

Abstract
Magnolia as an herbal material obtained from Magnolia officinalis has been found to play an important role in anti-inflammation, antioxidative stress, and antiapoptosis. This study was designed to investigate the effect of Magnolia extract (BL153) on obesity-associated lipid accumulation, inflammation, oxidative stress, and apoptosis in the heart. C57BL/6 mice were fed a low- (10 kcal% fat) or high-fat (60 kcal% fat) diet for 24 weeks to induce obesity. These mice fed with high-fat diet (HFD) were given a gavage of vehicle, 2.5, 5, or 10 mg/kg body weight BL153 daily. The three doses of BL153 treatment slightly ameliorated insulin resistance without decrease of body weight gain induced by HFD feeding. BL153 at 10 mg/kg slightly attenuated a mild cardiac hypertrophy and dysfunction induced by HFD feeding. Both 5 mg/kg and 10 mg/kg of BL153 treatment significantly inhibited cardiac lipid accumulation measured by Oil Red O staining and improved cardiac inflammation and oxidative stress by downregulating ICAM-1, TNF-α, PAI-1, 3-NT, and 4-HNE. TUNEL staining showed that BL153 treatment also ameliorated apoptosis induced by mitochondrial caspase-3 independent cell death pathway. This study demonstrates that BL153 attenuates HFD-associated cardiac damage through prevention of HFD-induced cardiac lipid accumulation, inflammation, oxidative stress, and apoptosis.
1. Introduction
In recent years, the number of the obese people increased significantly. Obesity typically results from overeating and lack of enough exercise, which is a growing global health problem [1]. Obesity is associated with many cardiovascular diseases (CVD). Hyperlipidemia, inflammation, oxidative stress, myocardial apoptosis, lipid metabolic disorder, and insulin resistance are all important pathological factors for the increased CVD in both diabetic and obese patients [2–4]. Currently, there are many therapeutic strategies for diabetes and its complications, such as glycemic management, diet control, and exercise [5]. However, no single approach is able to efficiently prevent obesity associated CVD.
Natural products with significant antidiabetes and antiobesity efficacy, but without toxic effects, are very attractive for the preventive and therapeutic purposes [6]. Magnolia officinalis is a natural product used for the empirical treatment of diabetes in Korea [7]. Honokiol (HON) and magnolol (MAG), 4-O-methylhonokiol and obovatol isolated from the stem bark of Magnolia plants, are considered as the major bioactive constituents [8]. Recently, the studies demonstrated that constituents of Magnolia such as HON and MAG have anti-inflammatory [9–11], -oxidative [12, 13], and -apoptotic effects [13, 14]. Moreover, another study showed that MAG reduced fasting blood glucose and plasma insulin levels in type 2 diabetic model without altering body weight [15] and increased glucose uptake in 3T3-L1 adipocytes [16]. However, the effects of Magnolia officinalis on heart of obesity induced by HFD still remain unclear.
In the present study, we investigated whether Magnolia officinalis extract prevents cardiac lipid accumulation, inflammation, oxidative stress, and apoptosis in the heart of obese mice induced by HFD.
2. Material and Methods
2.1. Magnolia Extract (BL153)
Magnolia extract (BL153) was prepared by Bioland Co., Ltd. (Chungnam, Korea) and dissolved in 0.5% ethanol as previously reported [17].
2.2. Animal Models
C57BL/6J male mice, 8 weeks of age, were purchased from the Jackson Laboratory (Bar Harbor, Maine) and housed in the University of Louisville Research Resources Center at 22°C with a 12 h light/dark cycle with free access to standard rodent chow and tap water. All experimental procedures for these animals were approved by the Institutional Animal Care and Use Committee of the University of Louisville, which is compliant with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85-23, revised in 1996).
A Total of 25 mice were randomly assigned into 5 groups with 5 mice per group as follows: Ctrl (control) group, fed control diet of 10 kcal% from fat (D12450B, Research Diets Inc. 3.85 kcal/g) and given a gavage of the vehicle (0.5% ethanol); HFD group, fed the high-fat diet (HFD) of 60 kcal% from fat (D12492, Research Diets Inc. 5.24 kcal/g) and given a gavage of the vehicle; HFD + 2.5 mg/kg group, fed HFD and given a gavage of 2.5 mg/kg body weight of BL153; HFD + 5 mg/kg group, fed HFD and given a gavage of 5 mg/kg body weight of BL153; HFD + 10 mg/kg group, fed HFD and given a gavage of 10 mg/kg body weight of BL153. All mice were fed the corresponding diet and treated with BL153 or vehicle as described above simultaneously for 24 weeks. Energy intake and body weight were monitored daily or weekly, respectively. After insulin tolerance test and blood pressure and echocardiography measurements, mice were sacrificed. The hearts were isolated and weighted, and blood plasma was collected. Average energy intake was calculated following the diet formula (D12450B or D12492, Research Diets Inc.): per mice/day (kcal) = (food (g) intake/cage/day) × 3.85 (control diet) or 5.24 (HFD)/(mice/per cage).
2.3. Intraperitoneal Insulin Tolerance Test (IPITT)
IPITT was conducted after 24 weeks of high-fat diet feeding. For IPITT [18], mice (n = 5 per group) were fasted overnight (14 h), weighed, and then injected with human insulin (Humulin R; Eli Lilly, Indianapolis, IN) intraperitoneally at a dose of 1 unit/kg body weight. Blood glucose levels at 0, 15, 30, 60, and 120 min after insulin injection were measured using a FreeStyle Lite glucometer (Abbott Diabetes Care, Alameda, CA). Area under the curve (AUC) was calculated by the trapezoid rule for the insulin tolerance curve using Origin 7.5 software (OriginLab Corporation, Northampton, MA).
2.4. Noninvasive Blood Pressure (BP)
BP was measured by tail-cuff plethysmography using a CODA6 noninvasive BP monitoring system (Kent Scientific, Torrington, CT) as previously reported [19]. Mice (n = 5 per group) were restrained in a plastic tube restrainer. Occlusion and volume-pressure recording (VPR) cuffs were placed over the tail, and the mice were allowed to adapt to the restrainer for 5 min prior to starting BP measurement. After a 5 min adaptation period, BP was measured for 10 acclimation cycles followed by 20 measurement cycles. Mice were warmed by heating pads during the acclimation cycles to ensure sufficient blood flow to the tail. The animals were monitored closely throughout the measurement protocol and removed from restraint as soon as possible upon completing the measurement protocol. After three days of training for the BP measurement, formal measurements were performed and systolic, mean, and diastolic BP and heart rate data were collected.
2.5. Echocardiography
Transthoracic echocardiography (Echo) was detected for Avertin anesthetized mice by high-resolution imaging system (Vevo 770, VisualSonics, Canada) equipped with a high-frequent ultrasound probe (RMV-707B) as described in previous study [20]. The chests of the mice (n = 5 per group) were treated with a chemical hair remover to reduce ultrasound attenuation. Parasternal long-axis and short-axis views were acquired. Left ventricular (LV) dimensions and wall thicknesses were determined from parasternal short axis M-mode images. The anesthetized heart rate was collected. Meanwhile, ejection fraction (EF), fractional shortening (FS), and LV mass were calculated by Vevo770 software. The final data represent averaged values of 10 cardiac cycles.
2.6. Oil Red O Staining for Lipid Accumulation in Heart
Cryosections (10 μm) from OCT-embedded heart tissue samples were fixed in 10% buffered formalin for 5 min and slides were washed in water. Then the slides were immersed in 60% isopropanol for 30 s and incubated in Oil Red O solution for 1 h at room temperature. The slides were washed with 60% isopropanol twice and then counterstained with hematoxylin (DAKO, Carpinteria, CA, USA) for 30 s. Excess hematoxylin was washed in water. A Nikon microscope (Nikon, Melville, NY) was used to capture the Oil Red O-stained tissue sections.
2.7. Plasma Triglyceride Quantification
Concentration of triglyceride in the plasma was measured according to the manufacturers' instruction provided in the triglyceride colorimetric assay kit (Cayman Chemical, CA).
2.8. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling (TUNEL) Assay
Apoptotic nuclei in the heart were examined by TUNEL staining using ApopTag In Situ kit (Chemicon, Temecula, CA). Heart tissue was fixed in buffered neutral 10% formalin, dehydrated in graded alcohol series, embedded in paraffin, and sectioned at 4 μm. Deparaffinized and hydrated slides were used for TUNEL staining according to the manufacturer's instructions. TUNEL positive nuclei cells were counted under high power field (HPF, 20x) in five random fields for each of three slides from one mouse with at least five mice (as indicated) in each group and presented as TUNEL positive nuclei per HPF as described in previous study [21].
2.9. Western Blot Assay
Briefly, heart tissues were homogenized with RIPA lysis buffer (Santa Cruz Biotechnology, CA). Total proteins were extracted and separated on SDS-PAGE gels, and then proteins were transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). The membrane was blocked with 5% nonfat dried milk for 1 h and then incubated overnight at 4°C with the following primary antibodies: intercellular adhesion molecule 1 (ICAM-1, Santa Cruz Biotechnology, CA), tumor necrosis factor alpha (TNF-α, Abcam, Cambridge, MA), plasminogen activator inhibitor-1 (PAI-1, BD Bioscience, Sparks, MD), 3-nitrotyrosine (3-NT, Millipore, Billerica, MA), 4-hydroxy-2-nonenal (4-HNE, Alpha Diagnostic International, San Antonio, TX), B-cell lymphoma 2 (BCL-2) and BCL2-associated X protein (BAX) (Cell Signaling, MA), cleaved-caspase-8 (Cell Signaling, MA), apoptosis inducing factor (AIF) (Cell Signaling, MA), caspase-12 (Exalpha Biologicals, MA), C/EBP homologous protein (CHOP; Santa Cruz Biotechnology, CA) GAPDH (Santa Cruz Biotechnology, CA), and cleaved-caspase-3 (Cell Signaling, MA). After three washes with Tris-buffered saline (pH 7.2) containing 0.1% Tween 20 (TBST), membranes were incubated with appropriate secondary antibodies for 1 h at room temperature. Antigen-antibody complexes were then visualized using an enhanced chemiluminescence kit (Thermo Scientific, Rockford, IL) [22].
2.10. Quantitative Analysis of Lipid Peroxides
The lipid peroxide concentration was measured by detecting thiobarbituric acid (TBA) reactivity reflected by the amount of malondialdehyde (MDA) formed during acid hydrolysis of the lipid peroxide compound. The reaction mixtures included 20 μL 8.1% sodium dodecyl sulfate, 50 μL protein sample, 210 μL 0.571% TBA, and 150 μL 20% acetic acid solution (pH 3.5). Each sample was duplicated. The reaction mixtures were incubated at 90°C for 1 h. After cooling down on ice, 100 μL distilled water was added and centrifuged at 4000 rpm for 15 min. Then the absorbance of each sample at 540 nm was measured by using 150 μL supernatant at a microplate reader.
2.11. Statistical Analysis
Data were collected from 5 mice per group and presented as means ± SD as indicated. We used Image Pro Plus 6.0 software (Media Cybernetics, Bethesda, MA) to identify the positive staining and Image Quant 5.2 software (GE Healthcare Bio-Sciences AB) to analyze Western blot. Comparisons were performed by one-way ANOVA for the different groups, followed by post hoc pairwise repetitive comparisons using Tukey's test with Origin 8.0 Lab data analysis and graphing software (Origin Lab Corporation, Northampton, MA). Statistical significance was considered as P < 0.05.
3. Results
3.1. The Effects of BL153 on Body Weight and Energy Intake in HFD Feeding Mice
HFD feeding gradually and significantly increased the body weight in HFD group compared to the control diet feeding group (Figure 1(a)). Low dose of BL153 (2.5 mg/kg) treatment had no significant effects on HFD feeding induced body weight increases compared to HFD group (Figure 1(a)). Both 5 mg/kg and 10 mg/kg of BL153 treatment slightly increased the body weight between 8 and 21 weeks compared to HFD group (Figure 1(a)). Energy intake for each group was calculated and it was found that the energy intake in HFD fed mice was significantly higher than that of control diet fed mice (Figure 1(b)). BL153 treatment had no significant effects on HFD-induced energy intake changes (Figure 1(b)). HFD feeding significantly increased heart weight (Figure 1(c)) and BL153 administration also had no significant effects on HFD increased heart weight (Figure 1(c)).
Figure 1.
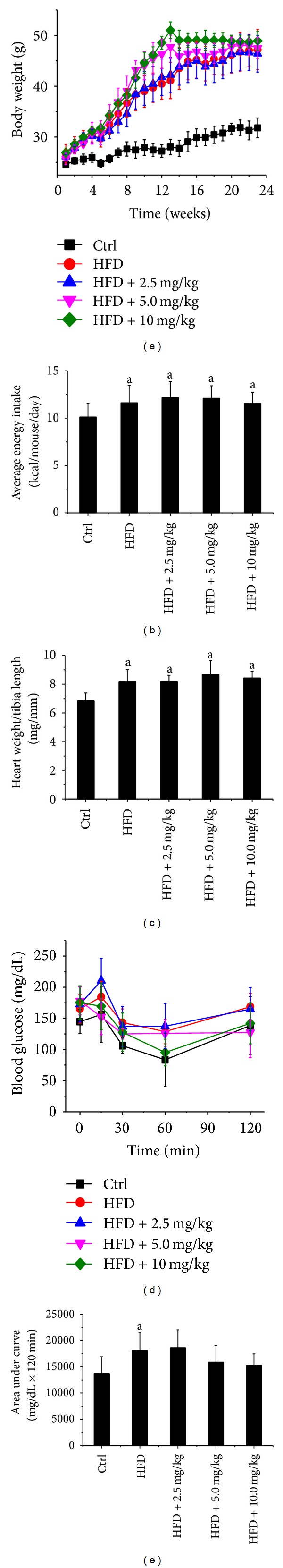
The effects of BL153 on body weight, energy intake, heart weight, and lung weight. Male C57BL/6J mice at 8 weeks of age were fed either a LFD (10% kcal as fat) or HFD (60% kcal as fat) with or without indicated dose of BL153 (2.5, 5, or 10 mg/kg body weight) for 24 weeks. (a) Body weight; (b) average energy intake for 12 weeks; (c) heart weight/tibia length ratio; and (d) insulin tolerance test (IPITT) as well as (e) area under the curves (AUC) after 20 weeks of control diet or HFD feeding. Data were presented as means ± SD (n = 5). a P < 0.05 versus control; b P < 0.05 versus HFD. Ctrl: control; HFD: high-fat diet.
3.2. The Effects of BL153 on HFD-Induced Insulin Resistance
To evaluate the effects of long term BL153 administration on whole body glucose metabolism, IPITT was performed at 24 weeks after HFD feeding. HFD feeding significantly attenuated the insulin sensitivity in HFD group, while 5 mg/kg and 10 mg/kg of BL153 slightly prevented HFD-induced insulin sensitivity attenuation (Figures 1(d) and 1(e)).
3.3. The Effects of BL153 on HFD-Induced Cardiac Function Changes
Echo examination found that HFD feeding slightly, but not significantly, decreased cardiac function, including increases in LVID; d (LV end diastolic diameter), LVID; s (LV end systolic diameter), LV vol; s (LV end systolic volume), LV vol; d (LV end diastolic volume), and LV mass and decreases in ejection faction and fraction shortening in HFD group compared to control diet group. BL153 (10 mg/kg) administration prevented HFD-induced cardiac function changes (Table 1). Both HFD feeding and BL153 treatments had no significant effects on the diastolic, systolic, and mean BP and heart rate compared to control diet feeding group (Table 1).
Table 1.
The effects of BL153 on cardiac function in HFD feeding mice.
| Control | HFD | HFD + 2.5 mg/kg | HFD + 5.0 mg/kg | HFD + 10 mg/kg | |
|---|---|---|---|---|---|
| IVS; d (mm) | 0.72 ± 0.03 | 0.71 ± 0.03 | 0.69 ± 0.02 | 0.70 ± 0.03 | 0.70 ± 0.0232 |
| LVID; d (mm) | 3.26 ± 0.18 | 3.61 ± 0.25 | 3.61 ± 0.17 | 3.68 ± 0.21 | 3.72 ± 0.23 |
| LVPW; d (mm) | 0.80 ± 0.10 | 0.85 ± 0.08 | 0.99 ± 0.08 | 0.89 ± 0.18 | 0.96 ± 0.10 |
| IVS; s (mm) | 1.10 ± 0.02 | 1.16 ± 0.15 | 1.03 ± 0.12 | 1.13 ± 0.17 | 1.19 ± 0.10 |
| LVID; s (mm) | 1.37 ± 0.23 | 1.72 ± 0.36 | 1.77 ± 0.35 | 1.82 ± 0.25 | 1.60 ± 0.19 |
| LVPW; s (mm) | 0.50 ± 0.24 | 1.48 ± 0.34 | 1.74 ± 0.25 | 1.55 ± 0.26 | 1.79 ± 0.20 |
| LV vol; d (μL) | 43.16 ± 5.55 | 55.49 ± 9.08 | 55.05 ± 5.90 | 57.83 ± 8.07 | 59.40 ± 8.56 |
| LV vol; s (μL) | 4.98 ± 2.04 | 9.40 ± 4.69 | 10.06 ± 4.73 | 10.54 ± 4.03 | 7.60 ± 2.29 |
| %EF | 88.76 ± 3.29 | 82.59 ± 10.14 | 81.71 ± 8.58 | 82.18 ± 4.45 | 87.44 ± 3.15 |
| %FS | 58.24 ± 4.59 | 52.25 ± 11.21 | 50.99 ± 10.22 | 50.80 ± 4.69 | 57.13 ± 4.15 |
| LV mass (mg) | 79.02 ± 3.31 | 97.10 ± 15.86 | 106.85 ± 13.03 | 101.68 ± 18.27 | 110.94 ± 18.34 |
| HR (beats/min) | 697.93 ± 53.40 | 677.98 ± 50.60 | 689.18 ± 34.90 | 675.86 ± 36.11 | 667.95 ± 27.90 |
| Systolic BP (mmHg) | 110.92 ± 5.13 | 113.09 ± 3.71 | 97.90 ± 11.34 | 98.79 ± 10.41 | 109.39 ± 5.48 |
| Diastolic BP (mmHg) | 79.69 ± 6.99 | 77.30 ± 5.65 | 69.75 ± 10.10 | 70.68 ± 10.27 | 79.34 ± 4.75 |
| Mean BP (mmHg) | 89.76 ± 6.35 | 88.92 ± 3.82 | 78.79 ± 10.46 | 79.73 ± 10.30 | 89.03 ± 4.84 |
Notes. IVS: interventricular septum; LVID; d: left ventricular end diastolic diameter; LVID; s: left ventricular end systolic diameter; LVPW: left ventricular posterior wall; EF: ejection fraction; FS: fractional shortening; LV vol; s: left ventricular end systolic volume; LV vol; d: left ventricular end diastolic volume; LV mass: left ventricular mass. HR: heart rate; BP: blood pressure. Data were presented as means ± SEM (n = 5).
3.4. BL153 Attenuated HFD-Induced Cardiac Lipid Accumulation
Chronic obesity is associated with whole body and organ specific lipid metabolic disorder [23]. As expected, HFD feeding significantly increased triglyceride level in plasma in HFD group compared to the control diet feeding group. BL153 treatment significantly attenuated plasma triglyceride levels only at the dose of 10 mg/kg compared to HFD feeding group (Figure 2(a)). Correspondingly, Oil Red O staining revealed that HFD significantly increased cardiac lipid accumulation compared to control diet feeding group. BL153 administration at all dose levels significantly, but not completely, prevented HFD feeding-induced cardiac lipid accumulation compared to HFD fed group (Figure 2(b)).
Figure 2.
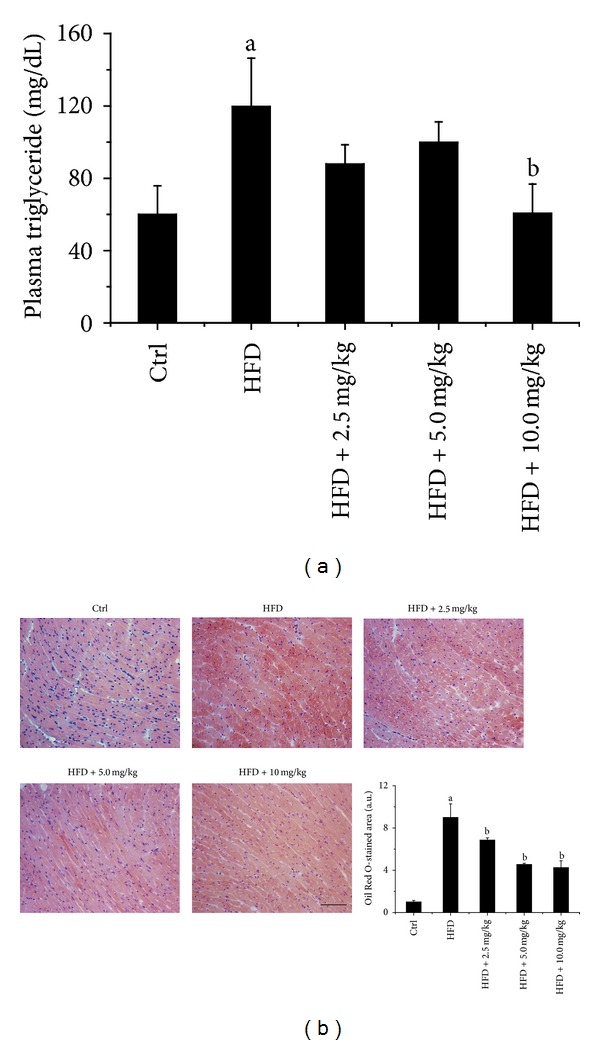
BL153 attenuated lipid accumulation induced by HFD in heart and plasma triglyceride. (a) Level of triglyceride in plasma was measured by a triglyceride colorimetric assay kit; (b) Lipid accumulation in heart was examined by Oil Red O staining (bar = 50 μm). Data were presented as means ± SD (n = 5). a P < 0.05 versus control; b P < 0.05 versus HFD group.
3.5. BL153 Attenuated HFD-Induced Cardiac Inflammation
Inflammation is elevated in obese rodents and humans, which is also an important contributor to the development of insulin resistance [24]. Thus, we detected the markers of inflammation in cardiac tissue. We found that HFD feeding significantly increased cardiac ICAM-1, TNF-α, and PAI-1 expression in HFD feeding mice (Figure 3), and BL153 significantly inhibited the inflammatory markers upregulation, while no significant differences between three dose levels of BL153 administration were observed (Figure 3).
Figure 3.
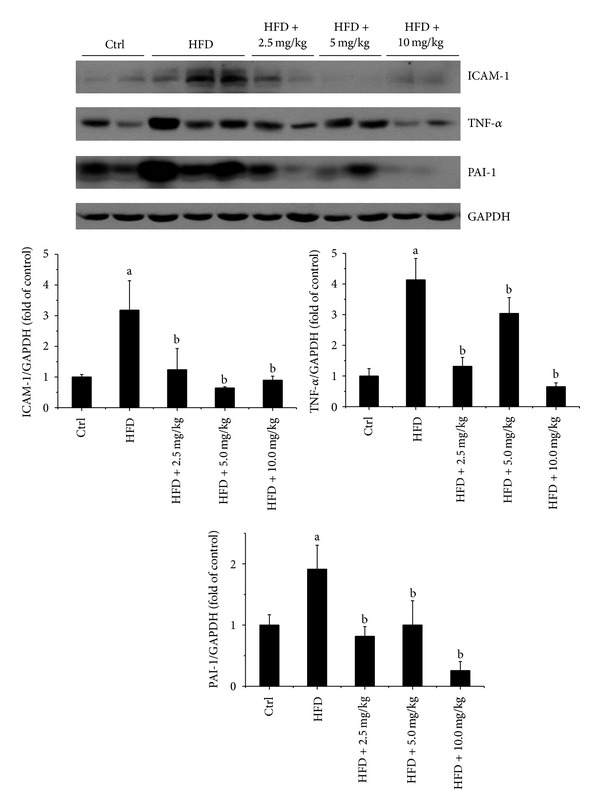
BL153 attenuated HFD-induced cardiac inflammation. The protein levels of inflammatory markers ICAM-1, TNF-α, and PAI-1 were examined by Western blot assay and quantified by densitometric analysis. GAPDH was used as loading control. Data were presented as means ± SD (n = 5). a P < 0.05 versus control group; b P < 0.05 versus HFD group.
3.6. BL153 Attenuated HFD-Induced Cardiac Oxidative Stress
Obesity is associated with oxidative stress in heart and liver [25]. So we investigated whether BL153 administration can prevent oxidative stress induced by HFD feeding. We measured the makers of oxidative stress including 3-NT, 4-HNE, and MDA, and found that the accumulation of 3-NT, 4-HNE (Figure 4(a)), and MDA (Figure 4(b)) in the heart was significantly enhanced by HFD feeding in HFD group, and both 5 mg/kg and 10 mg/kg of BL153 significantly prevented HFD-induced cardiac oxidative stress accumulation (Figures 4(a) and 4(b)).
Figure 4.
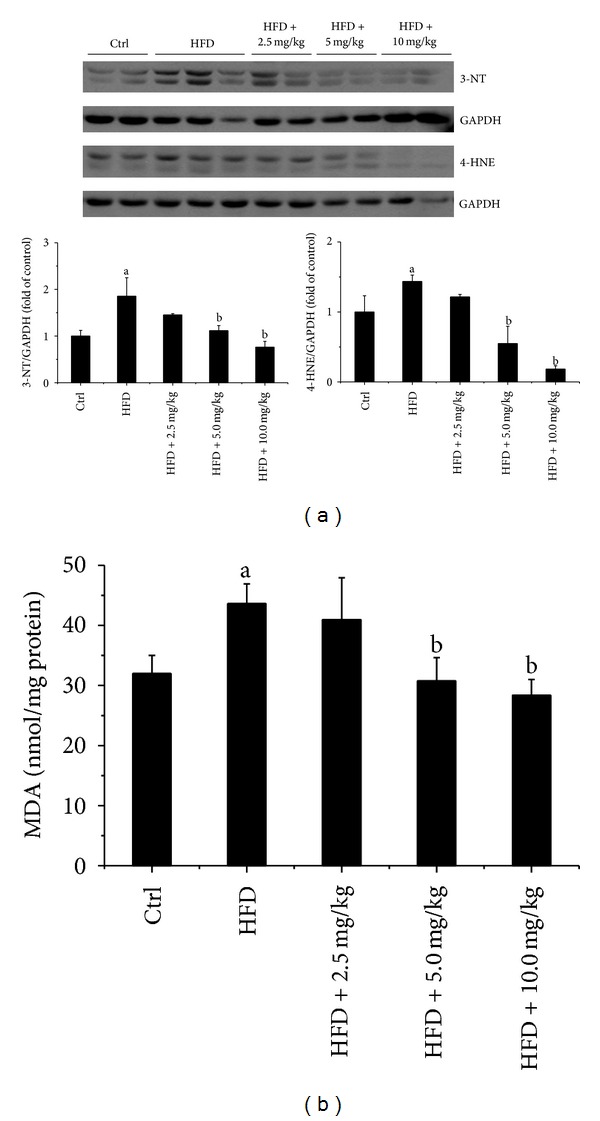
BL153 attenuated HFD-induced cardiac oxidative stress. (a) Cardiac oxidative stress was measured by Western blot examination of 3-NT and 4-HNE accumulation and quantified by densitometric analysis; (b) lipid peroxides (MDA) in cardiac tissue were evaluated by thiobarbituric acid reactivity assay. Data were presented as means ± SD (n = 5). a P < 0.05 versus control; b P < 0.05 versus HFD group.
3.7. BL153 Prevented HFD-Induced Cardiac Apoptotic Cell Death
Reportedly, obesity related cardiac lipid accumulation and oxidative stress are important contributors to cardiac cell death and dysfunction in rodent and human [26]. In the current study, the cardiac cell death was detected by TUNEL staining (Figure 5). We found that HFD feeding significantly induced cardiac cell death in HFD fed mice, and BL153 treatment significantly, even not completely, prevented HFD-induced cardiac apoptotic cell death in all of the three dose levels of BL153 treated mouse.
Figure 5.
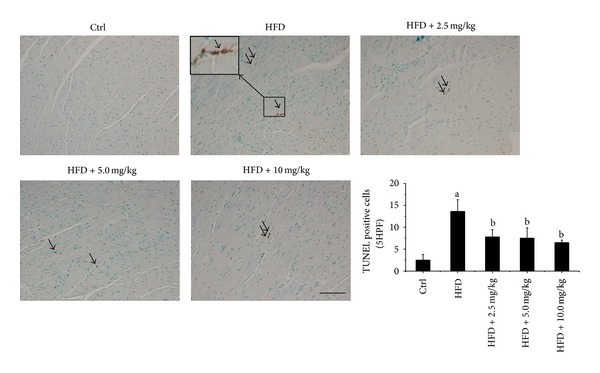
BL153 prevented HFD-induced cardiac apoptotic cell death. Myocardial apoptotic cell death was examined by TUNEL staining (Bar = 50 μm). Arrows indicate TUNEL positive nuclei. The results were expressed as average number of TUNEL positive nuclei per five high power field under a microscopy. Data were means ± SD (n = 5). a P < 0.05 versus control; b P < 0.05 versus HFD group.
Endoplasmic reticulum (ER) and/or mitochondrial-associated cell death pathways may be involved in cardiac cell death under different pathological conditions [27, 28]. In order to investigate the signaling changes that may be involved in HFD-induced cardiac apoptotic cell death, both ER stress and mitochondrial-associated cell death pathway related molecular markers were examined by Western blot assay (Figures 6 and 7). Interestingly, HFD feeding both with and without BL153 treatment had no significant effects on the expression of ER stress related cell death markers including caspase-12, CHOP, and cleaved-caspase-3 (Figure 6), which indicated that HFD-induced cardiac apoptosis was independent of ER stress associated cell death signals. While HFD feeding significantly increased mitochondrial-associated cardiac cell death signals including the upregulation of cleaved-caspase-8 and Bax to Bcl-2 expression ratio in HFD group, and BL153 treatment completely prevented HFD-induced these cardiac cell death signals (Figure 7). The release of AIF from mitochondrial activated apoptotic cell death is independent of caspase-3 pathway [29]. The Western blot results revealed that HFD feeding significantly elevated cardiac AIF expression in HFD fed hearts and BL153 completely prevented HFD induced cardiac AIF upregulation (Figure 7), which indicates that HFD-induced cardiac cell death is predominantly attributed to mitochondrial-associated pathway and AIF/caspase-8/Bax/Bcl-2 apoptotic cascade is a critical target for BL153 prevention of HFD-induced cardiac cell apoptosis.
Figure 6.
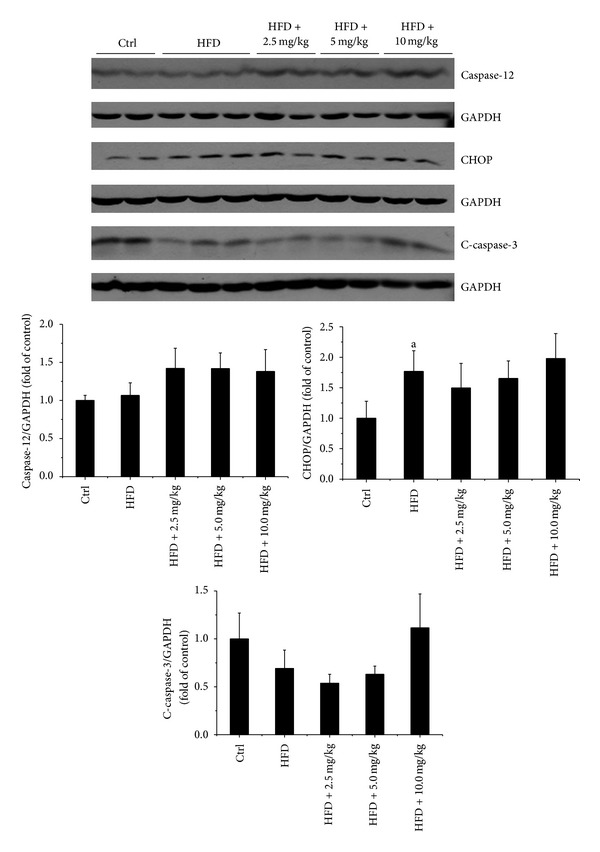
The effects of HFD and BL153 on ER stress-associated cell death makers. ER stress-associated cell death markers including CHOP, caspase-12, and C-caspase-3 (cleaved-caspase-3) were examined by Western blot assay and quantified by densitometric analysis. Data were presented as mean ± SD (n = 5). a P < 0.05 versus control; b P < 0.05 versus HFD group.
Figure 7.
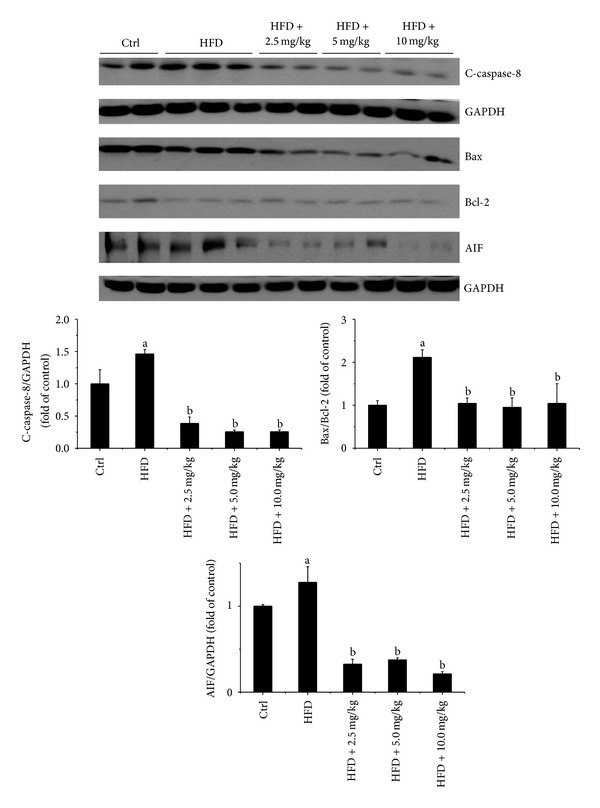
BL153 prevented HFD-induced cardiac apoptosis via activation of the mitochondrial cell death pathway. The expression of C-caspase-8 (cleaved-caspase-8), Bax, Bcl-2, and AIF in the heart was detected by Western blot assay and quantified by densitometric analysis. Data were presented as mean ± SD (n = 5). a P < 0.05 versus control. b P < 0.05 versus HFD group.
4. Discussion
The present study demonstrated that mice fed an HFD developed obesity phenotype, which was characterized by increased body weight, insulin resistance, and high level of triglyceride in the blood. The treatment of BL153 slightly ameliorated insulin resistance and decreased body weight. HFD feeding also resulted in significant lipid accumulation in the cardiac tissue accompanied by mild cardiac hypertrophy and slight cardiac function changes (Table 1), along with significant increases of cardiac inflammation (Figure 3), oxidative stress (Figure 4), and cardiac cell death (Figure 5), and the treatment of BL153 significantly prevented the above-mentioned cardiac detrimental changes (Figures 3–5).
Several studies have investigated cardiac lipotoxicity in HFD feeding induced and genetically manipulated rodent models [30–32]. Rats fed 60 kcal% fat diet for 6 weeks slightly elevated cardiac triglyceride levels [33]. We found a six fold increase in cardiac lipid accumulation by Oil Red O staining after 24 weeks of HFD feeding in mice, indicating that long term exposure to a HFD can induce significant cardiac lipid accumulation. Interestingly, treatment with BL153 significantly attenuated HFD feeding-induced cardiac lipid accumulation (Figure 2). This is the first study to show that BL153 exerts a function of cardiac lipid metabolic regulation in HFD-induced obesity.
Obesity is associated with cardiac hypertrophy and cardiac dysfunction, which is largely attributed to many pathophysiological conditions such as chronic inflammation, oxidative stress, mitochondrial dysfunction and apoptosis of cardiomyocytes [34–36]. HFD feeding-induced lipid accumulation evoked cardiac oxidative stress, inflammation and, eventually, cardiac dysfunction in animals [37]. It is known that the role of oxidative stress is postulated in many cardiovascular diseases [38]. Reactive oxygen species can promote inflammation, and vice versa, and both induce apoptotic cell death [39, 40]. Magnolia constituents including HON and MAG have been evaluated as antioxidants [41, 42] and anti-inflammation agents [10]. Our study also demonstrated for the first time that treatment with BL153 significantly ameliorated HFD-induced cardiac inflammatory responses including decreases of PAI-1, TNF-α, and ICAM-1 expression (Figure 3) and oxidative stress including decreases of 3-NT, 4-HNE, and MDA accumulation (Figure 4) and consequently prevented HFD-induced mild cardiac hypertrophy and cardiac function changes (Table 1). The anti-inflammatory and antioxidative mechanisms of Magnolia extract are proven to be associated with the decrease of nitric oxide (NO) production, the expression of inducible NO synthase (iNOS), TNF-α, and cyclooxygenases, and the activation of NF-κB [43, 44].
A commonly proposed mechanism for lipid-induced cardiomyocyte death is via activation of caspases by ceramide, a potent proapoptotic by product of lipid metabolism, which elevates the expression of iNOS, thereby enhancing NO and peroxynitrite formation [45]. Incubation of cultured neonatal rat cardiomyocytes with palmitate causes a decrease in the ability of these cells to oxidize fatty acids, an increase in cellular malonyl-CoA, and a decrease in the activity of AMP-activated protein kinase (AMPK) compared to myocytes incubated in the presence of oleate [46]. While palmitate decreases the oxidative metabolism of fatty acids, it increases the formation of intracellular triglyceride and ceramide [46]. Increased ceramide formation is associated with an increase in apoptosis along with an increase in caspase-3 activity and DNA-laddering in many cell systems [46]. Our TUNEL staining results revealed that cardiac lipid accumulation significantly increased cardiac apoptotic cell death (Figure 5), which is consistent with the previous reports [47]. However, the protein levels of CHOP and caspase-12, as makers of ER stress, were similar in all groups (Figure 6). We also did not observe any significant change of caspase-3 cleavage (Figure 6). Therefore, the cardiac apoptotic cell death pathway induced by HFD in the present study may be ER stress and caspase-3 independent [48]. Relling et al. have demonstrated that HFD feeding-induced cardiac dysfunction was mitochondria dependent [48]. Our previous study also demonstrated that hyperglycemia-derived oxidative stress mediates a mitochondrial cytochrome c-mediated caspase-3 activation pathway playing an important role in diabetes-induced cardiac cell death [49, 50]. With the consistence, here cardiac lipid accumulation was found to induce cardiac apoptotic cell death associated with mitochondrial cell death pathway, shown by increased expression ratio of Bax to Bcl-2 expression along with the increased AIF expression. Several studies also have shown the fact that induction of cell death is caspase-3 independent in vitro and in vivo [51–53]. Taken together, the previous studies and our findings indicate that cardiac lipid accumulation induced cell death through upregulating inflammation and oxidative stress is associated with caspase-3 independent pathway.
Jin et al. demonstrated that MAG, one of the major bioactive constituents in Magnolia extract, at the dose of 10 mg/kg via intraperitoneal injection prevented myocardial ischemia and reperfusion injury through prevention of cardiomyocytes apoptosis [54]. Consistent with this study, our study also demonstrated for the first time that BL153 significantly inhibited HFD feeding-induced cardiac apoptotic cell death by prevention of cardiac lipid accumulation-induced cardiac inflammation and oxidative stress.
In summary, our study demonstrated that BL153 attenuates HFD-associated cardiac damage through prevention of HFD induced cardiac lipid accumulation, inflammation, oxidative stress, and mitochondria caspase-3 independent cell death pathway.
Acknowledgments
This study was supported in part by Chungbuk Technopark Grant Bio-International Collaborating Research, funded by Chungbuk province, Republic of Korea, a collaborative project between University of Louisville and Bioland Co. Ltd. for “Screening Antidiabetes and/or Obesity Naturally Extracted Compounds,” funded by Bioland Co., Ltd. (10-0826 Bioland), a Research Development Fund of Wenzhou Medical University (QTJ 13007), a Junior Faculty Award (1-13-JF-53) from American Diabetes Association, and the National Natural Science Foundation of China and a Key Science and Technology Development Plan from Wenzhou City (81273509, 81370318, 20100001).
Conflict of Interests
The authors declare that they have no conflict of interests.
Authors' Contribution
Lu Cai, Qiang Chen, and Yi Tan conceived and designed the experiments. Weixia Sun, Zhiguo Zhang, and Qiang Chen performed the experiments. Weixia Sun, Qiang Chen, and Zhiguo Zhang analyzed the data. Yaowen Fu, Yang Zheng, Ki-Soo Kim, Ki Ho Kim, and Young Heui Kim contributed reagents/materials/analysis tools. Zhiguo Zhang, Yi Tan, Lu Cai, Yaowen Fu, Yang Zheng, Ki-Soo Kim, Ki Ho Kim, and Young Heui Kim wrote and reviewed the paper. Weixia Sun and Zhiguo Zhang equally contributed to this study.
References
- 1.Rössner S. Obesity: the disease of the twenty-first century. International Journal of Obesity. 2002;26(4):S2–S4. doi: 10.1038/sj.ijo.0802209. [DOI] [PubMed] [Google Scholar]
- 2.Jang H, Conklin DJ, Kong M. Piecewise nonlinear mixed-effects models for modeling cardiac function and assessing treatment effects. Comput Methods Programs Biomed. 2013;110(3):240–252. doi: 10.1016/j.cmpb.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Subramanian S, Turner MS, Ding Y, et al. Increased levels of invariant natural killer T lymphocytes worsens metabolic abnormalities and atherosclerosis in obese mice. Journal of Lipid Research. 2013;54(10):2831–2841. doi: 10.1194/jlr.M041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Younce CW, Burmeister MA, Ayala JE. Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis via inhibition of endoplasmic reticulum stress and activation of SERCA2a. American Journal of Physiology. 2013;304(6):C508–C518. doi: 10.1152/ajpcell.00248.2012. [DOI] [PubMed] [Google Scholar]
- 5.Ryden L, Grant PJ, Anker SD, et al. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: The Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD) European Heart Journal. 2013;34(39):3035–3087. doi: 10.1093/eurheartj/eht108. [DOI] [PubMed] [Google Scholar]
- 6.Li WL, Zheng HC, Bukuru J, de Kimpe N. Natural medicines used in the traditional Chinese medical system for therapy of diabetes mellitus. Journal of Ethnopharmacology. 2004;92(1):1–21. doi: 10.1016/j.jep.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 7.Kim YS, Jung DH, Kim NH, et al. Effect of magnolol on TGF-beta1 and fibronectin expression in human retinal pigment epithelial cells under diabetic conditions. European Journal of Pharmacology. 2007;562(1):12–19. doi: 10.1016/j.ejphar.2007.01.048. [DOI] [PubMed] [Google Scholar]
- 8.Maruyama Y, Kuribara H. Overview of the pharmacological features of honokiol. CNS Drug Reviews. 2000;6(1):35–44. [Google Scholar]
- 9.Lee J, Jung E, Park J, et al. Anti-inflammatory effects of magnolol and honokiol are mediated through inhibition of the downstream pathway of MEKK-1 in NF-κB activation signaling. Planta Medica. 2005;71(4):338–343. doi: 10.1055/s-2005-864100. [DOI] [PubMed] [Google Scholar]
- 10.Kane JS, Lee KH, Han MH, et al. Antiinflammatory activity of methanol extract isolated from stem bark of Magnolia kobus. Phytotherapy Research. 2008;22(7):883–888. doi: 10.1002/ptr.2386. [DOI] [PubMed] [Google Scholar]
- 11.Kassuya CAL, Cremoneze A, Barros LFL, et al. Antipyretic and anti-inflammatory properties of the ethanolic extract, dichloromethane fraction and costunolide from Magnolia ovata (Magnoliaceae) Journal of Ethnopharmacology. 2009;124(3):369–376. doi: 10.1016/j.jep.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka K, Hasegawa J, Asamitsu K, Okamoto T. Magnolia ovovata extract and its active component magnolol prevent skin photoaging via inhibition of nuclear factor κB. European Journal of Pharmacology. 2007;565(1–3):212–219. doi: 10.1016/j.ejphar.2007.01.095. [DOI] [PubMed] [Google Scholar]
- 13.Chang W-C, Yu Y-M, Hsu Y-M, et al. Inhibitory effect of Magnolia officinalis and lovastatin on aortic oxidative stress and apoptosis in hyperlipidemic rabbits. Journal of Cardiovascular Pharmacology. 2006;47(3):463–468. doi: 10.1097/01.fjc.0000211708.03111.6e. [DOI] [PubMed] [Google Scholar]
- 14.Wu X-N, Yu C-H, Cai W, Hua J, Li S-Q, Wang W. Protective effect of a polyphenolic rich extract from Magnolia officinalis bark on influenza virus-induced pneumonia in mice. Journal of Ethnopharmacology. 2011;134(1):191–194. doi: 10.1016/j.jep.2010.11.074. [DOI] [PubMed] [Google Scholar]
- 15.Sohn EJ, Kim C-S, Kim YS, et al. Effects of magnolol (5,5′-diallyl-2,2′-dihydroxybiphenyl) on diabetic nephropathy in type 2 diabetic Goto-Kakizaki rats. Life Sciences. 2007;80(5):468–475. doi: 10.1016/j.lfs.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 16.Choi S-S, Cha B-Y, Lee Y-S, et al. Magnolol enhances adipocyte differentiation and glucose uptake in 3T3-L1 cells. Life Sciences. 2009;84(25-26):908–914. doi: 10.1016/j.lfs.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Cui W, Wang Y, Chen Q, et al. Magnolia extract (BL153) ameliorates kidney damage in a high fat diet-induced obesity mouse model. Oxidative Medicine and Cellular Longevity. 2013;2013:9 pages. doi: 10.1155/2013/367040.367040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ize-Ludlow D, Lightfoot YL, Parker M, et al. Progressive erosion of β-cell function precedes the onset of hyperglycemia in the NOD mouse model of type 1 diabetes. Diabetes. 2011;60(8):2086–2091. doi: 10.2337/db11-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan Y, Li X, Prabhu SD, et al. Angiotensin II plays a critical role in alcohol-induced cardiac nitrative damage, cell death, remodeling, and cardiomyopathy in a protein kinase c/nicotinamide adenine dinucleotide phosphate oxidase-dependent manner. Journal of the American College of Cardiology. 2012;59(16):1477–1486. doi: 10.1016/j.jacc.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai Y, Cui WP, Xin Y, et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. Journal of Molecular and Cellular Cardiology. 2013;57:82–95. doi: 10.1016/j.yjmcc.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Jiang X, Zhang C, Xin Y, et al. Protective effect of FGF21 on type 1 diabetes-induced testicular apoptotic cell death probably via both mitochondrial-and endoplasmic reticulum stress-dependent pathways in the mouse model. Toxicology Letters. 2013;219(1):65–76. doi: 10.1016/j.toxlet.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 22.Zhou G, Li X, Hein DW, et al. Metallothionein suppresses angiotensin II-induced nicotinamide adenine dinucleotide phosphate oxidase activation, nitrosative stress, apoptosis, and pathological remodeling in the diabetic heart. Journal of the American College of Cardiology. 2008;52(8):655–666. doi: 10.1016/j.jacc.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 23.Després J-P, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444(7121):881–887. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- 24.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 25.Abbas AM, Sakr HF. Simvastatin and vitamin E effects on cardiac and hepatic oxidative stress in rats fed on high fat diet. Journal of Physiology and Biochemistry. 2013;69(4):737–750. doi: 10.1007/s13105-013-0250-y. [DOI] [PubMed] [Google Scholar]
- 26.Listenberger LL, Schaffer JE. Mechanisms of lipoapoptosis: implications for human heart disease. Trends in Cardiovascular Medicine. 2002;12(3):134–138. doi: 10.1016/s1050-1738(02)00152-4. [DOI] [PubMed] [Google Scholar]
- 27.Crow MT, Mani K, Nam Y-J, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circulation Research. 2004;95(10):957–970. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 28.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nature Reviews Drug Discovery. 2008;7(12):1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 29.Candé C, Vahsen N, Garrido C, Kroemer G. Apoptosis-inducing factor (AIF): caspase-independent after all. Cell Death and Differentiation. 2004;11(6):591–595. doi: 10.1038/sj.cdd.4401400. [DOI] [PubMed] [Google Scholar]
- 30.Ouwens DM, Boer C, Fodor M, et al. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia. 2005;48(6):1229–1237. doi: 10.1007/s00125-005-1755-x. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y-T, Grayburn P, Karim A, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(4):1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christoffersen C, Bollano E, Lindegaard MLS, et al. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144(8):3483–3490. doi: 10.1210/en.2003-0242. [DOI] [PubMed] [Google Scholar]
- 33.Unger RH. Lipotoxic diseases. Annual Review of Medicine. 2002;53:319–336. doi: 10.1146/annurev.med.53.082901.104057. [DOI] [PubMed] [Google Scholar]
- 34.Westermann D, van Linthout S, Dhayat S, et al. Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Research in Cardiology. 2007;102(6):500–507. doi: 10.1007/s00395-007-0673-0. [DOI] [PubMed] [Google Scholar]
- 35.Tan Y, Ichikawa T, Li J, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60(2):625–633. doi: 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pagano C, Calcagno A, Granzotto M, et al. Heart lipid accumulation in obese non-diabetic rats: effect of weight loss. Nutrition, Metabolism and Cardiovascular Diseases. 2008;18(3):189–197. doi: 10.1016/j.numecd.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. Journal of Clinical Investigation. 2005;115(3):500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ungvári Z, Gupte SA, Recchia FA, Bátkai S, Pacher P. Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Current Vascular Pharmacology. 2005;3(3):221–229. doi: 10.2174/1570161054368607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takano H, Zou Y, Hasegawa H, Akazawa H, Nagai T, Komuro I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: involvement of ROS in heart diseases. Antioxidants and Redox Signaling. 2003;5(6):789–794. doi: 10.1089/152308603770380098. [DOI] [PubMed] [Google Scholar]
- 40.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circulation Research. 1999;85(8):753–766. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- 41.Kang KA, Lee KH, Kim SY, Kim HS, Kim JS, Hyun JW. Cytoprotective effects of KIOM-79 on streptozotocin induced cell damage by inhibiting ERK and AP-1. Biological and Pharmaceutical Bulletin. 2007;30(5):852–858. doi: 10.1248/bpb.30.852. [DOI] [PubMed] [Google Scholar]
- 42.Haraguchi H, Ishikawa H, Shirataki N, Fukuda A. Antiperoxidative activity of neolignans from Magnolia obovata. Journal of Pharmacy and Pharmacology. 1997;49(2):209–212. doi: 10.1111/j.2042-7158.1997.tb06781.x. [DOI] [PubMed] [Google Scholar]
- 43.Oh JH, Kang LL, Ban JO, et al. Anti-inflammatory effect of 4-O-methylhonokiol, a novel compound isolated from Magnolia officinalis through inhibition of NF-κB. Chemico-Biological Interactions. 2009;180(3):506–514. doi: 10.1016/j.cbi.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 44.Chiang J, Shen Y-C, Wang Y-H, et al. Honokiol protects rats against eccentric exercise-induced skeletal muscle damage by inhibiting NF-κB induced oxidative stress and inflammation. European Journal of Pharmacology. 2009;610(1-3):119–127. doi: 10.1016/j.ejphar.2009.03.035. [DOI] [PubMed] [Google Scholar]
- 45.Szczepaniak LS, Victor RG, Orci L, Unger RH. Forgotten but not gone: the rediscovery of fatty heart, the most common unrecognized disease in America. Circulation Research. 2007;101(8):759–767. doi: 10.1161/CIRCRESAHA.107.160457. [DOI] [PubMed] [Google Scholar]
- 46.Hickson-Bick DLM, Buja ML, McMillin JB. Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. Journal of Molecular and Cellular Cardiology. 2000;32(3):511–519. doi: 10.1006/jmcc.1999.1098. [DOI] [PubMed] [Google Scholar]
- 47.Okere IC, Chandler MP, McElfresh TA, et al. Differential effects of saturated and unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose distribution, and serum leptin. American Journal of Physiology—Heart and Circulatory Physiology. 2006;291(1):H38–H44. doi: 10.1152/ajpheart.01295.2005. [DOI] [PubMed] [Google Scholar]
- 48.Relling DP, Esberg LB, Fang CX, et al. High-fat diet-induced juvenile obesity leads to cardiomyocyte dysfunction and upregulation of Foxo3a transcription factor independent of lipotoxicity and apoptosis. Journal of Hypertension. 2006;24(3):549–561. doi: 10.1097/01.hjh.0000203846.34314.94. [DOI] [PubMed] [Google Scholar]
- 49.Cai L, Wang Y, Zhou G, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. Journal of the American College of Cardiology. 2006;48(8):1688–1697. doi: 10.1016/j.jacc.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 50.Cai L, Li W, Wang G, Guo L, Jiang Y, James Kang Y. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. 2002;51(6):1938–1948. doi: 10.2337/diabetes.51.6.1938. [DOI] [PubMed] [Google Scholar]
- 51.Asmis R, Begley JG. Oxidized LDL promotes peroxide-mediated mitochondrial dysfunction and cell death in human macrophages: a caspase-3-independent pathway. Circulation research. 2003;92(1):e20–e29. doi: 10.1161/01.res.0000051886.43510.90. [DOI] [PubMed] [Google Scholar]
- 52.An D, Kewalramani G, Chan JKY, et al. Metformin influences cardiomyocyte cell death by pathways that are dependent and independent of caspase-3. Diabetologia. 2006;49(9):2174–2184. doi: 10.1007/s00125-006-0338-9. [DOI] [PubMed] [Google Scholar]
- 53.Jiang X, Zhang C, Xin Y, et al. Protective effect of FGF21 on type 1 diabetes-induced testicular apoptotic cell death probably via both mitochondrial- and endoplasmic reticulum stress-dependent pathways in the mouse model. Toxicology Letters. 2013;219(1):65–76. doi: 10.1016/j.toxlet.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 54.Jin YC, Kim KJ, Kim YM, et al. Anti-apoptotic effect of magnolol in myocardial ischemia and reperfusion injury requires extracellular signal-regulated kinase1/2 pathways in rat in vivo. Experimental Biology and Medicine. 2008;233(10):1280–1288. doi: 10.3181/0803-RM-79. [DOI] [PubMed] [Google Scholar]