles. (B) uba-5 and ufc-1 deletion mutants show wild type chemotaxis to diacetyl. (C) Deleting uba-5 does not alter the ODR-10-GFP phenotype in wild type or odr-8 mutants. (D) ODR-10::GFP expression driven by the ufm-1 promoter. (E) Expressing a ufm-1::SL2::mCherry operon from the ufm-1 promoter does not highlight AWA neurons marked with ODR-10::GFP.
(TIF)
ODR-4 and ODR-8 are not sufficient to promote efficient ODR-10-GFP expression at the cell surface in HeLa cells. (A) HeLa cells were transiently transfected with ODR-10-GFP for 3 days. After fixation, the cells were subjected to immunocytochemistry by using rabbit anti- TRAPα antibodies, to highlight the ER. (B) HeLa cells were transiently transfected with indicated plasmids for 3 days. After fixation, plasma membrane was stained by PKH26. Bar, 5 µm.
(TIF)
Sequence of the pEXPRESSION vector used for odr-8 mutant phenotype rescue. Green: odr-8 promoter; yellow: odr-8 genomic DNA; cyan: odr-8 3′UTR; grey: unc-54 3′UTR.
(PDF)
C. elegans strains used in this study.
(DOCX)
Acknowledgments
We thank James Hadfield and the staff at CRI Core Genomics for Next Generation Sequencing, the Caenorhabditis Genetics Center, and the Japanese knockout consortium for mutant strains, Sean Munro and de Bono lab members for critical reading of the manuscript, and Nick Barry and Jonathan Howe for help with microscopy and image analysis.
Funding Statement
Our funding includes a Long Term Fellowship to CC from the European Molecular Biology Organization (EMBO ALTF 1098-2011), a research Fellowship of the Japan Society for the Promotion of Science for young scientists to EI, a Marshall Scholarship and a National Science Foundation studentship to KPW, and funding from the Medical Research Council of the United Kingdom and the European Research Council (ERC project number 269058). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Buchberger A, Bukau B, Sommer T (2010) Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 40: 238–252. [DOI] [PubMed] [Google Scholar]
- 2. Rosenbaum EE, Brehm KS, Vasiljevic E, Liu CH, Hardie RC, et al. (2011) XPORT-dependent transport of TRP and rhodopsin. Neuron 72: 602–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Halevi S, McKay J, Palfreyman M, Yassin L, Eshel M, et al. (2002) The C. elegans ric-3 gene is required for maturation of nicotinic acetylcholine receptors. EMBO J 21: 1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6: 11–22. [DOI] [PubMed] [Google Scholar]
- 5. Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, et al. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494: 185–194. [DOI] [PubMed] [Google Scholar]
- 6. Park E, Rapoport TA (2012) Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu Rev Biophys 41: 21–40. [DOI] [PubMed] [Google Scholar]
- 7. Mizrachi D, Segaloff DL (2004) Intracellularly located misfolded glycoprotein hormone receptors associate with different chaperone proteins than their cognate wild-type receptors. Mol Endocrinol 18: 1768–1777. [DOI] [PubMed] [Google Scholar]
- 8. Morello JP, Salahpour A, Petaja-Repo UE, Laperriere A, Lonergan M, et al. (2001) Association of calnexin with wild type and mutant AVPR2 that causes nephrogenic diabetes insipidus. Biochemistry 40: 6766–6775. [DOI] [PubMed] [Google Scholar]
- 9. Lu M, Echeverri F, Moyer BD (2003) Endoplasmic reticulum retention, degradation, and aggregation of olfactory G-protein coupled receptors. Traffic 4: 416–433. [DOI] [PubMed] [Google Scholar]
- 10. Saito H, Kubota M, Roberts RW, Chi Q, Matsunami H (2004) RTP family members induce functional expression of mammalian odorant receptors. Cell 119: 679–691. [DOI] [PubMed] [Google Scholar]
- 11. Stamnes MA, Shieh BH, Chuman L, Harris GL, Zuker CS (1991) The cyclophilin homolog ninaA is a tissue-specific integral membrane protein required for the proper synthesis of a subset of Drosophila rhodopsins. Cell 65: 219–227. [DOI] [PubMed] [Google Scholar]
- 12. Bermak JC, Li M, Bullock C, Zhou QY (2001) Regulation of transport of the dopamine D1 receptor by a new membrane-associated ER protein. Nat Cell Biol 3: 492–498. [DOI] [PubMed] [Google Scholar]
- 13. Hurt CM, Ho VK, Angelotti T (2013) Systematic and quantitative analysis of G protein-coupled receptor trafficking motifs. Methods Enzymol 521: 171–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mendes HF, Zaccarini R, Cheetham ME (2010) Pharmacological manipulation of rhodopsin retinitis pigmentosa. Adv Exp Med Biol 664: 317–323. [DOI] [PubMed] [Google Scholar]
- 15. Morello JP, Bichet DG (2001) Nephrogenic diabetes insipidus. Annu Rev Physiol 63: 607–630. [DOI] [PubMed] [Google Scholar]
- 16. Maya-Nunez G, Janovick JA, Ulloa-Aguirre A, Soderlund D, Conn PM, et al. (2002) Molecular basis of hypogonadotropic hypogonadism: restoration of mutant (E(90)K) GnRH receptor function by a deletion at a distant site. J Clin Endocrinol Metab 87: 2144–2149. [DOI] [PubMed] [Google Scholar]
- 17. Troemel ER, Chou JH, Dwyer ND, Colbert HA, Bargmann CI (1995) Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans . Cell 83: 207–218. [DOI] [PubMed] [Google Scholar]
- 18. Thomas JH, Robertson HM (2008) The Caenorhabditis chemoreceptor gene families. BMC Biol 6: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sengupta P, Chou JH, Bargmann CI (1996) odr-10 encodes a seven transmembrane domain olfactory receptor required for responses to the odorant diacetyl. Cell 84: 899–909. [DOI] [PubMed] [Google Scholar]
- 20. Dwyer ND, Troemel ER, Sengupta P, Bargmann CI (1998) Odorant receptor localization to olfactory cilia is mediated by ODR-4, a novel membrane-associated protein. Cell 93: 455–466. [DOI] [PubMed] [Google Scholar]
- 21. de Bono M, Tobin DM, Davis MW, Avery L, Bargmann CI (2002) Social feeding in Caenorhabditis elegans is induced by neurons that detect aversive stimuli. Nature 419: 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rogers C, Persson A, Cheung B, de Bono M (2006) Behavioral motifs and neural pathways coordinating O2 responses and aggregation in C. elegans. Curr Biol 16: 649–659. [DOI] [PubMed] [Google Scholar]
- 23. Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, et al. (2004) A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J 23: 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sasakawa H, Sakata E, Yamaguchi Y, Komatsu M, Tatsumi K, et al. (2006) Solution structure and dynamics of Ufm1, a ubiquitin-fold modifier 1. Biochem Biophys Res Commun 343: 21–26. [DOI] [PubMed] [Google Scholar]
- 25. Ha BH, Jeon YJ, Shin SC, Tatsumi K, Komatsu M, et al. (2011) Structure of ubiquitin-fold modifier 1-specific protease UfSP2. J Biol Chem 286: 10248–10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang SH, Kim GR, Seong M, Baek SH, Seol JH, et al. (2007) Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J Biol Chem 282: 5256–5262. [DOI] [PubMed] [Google Scholar]
- 27. Ha BH, Ahn HC, Kang SH, Tanaka K, Chung CH, et al. (2008) Structural basis for Ufm1 processing by UfSP1. J Biol Chem 283: 14893–14900. [DOI] [PubMed] [Google Scholar]
- 28. Rolls MM, Hall DH, Victor M, Stelzer EH, Rapoport TA (2002) Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neurons. Mol Biol Cell 13: 1778–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Witte K, Schuh AL, Hegermann J, Sarkeshik A, Mayers JR, et al. (2011) TFG-1 function in protein secretion and oncogenesis. Nat Cell Biol 13: 550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grant B, Hirsh D (1999) Receptor-mediated endocytosis in the Caenorhabditis elegans oocyte. Mol Biol Cell 10: 4311–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Treusch S, Knuth S, Slaugenhaupt SA, Goldin E, Grant BD, et al. (2004) Caenorhabditis elegans functional orthologue of human protein h-mucolipin-1 is required for lysosome biogenesis. Proc Natl Acad Sci U S A 101: 4483–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hermann GJ, Schroeder LK, Hieb CA, Kershner AM, Rabbitts BM, et al. (2005) Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol Biol Cell 16: 3273–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaplan OI, Molla-Herman A, Cevik S, Ghossoub R, Kida K, et al. (2010) The AP-1 clathrin adaptor facilitates cilium formation and functions with RAB-8 in C. elegans ciliary membrane transport. J Cell Sci 123: 3966–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dwyer ND, Adler CE, Crump JG, L'Etoile ND, Bargmann CI (2001) Polarized dendritic transport and the AP-1 mu1 clathrin adaptor UNC-101 localize odorant receptors to olfactory cilia. Neuron 31: 277–287. [DOI] [PubMed] [Google Scholar]
- 35. Mizuno-Yamasaki E, Rivera-Molina F, Novick P (2012) GTPase networks in membrane traffic. Annu Rev Biochem 81: 637–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Duvernay MT, Filipeanu CM, Wu G (2005) The regulatory mechanisms of export trafficking of G protein-coupled receptors. Cell Signal 17: 1457–1465. [DOI] [PubMed] [Google Scholar]
- 37. Sumakovic M, Hegermann J, Luo L, Husson SJ, Schwarze K, et al. (2009) UNC-108/RAB-2 and its effector RIC-19 are involved in dense core vesicle maturation in Caenorhabditis elegans. J Cell Biol 186: 897–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edwards SL, Charlie NK, Richmond JE, Hegermann J, Eimer S, et al. (2009) Impaired dense core vesicle maturation in Caenorhabditis elegans mutants lacking Rab2. J Cell Biol 186: 881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chun DK, McEwen JM, Burbea M, Kaplan JM (2008) UNC-108/Rab2 regulates postendocytic trafficking in Caenorhabditis elegans. Mol Biol Cell 19: 2682–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hertel P, Daniel J, Stegehake D, Vaupel H, Kailayangiri S, et al. (2013) The Ubiquitin-fold Modifier 1 (Ufm1) Cascade of Caenorhabditis elegans. J Biol Chem 288: 10661–10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen C, Fenk LA, de Bono M (2013) Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Res 41 (20) e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brodsky JL (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151: 1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL (2008) Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 132: 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang ZR, Bonifacino JS, Hegde RS (2013) Deubiquitinases sharpen substrate discrimination during membrane protein degradation from the ER. Cell 154: 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sulston J, Hodgkin J (1988) Methods. In: Wood WB, editor. The nematode Caenorhabditis elegans. Cold Spring Harbor: CSHL Press. pp. 587–606. [Google Scholar]
- 46. de Bono M, Bargmann CI (1998) Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans . Cell 94: 679–689. [DOI] [PubMed] [Google Scholar]
- 47. Bargmann CI, Hartwieg E, Horvitz HR (1993) Odorant-selective genes and neurons mediate olfaction in C. elegans . Cell 74: 515–527. [DOI] [PubMed] [Google Scholar]
- 48. Cheung BH, Cohen M, Rogers C, Albayram O, de Bono M (2005) Experience-dependent modulation of C. elegans behavior by ambient oxygen. Curr Biol 15: 905–917. [DOI] [PubMed] [Google Scholar]
- 49. Tatsumi K, Sou YS, Tada N, Nakamura E, Iemura S, et al. (2010) A novel type of E3 ligase for the Ufm1 conjugation system. J Biol Chem 285: 5417–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Ufm1 conjugation pathway. (A) The Ufm1 conjugation pathway highlighted to date by biochemistry [49]. Pro-Ufm1 is activated by UfSP1 or UfSP2 cysteine proteases. Mature Ufm1 is activated by the Uba5 E1 like enzyme, and then transferred to the E2 like enzyme Ufc1. The E3 like enzyme Ufl1 conjugates Ufm1 onto target substrate. Ufmylated substrates can be de-ufmylated by UfSP1 or UfSP2. (B) Alignment of Ufm1 sequences from different species, indicating where we generated frame shift mutations using CRISPR. Colors refer to amino acid types.
(TIF)
ODR-8 UfSP2 across phylogeny. (A) Alignment of UfSP2 from mouse, human, C. elegans and A. thaliana. Colors refer to amino acid types. (B) Phylogenetic tree of UFSP2/ODR-8 orthologues, showing that this protein is found in many eukaryotes, including sponges, plants and most metazoan phyla.
(TIF)
ODR-4 across phylogeny. (A) Alignment of ODR-4 from mouse, human, C. elegans and fish. Colors refer to amino acid types. (B) Phylogenetic tree of ODR-4 orthologues, showing that this protein, like UfSP2, is found in many eukaryotes.
(TIF)
Quantification of ODR-10-GFP localization in the cilia of AWA neurons. A region of interest (ROI) was cropped and thresholded with 1200 signal intensity. Pixels with intensities over 1200 were computed and summed.
(TIF)
Co-localization of ODR-10-GFP with various markers in AWA neurons. (A–D) Examples of co-localization analysis of ODR-10-GFP with different markers in wild type (A–B) and odr-8 mutants (C–D). See Methods for details. (E–F). Quantification of ODR-10-GFP co-localization with different markers in wild type (E), and odr-8 mutants (F). ** indicates p<0.01 by ANOVA, Bonferroni's multiple comparisons test.
(TIF)
ODR-10-GFP localization in mutants expressing defective rab proteins. (A) Localization of ODR-10-GFP in animals expressing defective rab proteins in AWA neurons of npr-1 and odr-8(ky31); npr-1 animals. For rab-1, rab-6.1 and rab-6.2, the indicated dominant active and dominant negative versions of the RAB proteins were transgenically expressed in AWA from the odr-10 promoter. For rab-2, we used the null allele nu415 and the dominant active allele n777 [37] [39]. (B) Quantitation of ODR-10-GFP accumulation in cilia in different rab backgrounds. **, p<0.01 (ANOVA, Bonferroni's multiple comparisons test).
(TIF)
Deleting ufc-1 or uba-5 does not alter ODR-10 function. (A) Molecular nature of mutations in ufc-1 and uba-5 deletion alleles. (B) uba-5 and ufc-1 deletion mutants show wild type chemotaxis to diacetyl. (C) Deleting uba-5 does not alter the ODR-10-GFP phenotype in wild type or odr-8 mutants. (D) ODR-10::GFP expression driven by the ufm-1 promoter. (E) Expressing a ufm-1::SL2::mCherry operon from the ufm-1 promoter does not highlight AWA neurons marked with ODR-10::GFP.
(TIF)
ODR-4 and ODR-8 are not sufficient to promote efficient ODR-10-GFP expression at the cell surface in HeLa cells. (A) HeLa cells were transiently transfected with ODR-10-GFP for 3 days. After fixation, the cells were subjected to immunocytochemistry by using rabbit anti- TRAPα antibodies, to highlight the ER. (B) HeLa cells were transiently transfected with indicated plasmids for 3 days. After fixation, plasma membrane was stained by PKH26. Bar, 5 µm.
(TIF)
Sequence of the pEXPRESSION vector used for odr-8 mutant phenotype rescue. Green: odr-8 promoter; yellow: odr-8 genomic DNA; cyan: odr-8 3′UTR; grey: unc-54 3′UTR.
(PDF)
C. elegans strains used in this study.
(DOCX)