

Abstract
Severe seizure activity is associated with reoccurring cycles of excitotoxicity and oxidative stress that result in progressive neuronal damage and death. Intervention with these pathological processes is a compelling disease-modifying strategy for the treatment of seizure disorders. We have optimized a series of small molecules for neuroprotective and anticonvulsant activity as well as altered their physical properties to address potential metabolic liabilities, to improve CNS penetration and to prolong the duration of action in vivo. Utilizing phenotypic screening of hippocampal cultures with nutrient medium depleted of antioxidants as a disease model, cell death and decreased neuronal viability produced by acute treatment with glutamate or hydrogen peroxide were prevented. Modifications to our previously reported proof of concept compounds have resulted in a lead which has full neuroprotective action at < 1 nM and antiseizure activity across six animal models, including the kindled rat, and displays excellent pharmacokinetics including high exposure to the brain. These modifications have also eliminated the requirement for a chiral molecule, removing the possibility of racemization and making large scale synthesis more easily accessible. These studies strengthen our earlier findings which indicate that potent, multifunctional neuroprotective anticonvulsants are feasible within a single molecular entity which also possesses favorable CNS-active drug properties in vitro and in vivo.
Keywords: neuroprotection, glutamate toxicity, oxidative stress, hippocampal cultures, epilepsy, anticonvulsant, pharmacokinetics
Introduction
Although effective anticonvulsant drugs have been available since the early 1900’s, the single most significant unmet medical need remains disease modification to slow or stop the progression of seizure disorders. No existing commercial antiepileptic drug (AED) is disease-modifying. Neuroprotection from seizure-related neural damage may be a key strategy for disease modification (Acharya et al., 2007). To date, this strategy has lacked an effective probe compound of a highly potent neuroprotective anticonvulsant. We recently reported (Brenneman, et al., 2012) a “proof of concept” small molecule, AND-287, that exhibits both broad spectrum anticonvulsant activity and high potency neuroprotection against toxic processes that are relevant to the pathology of seizure disorders.
The rationale for treating epileptogenesis through neuroprotection resides in our understanding of the multiple factors that contribute to neuropathology in this disease (Bengzon et al., 2002). These factors include genetic mutations, glutamate-induced excitotoxicity, mitochondrial dysfunction, oxidative stress, growth factor loss and increases in cytokine concentration (Ferriero, 2005). Further, intense seizure activity produces large increases in NMDA-mediated calcium influx (Van Den Pol et al., 1996). High levels of calcium lead to apoptotic cascades that result in acute neuronal cell death. Elevated calcium levels can also generate reactive oxygen species that can produce cell damage and death. With this background in mind, our neuroprotective anticonvulsant program focused on structural optimization to provide potent protection from excitotoxicity and oxidative stress.
Previous studies employing animal models of seizure disorders have indicated neurodegenerative and behavioral abnormalities result from treatment with proconvulsant agents. For example, the cholinomimetic convulsant pilocarpine induced a status epilepticus that was accompanied by hippocampal damage and the development of spontaneous, recurrent seizures (Muller et al., 2009). In this epileptic model, increases in anxiety-related behavior and impaired performance on learning and memory tests were observed. Intrahippocampal injections of kainic acid produce both neural damage and alteration in cognitive performance (Groticke et al., 2008). Further, pentylenetetrazol kindling has been used to produce a convulsive component of epilepsy that resulted in impairment of learning of memory tests which included social discrimination, acoustic fear conditioning, water maze and passive avoidance (Jia et al., 2006). Studies in human seizure disorder have shown that chronic uncontrolled epilepsy can result in neurodegeneration to specific brain areas (Houser, 1992). Because seizure disorders have been associated with these indications of neural deficits and accompanying behavior problems, the concept has emerged that neuroprotection from these events might lead to disease modification to either slow or stop the progression of the disease.
The core probe compound we selected for these exploratory studies was AND-171, a molecule that included a sulfamide moiety combined with features of a recently described anticonvulsant that had broad activity in animal models coupled with low potency (mM) neuroprotection (Deshpande et al., 2008). The fundamental choice of the sulfamide functionality was supported by reports on anticonvulsant compounds that indicated a sulfamide group with N,N′-disubstitution or N,N,N′-trisubstitution (Aeberli et al., 1967; Gavernet et al., 2007). Further, two primary sulfamides PhCH2NHSO2NH2 and BuNHSO2NH2 had anticonvulsant activity. Additionally, other groups have described anticonvulsant activity associated with sulfamides (Thiry et al., 2008; Parker et al., 2009). Initial testing of the core compound indicated both anticonvulsant activity and micromolar potency for neuroprotection. Our initial efforts provided a series of compounds which possessed both neuroprotective and anticonvulsant activities in a single entity as proof of concept probes. We set out to improve potency and efficacy as well as improve drug-like properties to increase their duration of action, CNS penetration and exposure following oral administration.
Based on these initial findings, we have discovered a series of anticonvulsant agents, most notably AND-302, which can completely protect hippocampal neurons from excitotoxicity and oxidative stress with high potency in vitro. These studies have provided an optimized neuroprotective anticonvulsant with suitable ADME properties and a favorable PK/PD profile for which further preclinical development is warranted.
Materials and Methods
Materials
The following reagents were obtained from Sigma-Aldrich (Milwaukee, WI): L-glutamic acid monosodium salt monohydrate (49621); hydrogen peroxide solution, 30 wt % (216763); propidium iodide solution (1.0 mg/ml in water; P4864); and 6-carboxyfluoresceine diacetate (CFDA, C5041).
Chemical synthesis and structure verification
All reagents used in chemical synthesis were purchased from Aldrich Chemical Co. (Milwaukee, WI), Alfa Aesar (Ward Hill, MA) Thermo Fisher Scientific (Pittsburgh, PA) or Oakwood Products (West Columbia, SC) and were used without purification. 1H-NMR spectra were obtained on a Varian Mercury 300-MHz NMR spectrometer. Chemical shifts were reported in parts per million (ppm, δ) using various solvents as internal standards (CDCl3, δ 7.26 ppm; DMSO-d6, δ 2.50 ppm). 1H-NMR splitting patterns were designated as singlet (s), doublet (d), triplet (t), or quartet (q). Splitting patterns that could not be interpreted or easily visualized were recorded as multiplet (m) or broad (br). Coupling constants were reported in hertz (Hz). UV Purity and electrospray mass spectral data were determined using a Waters Alliance 2695 HPLC/MS (Waters Symmetry C18, 4.6 × 75 mm, 3.5 μm) with a 2996 diode array detector from 210–400 nm. All compounds were determined to be >95% pure by HPLC at 225 nm.
Synthesis of 2,2-difluoro-2-arylethylsulfamides
The synthetic details are summarized in Scheme 1. Aryl bromides or iodides 1 were treated with n-butyllithium at low temperature and the resulting aryllithium was added to a solution of diethyl oxalate which, upon quenching and workup, yielded ketoesters 2 which were then fluorinated with Deoxo-Fluor [bis(2-methoxyethyl)aminosulfur trifluoride] to give difluoroethyl esters 3. Alternatively, aryl halides 1 (X = I) were directly converted to difluoroethyl esters 3 by copper mediated reaction with bromodifluoroethyl acetate. Difluoroethyl esters 3 were then reduced with sodium borohydride to the difluoroethyl alcohols 4 which were subsequently converted to the difluoroethyl amines 5 via activation of the alcohol to the triflate followed by displacement with ammonia. Finally, heating the corresponding difluoroethyl amines 5 with sulfamide in 1,4-dioxane provided the desired difluoroethylsulfamides 6.
Scheme 1.
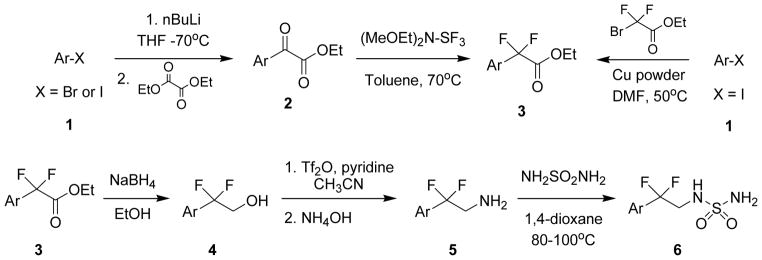
Synthesis of 2,2-difluoro-2-arylethylsulfamides.
Culture model
Dissociated hippocampal cultures derived from embryonic day 18 rats were employed as the primary screening system to test for toxicity as well as neuroprotection. This cellular system was chosen because a limited number of compounds were to be tested thereby permitting the use of a low throughput screening system that is derived from a brain area that is highly susceptible to seizures (Heineman et al., 1992). In brief, hippocampal tissue was obtained commercially through Brain Bits (Springfield, IL) and cultures prepared with slight modifications to methods previously described (Brewer et al., 1993). Tissue was dissociated with a papain-based kit from Worthington Biochemical Corporation (Lakewood, NJ) that is modeled after the method described by Huettner and Baughman (1986). The hippocampal neurons were plated at low density (10,000 cell/well) in a 96-well format and maintained in serum-free medium consisting of Neurobasal Medium supplemented with B27 and GlutaMAX (Gibco). Poly-L-lysine coated plates (BD Biosciences, Franklin Lakes, NJ) were used because of the preferential adherence and survival of neurons on this matrix support. All assays were conducted between days 12 and 22 of age. This period of experimentation was chosen so that the cultures were mature enough to exhibit a response to either glutamate or hydrogen peroxide and to ensure a consistent reproducible response to the toxins. Prior to the initiation of experiments between days 11 and 21 in vitro, a complete change of medium was performed in a working volume of 100 μL. This medium differed from the medium utilized for plating in that the B27-AO did not contain the five antioxidants present in the standard B27 (Gibco, product #10889). This change of medium was designed to decrease the background levels of antioxidants to facilitate the damage and death with glutamate or hydrogen peroxide treatment. The rationale for this change of medium was twofold: (1) because loss of antioxidant control may also be a component of epileptogenesis (Waldbaum and Patel, 2010; Wu et al., 2010); and (2) because the goal was to obtain a significant and reproducible toxicity signal in hippocampal neurons. Thus, this decrease in the background antioxidant concentration was used to recapitulate a disease model for epilepsy as well as a means to produce neurotoxicity at the lowest concentration of toxin. Both the amount of glutamate and hydrogen peroxide used in the assays, as well as the time of treatment and duration of the experiment were designed to be relevant to the disease. Further, all time parameters employed in these studies were empirically determined to be within the limits of reversible toxic events. A critical feature in the glutamate studies was the duration of treatment of the hippocampal neurons. The rationale for using a 5 min treatment with glutamate was the previous observation that indicated 5 min glutamate exposure produced significant toxicity (Choi, 1986) as well as a demonstrated delay of increased intracellular calcium that overloaded neurons and produced cell death (Randall and Thayer, 1992). The amount of glutamate employed in our screen (30 μM) was selected for two reasons: (1) because this concentration was the basal level of glutamate observed in microdialysis measurements of the hippocampus from epileptogenic patients (Cavus et al., 2008); and (2) glutamate at this concentration produced a reliable and reproducible toxicity that could be prevented by pharmacological intervention in hippocampal cultures. The concentration of hydrogen peroxide employed (10 μM) was also selected because this level was detected in the hippocampus of rats after kainate-induced status epilepticus (Jarrett et al., 2008).
Neuroprotection from oxidative stress
All the experimental compounds were dissolved to 10 mM stock solutions in Dulbecco’s phosphate buffered saline (DPBS; Sigma:D-5780) prior to testing. To evaluate the compounds for neuroprotection from hydrogen peroxide, hippocampal cultures between day 11 and 21 of age were given a complete change of medium containing 100 μL of Neurobasal medium with B27 that contained no antioxidants. Twenty four hours after the change in medium, the neuroprotective studies against hydrogen peroxide were started. The test compounds were added to the hippocampal cultures for a 4 hour test period in concentrations that ranged from 1 pM to 300 μM with 8 replications. Immediately after treatment with test compound, 10 μM hydrogen peroxide was added for the 4 hour test period.
Neuroprotection from excitotoxicity
For the neuroprotection studies using glutamate excitotoxicity, several modifications were made from the method described for the hydrogen peroxide assay. For the glutamate neuroprotection assay, hippocampal cultures between day 11 and 21 of age were given a complete change of medium containing 100 μL of Neurobasal medium with B27 that contained no antioxidants. Twenty four hours after the change in medium, the glutamate neuroprotection studies were started. The cultures were treated for 5 min with 30 μM glutamate dissolved in DPBS. For this treatment, a 900 μM solution of glutamate was prepared and then 3.3 μL of this solution added to the culture well containing 100 μL of media. After this acute treatment, the medium containing the glutamate was removed from the cultures and fresh medium without antioxidants added. The test compound was then added to the hippocampal cultures for a 4 hour test period in concentrations that ranged from 1 pM to 300 μM, with 8 replications per concentration tested.
Fluorescence-based assays
At the conclusion of the test period, the cultures were evaluated with fluorescent dye-based assays for cell death (propidium iodide) and for neuronal viability (6-carboxyfluorescein diacetate, CFDA). These two standard assays were chosen because they could be measured as multiplexed determinations within a single well thereby monitoring both an increasing (cell death) parameter and a decreasing parameter (neuronal viability) after a toxic treatment of the hippocampal cultures. For the cell death assessment with the propidium iodide, slight modification to a method previously described was used (Sarafian et al., 2002). Propidium iodide (PI) stock solution of 1 mg/mL (1.5 mM) was diluted 1:30 in DPBS for a final working concentration of 50 μM. After removal of the growth medium, 50 μL of the 50 μM PI solution was added to cultures and allowed to incubate in the dark at room temperature for 15 min. On every plate, wells without cells were used to provide a blank reading that was used to subtract background fluorescence. The cultures were assessed for fluorescence intensity at Ex536/Em590 nm in a CytoFluor fluorimeter (Perceptive Biosystems). Results were expressed in relative fluorescent units and EC50s calculated from the dose responses of the test compound and compared to values obtained from controls and wells treated with toxin alone.
After the assessment of cell death, cultures were then further assayed for neuronal viability by the CFDA method (Petroski and Geller, 1994). For the neuronal viability assay, 1 mg of 6-carboxyfluorescein diacetate (CFDA) dye was dissolved in 100 mL of DPBS (Gibco:D-5780) and kept in the dark until added to the hippocampal cultures. After a complete change of medium, 100 μL CFDA dye solution was added for 15 min of incubation at 37°C in the dark. At the conclusion of the incubation period, the dye was removed from the cultures and washed once with 100 μL of DPBS. After removal of the first wash, a second wash of DPBS was added to the culture and then incubated for 30 min to allow the efflux of dye out of glia in the cultures. At the conclusion of the 30 min efflux period, the culture efflux medium was removed and 100 μl of 0.1% triton-X100 in water was added to the cultures before reading at Ex490/Em517 in a CytoFluor fluorimeter. Results were expressed in relative fluorescent units (RFU) and EC50’s calculated from the dose response of the test compound. All statistical comparisons were made by ANOVA, with normality of values tested by the Shapiro-Wilk test followed by a multiple comparison of means test with the Holm-Sidak method as performed through Sigmaplot (v. 11).
Aqueous solubility determination
Aqueous solubility was determined using the shaken flask method as previously described (Alsenz and Kansy, 2007) at desired pHs of 1.2, 7.4 and 9.2 to mimic the stomach, plasma, and intestinal environments, respectively. Concentrations were determined using HPLC where a standard curve was used for reference.
Metabolic stability assay
The stability of test compound was determined by incubation with liver microsomes derived from rat, mouse or human. All assays were conducted by Absorption Systems (Exton, PA) by methods previously described (Jeffrey et al, 2001). Test compound at 1 μM was assayed at various times of incubation up to 60 min. Samples were analyzed using a LTQ-Orbitrap XL mass spectrometer. The assay was a single determination at each time point. As a metabolically labile control, 1 μM testosterone was also tested.
Caco-2 permeability assay
The human colon adenocarcinoma cell line Caco-2 has been widely used as an in vitro absorption model. Caco-2 in vitro permeability correlates well with human drug absorption (Artursson et al., 2001). Bidirectional cell permeability was estimated from Caco-2 cells by methods described in a commercial protocol by Absorption Systems (Exton, PA). Assays were performed in duplicate in 25-day-old cultures, and test compounds were tested at 5 μM using LC-MS/MS to determine concentrations.
Cytochrome P-450 inhibition assays
Inhibition of Cytochrome P-450 enzymes is a potential liability for drug candidates. High throughput assays to determine inhibition of these enzymes have been described (Trubetskoy and Gibson, 2005). Five of the most commonly screened isoforms are 3A4, 1A2, 2C9, 2D6 and 2C19. This assay was performed by Anthem Biosciences (Bangalore, India) using Vivid® CYP450 screening kits obtained from Life Technologies (Carlsbad, CA). Each of these was performed using a specific, known inhibitor as a reference standard to insure viability of the assay. Assay readouts were performed using a fluorescence plate reader.
Plasma protein binding assay
Compounds which are highly (>98%) bound to plasma proteins can have issues including poor in vivo efficacy and highly variable pharmacokinetics given a low free fraction (Jusko and Gretch, 1976). This assay was performed by Anthem Biosciences using a Rapid Equilibrium Dialysis™ (RED) device from Pierce/Thermo Fisher Scientific (Pittsburgh, PA) Warfarin and propranolol were used as standards. Percent of compound unbound was determined by LC/MS/MS determination of compound remaining in solution after incubation with plasma (human or mouse). Percent bound to plasma protein was reported.
hERG assay
The in vitro effects on the hERG (human ether-à-go-go-related gene) potassium channel current (a surrogate for IKr, the rapidly activating, delayed rectifier cardiac potassium current) expressed in mammalian cells were evaluated at room temperature using the PatchXpress 7000A (Molecular Devices), an automatic parallel patch clamp system, at ChanTest Corp (Cleveland, OH). The test article was evaluated at 1, 10, and 30 μM with each concentration tested in two cells (n = 2). The duration of exposure to each test article concentration was 5 min. The positive control (E-4031) confirms the sensitivity of the test system to hERG inhibition.
Pharmacokinetics study
This protocol was conducted by DavosPharma (Upper Saddle River, NJ), in collaboration with Anthem Biosciences (Bangalore, India) after obtaining Institutional Animal Ethics Committee (IAEC) permission. Male C57BL/6 mice aged 6–8 weeks and weighing 18 to 22 g were used for experimentation after a minimum of 5 days of acclimation. Fasted animals were administered a single dose of test substance by oral gavage with a dose of 5 mg/kg body weight in 0.5% carboxymethylcellulose (CMC) in water at a dose volume 10 mL/kg body weight or intravenously (via tail vein) with a dose of 5 mg/kg body weight in normal saline at a dose volume of 10 mL/kg body weight.
Under mild anesthesia, blood samples were collected the retro-orbital route into labeled centrifuge tubes containing anticoagulant (K2EDTA) during the next 24 hours post dose. After blood collection, animals were euthanized and brain samples were collected. Brain samples were washed in milli-Q water to remove any blood clots. Excess water was blotted and dry weight of the brain was taken and 1 ml of milli-Q water was added in test tube for homogenization. Collected blood samples were centrifuged at 3500 rpm for 10 minutes and plasma were separated and stored at −80°C until analysis. Homogenized brain samples were stored at −80°C until analysis by LC-MS\MS. Drug concentration in plasma was quantified with an API 3200 Q-trap LC-MS/MS system and these data were analyzed using WinNonlin version 5.3 (Pharsight).
Antiseizure testing
All in vivo efficacy testing was conducted by the Anticonvulsant Screening Program (ASP) of the National Institute of Neurological Disease and Stroke at the National Institutes of Health. As previously described (White, 2009), compounds were evaluated in a series of antiseizure tests that are highly predictive of efficacy in human epilepsy. This testing began with the maximal electroshock test (MES) that is conducted in both mice and rats by methods previously described (Swinyard, 1969; Rowley and White, 2010). The route of administration was by intraperitoneal (i.p.) injection for the mouse MES and by oral gavage for the rat MES test. The MES testing was followed by the 6 Hz seizure test that further evaluated compounds in this model of psychomotor seizures (Barton et al., 2001). This model was used to detect seizures that may be useful for the treatment of therapy-resistant partial seizures. As a third test in these initial screens, the subcutaneous pentylenetetrazol (scPTZ) test was used as a model to identify compounds that raise seizure threshold (Swinyard et al., 1993). For this model, the amount of test compound required to protect against threshold seizures (5 seconds of clonic activity) induced by subcutaneous injection of PTZ (85 mg/kg) was determined.
Hippocampal Kindled Rat Model
The kindling model is a useful test to identify compounds for treating pharmacoresistant limbic epilepsy exhibited by complex partial seizures with secondarily generalized seizures. In studies conducted by the ASP, the rapid hippocampal kindling mode by Lothman et al. (1988) was employed. Adult male Sprague-Dawley rats (300–400 g) were surgically implanted with bipolar electrodes placed in the hippocampus. Rats were kindled by repetitive electrical stimulation (50 Hz, 10 s train of 1 ms, biphasic 200 μA pulses every 30 min for 6 h every other day for a total of 60 stimulations) resulting in stage 5 bilateral motor seizures. One week later, the rats received 2–3 supra-threshold stimulations delivered every 30 min before compound treatment to ensure stability of the behavioral seizure stage and after-discharge duration. Fifteen minutes after the last stimulation, a single dose of vehicle or test compound was administered i.p. After 15 min, each rat was then stimulated every 30 min for 3 to 4 h. After each stimulation, individual seizure scores and after-discharge durations were recorded. The group mean ± SEM were calculated for each parameter. Seven rats per dose and a minimum of four doses were used to establish an ED50 value. Efficacy was measured as the ability of a compound to modify the seizure score (severity of spread) and after-discharge duration (ADD, excitability) of the generalized seizures.
Frings Mouse Model
The Audiogenic Seizure (AGS)-susceptible Frings mouse model was used to assess antiseizure activity (White at al., 1992). At the effective testing duration determined in the MES test, individual mice (eight mice per dose) were placed into a plexiglass cylinder (diameter, 15 cm; height, 18 cm) fitted with an audio transducer (Model AS-ZC; FET Research and Development, Salt Lake City, UT) and exposed to a sound stimulus of 110 decibels (11 KHz) delivered for 20 sec. Mice were observed for 25 sec for the presence or absence of hindlimb tonic extension. Sound-induced seizures are characterized by wild running followed by loss of righting reflex with forelimb and hindlimb tonic extension. Mice not displaying hindlimb tonic extension were considered protected.
Results
In vitro neuroprotection
Neuroprotection from toxicity associated with glutamate or hydrogen peroxide treatment of hippocampal cultures was used as a primary screen to evaluate AND-302 and its analogs. Two multiplexed assays were used to assess cell culture viability: a propidium iodide (PI) assay used to estimate changes in the amount of cell death and a CFDA assay used to estimate changes in neuronal viability. With these multiplexed assays that were measured in the same culture well, neuroprotection was observed as an increase in fluorescence for CFDA and a decrease in fluorescence for the PI assay. Representative experiments for the most studied compound (AND-302) of this chemical series against glutamate toxicity are shown in Figure 1A for the CFDA and PI assays. For experiments conducted in day 20 cultures, the 30 μM glutamate treatment produced a decrease in the CFDA fluorescence to 64 ± 2 % of that for control cultures. This decrease in neuronal viability signal was contingent on the removal of antioxidant substances in the growth medium. With the PI assay, the increase in fluorescence associated with cell death after treatment with 30 μM glutamate was 182 ± 10 % of that observed with control cultures. As shown in Figure 1A, EC50s for AND-302-mediated neuroprotection as determined by the two viability assays were similar, 0.07–0.13 nM. With both assays, AND-302 treatment resulted in changes in fluorescence levels that were not different from that of control cultures (see dashed lines for respective control levels), indicating full efficacy protection under these conditions. AND-302 was more than 10-fold more potent than our best previously described neuroprotective agent, AND-287.
Figure 1. Effect of AND-302 on neuroprotection from glutamate (A) and hydrogen peroxide (B) in hippocampal cultures tested in nutrient medium without antioxidants.

For all neuroprotective studies, fluorescent assays for neuronal viability [6-carboxyfluorescein diacetate (CFDA), closed circles] and cell death (propidium iodide, open circles) were measured in the same culture well. For each of the fluorescent assays, the relative fluorescence units (RFU) are displayed, as wells as the excitation and emission wavelengths that were utilized. For protection assays against 30 μM glutamate, hippocampal cultures between 12 and 22 days of age were utilized. Glutamate treatment was acute with a 5 minute incubation followed by removal of the glutamate and replacement with growth medium. Treatment with AND-302 was then initiated four hours, followed by the viability assays. For protective assays against 10 μM hydrogen peroxide, hippocampal cultures between 12 and 22 days of age were utilized. Cultures were treated with various concentrations of AND-302 followed by the addition of hydrogen peroxide (HP). The duration of co-treatment with drug and HP was for four hours followed by the viability assays. Each point for all assays is the mean of 8 determinations from two separate experiments. The error bars are the S.E.M. The dotted lines for all assays represent the fluorescence levels from respective control cultures. In Figure 1A, significant changes (P< 0.001) from cultures treated only with glutamate are shown as a single asterisk for propidium iodide and those measured with CFDA with a double asterisk. Similarly in Figure 1B, significant changes (P< 0.001) from cultures treated only with hydrogen peroxide are shown as a single asterisk for propidium iodide and those measured with CFDA with a double asterisk.
Representative experiments for the effects of AND-302 on neuroprotection from hydrogen peroxide are shown in Figure 1B. For these experiments conducted in day 13 cultures, the 10 μM hydrogen peroxide treatment produced a decrease in CFDA fluorescence to 67 ± 3 % of that for control cultures, very similar to the toxic signal observed with 30 μM glutamate in day 20 cultures (Figure 1A). With the PI assay, the increase in fluorescence associated with cell death after treatment with 10 μM hydrogen peroxide was 202 ± 5 % of that observed with control cultures. AND-302 also exhibited potent neuroprotective activity hydrogen peroxide toxicity in the day 13 hippocampal cultures with EC50s of 0.09–3 nM observed from the two assays. As in the glutamate assays, AND-302 was more active than AND-287 by more than 10-fold.
A summary of the multiplexed assays for neuroprotection against both 30 μM glutamate- and 10 μM hydrogen peroxide-associated toxicity for the compounds reported here is shown in Table 1. As previously reported, the R-enantiomer, AND-287, was very active in these assays while the analog where the benzylic position was unsubstituted, AND-319, showed no activity in any of the neuroprotection assays. Replacement of the hydroxyl group of AND-287 with a difluoro group yielded a compound which ranges from 10 to 100 fold more active in all assays of neuroprotection. A series of difluoroethylsulfamides were synthesized and screened. Several analogs of AND-302 in this series showed very similar neuroprotection in all assays including AND-381, -383, -404 and -1432. Several compounds showed protection in some, but not all assays. AND-379, the 2-methoxphenyl analog, was more potent in protection from hydrogen peroxide than from glutamate while the 2-pyridyl analog, AND-382, lost activity relative to AND-302 in all assays. The addition of another halogen had mixed results. AND-1432, the 2,6-difluoro analog, showed very similar neuroprotection to AND-302, while the other di-halogenated analogs (AND-1433, -1434, -1435, -1436) lost significant if not all neuroprotection in all assays. Replacement of the benzylic difluoro with other substituents including AND-479 (gem-dimethyl), AND-480 (spiro-cyclohexyl), AND-482 (spiro-tetrahydropyranyl) and AND-483 (spiro-cyclopropyl) groups all resulted in significant or total loss of neuroprotective action in all assays. Additionally, replacement of the sulfamide with sulfamate or carbamate produces analogs of greatly diminished or complete loss of activity.
Table 1.
Comparison of Compounds for Neuroprotection against Acute Glutamate or Hydrogen Peroxide toxicity in Hippocampal Cultures after 4 hours Treatment.
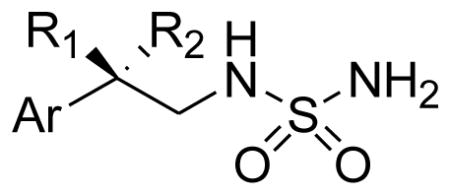
| |||||||
|---|---|---|---|---|---|---|---|
| AND ID # | Aryl Group (Ar) | R1 | R2 | CFDA* Glutamate | PI** Glutamate | CFDA* HP | PI** HP |
| 287 | 2-fluorophenyl | OH | H | 3 nM | 4 nM | 40 nM | 30 nM |
| 319 | 2-fluorophenyl | H | H | > 100 μM | > 100 μM | 100 μM | > 100 μM |
| 302 | 2-fluoropheny | F | F | 0.07 nM | 0.13 nM | 3 nM | 0.09 nM |
| 379 | 2-methoxyphenyl | F | F | 0.3 μM | 0.5 μM | 30 nM | 10 nM |
| 381 | phenyl | F | F | 0.01 nM | 0.01 nM | 0.01 nM | 0.01 nM |
| 382 | 2-pyridyl | F | F | 0.4 μM | 0.4 μM | 3 μM | 0.5 μM |
| 383 | 2-chlorophenyl | F | F | 1 nM | 6 nM | 0.02 nM | 0.05 nM |
| 404 | benzo[b]thiophen-3-yl | F | F | 0.09 nM | 0.10 nM | 0.10 nM | 0.5 nM |
| 1432 | 2,6-difluorophenyl | F | F | 0.1 nM | 0.4 nM | 0.02 nM | 0.01 nM |
| 1433 | 2,5-difluorophenyl | F | F | 30 μM | 50 μM | 7 μM | 6 nM |
| 1434 | 2,3-difluorophenyl | F | F | 11 μM | 20 μM | 2 nM | 5 nM |
| 1435 | 2,4-difluorophenyl | F | F | > 100 μM | > 100 μM | >100 μM | > 100 μM |
| 1436 | 2-chloro-6-fluorophenyl | F | F | > 100 μM | > 100 μM | >100 μM | 70 nM |
| 479 | 2-fluorophenyl | Me | Me | > 100 μM | 0.1 μM | >100 μM | > 100 μM |
| 480 | 2-fluorophenyl | cyclohexyl | > 100 μM | > 100 μM | >100 μM | > 100 μM | |
| 482 | 2-fluorophenyl | tetrahydropyranyl | 40 μM | 20 μM | >100 μM | 40 μM | |
| 483 | 2-fluorophenyl | cyclopropyl | > 100 μM | 0.3 μM | >100 μM | 0.3 μM | |
| 526 |
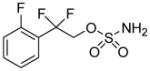
|
- | - | > 100 μM | > 100 μM | >100 μM | > 100 μM |
| 542 |
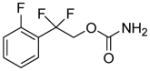
|
- | - | > 100 μM | 50 μM | 2 μM*** | 1 μM*** |
Carboxyfluorescein diacetate
Propidium Iodide
Low efficacy
In vivo anticonvulsant activity
A summary comparison of anticonvulsant activity for study compounds is presented in Table 2. All of the animal evaluation for antiseizure activity was performed by the Anticonvulsant Screening Program of NINDS/NIH. In general, the most potent of the neuroprotective compounds were tested for antiseizure activity. Three of the most potent neuroprotective difluoro analogs (AND-302, -381, and -383) showed significant activity in four antiseizure assays: maximal electroshock model (in both rats and mice), the 6 Hz test and mouse pentylenetetrazol model, with the most potent activity in the 6 Hz mouse model. These three compounds were more potent than AND-287 in all four of these seizure models. For the 6 Hz mouse model, AND-387 showed 3 out of 4 mice protected at a high dose of 100 mg/kg, whereas AND-302 protected 4 out of 4 mice at every time point for the duration of the preliminary experiment and yielded an ED50 = 19±1 mg/kg upon quantitation. Additionally, AND-302 was screened in several other antiseizure models. In the challenging hippocampal kindled rat study, AND-302 showed efficacy with an ED50 = 44±3 mg/kg (Figure 3). In the Audiogenic Seizure (AGS)-susceptible Frings mouse model, AND-302 protected mice from sound-induced seizures with an ED50 = 19±2 mg/kg. Treated mice were also evaluated for rotarod performance as a means of evaluating frank toxicity. While no rotarod impairment was observed for AND-287 up to a dose of 500 mg/kg, AND-302 showed modest impairment with an ED50 = 217±7 mg/kg or a Therapeutic Index (TI, TD50/ED50) for mouse MES and 6 Hz tests equal to 4.3 and 11.4 mg/kg, respectively.
Table 2.
Comparison of anticonvulsant activity of test compounds in four animal models.
| AND ID# | Rat MES (oral) ED50 (mg/kg) |
Mouse MES (i.p.) ED50 (mg/kg) |
Mouse 6 Hz (i.p.) ED50 (mg/kg) |
Mouse scPTZ (s.c.) ED50 (mg/kg) |
Mouse Rotarod Impairment (i.p.) TD50 (mg/kg) |
|---|---|---|---|---|---|
| 287 | 50±2 | 75±1 | 3/4 at 100* | > 250 | > 500 |
| 319 | 36±1 | 45±2 | 50±1 | 74±3 | 170±5 |
| 302 | 44±1 | 50±1 | 19±1 | 108±2 | 217±7 |
| 381 | 26±4 | 49±1 | 16±1 | 41±1 | 141±4 |
| 383 | 30±2 | 55±5 | 29±1 | 99±3 | 168±2 |
Preliminary data indicated 3 out of 4 mice were protected at 100 mg/kg. ED50 has not been determined.
Figure 3. ED50 of 44 ± 3 (mg/kg) for AND-302 in the Hippocampal Kindling Test.

The drop lines depict the ED50. The measures were taken at 0.25 hours after stimulation. Each point is the mean value from seven rats.
ADME and Pharmacokinetics
In addition to measures of neuroprotective and anticonvulsive efficacy, several tests of early ADME (Absorption, Distribution, Metabolism and Elimination) properties were conducted for the most potent and efficacious neuroprotective anticonvulsant of this chemical series, AND-302, and are summarized in Table 3. AND-302 has high water solubility of 7.6, 11.1 and 9.2 mg/mL at relevant physiological pHs of 1.2, 7.4 and 9.2, respectively. The permeability of AND-302 to the Caco-2 cell line was determined to be high as it was shown to have an apparent permeability (Papp) of 50 × 10−6 cm/s. The stability of AND-302 in liver microsomes from human, rat and mice was determined. In these experiments, the stability of the compound was monitored by mass spectrometry over a time course of 60 minutes of incubation. AND-302 was highly stable in this test, with > 70% of compound remaining unchanged during the test period with microsomes from all three species. Plasma protein binding (PPB) was also measured for AND-302, which exhibits low to moderate PPB of 57 and 39% in human and mouse, respectively. In addition, inhibition of CYP-450 enzymes was measured for AND-302 and IC50 values were determined to be greater than 74 μM for five major isoforms. Lastly, the effect of AND-302 was evaluated on the hERG channel and was shown to have IC50 > 30 μM, indicating a low probability of causing arrhythmias associated with Q-T prolongation.
Table 3.
Drug Suitability Profile of AND-302.
| Assay | AND-302 | |
|---|---|---|
| Aqueous Solubility | pH 1.2 | 7.6 mg/mL |
| pH 7.4 | 11.1 mg/mL | |
| pH 9.2 | 9.2 mg/mL | |
|
| ||
| Caco-2 cell permeability (Papp × 10−6 cm/s) | 50 (high) | |
|
| ||
| Liver Microsome Stability (% remaining after 60 min) | Rat: 100% | |
| Mouse: 89% | ||
| Human: 73% | ||
|
| ||
| Plasma Protein Binding | -Human | 57% |
| -Mouse | 39% | |
|
| ||
| Cytochrome P450 Inhibition (IC50) |
3A4 | 74 μM |
| 1A2 | >120 μM | |
| 2C9 | >120 μM | |
| 2D6 | >120 μM | |
| 2C19 | 96 μM | |
|
| ||
| hERG (IC50) | >30 μM | |
Pharmacokinetic (PK) properties were determined after administering AND-302 both orally (p.o.) and intravenously (i.v.) to mice. Figure 2 shows the relationship of drug concentration vs. time in both plasma and brain after oral administration and Table 4 shows the accompanying measured and calculated PK parameters. AND-302 displays rapid absorption into plasma and brain, with the time to maximum concentration (Tmax) being the earliest time point measured in both plasma and brain. The concentration at Tmax (Cmax) for AND-302 is 7.54 g/g and 3.13 g/mL for brain tissue and plasma, respectively. AND-302 distributes preferentially to brain tissue over plasma at a ratio of 2.4:1 at Cmax and 2.8:1 for total exposure (Area-Under-the-Curve, AUC). The high Cmax and AUC, and brain/plasma ratio ≫ 1 are quite desirable for a CNS acting drug. The terminal half-life was determined to be 4.5 hours for both brain and plasma after oral administration. Coupled with the i.v. arm of the experiment, clearance (CL) was shown to be low, in brain and plasma, at 1.41 and 6.43 mL/min/kg, respectively and bioavailability (%F) was calculated to be quite high, essentially 100%.
Figure 2. Pharmacokinetics: Distribution of AND-302 in Plasma and Brain after Oral Administration in Mice.

The time course of concentration of AND-302 vs. time. Male C57BL/6 mice aged 6–8 weeks and weighing 18 to 22 g were used for experimentation after a minimum of 5 days of acclimation. Fasted animals were administered a single dose of test substance by oral gavage with a dose of 5 mg/kg body weight in 0.5% carboxymethylcellulose (CMC) in water at a dose volume 10 mL/kg body weight. Blood and brain samples were collected as per the Materials and Methods described above. Drug concentration was quantified with an API 3200 Q-trap LC-MS/MS system and these data were analyzed using WinNonlin version 5.3 (Pharsight).
Table 4.
Pharmacokinetic Parameters of AND-302 after Oral and IV Administration to Mice.
| Pharmacokinetic Parameters | Brain | Plasma |
|---|---|---|
| Cmax (μg/g or μg/mL) | 7.54 | 3.13 |
| Tmax (h) | 1.00 | 0.50 |
| AUClast (h* μg/g or μg/mL) (0 to 24 h) | 47.77 | 17.04 |
| T1/2 (h) | 4.5 | 4.5 |
| CL (mL/min/kg)** | 1.41 | 6.43 |
| F% (oral bioavailability)** | 82 | ~100 |
μg/g determined for brain tissue and μg/mL determined for plasma.
Determined from i.v. administration at 5 mg/kg
Discussion
Epilepsy is a common chronic neurological condition that affects at least 50 million people worldwide. It has been estimated that 25% of people suffering from epilepsy receive no effective treatment for their seizures from available drugs. Further, no currently marketed antiepileptic drug has been shown to be antiepileptogenic, indicating that the available therapies do not address the underlying causes of the disease, only the symptomatic effect of seizures. The long-term purpose of our studies was to develop new antiepileptic drugs that are both effective in treating patients which are refractory to current treatments and that are disease-modifying by addressing the long term causes of reoccurring cycles of seizure episodes. Our underlying hypothesis is that neurotoxicity associated with elevated glutamate and oxidative stress are at the center of the neural damage (Bonilha et al., 2010; Proper et al., 2000) and the neuronal cell death (Mathern et al., 1995) that contributes to the neuropathology and etiology of epilepsy. Neuroprotection from these damaging and cyclical processes was the strategy for discovering disease-modifying agents, along with the possibility of providing an alternative strategy for seizure control through multiple mechanisms. With these unmet medical needs in mind, the central concept was to discover a proof of concept agent that provided both broad spectrum antiseizure activity and potent neuroprotection from excitotoxicity and oxidative stress within a single molecular entity.
Commercial antiepileptic drugs have been reported to have neuroprotective properties as summarized in a review of progress for neuroprotective strategies in preventing epilepsy (Acharya et al., 2008), which suggests that the extent of the neuroprotective and antiepileptogenic effects of current individual antiepileptic drugs is still unclear in part due to a lack of long-term animal studies as well as clinical trials to fully evaluate the impact of putatively neuroprotective compounds. Among the drugs with reported low-potency neuroprotective properties are valproate (Yamauchi et al., 2008), topiramate (Noh et al., 2006), felbamate (Longo et al., 1995), gabapentin (Cilio et al., 2001), lamotrigine (Papazisis et al., 2008), carbamazepine (Costa et al., 2008) and vigabatrin (Andre et al., 2001). In all cases, the neuroprotection was apparently incidental to the antiseizure properties in the chemical design of the anticonvulsants. The end result has been that anticonvulsants in general possess low potency neuroprotective activity. It is known that some anticonvulsants show activity against carbonic anhydrase II (CAII). Our lead series of compounds have also shown CAII inhibition in the micromolar range (screened at Cerep, Celle l’Evescault, France following Iyer, et al., 2006). While this activity may account, in part, for their anticonvulsant activity, the neuroprotective activity is likely multi-factorial. In the present study, optimization of neuroprotective activity was the primary goal and design strategy for the antiseizure molecule.
With neuroprotection set as the defining strategy for the anticonvulsant lead, the central objective of all neuroprotective assays was their relevancy to excitotoxicity and oxidative stress. Both the amount of glutamate and hydrogen peroxide used in the assays, as well as the time of treatment and duration of the experiment, were designed to be relevant to the disease. Time parameters employed in these studies were empirically determined to be within limits (4 hours) that would permit preventable amounts of neural damage and death. To produce neural damage and death with glutamate and hydrogen peroxide, the cultures were changed to a medium with significant depletion of antioxidant components in the defined medium supplement B-27. This was performed to obtain a significant and reproducible toxic signal in the hippocampal neurons and because loss of antioxidant control may be a component of epileptogenesis (Waldbaum and Patel, 2010; Wu et al., 2010). In particular, this removal of antioxidants from the medium was hypothesized to be a disease model for epilepsy.
With excitotoxicity associated with excessive glutamate as a focus for epilepsy-related damage and death, the details of neuroprotection from this toxic process are important to the rationale of the screening process. A critical feature of the glutamate-related studies was the duration of exposure to the hippocampal neurons. The rationale for using a short (5 min) exposure with glutamate was based on the observation of Randall and Thayer, 1992, which produced a delayed but substantial increase in intracellular calcium that overloaded neurons and produced cell death. This intense burst of glutamate and resulting calcium overload may be relevant to seizures and therefore represents an important feature to be captured in in vitro assays.
After our initial studies with AND-287, we sought to alter its chemical structure to improve its drug like properties as well as to further optimize both neuroprotective and antiseizure activities. The low molecular weight of AND-287 makes it attractive as a CNS active compound as these compounds have lower molecular weights on average than other drugs. We sought to replace the benzylic hydroxyl group for several reasons. First, a terminal sulfamide is a rather polar substituent. The combination of both the hydroxyl group and the sulfamide result in a compound which has a low partition coefficient (cLogP = −0.25) and somewhat high topological polar surface area (tPSA = 92 Å2), both of which have a negative impact on optimal CNS penetrance. Second, the benzylic hydroxyl group is known to be a site of metabolism through multiple pathways including sulfation and glucuronidation. Both of these pathways would make the compound less active or inactive and cause it to be cleared rapidly. In addition, the benzylic hydroxyl group may be prone to racemization, i.e., in the case of AND-287, from the R enantiomer to a mixture of R and S enantiomers. Our previous work showed that the S enantiomer, AND-286, was essentially inactive in our neuroprotection assays and, hence, racemization would be detrimental to activity and duration of action. Fluorine has been used in medicinal chemistry as a bioisosteric replacement for the hydroxyl group. Table 5 shows a comparison of calculated physical properties of AND-287 and the analog where the benzylic hydroxyl has been replaced with difluoro, AND-302. First, the difluoro group is more lipophilic and would increase the calculated LogP as well as lower the tPSA. Second, the difluoro group is not susceptible to the same metabolic pathways as the hydroxyl group and is resistant to other known oxidative pathways involved in drug clearance. In addition, the difluoro containing compound is achiral so racemization would no longer be an issue.
Table 5.
Comparison of Calculated Physical Properties of AND-287 and AND-302.
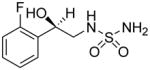
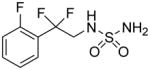
CLogP (calculated partition coefficient) and tPSA (topological polar surface area) were calculated using Instant JChem (v. 5.9.4) under license from ChemAxon.
The data in the neuroprotection assays against glutamate and hydrogen peroxide shown in Table 1 allow for some structure-activity relationships (SAR) to be drawn for AND-302 and related analogs. As previously reported, deletion of the hydroxyl group at the benzylic carbon of AND-287 rendered that compound, AND-319, devoid of neuroprotective activity. Introduction of the difluoro group provides compounds which are 10-100-fold more potent than AND-287. Some small changes in the substitution of the aryl ring result in retention of all activity. Replacement of the 2-fluoro group with a 2-chloro (AND-383) or hydrogen (and AND-381) result in compounds which are nearly as potent as AND-302 in all four neuroprotection assays. The change to a 2-methoxy group (AND-379) causes modest losses in potency. Replacement of the phenyl ring with a 2-pyridyl ring (AND-382) causes similar loss in potency, albeit still sub-micromolar in three of the four assays. Surprisingly, other simple changes, such as the addition of an another halogen, cause substantial loss in potency, except for the 2,6-difluorophenyl analog, AND-1432, which is nearly as potent as AND-302 in all four assays. The benzylic difluoro coupled with the sulfamide appear to be required for neuroprotection. Analogs where the difluoro is replaced with a gem-dimethyl or spirocyclic groups (AND-479, -480, -482 and -483) lose most if not all neuroprotective activity. Further, replacement of the sulfamide with either a sulfamate (AND-526) or a carbamate (AND-542) result in compounds with greatly reduced or complete loss in neuroprotective activity.
A clear advantage for drug discovery against seizure disorders is the demonstrated validity of animal models that are predictive of efficacy for various types of epilepsy in humans (Rowley and White, 2010). This in vivo advantage is particularly important in that validated molecular targets which permit screening of compounds in vitro for antiseizure properties remain elusive. Based on the predictive quality of the maximal electroshock model, it is likely that our lead compound, AND-302 and close analogs, AND-381 and -383 would be active against generalized tonic-clonic seizures in patients (Swinyard, 1969). With a rather high ED50 of 108±2 mg/kg, AND-302 is active in the subcutaneous pentylenetetrazol test, a model that identifies compounds that raise seizure threshold. Both AND-381 and -383 were also active in this model, with AND-381 being significantly more active with an ED50 of 41±1 mg/kg s.c. In addition, all three of these compounds were quite active in the 6 Hz psychomotor test, a model that predicts activity against complex partial seizures. Further, because the 6 Hz test possesses a differential pharmacological profile in comparison to the MES and scPTZ tests, this screen may be useful in identifying compounds that could be useful for pharmacoresistant epilepsy (Rowley and White, 2010). As such, AND-302 was chosen as a candidate for further evaluation in the hippocampal kindled rat model, where it had an ED50 = 44±3 mg/kg i.p. as shown in Figure 3. Many currently marketed anticonvulsants are inactive in this challenging seizure model. Finally, AND-302 was shown to be active in the (AGS)-susceptible Frings mouse model, with an ED50 = 19±2 mg/kg i.p. All together, AND-302 was active in six different antiseizure models making it an effective broad spectrum anticonvulsant. Additionally, rotarod performance evaluation shows that AND-302 has a reasonable Therapeutic Index (TI) for MES and 6 Hz tests equal to 4.3 and 11.4, respectively. This therapeutic window is larger than some known anticonvulsants, including valproic acid, whose TI is essentially 1.
The structural change that we employed, namely replacement of the benzylic hydroxyl with two fluorines, has yielded a compound with very favorable drug-like properties. In addition to measures of neuroprotective and anticonvulsive efficacy, several tests of early ADME (Absorption, Distribution, Metabolism and Elimination) properties were conducted for the most potent and efficacious neuroprotective anticonvulsant of this chemical series, AND-302 and are summarized in Table 3. These tests included determination of the following: (1) aqueous solubility, (2) bidirectional permeability in the human colon adenocarcinoma cell line, Caco-2, (3) metabolic stability in liver microsomes, (4) plasma protein binding, (5) cytochrome P450 (CYP) inhibition, and (6) binding to the hERG channel. Known agents that act as positive controls were also tested, indicating that the model systems were fully functional.
Poor aqueous solubility can be a major issue in both drug discovery and development. Insoluble compounds make screening difficult at higher concentrations as well as cause many compounds to be dropped from development because of poor absorption and hence, poor PK properties. Compounds with aqueous solubility greater than 1 mg/mL are considered highly soluble. As shown in Table 3, AND-302 has high water solubility of 7.6, 11.1 and 9.2 mg/mL at relevant physiological pHs of 1.2, 7.4 and 9.2, respectively. The permeability of compounds to the Caco-2 cell line is often used as a model for intestinal absorption of drugs. AND-302 is considered to be highly permeable as it was shown to have an apparent permeability (Papp) of 50 × 10−6 cm/s; compounds with Papp of 1 × 10−6 cm/s or higher are considered highly permeable. Because drug metabolism can be a significant factor in the agent evaluation process, the stability of AND-302 in liver microsomes from human, rat and mice was determined. In these experiments, the stability of the compound was monitored by mass spectrometry analysis over a time course of 60 minutes of incubation. As indicated, AND-302 was highly stable in this test, with > 70% of compound remaining unchanged during the test period with microsomes from all three species. Compounds which have high (>99%) plasma protein binding (PPB) often have poor efficacy and highly variable patient-to-patient exposure and hence, highly variable safety profiles. AND-302 exhibits low to moderate PPB of 57 and 39% in human and mouse, respectively. Inhibition of CYP isoforms can cause variable and unwanted metabolism and drug-drug interactions. With IC50 values greater than 74 μM for five major isoforms, AND-302 appears to be devoid of CYP inhibition issues. Finally, many preclinical compounds fail due to cardiovascular toxicities which can result from interaction with the hERG (human Ether-à-go-go-Related Gene) potassium channel which transmits IKr, the rapidly activating, delayed rectifier cardiac potassium current. The effect of AND-302 was evaluated on the hERG channel and was shown to have an IC50 > 30 μM, indicating a low probability of arrhythmias associated with Q-T prolongation.
Given the positive ADME profile for AND-302, PK studies were performed. The results in Figure 2 and Table 4 show AND-302 to have very favorable pharmacokinetics in mouse. The half-life of 4.5 hours, high AUC, low clearance and high bioavailability (near 100%) are all favorable traits of a drug candidate. In addition, the high brain to plasma ratio shown for AND-302, 2.4 for Cmax and 2.8 for AUC, is highly desired for a CNS acting drug. In summary, AND-302 is a potent neuroprotective anticonvulsant drug candidate with an attractive ADME and pharmacokinetic profile suited for further pre-clinical development activities.
Acknowledgments
We wish to thank the Anticonvulsant Screening Program from the National Institute of Neurological Disorders and Stroke for in vivo testing. This work was supported by NIH grant R43NS066537.
References
- Aeberli P, Gogerty J, Houlihan WJ. Neuropharmacological investigation of N-benzylsulfamides. J Med Chem. 1967;10:636–642. doi: 10.1021/jm00316a025. [DOI] [PubMed] [Google Scholar]
- Acharya MM, Hattiangady B, Shetty AK. Progress in neuroprotective strategies for preventing epilepsy. Prog Neurobiol. 2007;84:363–404. doi: 10.1016/j.pneurobio.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsenz J, Kansy M. High throughput solubility measurement in drug discovery and development. Adv Drug Delivery Rev. 2007;59:546–567. doi: 10.1016/j.addr.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Delivery Rev. 2001;46:27–43. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- Bonilha L, Edwards JC, Kinsman SL, Morgan PS, Fridriksson J, Rorden C, Rumboldt Z, Roberts DR, Eckert MA, Halford JJ. Extrahippocampal gray matter loss and hippocampal deafferentation in patients with temporal lobe epilepsy. Epilepsia. 2010;51:519–528. doi: 10.1111/j.1528-1167.2009.02506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Smith GR, Zhang Y, Du Y, Kondaveeti SK, Zdilla MJ, Reitz AB. Small-molecule anticonvulsant agents with potent in vitro neuroprotection. J Mol Neurosci. 2012;47:368–379. doi: 10.1007/s12031-012-9765-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27 supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SW, Jeffrey P. Utility of metabolic stability screening: comparison of in vitro and in vivo clearance. Xenobiotica. 2001;31:591–598. doi: 10.1080/00498250110057350. [DOI] [PubMed] [Google Scholar]
- Deshpande LS, Nagarkatti N, Ziobro JM, Sombati S, DeLorenzo RJ. Carisbamate prevents the development and expression of spontaneous recurrent epileptiform discharges and is neuroprotective in cultured hippocampal neurons. Epilepsia. 2008;49:1795–1802. doi: 10.1111/j.1528-1167.2008.01667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Baughman RW. Primary culture of identified neurons from the visual cortex of postnatal rats. J Neurosci. 1986;6:3044–3060. doi: 10.1523/JNEUROSCI.06-10-03044.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavernet L, Barrios IA, Cravero MS, Bruno-Blanch LE. Design, synthesis and anticonvulsant activity of some sulfamides. Bioorg Med Chem. 2007;15:5604–5614. doi: 10.1016/j.bmc.2007.05.024. [DOI] [PubMed] [Google Scholar]
- Iyer R, Barrese AA, III, Parakh S, Parker CN, Tripp BC. Inhibition profiling of human carbonic anhydrase II by high-throughput screening of structurally diverse, biologically active compounds. J Biol Screen. 2006;11:782–791. doi: 10.1177/1087057106289403. [DOI] [PubMed] [Google Scholar]
- Jia F, Kato M, Dai H, Xu A, Okuda T, Sakurai E, Okamura N, Lovenberg TW, Barbier A, Carruthers NI, Iinuma K, Yanai K. Effects of histamine H3 antagonists and donepezil on learning and mnemonic deficits induced by pentylenetetrazol kindling in weanling mice. Neuropharmacology. 2006;50:404–411. doi: 10.1016/j.neuropharm.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Jusko WJ, Gretch M. Plasma and tissue protein binding of drugs in pharmacokinetics. Drug Metab Rev. 1976;5:42–139. doi: 10.3109/03602537608995839. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Salerno RA, Perlin JB, Kaiser DL. Screening and characterization of antiepileptic drugs with rapidly recurring hippocampal seizures in rats. Epilepsia. 1988;2:367–379. doi: 10.1016/0920-1211(88)90048-4. [DOI] [PubMed] [Google Scholar]
- Lundquist S, Renftel M, Brillault J, Fenart L, Cecchelli R, Dehouck M-P. Prediction of drug transport through the blood-brain barrier in vivo: a comparison between two in vitro models. Pharma Res. 2002;19:976–981. doi: 10.1023/a:1016462205267. [DOI] [PubMed] [Google Scholar]
- Mamidi RNVS, Mannens G, Annaert P, Hendrickx J, Goris I, Bockx M, Janssen CGM, Kao M, Kelley MF, Meuldermans W. Metabolism and excretion of RWJ-333369 [1,2-ethanediol, 1-(2-chlorophenyl)-, 2-carbamate, (S)-] in mice, rats, rabbits, and dogs. Drug Met and Dist. 2007;35:566–575. doi: 10.1124/dmd.106.012336. [DOI] [PubMed] [Google Scholar]
- Mannens GSJ, Hendrickx J, Janssen CGM, Chien S, Van Hoof B, Verhaeghe T, Kao M, Kelley MF, Goris I, Bockx M, Verreet B, Bialer M, Meuldermans W. The Absorption, metabolism, and excretion of the novel neuromodulator RWJ-333369 (1,2-ethanediol, [1-2-chlorophenyl]-,2-carbamate, [S]-) in humans. Drug Met and Dist. 2007;35:554–565. doi: 10.1124/dmd.106.011940. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Pretorius JK, Melendez M, Levesque MF. The pathophysiologic relationships between lesion pathology, intracranial ictal EEG onsets, and hippocampal neuron losses in temporal lobe epilepsy. Epilepsy Res. 1995;21:133–147. doi: 10.1016/0920-1211(95)00014-2. [DOI] [PubMed] [Google Scholar]
- Muller CJ, Bankstahl M, Grotick I, Loscher W. Pilocarpine vs lithium-pilocarpine for induction of status epilepticus in mice: development of spontaneous seizures, behavioral alterations and neuronal damage. Eur J Pharmacol. 2009;619:15–24. doi: 10.1016/j.ejphar.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Parker MH, Smith-Swintosky VL, McComsey DF, Huang Y, Brenneman D, Klein B, Malatynska E, White HS, Milewski ME, Herb M, Finley MF, Lubin ML, Qin N, Iannucci R, Leclercq L, Cuyckens F, Reitz AB, Maryanoff BE. Novel, broad-spectrum anticonvulsants containing a sulfamide group: advancement of N-((benzo[b]thien-3-yl)methyl)sulfamide (JNJ-26990990) into human clinical studies. J Med Chem. 2009;52:7528–7536. doi: 10.1021/jm801432r. [DOI] [PubMed] [Google Scholar]
- Petroski RE, Geller HM. Selective labeling of embryonic neurons cultures on astrocyte monolayers with 5(6)-carboxyfluorescein diacetate (CFDA) J Neurosci Methods. 1994;52:23–32. doi: 10.1016/0165-0270(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Piredda SG, Woodhead JH, Swinyard EA. Effect of stimulus intensity on the profile of anticonvulsant activity of phenytoin, ethosuximide and valproate. J Pharmacol Exp Ther. 1985;232:741–745. [PubMed] [Google Scholar]
- Proper EA, Oestreicher AB, Jansen GH, Veelen CW, van Rijen PC, Gispen WH, deGraan PN. Immunohistochemical characterization of mossy fibre sprouting in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 2000;123:19–30. doi: 10.1093/brain/123.1.19. [DOI] [PubMed] [Google Scholar]
- Randall RD, Thayer SA. Glutamate-induced calcium transient triggers calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafian TA, Kouyoumjian S, Tashkin D, Roth MD. Synergistic cytotoxicity of 9-tetrahydrocannabianol and butylated hydroxyanisole. Tox Letters. 2002;133:171–179. doi: 10.1016/s0378-4274(02)00134-0. [DOI] [PubMed] [Google Scholar]
- Swinyard EA. Laboratory evaluation of antiepileptic drugs: review of laboratory methods. Epilepsia. 1969;10:107–119. doi: 10.1111/j.1528-1157.1969.tb03838.x. [DOI] [PubMed] [Google Scholar]
- Thiry A, Dogne JM, Supuran CT, Masereel B. Anticonvulsant sulfonamides / sulfamates /sulfamides with carbonic anhydrase inhibitory activity: drug design and mechanism of action. Curr Pharm Des. 2008;14:661–671. doi: 10.2174/138161208783877956. [DOI] [PubMed] [Google Scholar]
- Trubetskoy OV, Gibson JR, Marks BD. Highly miniaturized formats for in vitro drug metabolism assays using Vivid® fluorescent substrates and recombinant human cytochrome P450 enzymes. J Biomol Screen. 2005;10:56–66. doi: 10.1177/1087057104269731. [DOI] [PubMed] [Google Scholar]
- Waldbaum S, Patel M. Mitochondria, oxidative stress and temporal lobe epilepsy. Epilepsy Res. 2010;88:23–45. doi: 10.1016/j.eplepsyres.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M. Neuroprotection in epilepsy. Epilepsia. 2007;48:2384. doi: 10.1111/j.1528-1167.2007.01354.x. [DOI] [PubMed] [Google Scholar]
- White HS. Animal models of epileptogenesis. Neurology. 2002;59:S7–S14. doi: 10.1212/wnl.59.9_suppl_5.s7. [DOI] [PubMed] [Google Scholar]
- White HS, Patel S, Meldrum BS. Anticonvulsant profile of MDL27,266: an orally active, broad-spectrum anticonvulsant agent. Epilepsy Res. 1992;12:217–226. doi: 10.1016/0920-1211(92)90076-6. [DOI] [PubMed] [Google Scholar]
- Willmore LJ. Antiepileptic drugs and neuroprotection: current status and future roles. Epilepsy Behav Suppl. 2005;3:S25–S28. doi: 10.1016/j.yebeh.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Wu SB, Ma YS, Wu YT, Chen YC, Wei YH. Mitochondrial DNA mutation-elicited oxidative stress, oxidative damage, and altered gene expression in cultured cells of patients with MERRF syndrome. Mol Neurobiol. 2010;41:256–266. doi: 10.1007/s12035-010-8123-7. [DOI] [PubMed] [Google Scholar]
- Vieth M, Siegel MG, Higgs RE, Watson IA, Robertson DH, Savin KA, Durst GL, Hipskind PL. Characteristic Physical Properties and Structural Fragments of Marketed Oral Drugs. J Med Chem. 2004;47:224–232. doi: 10.1021/jm030267j. [DOI] [PubMed] [Google Scholar]
- Chico LK, Van Eldik LJ, Watterson DM. Targeting protein kinases in central nervous system disorders. Nat Rev Drug Disc. 2009;8:892–909. doi: 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]