

Abstract
A metabolomics study demonstrated a decrease in glutathione and an increase in cysteine (Cys) levels in human prostate cancer (PCa) tissues as Gleason scores increased, indicating redox imbalance with PCa progression. These results were extended in the present study by analyzing redox state of the protein thioredoxin 1 (Trx1) and sulfinylation (SO3) of peroxiredoxins (Prxs) (PrxsSO3) in PCa tissues and cell lines. Lysates of paired human PCa tissues with varying degree of aggressiveness and adjacent benign (BN) tissues were used for analysis. Redox western blot analysis of Trx1 demonstrated low levels of reduced and high levels of oxidized Trx1 (functional and non-functional, respectively) in high grade PCa (Gleason scores 4+4 to 4+5) in comparison to intermediate grade PCa (Gleason scores 3+3 to 3+4) or BN tissues. PrxsSO3 were increased in high grade PCa. Oxidized Trx1 and PrxsSO3 are indicators of oxidative stress. To study whether redox imbalance may potentially affect enzyme activities of antioxidant proteins (AP), we determined levels of selected AP in PCa tissues by western blot analysis and found that mitochondrial manganese superoxide dismutase (MnSOD), Prx 3, and Trx1 were increased in high grade PCa tissues when compared with BN tissues. Enzyme activities of MnSOD in high grade PCa tissues were significantly increased but at a lower magnitude when compared with the levels of MnSOD protein (0.5 folds vs. 2 folds increase). Trx1 activity was not changed in high grade PCa tissues despite a large increase in Trx1 protein expression. Further studies demonstrated a significant increase in posttranslational modifications of tyrosine and lysine residues in MnSOD protein and oxidation of Cys at active site (Cys 32 and Cys 35) and regulatory site (Cys 62 and Cys 69) of Trx1 in high grade PCa compared to BN tissues. These discordant changes between protein levels and enzyme activities are consistent with protein inactivation by redox imbalance and/or posttranslational modifications. In contrast, the protein level and activity of extracellular superoxide dismutase (ECSOD) were significantly decreased in high grade PCa when compared with adjacent BN tissues. Results from cell lines mirror those from PCa tissues. Knowledge of redox state profiles in specific cancers may help to predict the behavior and response of each cancer to chemotherapeutic drugs and radiation.
Keywords: ECSOD, MnSOD, Prx, Trx1, prostate cancer, posttranslational modifications, redox balance
Introduction
Alteration of redox status is an important feature of carcinogenesis and has been exploited therapeutically in the treatment of cancer. For example, cancer cells are frequently in oxidative stress conditions as demonstrated by higher levels of intracellular reactive oxygen species (ROS), increased hydrogen peroxide (H2O2) release as well as being susceptible to prooxidant-induced cell death [1-3]. Radiation and many other therapeutic agents such as doxorubicin or mitomycin C may exhibit their cancer killing effects by increased ROS production, pushing already stressed cancer cells beyond their limits of tolerance [4-5]. On the other hand, up-regulation of antioxidant enzymes has been frequently detected in cancers and has been suggested to confer resistance of cancer cells to ROS-generating therapeutic agents [6]. These contradictory results emphasize the importance of defining the redox state of each type of cancer.
Prostate cancer (PCa) is the most common and second leading cause of cancer deaths of men in the USA. Previous studies in our laboratory have demonstrated an increase in oxidative/nitrative damage in human PCa tissues and cells [7-8], implicating a role of oxidative stress in cancer development. In addition, we demonstrated that intra-and extracellular redox states were altered in aggressive PCa cells [9-10] and subsequently regulated PCa cell behavior [11]. It is increasingly recognized that subcellular redox systems have distinct functions and are subject to independent regulation [12]. The redox status of proteins involved in the transmission of redox signals in distinct suborganelles such as thioredoxin 1(Trx1), peroxiredoxin(s) (Prxs), manganese superoxide dismutase (MnSOD), or extracellular superoxide dismutase (ECSOD) have not been previously studied in human PCa tissues.
Trx1 is a redox modulating protein, providing reducing equivalents to numerous redox-sensitive proteins, such as ribonucleotide reductase [13] and redox-related transcriptional factors [6]. Trx1 contains five cysteine residues and reduces target proteins using its two vicinal cysteine (Cys) residues (Cys32 and Cys35) in the active site, where the molecule itself becomes oxidized, forming an intramolecular disulfide bond. Further oxidation of Trx1 results in the formation of a second intramolecular disulfide bond between Cys62 and Cys69, and then an intermolecular disulfide bond between Cys73 of two different Trx1 molecules [14]. Our previous study using PCa cell lines suggested that Trx1 redox states may function as a biomarker of redox imbalance and play an important role in modulating the response of PCa cells to prooxidant treatments [15]. One of the Trx1-target proteins is the Prx family. Trx(s) reduces Prx(s) by transferring disulfide bonds to the Cys pair, and without Trx activity Prxs will mainly be in oxidized state [16]. Sulfinylation of peroxiredoxin (PrxsSO3 or hyperoxidized-Prx) is one of oxidative stress indicator. Thus, Trx1 and PrxsSO3 are important cellular redox sensitive proteins.
MnSOD and ECSOD convert superoxide radical (O2•− ) to H2O2 and can have both antioxidant (reduction of O2•− ) and prooxidant (production of H2O2) effects. MnSOD is located in mitochondria and ECSOD is found in cell membranes/extracellular spaces. The ultimate effects of SODs on redox state depend on cell milieu, environmental factors, reducing equivalents, and/or the balance of other APs inside/outside of the cells. Recent studies in our laboratory demonstrated that modulation of MnSOD inhibited PCa growth, possibly via H2O2 production [17] whereas modulation of extracellular redox state by overexpression of ECSOD resulted in inhibition of prostate cell invasion, possibly by inhibition of matrix metalloproteinase (MMP) and membrane type 1-MMP enzymatic activities [18].
While many studies have focused on oxidative stress mediated-gene mutation, we hypothesize that redox imbalance-mediated alterations of protein function, specifically synthesis of inactive proteins or posttranslational modifications, may contribute to cancer progression. In the present study, Trx1, Prxs, MnSOD, and ECSOD were analyzed using two systems: human PCa tissues with varying degree of progression compared to their adjacent benign prostate tissues (BN) and normal prostate epithelial PrEC cells compared with androgen-responsive LNCaP and LNCaP-C4-2B and/or androgen-independent PC3 and PC3M prostate cancer cell lines.
Our results from both human PCa tissues and cell lines consistently demonstrated that redox imbalance-mediated posttranslational modifications are more extreme in highly aggressive PCa than less aggressive cancer as demonstrated by oxidation of Trx1, sulfinylation of Prxs, and nitration and methylation of MnSOD. These significant differences in cancer redox states provide new insights into the possible usefulness or dangers of prooxidant or antioxidant related cancer therapeutic drugs and radiation therapy.
Materials and Methods
Chemicals and antibodies
Microspin G-25 columns were purchased from GE Healthcare (Little Chalfont, Bucks, UK). Human purified Trx proteins, anti-GAPDH, anti-Prxs 1 to 4, anti-PrxsSO3, anti-Trx1 and anti-thioredoxin reductase 1 (TR1) antibodies were purchased from AbFrontier Co., Ltd. (Geumcheon-gu, Seoul, Korea). Anti-thioredoxin interacting protein (TXNIP) antibody was purchased from Invitrogen Corporation (Camarillo CA). Anti-CuZnSOD, MnSOD, and ECSOD antibodies were purchased from Enzo Life Sciences, Inc. (Farmingdale, NY). Anti-3-nitrotyrosine (NT) antibody was purchased from EMD-Millipore (Darmstadt, Germany). Anti-methylated lysine antibody was purchased from Abcam (Cambridge, MA). Anti-β-actin antibody was purchased from Santacruz Inc. IRDye 800CW and 700CW goat anti-rabbit IgG antibodies were purchased from Li-COR Biosciences (Lincoln, NE). All other chemicals, antibodies, and reagents were purchased from Sigma Chemical Co., unless otherwise specified.
Human cancer tissue specimens
Paired human PCa and adjacent benign tissues (BN) were purchased from the UW tissue bank (Madison, WI) with approval of the University of Wisconsin Institutional Review Board (IRB). Both human PCa and BN tissues composed of epithelial and stromal cells. Specimens were immediately frozen in liquid nitrogen and then stored at −80°C for further use. Twenty two paired human PCa tissues were included in the studies. Human prostate tissues in different stages of progression, Gleason score 4+4 to 4+5 (10 pairs) and 3+3 to 3+4 (12 pairs) were used. Gleason score represents degree of deviation from normal behavior in tumors. Gleason scores 3+3 to 3+4 is intermediate PCa grade 3 with a secondary pattern of grade 3 or 4. Gleason scores 4+4 to 4+5 is high grade PCa of Gleason pattern 4 with a secondary Gleason pattern 4 or 5, with high metastatic potential. Cancer scoring was provided by board certified pathologists. Details of each sample used in the experiment were shown in Supplementary Table 1.
Prostate cancer cells
LNCaP, PC3, and WPMY-1 cell lines were obtained from ATCC (Manassas, VA). LNCaP and PC3 cells were tested and confirmed for authenticity using short tandem repeats DNA typing by Biosynthesis Cell Inc. (Lewisville, TX). LNCaP-C4-2B (C4-2B) was obtained from ViroMed Laboratories (Minnetonka, MN). Benign prostate epithelial cells (PrEC) were obtained from Lifeline Cell Technology (Walkersville, MD). PC3M cells were obtained from Dr. Ajit Verma, University of Wisconsin-Madison. LNCaP, C4-2B, PC3, PC3M, and WPMY-1 cells were cultured in RPMI 1640 medium supplemented with 5% serum (Hyclone Laboratories Inc., Logan, Utah) and 100 mg/L kanamycin sulfate. PrEC cells were cultured in prostate epithelial growth medium (Lifeline Cell Technology). Supplies and reagents for cell cultures were purchased from BD Falcon and Thermo Fisher Scientific, unless otherwise specified.
Western Blotting and Redox Western Blotting Analysis
Cell pellets were lysed with M-PER lysis buffer containing protease inhibitor cocktails (Pierce Biotechnology, Rockford, IL) for 15 min on ice. Tissue specimens were cut into small pieces and then lysed with T-PER tissue protein extraction reagent containing protease inhibitor cocktails (Pierce Biotechnology) using a pellet pestle to homogenize tissues (Kimble Kontes LLC, Vineland, NJ). Crude supernatants were collected after centrifugation at 10,000 rpm, 4°C, for 10 min. Protein concentrations were determined using the Bradford assay (Bio-Rad, Philadelphia, PA). Equal amounts of proteins were loaded and electrophoresed in SDS polyacrylamide gels and then transferred onto nitrocellulose membranes. The following primary antibodies were used: anti-CuZnSOD (1:1,000), anti-ECSOD (1:1,000), anti-MnSOD (1:1,000), anti-Prxs 1 to 4 (1:1,000), anti-PrxsSO3 (1:1,000), anti-Trx1 (1:1,000), anti-TR1 (1:1,000), and anti-TXNIP (1:1,000). Anti-GAPDH (1:5,000) or anti-β-actin (1:1,000) antibodies were used as protein loading controls. IRDye 800CW or 680CW-conjugated goat anti-rabbit secondary antibodies (Li-Cor Biosciences, Lincoln, NE) were used. Bands were visualized using an Odyssey scanner and quantified using Odyssey analysis software (Li-Cor Biosciences). Data are presented as density of selected bands normalized by the density of GAPDH or β-actin bands. GAPDH was used to normalize protein since there are no significantly difference in protein expression levels between BN and prostate cancer tissues that were used in the experiment (Supplementary Fig 1).
Redox western blotting was previously described in Chaiswing et.al, [11]. Briefly, cells were washed with ice-cold PBS and then were immediately lysed in G-lysis buffer (6 M guanidinium chloride, 50 mM Tris–HCl, 3 mM EDTA, protease inhibitors, 0.5% Triton X-100, 50 mM iodoacetic acid (IAA), pH 8.3). For tissue specimens, tissues were cut into small pieces, lysed in T-PER tissue protein extraction reagent supplemented with 50 mM IAA, and incubated with G-lysis buffer containing 100 mM IAA. After 30 min incubation at 37°C, excess IAA from crude supernatants of cells or tissues were removed using microspin G-25 columns (GE Health Care Life Science, Piscataway, NJ). Trx1 redox isoforms were separated using native polyacrylamide gels. IRDye 800CW-conjugated goat anti-rabbit secondary antibody (Li-Cor Biosciences) was used. Bands were visualized using an Odyssey scanner and quantified using Odyssey analysis software. Data from the Trx1 redox western blotting are presented as the percentage of each Trx1 form (fully reduced (R), mixture of reduced and oxidized (Oxy1, one disulfide), and fully oxidized (Oxy2, two disulfides) forms) of the total Trx1. Dithiothreitol (DTT; 100 mM) and H2O2 (10 mM) were incubated with the lysates PCa tissues (Fig. 1A) for 30 min and were used as reduced Trx1 and oxidized Trx1 controls, respectively.
Figure 1. Redox state and levels of Trx1 and PrxsSO3 proteins in human PCa tissues.

(A) Representative and quantitative analysis of Trx1 redox western blot (intermediate grade N = 7, high grade N = 8). R, fully reduced form; Oxy1, partial oxidized forms (one disulfide); Oxy2, fully oxidized forms (two disulfides). DTT (100 mM) and H2O2 (10 mM) treated PCa samples were used as controls (reduced and oxidized forms, respectively). (B) Representative and quantitative analysis of PrxsSO3 western blot. β-actin was used as internal control (high grade N = 5). BN, adjacent benign prostate tissues; 4+4 to 4+5, high grade PCa with Gleason scores 4+4 to 4+5; 3+3 to 3+4, intermediate PCa with Gleason scores 3+3 to 3+4.
Immunoprecipitation
Cells and tissues lysates were prepared as indicated in western blot analysis. Briefly, 500 μg of total protein was used and pre-incubated for 1 hr at 4°C with 5 μl of agarose beads (ImmunoPure Immobilized Protein A/G, Pierce, Thermo Scientific) [19]. Prewashed cell lysates were incubated overnight at 4°C with either 5 μg of 3-nitrotyrosine antibody or methylated lysine antibody or normal rabbit non-immune IgG. Fifty microliters of agarose beads Protein A (Pierce) was added to antibody-treated cell lysates and incubated for 6 hr at 4°C. Agarose beads were collected by pulse centrifugation at 14,000 rpm and washed twice with PBS. Samples were analyzed according to a standard Western blot analysis procedure. Anti-MnSOD antibody was used at a 1:1,000 dilution.
MnSOD and ECSOD activity gels
SOD activity gels were performed as previously described [20]. The principle of this assay is based on the ability of O2•− to interact with nitroblue tetrazolium (NBT), reducing yellow tetrazolium within the gel to a blue precipitate. Areas where SOD is active develop a clear area (achromatic bands) as a result of competition of NBT with O2•−.
Sample preparation of cell cultures
cells were used for MnSOD and conditioned media were used for the ECSOD activity gel assays. Briefly, cells were washed with ice-cold 50 mM phosphate buffer (PB, pH 7.8) and pelleted for 5 min at 200 × g, 4°C, in 1.5 ml microcentrifuge tubes. The supernatants were removed, cells were resuspended three times in 50 mM PB, and sonicated at 40% power on ice for 45 sec (3× 15 sec) using a Branson cup horn (Danbury, CN) and Sonic Dismember sonicator model 500 (Fisher Inc.). For ECSOD activity gel assay, conditioned media were treated with 500 μg/ml heparin and concentrated using Amicon Ultra-15 (Millipore Co.).
Sample preparation for tissues
tissue specimens were cut into small pieces and were homogenized in cold PB on ice three times for 30 sec using a Polytron homogenizer (Kinematica Ag Inc., Bohemia, NY). For ECSOD activity gels, supernatants were incubated with 500 μg/ml heparin (2:1) for 15 min. The supernatants were sonicated for 1 min (2× 30 sec) using a Branson cup horn (Danbury, CN) and sonicator at 40% power.
Supernatants and media were subjected to electrophoresis on 12% (MnSOD) or 8% (ECSOD) PAGE native gels. After electrophoresis, gels were stained with NBT as previously described [20]. MnSOD can be differentiated by the presence of sodium cyanide in the staining solution, which inhibits CuZnSOD and ECSOD. The presence of a white band on the purple background with a molecular weight around 88 kDa indicates MnSOD activity, whereas a white band with molecular weight higher than 130 kDa indicates ECSOD activity. Band intensity was calculated by using Image J and was reported as relative density (RD).
Tissues/tissue microarray (TMA) construction and immunohistochemistry
Paraffin-embedded tissue blocks of prostatectomies were obtained from the Department of Pathology and Laboratory Medicine, University of Wisconsin School of Medicine and Public Health (N = 3 for each stage of prostate cancer). Approval for use of human prostate tissue was obtained from the University of Wisconsin IRB. Five micrometer paraffin sections on glass slides were deparaffinized in a 60°C oven for 1 hr and then rinsed in 3 changes of xylene for 10 min each. Slides were blocked for endogenous peroxidases with 0.3% methanol/hydrogen peroxide for 20 min and then rehydrated in 1 min changes each of 100%, 95%, 75%, and 50% ethanol, then double-distilled H2O. Heat-induced epitope retrieval was performed using a digital decloaking chamber (BioCare Medical, Concord, CA) for 30 seconds at 120°C in Tris urea buffer, pH 9.5. Slides were then blocked in 2.5% normal horse serum for 20 min at room temperature followed by incubation with primary antibodies (MnSOD 1:100) overnight at 4°C in a humidified chamber. The slides were then rinsed in 3 changes of 0.5 M Tris-buffered saline and incubated with the biotinylated universal secondary antibody (Vector Laboratories Inc., Burlingame, CA) for 30 min, followed by Immunopure Metal-enhanced DAB substrate for 3 min. After rinsing in distilled H2O, slides were counterstained with hematoxylin, dehydrated, and mounted with coverslips followed by microscopic analysis.
Trx activity assay
The insulin disulfide reduction assay was used to measure total Trx activity as previously described [21]. Briefly, equal amounts (100 μg) of protein extracts were incubated at 37°C for 15 min with 1 μl of activation buffer containing 50 mM HEPES (pH 7.6), 1 mM EDTA, 1 mg/ml bovine serum albumin, and 2 mM DTT in a total volume of 35 μL to reduce Trx. Reaction buffer (20 μl) containing 200 μl of 1 M HEPES (pH 7.6), 40 μl of 0.2 M EDTA, 40 μl of NADPH (40 mg/ml), and 500 μl of insulin (10 mg/ml) was then added to the samples. The reaction was started by the addition of 5 μl bovine TR or 5 μl water as control. The samples were incubated for 20 min at 37°C. The reaction was terminated by adding 250 μl of stop solution containing 6 M guanidine hydrochloride and 0.4 mg/ml 5, 5′-dithiobis nitrobenzoic acid in 0.2 M Tris-HCl (pH 8.0). Absorption at 412 nm was measured.
Statistics
Each experiment was performed at least three times. Statistical analysis was performed with student’s t test using SPSS10 software (SPSS Inc., Chicago, IL, USA). Mean differences were considered significant at P-value ≤ 0.05 unless otherwise specified. All data were presented as Mean ± SEM.
Results
Increased Trx1 oxidation and PrxsSO3 levels indicate alteration of intracellular redox state in high grade human PCa
We used redox western blot to analyze active-site Cys of Trx1 and to characterize cellular redox state in human PCa and corresponding BN tissues from the same patients. Trx1 redox western assay measures the redox state of two active-site Cys residues (Cys32 and Cys35) and two Cys residues outside the active site (Cys62 and Cys69), with possible results being both reduced (i.e. present as sulhydryls), both oxidized (i.e. present as disulfides), or one pair oxidized and the other reduced. Performing a Trx1 redox western thus provides two important pieces of information: i) the redox status of cells/tissues, and ii) the activity of Trx1. If both cysteines are reduced, the cells/tissues are in a reduced environment and Trx1 is active. If both cysteines are oxidized, the cells/tissues are in an oxidized environment and Trx1 is inactive.
As demonstrated in Fig. 1A, Trx1 in human PCa tissues showed multiple bands. To verify these bands as reduced and oxidized forms of Trx1, PCa tissue lysates were treated with DTT or H2O2. Both DTT treated- and H2O2 treated-samples showed multiple bands, but DTT-treated samples showed increased levels of the lowest and middle bands with decreased levels of the upper bands compared to sample with H2O2 treatment (Fig. 1A), confirming that the lowest band is reduced Trx1 and upper bands are oxidized Trx1. To completing reverse upper and middle bands to bottom band, an additional catalytic enzyme such as TR1, may be needed. Both high grade human PCa tissues (Gleason scores 4+4 to 4+5) and intermediate grade human PCa tissues (Gleason scores 3+3 to 3+4) demonstrated lower levels of reduced Trx1 compared with their adjacent BN tissues (Fig. 1A). Additionally, levels of reduced Trx1 were lower in high grade PCa tissues when compared with intermediate grade PCa tissues, implying a higher oxidized state in aggressive PCa. Notably, reduced Trx1 levels were slight decreased in BN tissues of high grade PCa tissues.
PrxsSO3 are oxidized forms of Prx proteins; an active site cysteine (Cys51) becomes oxidized mostly by H2O2 to sulfinic or sulfonic forms. Western blot analysis showed that high grade PCa tissues had higher levels of PrxsSO3 when compared to BN tissues (Fig. 1B). These data correlated with lower levels of reduced Trx1, implying progressive oxidative stress as PCa become more aggressive. To study whether redox imbalance may potentially affect selected APs, western blot analyses were performed in human PCa tissues and cells (Supplementary Fig. 2). High grade human PCa tissues demonstrated significantly higher levels of intracellular antioxidant proteins including CuZnSOD, MnSOD, Prx3, and Trx1 and 2 and lower levels of TR1 and TR2 when compared with adjacent BN tissues (Supplementary Fig. 2A). Based on western blots data, we selected to further study MnSOD, Trx1, TR, Prxs, and ECSOD.
Increased MnSOD protein levels but not enzymatic activities in high grade human PCa
Mitochondria are the major source of cellular ROS and MnSOD is known to regulate mitochondrial ROS levels. To determine if MnSOD varies in PCa tissues with varying degree of aggressiveness, MnSOD protein expression was analyzed using immunohistochemistry in human prostate tissues. Immunohistochemistry demonstrated a moderate degree of finely granular staining for MnSOD in benign epithelium, high grade prostate intraepithelial neoplasia (HGPIN), and low grade prostate adenocarcinoma cells (Fig. 2A-C). High-grade prostatic adenocarcinoma showed strong staining for MnSOD (Fig. 2D). Stromal tissues show light staining in both BN and PCa tissues. No significant staining was detected in control samples without primary antibody added (data not shown).
Figure 2. Immunohistochemistry analysis of human prostate tissues stained with anti-MnSOD.

(A) Benign prostate tissues, (B) high grade prostatic intraepithelial neoplasia (PIN), and (C) well-differentiated prostate adenocarcinoma (WCa), all showed light granular staining (arrows) for MnSOD. (D) Poorly-differentiated adenocarcinoma (PCa) showed strong cytoplasmic (arrows) staining for MnSOD. Stromal tissues show light staining in both BN and PCa with varying degree of progression (A-D). Results are representative of N = 3 for each histologic category. Ep, epithelium.
Western blot analysis showed strong correlation with MnSOD immunohistochemical staining. MnSOD protein expression levels were significantly increased in high grade PCa (~ 2 fold increase) but were not increased in intermediate grade PCa when compared to BN tissues (Fig. 3A). In order to determine if increased MnSOD protein expression resulted in increased enzymatic activities, MnSOD activity gel assay was performed. MnSOD activity was significantly increased in high grade PCa (~0.5 fold increase) but was not increased in intermediate grade PCa when compared to adjacent BN tissues (Fig. 3B). Ratios of MnSOD activity/protein were calculated. The data showed a decreased MnSOD activity/protein ratio in high grade PCa tissues when compared to BN tissues (Fig. 3D). The lower ratio of MnSOD activity/protein in high grade PCa tissues indicates an increase in nonfunctional MnSOD proteins (Supplementary Fig. 3A), which was more prominent in high grade PCa tissues.
Figure 3. Expression, enzymatic activities, and post-translational modifications of MnSOD protein in human prostate tissues.

(A) MnSOD protein expression (intermediate grade N = 8, high grade N = 6) and (B) MnSOD enzyme activity (intermediate grade N = 7, high grade N = 6) in prostate cancer tissues. (C) Representative western blots and activity gels of MnSOD. (D) Lower ratio of MnSOD activity/protein expression indicates nonfunctional protein in high grade prostate cancer tissues (intermediate grade N = 7, high grade N = 6). (E) Representative immunoprecipitation and western blots of nitrotyrosine in MnSOD protein (N = 3) of prostate cancer tissues. (F) Representative immunoprecipitation and western blots of methylated lysine in MnSOD protein (N = 3) of prostate cancer tissues. BN, adjacent benign prostate tissues; 4+4 to 4+5, high grade PCa with Gleason scores 4+4 to 4+5; 3+3 to 3+4, intermediate PCa with Gleason scores 3+3 to 3+4. RD = Relative density.
Increased nitrotyrosine and methylated lysine of MnSOD in high grade human PCa
Despite large increases in the amounts of MnSOD protein in high grade PCa tissues, the levels of MnSOD activity did not increase correspondingly compared to its protein levels (Fig. 3 D). We postulated that posttranslational modifications of MnSOD protein may be responsible for these discordant changes. This possibility was tested using a standard immunoprecipitation approach with specific antibodies against various possible posttranslational modifications. As demonstrated in Figs. 3E-F, we identified two different posttranslational modifications in MnSOD protein of PCa tissues: nitrotyrosine and methylation of lysine. These two modifications significantly increased in human PCa when compared to BN tissues.
The relative amount of methylated lysine in MnSOD protein was significantly increased (Fig. 3F) whereas the relative amount of nitration in MnSOD (Fig. 3E) was slightly decreased in high grade PCa tissues when compared to intermediated grade PCa tissues. The specificity of MnSOD-immunoprecipitation was shown by the absence of detectable nitrotyrosine-modified protein and methylated lysine of MnSOD protein in the IgG control (data not show). Notably, immunoprecipitation analysis with anti-nitrotyrosine antibody showed two bands in the region of MnSOD in both BN and PCa tissues whereas immunoprecipitation analysis with anti-methylated lysine antibody showed single band for both BN and PCa tissues.
Since nitrotyrosine [22] and methylated lysine [23] modifications in MnSOD protein have been demonstrated to lead to decreased MnSOD activity, increased nitrotyrosine residue(s) and methylated lysine in MnSOD proteins may be partially accountable for discordant MnSOD activity in PCa tissues. The coefficient of determination (R2) of nitrotyrosine or methylated lysine in MnSOD versus activity of MnSOD in high grade PCa tissues was calculated (Supplementary Table 2). The results demonstrated a greater regression relation in high grade PCa tissues than in BN tissues (R2 = 0.99 vs. 0.60 for nitrotyrosine in MnSOD and R2 = 0.95 vs. 0.01 for methylated lysine in MnSOD in high grade PCa vs. BN tissues, respectively), thus suggesting that posttranslational modifications of MnSOD may be responsible for inactive activity of MnSOD in PCa tissues.
Overexpression of Trx1 protein levels but not enzymatic activities in high grade human PCa
High grade PCa tissues demonstrated significantly higher levels of Trx1 protein (~1.5 fold increase), whereas Trx1 protein levels in intermediate grade human PCa tissues were not changed compared to their adjacent BN tissues (Fig. 4A). Analysis of Trx activity demonstrated no statistically significant difference in Trx activities in high grade PCa tissues, although the activity of Trx was increased. In contract, there was no difference in Trx activity in intermediate grade PCa tissues in comparison to their adjacent BN tissues (Fig. 4B), despite high levels of Trx protein expression in high grade PCa (Fig. 4A). This paradox may be partially explained by enzyme inactivation due to posttranslational modification (Trx1 oxidation).
Figure 4. Increased Trx1 protein levels but not enzyme activities in prostate cancer tissues of varying degrees of progression.

(A) Representative western blots and quantitative analysis of Trx1 (intermediate grade N = 11, high grade N = 10). (B) Trx activities of PCa tissues compared with BN tissues (intermediate grade N = 3, high grade N = 4). (C) Quantitative analysis of TR1 protein (intermediate grade N = 3, high grade N = 4) and (D) TXNIP protein of PCa tissues compared with BN tissues (intermediate grade N = 3, high grade N = 4). GAPDH was used as housekeeping protein since there was no significantly difference in protein expression levels between BN and prostate cancer tissues that were used in these experiments. BN, adjacent benign prostate tissues; 4+5, high grade PCa with Gleason scores 4+4 to 4+5; 3+3, intermediate PCa with Gleason scores 3+3 to 3+4.
Decreased TR1 with no change in TXNIP protein expression levels in high grade human PCa
Levels of TR1 and TXNIP expression were analyzed in PCa and BN tissues. There was a significant decrease in TR1 protein levels in high grade PCa and TR1 was also decreased (but not significantly different) in intermediate grade PCa tissues in comparison to BN tissues (Fig. 4C). However, TXNIP levels did not show significant differences in either high grade or intermediate grade PCa when compared to their adjacent BN tissues (Fig. 4D).
Prx3 protein levels were increased in high grade PCa
Levels of Prxs (Prxs 1 to 4) were also analyzed. As shown in Fig. 5, protein levels of Prxs1 and 2 were not different, but levels of Prx3 protein were significantly increased in high grade PCa tissues compared to BN tissues. In contrast, levels of Prx4 protein in high grade PCa tissues were slightly decreased compared to adjacent BN tissues. The extra band of Prx3 in high grade PCa tissues may be the hyperoxidized Prx3.
Figure 5. Redox imbalance leads to modulation of Prxs protein levels in highly aggressive prostate tissues.
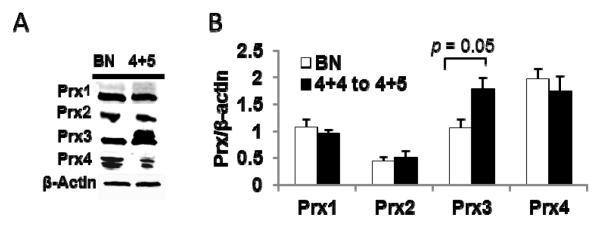
(A) Representative western blots of Prxs 1 to 4. (B) Quantification of Prxs 1 to 4 protein expression in high grade (N = 5) PCa tissues. BN, adjacent benign prostate tissues; 4+4 to 4+5, high grade PCa with Gleason scores 4+4 to 4+5.
ECSOD protein levels and activities were decreased in high grade human PCa
Protein levels and activities of ECSOD were significantly decreased in high grade PCa tissues (~ 3 and 2 folds decrease, respectively) (Fig. 6A-C). In contrast, ECSOD activities, but not protein levels, were significantly decreased (0.4 fold) in intermediate grade PCa tissues. The ratio of ECSOD activity/protein shows a slight increase in intermediate grade but not in high grade PCa tissues (Fig. 6D). Comparison of the activity/protein ratio of ECSOD in high grade vs. intermediate grade PCa tissues demonstrated a significant loss of ECSOD function with progression (Supplementary Fig. 3B). Notably, ECSOD protein levels and activities were increased in BN tissues adjacent to high grade PCa tissues.
Figure 6. Decreased ECSOD protein levels and enzyme activities in high grade prostate cancer tissues with increased ECSOD protein levels and enzyme activity in BN tissues adjacent to high grade prostate cancer.

(A) ECSOD protein expression (intermediate grade N = 4, high grade N = 4) and (B) ECSOD enzyme activities (intermediate grade N = 7, high grade N = 6) in prostate cancer tissues. (C) Representative western blots and activity gels of ECSOD. (D) Ratio of ECSOD activity/protein expression in high grade PCa compared to adjacent benign tissues (intermediate grade N = 4, high grade N = 4). BN, adjacent benign prostate tissues; 4+4 to 4+5, high grade PCa with Gleason scores 4+4 to 4+5; 3+3 to 3+4, intermediate PCa with Gleason scores 3+3 to 3+4. RD = Relative density.
Cellular redox state imbalance and decreases in protein levels and activities of MnSOD and ECSOD in highly aggressive PCa cell lines
We analyzed a tissue culture model in which normal prostate epithelial cells were compared to PCa cell lines with increasing degrees of aggressiveness. We used PrEC cells to represent normal prostate epithelial cells, less aggressive LNCaP cells to represent low grade PCa, C4-2B, to simulate intermediate grade PCa, more aggressive PC3 and PC3M cells as examples of highly aggressive PCa, and WPMY1 as an example of stromal cells.
Levels of selected antioxidant proteins were analyzed using western blot analysis. Trx1, oxidized Trx1, and Trx activity were previously studied in PrEC, LNCaP, C4-2, and PC3 cells [15] and demonstrated similar results as indicated in human PCa tissues: higher levels of Trx1 and oxidized Trx1 proteins whereas lower levels of Trx activity in PCa cell lines when compared to PrEC cells [15]. Herein, we demonstrated that Prx3 expression was significantly increased in highly aggressive PC3 cells but not low aggressive LNCaP and intermediate aggressive C4-2B cells (Supplementary Fig. 2B). Additionally, increases in MnSOD protein levels in highly aggressive PC3 and PC3M cells but not in LNCaP and C4-2B cells (Fig. 7A, left panel) were observed. MnSOD enzyme activities in PC3 and PC3M cells were significantly increased (Fig. 7B, left panel), but the ratios of MnSOD activity/protein were significantly decreased (Fig. 7C, left panel). WPMY1 cells demonstrated high levels of MnSOD protein and activity but low ratios of MnSOD activity/protein. Posttranslational modifications of MnSOD protein were analyzed in PrEC, PC3, PC3M, and WPMY1 cells using standard immunoprecipitation approach with specific antibodies to nitrotyrosine or methylated lysine. As demonstrated in Fig. 7D, immunoprecipitation with western blot analysis indicated significantly increased levels of nitrotyrosine or methylated lysine in MnSOD protein in PC3, PC3M, and WPMY1 cells when compared to PrEC cells. PC3M cells demonstrated greater levels of methylated lysine in MnSOD, whereas levels of nitrotyrosine in MnSOD were not significantly different between PC3, PC3M, and WPMY1 cells.
Figure 7. MnSOD and ECSOD protein levels and enzyme activities in prostate cancer cell lines of varying degrees of aggressiveness.

(A) Representative western blots and quantifications of MnSOD (left) and ECSOD (right) protein expression. (B) Representative activity gels and quantifications of MnSOD (left) and ECSOD (right) enzyme activity. (C) Ratios of MnSOD (left) and ECSOD (right) activity/protein expression in prostate cell lines. (D) Representative immunoprecipitation and western blots of nitrotyrosine and methylated lysine in MnSOD protein in prostate cancer cells. Numbers indicate fold increase compared to PrEC cells. RD = Relative density
ECSOD protein expression and activity were analyzed in PCa cell lines (Figs. 7A and B, right panel) and data showed that both ECSOD protein expression and activity were significantly decreased in PCa cell lines (LNCaP, C4-2B, PC3, and PC3M) when compared to PrEC cells (~5 and 2 fold decrease, respectively). Ratios of ECSOD activity/protein indicated no significant changes in comparison of cell lines (Figure 7C, right panel).
Discussion
PCa progression is a multistep process that involves genetic, environmental, aging, androgen-induced, epigenetic, and metabolic alterations. In general, treatment of metastatic cancer in humans is unsuccessful. This fact suggests that a basic understanding of cancer progression is lacking, and there may, in fact, be an underlying mechanism that causes all of the biochemical changes observed in cancer progression.
Based on studies in our laboratory [7-11] [15, 17-18] and metabolomics studies by others [24], we believe that redox imbalance-mediated posttranslational modifications occur during PCa progression. It is well known that oxidative damage products increase with age. Our laboratory documented a linear increase in 4-hydroxy-2-nonenal (4HNE) protein adducts in skeletal muscle of rhesus monkeys as a function of age [25]. 4HNE protein adducts arise largely from the reaction of superoxide anion with polyunsaturated fatty acids, with the 4HNE subsequently reacting to form covalent bonds with specific amino acid residues in proteins. Additionally, we demonstrated that reactive damage products including 4HNE protein adduct, 3-nitrotyrosine, and 8-hydroxy-2′-doxyguanosine (8OHdG) were significantly increased in PCa tissues [7-8, 26]. These results indicate oxidative stress occurred during aging and in PCa, suggesting the possibility that redox imbalance may cause posttranslational modifications in aging and cancer.
Trx1 is an oxidoreductase that produces cellular reducing equivalents for ribonucleotide reductase, Prxs, and several redox-regulated transcription factors [6]. We previously demonstrated increased nuclear Trx1 levels in high- vs. low-grade human PCas using a PCa tissue microarray [15]. PCa with high Gleason score and metastatic PCa showed intense labeling in nuclei of malignant epithelial cells, while benign or PCa with low Gleason scores showed only occasional nuclei with light staining. Additionally, PC3, LNCaP, and C4-2B cell lines demonstrated increased levels of nuclear Trx1 compared to PrEC cells [15]. Regardless of increased protein levels, the oxidized forms of nuclear Trx1 were higher in PCa cells, suggesting that nuclear redox imbalance occurred selectively in cancer cells. Thus, the data suggest the possibility of oxidation in nuclei of PCa cancer cells, results which are consistent with our previous study demonstrating increased nuclear 8OHdG in PCa tissues [8, 26].
Herein, we first analyzed tissue Trx1 protein levels in intermediate or high grade PCa vs. their BN tissue counterparts. Increasing levels of Trx1 were demonstrated in high grade and intermediate grade PCas when compared to benign tissues. To better understand how Trx1 levels are altered, protein levels of Nrf2 were determined. We found that Nrf2 protein expression was up-regulated in high grade PCa tissues (Supplementary Fig. 4A), suggesting the possibility that regulation of Trx1 occurred at the transcriptional level via Nrf2 activation. Additionally, Nrf2 and its downstream targets; Prx1, quinone oxidoreductase (NQO1), glutathione-s-transferase pi (GSTpi), TR1 and 2, and glutamate-cysteine ligase catalytic subunit (GCLC) were analyzed in PCa cells. The results showed that Nrf2 target genes were increased in highly aggressive cell lines (Supplementary Fig. 4B).
Trx1 redox western blots showed low levels of reduced Trx1 in intermediate grade PCa tissues and significantly decreased levels of reduced Trx1 in high grade PCa tissues when compared to benign tissues, suggesting significant oxidative stress in high grade PCa tissues. Analysis of Trx1 redox western blots in PCa tissues showed more bands than in PCa cell lines [27-29], indicating that redox modifications of Trx1 may be more complicated in tissues than cultured cells. Modifications on a single cysteine residue such as nitrosylation on Cys69 or Cys73 or glutathionylation of Cys73 [30] may cause different exposures of free thiol residues that can be potentially modified by IAA, resulting in additional bands in redox western blot analysis. Studies by Watson et al. showed that diamide-treated Trx1 had only one band in the redox western blot analysis, but subsequent mass spectrometry analysis demonstrated the presence of both unmodified and carboxymethylated Cys73, indicating a mixture of redox states [27]. Thus, the additional bands in our tissue redox western blots may be a result of detection of additional Trx1 redox states in tissues. Lastly, the extra bands observed in redox western blot may be derived from the stromal tissues, since PCa tissues are a mixture of epithelial and stromal cells (as indicated in Fig. 2). Further studies using mass spectrometry would be necessary to identify the specific modifications of Trx1 in these bands.
Further, we found that Trx activity was slightly but not significantly decreased in intermediate grade PCa tissues and slightly but not significantly increased in high grade PCa tissues. The absence of significant changes in Trx1 activity despite increase in protein levels may be due to active-site oxidation, changes in proteins regulating Trx1, or ischemic changes following surgery [31].
Trx1 function is regulated by TR1 and TXNIP. TR1 activates Trx1 by conversion of reversibly oxidized Trx1 to its reduced form, while TXNIP has an inhibitory effect on Trx1 activity by directly binding to Trx1 [6, 32]. Therefore, the balance of these two systems is necessary to maintain redox homeostasis of Trx1. To better understand the underlying mechanism of altered Trx1 redox states in PCa tissues, we analyzed TR1 and TXNIP protein expression levels, which showed a consistent decrease in TR1 protein levels and no significant change in TXNIP protein levels in low grade and high grade PCa tissues. A decrease in TR1 may result in increasing levels of oxidized Trx1. Santamarina et al, demonstrated that deletion of the TR gene resulted in oxidized Trx and massive thiol oxidation [33] which correlated with our data that Trx1 becomes a powerful oxidant and triggers general thiol oxidation when TR is decreased. The cause of decrease in TR protein levels is not known and needs to be further studied. Although TNXIP levels were unchanged, Trx1 conformation is altered by oxidative/nitrosative modifications of cysteine residues and may interfere with the binding capacity of TNXIP.
Prx is an antioxidant enzyme detoxifying reactive oxygen species and has a cysteine in its active site. Mammalian Prxs (Prxs 1 to 4) have two conserved Cys residues corresponding to Cys51 and Cys172 [28]. During oxidative stress or higher levels of H2O2, the active site cysteine (Cys51) is oxidized to cysteine sulfenic acid (Cys51-SOH). Because Cys51-SOH is unstable, it forms a disulfide with Cys172-SH which comes from the other subunit of the homodimer. The disulfide is then reduced back to the Prx active thiol form by the Trx-TR system. However, the formation of the disulfide is a slow process. Thus, the sulfenic intermediate (Cys51-SOH) can be easily over-oxidized to cysteine sulfinic acid (Cys-SO2H) or cysteine sulfonic acid (Cys-SO3H) before it is able to form a disulfide [34]. There are six isoforms of Prxs in mammals, with distinct cellular locations, including the cytosol (Prxs 1, 2, and 6), nucleus (Prx1), mitochondria (Prxs 3, 6), peroxisomes (Prx5), and endoplasmic reticulum/extracellular spaces (Prx4) [35]. The different Prx subcellular locations indicate possible distinct cellular functions. The typical 2-Cys Prxs (Prxs 1 to 4) are the best-characterized members of the Prx family. In addition to scavenging free radicals and ROS through the conserved cysteine residues, these Prxs also participate in multiple cell-signaling pathways, including those involving tumor suppressor proteins [36]. Since evidence implicates Prx1 as a tumor suppressor and Prxs are known to scavenge excess ROS [37-38], sulfinylation of Prxs may indicate persistent oxidative stress in high grade PCa tissues, which may contribute to redox imbalance and promote PCa progression. It has been shown that Trx1 directly transnitrosylates peroxiredoxin 1 at Cys173 and Cys83 and protects it from H2O2-induced overoxidation [39], thus, increased amounts of PrxSO3 could possibly be due to the presence of oxidized Trx1 (non-functional form), which cannot reduce PrxsSO3 back to active enzymes.
Under physiologic conditions, cells continuously produce O2•− or are exposed to H2O2. Protective antioxidant and protein repair mechanisms keep cells in a homeostatic steady state. MnSOD (located in mitochondria) and ECSOD (located in extracellular spaces) convert O2•− to H2O2. During PCa progression, we found that MnSOD protein levels were increased in human PCa tissues and in cell lines. The degree of MnSOD induction correlated with PCa degree of aggressiveness, increasing most in high grade PCa tissues and increasing less in low grade PCa tissues. The induction of MnSOD, which has both antioxidant (reduction of O2•−) and prooxidant effects (production of H2O2), is probably due to induction of oxidative stress or redox imbalance in PCa tissues. It has been shown by several investigators that MnSOD is redox-regulated both at transcriptional and translational levels [40]. Herein, we are the first to demonstrate that discordant MnSOD activity/protein ratios in highly aggressive PCa may be due to oxidative stress/redox imbalance-mediated inactivation of MnSOD protein or via posttranslational modifications of tyrosine and lysine by nitration or methylation. Notably, the levels of nitrotyrosine and methylated lysine of MnSOD appeared proportional to the protein levels in BN and high grade prostate cancer tissues, but not in intermediate grade prostate cancer tissues (3+3). As shown in Fig. 3A, levels of MnSOD in intermediate grade PCa were actually decreased compared to the BN. Thus, the increases in nitrotyrosine and methylated lysine in MnSOD are not absolutely due to the increase in MnSOD protein levels. Furthermore, our relative regression analysis indicated a greater correlation of nitrotyrosine and methylated lysine with decreased MnSOD activity in PCa tissues but not in BN tissues (Supplementary Table 2). Thus, we conclude that posttranslational modifications of MnSOD are, at least in part, responsible for inactivation of MnSOD in PCa tissues.
Among relevant oxidative modifications of MnSOD, the nitration of the amino acid tyrosine to 3-nitrotyrosine has been recognized in vitro and in vivo. Despite the protein containing nine tyrosine residues per monomer, posttranslational modifications promoted by peroxynitrite and other nitrating species in MnSOD occur site-specifically at Tyrosine 34 and inactivates its enzyme activity [40]. Nitrated MnSOD has been found in several pathophysiologic processes, including ischemia/reperfusion, inflammation, and apoptosis [19, 41]. It is worth noting that immunoprecipitation analysis of nitrotyrosine on MnSOD revealed two bands in region of MnSOD in both BN and PCa tissues (Fig. 3E). The significant of these results is not clear; it could represent proteolysis, splicing variants, mixtures of PCa epithelial and prostate stromal cells, and/or variables of posttranslational modifications in MnSOD. Alternatively, our antibody may recognize two proteins with one of them being MnSOD. However cross reaction of the antibody is unlikely since nitrotyrosine of MnSOD in PCa cell lines only showed a single band (Fig. 7D).
Epigenetic silencing of MnSOD has been demonstrated in breast cancer through hypoacetylation and hypomethylation of histones, thus forming a closed chromatin structure and inhibiting transcription factor function [42]. Herein, methylation of lysine residues in MnSOD was significantly increased in highly aggressive PCa. Methylation-dependent changes in MnSOD conformation have been demonstrated to regulate its enzymatic activity during transitions between quiescence and proliferation and influences glucose consumption of mouse embryonic fibroblasts. Mass spectrometry analysis of fibroblast cells demonstrated methylation of lysine (68, 89, 122, and 202) and arginine (197 and 216) residues of MnSOD may induce distinct conformational changes in the accessible and the electrostatic potential around the active site, regulating enzyme activities [23]. Searching for lysine residues that modulate MnSOD activity in PCa will be necessary in future studies to better understand PCa progression.
Our study demonstrated that ECSOD protein expression and activity were significantly decreased in intermediate grade and high grade PCa tissues and cell lines. Levels of mRNA SOD3 were slightly but not significantly increased in PCa and BN (data not shown); analysis of more samples is needed to confirm this result. It has been shown that ECSOD expression in lung cancer was decreased due to promoter methylation and loss of heterozygosity. ECSOD loss may contribute to extracellular matrix remodeling and malignant progression of lung cancer [43]. Treatment of PC3 cells with a DNA methyltransferase inhibitor, 5-aza-2′-deoxycytidine (5 μM), for 96 hr resulted in a moderate induction of ECSOD protein expression (Supplementary Fig. 4C), suggesting that DNA methylation of the ECSOD promoter could be at least contributing to the loss of ECSOD expression in PCa cell lines. Epigenetic (methyltransferase/acetyltranferase) and posttranslational modifications due to redox imbalance could possibly develop at varying stages of cancer progression depending on the unique biochemistry of each specific cancer type [44]. Overexpression of ECSOD enhanced the growth- and invasion-inhibitory effects of heparin/low molecular weight heparin in breast cancer cells [45]. We previously demonstrated that NADPH oxidase-mediated O2•− was overexpressed in PCa cell lines PC3 and WPE1-NB26 [18]. Overexpression of ECSOD significantly decreased PC3 and WPE1-NB26 migration and invasion via inhibition of MMP2 and MT1-MMP activities [18]. We hypothesize that PCa progression involves an increased flux of O2•− in cell membrane/extracellular spaces, thus postulating a central for ECSOD in the development of PCa metastasis.
Interestingly, ECSOD protein levels and enzyme activities were significantly higher in BN tissues adjacent to high grade PCa tissues than in BN tissues adjacent to intermediate grade PCa and both intermediate and high grade PCa tissues. These changes are correlated with the increasing of oxidized Trx1 in BN tissues adjacent to high grade PCa (Fig. 1A). Recent study demonstrated that the noncancerous tissues and extracellular matrix of neuroblastoma-bearing mice became highly oxidized and sensitive to oxidative damage [3]. In fact, prostate stromal cells, WPMY 1 demonstrated similar redox imbalance found in PCa cells, suggesting a possibility of PCa cells inducing redox imbalance in neighboring cells. Alternatively, redox imbalance in WPMY1 may precede malignancy of neighboring cancer cells (Fig. 7A-D). More change of redox state in nearby high grade prostate cancer may induce DNA methylation in BN tissues, thus subsequently alters redox state in BN tissues as a preventive mechanism. Nevertheless, the significances and mechanisms behind this finding are still unknown, and future studies will be necessary to better explain this phenomenon.
In summary, our study is the first to identify redox imbalance (oxidative stress) mediated-posttranslational modification of selected APs in human PCa tissues with varying degrees of aggressiveness. Trx1 and MnSOD showed increased protein levels without corresponding increase in enzyme activities in high grade PCa. The loss of Trx1 function was at least partially due to oxidation of active-site cysteines. The loss of MnSOD function in high PCa was at least partially due to nitrotyrosine and methylated lysine. Sulfinylation of Prxs in high grade PCa was demonstrated and may reflect and/or contribute to redox imbalance. ECSOD protein and activity levels were decreased in PCa tissues, possibly via an epigenetic mechanism, creating a redox imbalance between cancer cells and extracellular spaces. Epigenetic changes may be caused by and contribute to redox imbalance [46]. The proposed mechanisms of how these changes driven cancer progression are proposed in Figure 8. Nevertheless, our results suggest that redox imbalance may contribute to PCa progression and PCa cell culture models can be used to study redox imbalance mediated cancer progression since their relative redox states correlate with human PCa tissues.
Figure 8. Propose mechanism(s) of how alterations in intra- and extra-cellular redox states induce PCa progression.

In general, intracellular redox state is more reduced in benign prostate tissues (BN). At the intermediate grade stage, environmental, metabolic, androgens, aging, and epigenetic stressors induce influx of ROS/RNS inside the cells which subsequently up-regulates MnSOD, Trx1, and Prxs as protective mechanisms against oxidative stress. Persistent influx of ROS/RNS pushes these antioxidant proteins beyond their limits, which results in oxidative stress-mediated posttranslational modifications of cysteine residues in Trx1 and Prxs, and nitration of tyrosine and methylation of lysine in MnSOD. At the high grade stage, Trx1, Prxs, and MnSOD become non-functional, resulting in a greater intracellular oxidized state and subsequent activation of Nrf2 and its downstream targets e.g. HO-1 and others which are involved in cancer progression. On the other note, influx of ROS/RNS causes DNA methylation which inhibits the ECSOD activity and increases levels of O2•− in high grade cancers. During metastasis, these key antioxidant proteins may play roles in maintaining oxidized state for cancer progression (1) or protecting against oxidative stress for the survival of metastatic cancer cells (2).
Supplementary Material
Acknowledgments
The contents do not represent the views of the U.S. Department of Veterans Affairs. This work was supported in part by funds from the University of Wisconsin, Department of Pathology Research and Development Committee, University of Wisconsin-Madison Graduate School, UW Carbone Cancer Center (NIH grant P30 CA014520), NIH grants RO1 CA07359902 and RO1 CA09485301 to Daret St. Clair (TDO; Co-I), a 2010 mini-fellowship award from the Society for Free Radical Biology & Medicine, Weihua Shan for her technical assistance in redox western experiments, and resources and facilities at the William S. Middleton Memorial Veterans Hospital (Madison, WI, USA).
List of Abbreviations
- 4HNE
4-hydroxy-2-nonenal
- 8OHdG
8-hydroxy-2′-doxyguanosine
- AP
Antioxidant protein
- Cys
Cysteine
- CySS
Cystine
- DTT
Dithiothreitol
- ECSOD
Extracellular superoxide dismutase
- GSH
Glutathione
- IAA
Iodoacetic acid
- MnSOD
Manganese superoxide dismutase
- MMP
Matrix metalloproteinase
- Nrf2
Nuclear factor-erythoid 2-related factor 2
- PCa
Prostate cancer
- Prx
Peroxiredoxin
- PrxsSO3
Peroxiredoxins sulfinylation
- Redox
Reduction/oxidation
- ROS/RNS
Reactive oxygen species/Reactive nitrogen species
- Trx1
Thioredoxin 1
- TR1
Thioredoxin reductase 1
- TXNIP
Thioredoxin interacting protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J. 2007;401:1–11. doi: 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- [2].Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol. 2010;38:96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- [3].Bakalova R, Zhelev Z, Aoki I, Saga T. Tissue redox activity as a hallmark of carcinogenesis: from early to terminal stages of cancer. Clin Cancer Res. 2013;19:2503–2517. doi: 10.1158/1078-0432.CCR-12-3726. [DOI] [PubMed] [Google Scholar]
- [4].Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- [5].Brown NS, Bicknell R. Hypoxia and oxidative stress in breast cancer. Oxidative stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res. 2001;3:323–327. doi: 10.1186/bcr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mukherjee A, Martin SG. The thioredoxin system: a key target in tumour and endothelial cells. Br J Radiol. 2008;81(Spec No 1):S57–68. doi: 10.1259/bjr/34180435. [DOI] [PubMed] [Google Scholar]
- [7].Oberley TD, Zhong W, Szweda LI, Oberley LW. Localization of antioxidant enzymes and oxidative damage products in normal and malignant prostate epithelium. Prostate. 2000;44:144–155. doi: 10.1002/1097-0045(20000701)44:2<144::aid-pros7>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- [8].Bostwick DG, Alexander EE, Singh R, Shan A, Qian J, Santella RM, Oberley LW, Yan T, Zhong W, Jiang X, Oberley TD. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer. Cancer. 2000;89:123–134. [PubMed] [Google Scholar]
- [9].Chaiswing L, Bourdeau-Heller JM, Zhong W, Oberley TD. Characterization of redox state of two human prostate carcinoma cell lines with different degrees of aggressiveness. Free Radic Biol Med. 2007;43:202–215. doi: 10.1016/j.freeradbiomed.2007.03.031. [DOI] [PubMed] [Google Scholar]
- [10].Chaiswing L, Zhong W, Oberley TD. Distinct Redox Profiles of Selected Human Prostate Carcinoma Cell Lines: Implications for Rational Design of Redox Therapy. Cancers (Basel) 2011;3:3557–3584. doi: 10.3390/cancers3033557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chaiswing L, Zhong W, Liang Y, Jones DP, Oberley TD. Regulation of prostate cancer cell invasion by modulation of extra- and intracellular redox balance. Free Radic Biol Med. 2012;52:452–461. doi: 10.1016/j.freeradbiomed.2011.10.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Meyer AJ, Dick TP. Fluorescent protein-based redox probes. Antioxid Redox Signal. 2010;13:621–650. doi: 10.1089/ars.2009.2948. [DOI] [PubMed] [Google Scholar]
- [13].Holmgren A, Sengupta R. The use of thiols by ribonucleotide reductase. Free Radic Biol Med. 2010;49:1617–1628. doi: 10.1016/j.freeradbiomed.2010.09.005. [DOI] [PubMed] [Google Scholar]
- [14].Go YM, Jones DP. Thioredoxin redox western analysis. Curr Protoc Toxicol. 2009 doi: 10.1002/0471140856.tx1712s41. Chapter 17:Unit17 12. [DOI] [PubMed] [Google Scholar]
- [15].Shan W, Zhong W, Zhao R, Oberley TD. Thioredoxin 1 as a subcellular biomarker of redox imbalance in human prostate cancer progression. Free Radic Biol Med. 2010;49:2078–2087. doi: 10.1016/j.freeradbiomed.2010.10.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Miki H, Funato Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J Biochem. 2012;151:255–261. doi: 10.1093/jb/mvs006. [DOI] [PubMed] [Google Scholar]
- [17].Li N, Zhai Y, Oberley TD. Two distinct mechanisms for inhibition of cell growth in human prostate carcinoma cells with antioxidant enzyme imbalance. Free Radic Biol Med. 1999;26:1554–1568. doi: 10.1016/s0891-5849(99)00024-6. [DOI] [PubMed] [Google Scholar]
- [18].Chaiswing L, Zhong W, Cullen JJ, Oberley LW, Oberley TD. Extracellular redox state regulates features associated with prostate cancer cell invasion. Cancer Res. 2008;68:5820–5826. doi: 10.1158/0008-5472.CAN-08-0162. [DOI] [PubMed] [Google Scholar]
- [19].Kim A, Oberley LW, Oberley TD. Induction of apoptosis by adenovirus-mediated manganese superoxide dismutase overexpression in SV-40-transformed human fibroblasts. Free Radic Biol Med. 2005;39:1128–1141. doi: 10.1016/j.freeradbiomed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- [20].Weydert CJ, Cullen JJ. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat Protoc. 2010;5:51–66. doi: 10.1038/nprot.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Y, De Keulenaer GW, Lee RT. Vitamin D(3)-up-regulated protein-1 is a stress-responsive gene that regulates cardiomyocyte viability through interaction with thioredoxin. J Biol Chem. 2002;277:26496–26500. doi: 10.1074/jbc.M202133200. [DOI] [PubMed] [Google Scholar]
- [22].MacMillan-Crow LA, Crow JP, Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998;37:1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]
- [23].Sarsour EH, Kalen AL, Xiao Z, Veenstra TD, Chaudhuri L, Venkataraman S, Reigan P, Buettner GR, Goswami PC. Manganese superoxide dismutase regulates a metabolic switch during the mammalian cell cycle. Cancer Res. 2012;72:3807–3816. doi: 10.1158/0008-5472.CAN-11-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, Nyati MK, Ahsan A, Kalyana-Sundaram S, Han B, Cao X, Byun J, Omenn GS, Ghosh D, Pennathur S, Alexander DC, Berger A, Shuster JR, Wei JT, Varambally S, Beecher C, Chinnaiyan AM. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- [25].Zainal TA, Oberley TD, Allison DB, Szweda LI, Weindruch R. Caloric restriction of rhesus monkeys lowers oxidative damage in skeletal muscle. FASEB J. 2000;14:1825–1836. doi: 10.1096/fj.99-0881com. [DOI] [PubMed] [Google Scholar]
- [26].Oberley TD. Oxidative damage and cancer. Am J Pathol. 2002;160:403–408. doi: 10.1016/S0002-9440(10)64857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem. 2003;278:33408–33415. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- [28].Halvey PJ, Watson WH, Hansen JM, Go YM, Samali A, Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem J. 2005;386:215–219. doi: 10.1042/BJ20041829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol. 2006;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- [30].Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Biophys Biomol Struct. 2001;30:421–455. doi: 10.1146/annurev.biophys.30.1.421. [DOI] [PubMed] [Google Scholar]
- [31].Best S, Sawers Y, Fu VX, Almassi N, Huang W, Jarrard DF. Integrity of prostatic tissue for molecular analysis after robotic-assisted laparoscopic and open prostatectomy. Urology. 2007;70:328–332. doi: 10.1016/j.urology.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Watson WH, Yang X, Choi YE, Jones DP, Kehrer JP. Thioredoxin and its role in toxicology. Toxicol Sci. 2004;78:3–14. doi: 10.1093/toxsci/kfh050. [DOI] [PubMed] [Google Scholar]
- [33].Garcia-Santamarina S, Boronat S, Calvo IA, Rodriguez-Gabriel M, Ayte J, Molina H, Hidalgo E. Is oxidized thioredoxin a major trigger for cysteine oxidation? Clues from a redox proteomics approach. Antioxid Redox Signal. 2013;18:1549–1556. doi: 10.1089/ars.2012.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rhee SG, Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H(2)O(2), and protein chaperones. Antioxid Redox Signal. 2011;15:781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- [35].Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- [36].Immenschuh S, Baumgart-Vogt E. Peroxiredoxins, oxidative stress, and cell proliferation. Antioxid Redox Signal. 2005;7:768–777. doi: 10.1089/ars.2005.7.768. [DOI] [PubMed] [Google Scholar]
- [37].Hoshino I, Matsubara H, Akutsu Y, Nishimori T, Yoneyama Y, Murakami K, Sakata H, Matsushita K, Ochiai T. Tumor suppressor Prdx1 is a prognostic factor in esophageal squamous cell carcinoma patients. Oncol Rep. 2007;18:867–871. [PubMed] [Google Scholar]
- [38].Brigelius-Flohe R, Flohe L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal. 2011;15:2335–2381. doi: 10.1089/ars.2010.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wu C, Liu T, Chen W, Oka S, Fu C, Jain MR, Parrott AM, Baykal AT, Sadoshima J, Li H. Redox regulatory mechanism of transnitrosylation by thioredoxin. Mol Cell Proteomics. 2010;9:2262–2275. doi: 10.1074/mcp.M110.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yamakura F, Kawasaki H. Post-translational modifications of superoxide dismutase. Biochim Biophys Acta. 2010;1804:318–325. doi: 10.1016/j.bbapap.2009.10.010. [DOI] [PubMed] [Google Scholar]
- [41].Cruthirds DL, Novak L, Akhi KM, Sanders PW, Thompson JA, MacMillan-Crow LA. Mitochondrial targets of oxidative stress during renal ischemia/reperfusion. Arch Biochem Biophys. 2003;412:27–33. doi: 10.1016/s0003-9861(03)00039-0. [DOI] [PubMed] [Google Scholar]
- [42].Hitchler MJ, Oberley LW, Domann FE. Epigenetic silencing of SOD2 by histone modifications in human breast cancer cells. Free Radic Biol Med. 2008;45:1573–1580. doi: 10.1016/j.freeradbiomed.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Teoh-Fitzgerald ML, Fitzgerald MP, Jensen TJ, Futscher BW, Domann FE. Genetic and epigenetic inactivation of extracellular superoxide dismutase promotes an invasive phenotype in human lung cancer by disrupting ECM homeostasis. Mol Cancer Res. 2012;10:40–51. doi: 10.1158/1541-7786.MCR-11-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jeronimo C, Bastian PJ, Bjartell A, Carbone GM, Catto JW, Clark SJ, Henrique R, Nelson WG, Shariat SF. Epigenetics in prostate cancer: biologic and clinical relevance. Eur Urol. 2011;60:753–766. doi: 10.1016/j.eururo.2011.06.035. [DOI] [PubMed] [Google Scholar]
- [45].Teoh ML, Fitzgerald MP, Oberley LW, Domann FE. Overexpression of extracellular superoxide dismutase attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion. Cancer Res. 2009;69:6355–6363. doi: 10.1158/0008-5472.CAN-09-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal. 2011;15:551–589. doi: 10.1089/ars.2010.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.