

Background: Helicobacter pylori SabA outer membrane protein is crucial for the bacteria to adhere to host stomach surface during chronic infection.
Results: The structure of the extracellular SabA adhesion region is determined by x-ray crystallography.
Conclusion: SabA adhesion structure may provide insight into binding sites important for ligand receptor interactions.
Significance: SabA is the first reported extracellular domain structure of the H. pylori outer membrane protein family.
Keywords: Bacterial Adhesion, Carbohydrate Binding Protein, Helicobacter pylori, Membrane Proteins, X-ray Crystallography, Outer Membrane Protein
Abstract
The gastric pathogen Helicobacter pylori is a major cause of acute chronic gastritis and the development of stomach and duodenal ulcers. Chronic infection furthermore predisposes to the development of gastric cancer. Crucial to H. pylori survival within the hostile environment of the digestive system are the adhesins SabA and BabA; these molecules belong to the same protein family and permit the bacteria to bind tightly to sugar moieties LewisB and sialyl-LewisX, respectively, on the surface of epithelial cells lining the stomach and duodenum. To date, no representative SabA/BabA structure has been determined, hampering the development of strategies to eliminate persistent H. pylori infections that fail to respond to conventional therapy. Here, using x-ray crystallography, we show that the soluble extracellular adhesin domain of SabA shares distant similarity to the tetratricopeptide repeat fold family. The molecule broadly resembles a golf putter in shape, with the head region featuring a large cavity surrounded by loops that vary in sequence between different H. pylori strains. The N-terminal and C-terminal helices protrude at right angles from the head domain and together form a shaft that connects to a predicted outer membrane protein-like β-barrel trans-membrane domain. Using surface plasmon resonance, we were able to detect binding of the SabA adhesin domain to sialyl-LewisX and LewisX but not to LewisA, LewisB, or LewisY. Substitution of the highly conserved glutamine residue 159 in the predicted ligand-binding pocket abrogates the binding of the SabA adhesin domain to sialyl-LewisX and LewisX. Taken together, these data suggest that the adhesin domain of SabA is sufficient in isolation for specific ligand binding.
Introduction
Helicobacter pylori is a Gram-negative bacterium that chronically infects more than half of the world population and is a major etiological factor of peptic ulcer, atrophic gastritis and gastric adenocarcinoma, and mucosa-associated lymphoid tissue lymphoma (1). The ability to attach to the gastric epithelium is crucial for H. pylori to colonize and establish lifelong chronic infection in the host. The sialic acid binding adhesin (SabA)5 is one of the best characterized H. pylori adhesins (2, 3). It belongs to the Hop superfamily of outer membrane proteins, which also includes the LewisB antigen-binding adhesin, BabA, and the glycoprotein-binding adhesins AlpA and AlpB. The four proteins all comprise a similar domain structure: an N-terminal extracellular domain of unknown structure and a C-terminal trans-membrane domain that is predicted to adopt an integral eight stranded β-barrel similar to the outer membrane protein-like family of integral membrane proteins (see Fig. 1A).
FIGURE 1.

Domain organisation of SabA. A, schematic illustrating the predicted domain structure of SabA. B, crystal structure of SabA N-terminal adhesion region. The handle is in blue, the tetratricopeptide repeat region is colored in magenta and the remainder of the molecule is in orange. Dashed lines indicate disordered regions. The two conserved disulfide bonds are represented as cyan sticks. The short loop interruption in Helix-1 is marked with an asterisk. The C-terminal helix is connected to a trans-membrane β-barrel domain, which anchors the SabA adhesion region to the outer membrane of H. pylori.
SabA interacts with sialyl-LewisX antigen, an important blood group antigen that is rarely expressed in healthy gastric epithelium but is present in abundance in inflamed and cancerous gastric epithelium (3). The SabA-sialyl-LewisX interaction is therefore likely to play a pivotal role in H. pylori colonization during chronic infection, as chronic H. pylori infection is almost always associated with chronic active gastritis (1). Epidemiological data suggest that interaction between SabA and gastric sialyl-LewisX is also important for H. pylori colonization in patients with no or weak LewisB antigen expression (4).
SabA-mediated binding of H. pylori to sialyl glycoconjugates and sialyl gangliosides requires NeuAcα2–3Gal disaccharide as the minimal binding epitope and is favored by extended and flexible glycan core chains (5). Furthermore, SabA variants in different H. pylori clinical isolates exhibit different affinities and specificities for the sialylated glycans, sialyl dimeric LewisX antigen, sialyl-LewisA and dialyllactosamine (5). Such variable SabA ligand-binding specificity might be a host adaptation mechanism by which H. pylori rapidly modulates its adherence properties to achieve optimal colonization and concomitantly evade host immune responses.
Apart from binding to gangliosides, SabA also mediates binding of H. pylori to laminin in a manner that is dependent on the terminal α2–3-sialic acid of the glycan moiety on laminin (6). The physiological significance of this interaction remains to be elucidated. SabA is also essential for binding of H. pylori to sialylated glycoconjugates on erythrocytes and neutrophils (5, 7, 8). The latter interaction leads to non-opsonic oxidative burst involving G protein signaling and activation of phosphatidylinositol 3-kinase (8).
Our knowledge of the various important functions of SabA has so far been based on the results of competitive inhibition of H. pylori adherence using soluble sialyl-LewisX conjugates or the loss of functions observed with H. pylori sabA deletion mutants. The molecular mechanisms underpinning the various functions of SabA protein therefore remain unknown. Currently, however, it is predicted that the extracellular adhesin domain of SabA contains key determinants for interaction with the host cell surface (9). Accordingly, to investigate the structural basis of SabA adhesin function and furthermore to provide structural insights across the SabA/BabA superfamily, we set out to determine the three-dimensional structure of the soluble SabA adhesin domain.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification
The full-length sabA gene was amplified and isolated from the genome library of H. pylori strain 26695 (5′-ACAACAAAAACATTACTTTAAGG-3′ (forward primer) and 5′-CAAGCTCTCTTCTTTAAGGG-3′ (reverse primer)). The mature SabA protein contains an N-terminal extracellular adhesin region (residues 1–460), which shows no sequence homology to any known protein structures, and a predicted C-terminal β-barrel domain (residues 461–635), which anchors SabA to the outer membrane of H. pylori. For this structural study, the N-terminal soluble domain of SabA was cloned into pET15 vector for overexpression. The corresponding mutants with Tyr-148, Lys-152, Gln-159, and Gln-162 substituted by alanine were generated by site-directed mutagenesis using primers shown in supplemental Table 1. A hexahistidine tag and a tobacco etch virus protease site were introduced at the start of the constructs to facilitate purification and removal of the tag as required.
The wild-type and mutant recombinant SabA proteins were expressed in Escherichia coli Rosetta 2 (DE3) as insoluble inclusion bodies. The inclusion bodies were isolated, washed, and solubilized in 50 mm Tris-HCl, pH 8, 300 mm NaCl, and 8 m guanidinium chloride. Protein purity and integrity were checked by SDS-PAGE. SabA was refolded by dialysis against 20 mm Tris-HCl, pH 8, and 50 mm NaCl. The refold solution was clarified by centrifugation and further purified by size-exclusion chromatography. The success of the refolding of wild-type SabA and SabA mutants was ascertained by circular dichroism (see supplemental Fig. 2).
Selenomethionine-substituted SabA (SeMet-SabA) was expressed in E. coli BL21(DE3) grew in M9 minimal media supplemented with 20% glucose, 2 mm MgSO4, 100 μm CaCl2, 100 mg/liter lysine, phenylalanine, threonine each, 50 mg/liter isoleucine, leucine, valine each, and 60 mg/liter dl-selenomethionine. The expression, isolation, refolding and purification of SeMet-SabA were similar to those of the unlabeled protein.
Crystallization
For SabA crystallization, the N-terminal purification tag was first removed by digestion with the tobacco etch virus protease. SabA protein was buffer-exchanged into 20 mm Tris-HCl, pH 8, 50 mm NaCl, and concentrated by ultra-filtration to 5 mg/ml. Initial SabA crystallization conditions were screened by the sparse matrix approach using the hanging-drop vapor diffusion technique. A single hit (100 mm sodium acetate, pH 4.6, 200 mm ammonium acetate, 30% PEG 4000) was identified after one month. SabA crystallization was optimized by varying pH and PEG concentrations of hit condition, drop size, and by microseeding. Typically, using the optimized conditions (200 mm sodium acetate, pH 4.6, 18–20% PEG 4000), SabA crystals were visible within 2 days and grew to maximum size in 2 weeks. For cryo-protection, SabA crystals were equilibrated in mother liquor supplemented with 30% glycerol or PEG 4000 for at least 30 min prior to flash-cooling in liquid nitrogen.
Data Collection and Processing
X-ray diffraction data for both the native SabA and SeMet-SabA were collected on beamline MX2 at the Australian Synchrotron (Table 1). Diffraction data were indexed, integrated, and scaled using HKL2000 (10), MOSFLM (11), and SCALA (12) from the CCP4 package (13).
TABLE 1.
SabA X-ray data collection and structure refinement statistics
r.m.s.d., root mean square deviation.
| Data collection | SetMet derivative |
Native | |
|---|---|---|---|
| Peak | Inflection point | ||
| Temperature (K) | 100 | 100 | 100 |
| Space group | P3121 | P3121 | P3121 |
| Unit cell parameters | a = b = 179.08; c = 64.53 Å | a = b = 178.90; c = 64.42 Å | a = b = 180.04; c = 64.47 Å |
| Wavelength (Å) | 0.9690 | 0.9795 | 0.9537 |
| Resolution (Å) | 89.5–2.9 (3.06–2.9) | 89.5–3.3 (3.4–3.3) | 90.0–2.2 (2.3–2.2) |
| Completeness (%) | 100 (100) | 100 (100) | 100 (100) |
| Total reflections | 239,968 (35,408) | 164,853 (24,138) | 556,070 (81,900) |
| Unique reflections | 26,604 (3843) | 18,899 (2707) | 60,966 (8825) |
| Multiplicity | 9.0 (9.2) | 8.7 (8.9) | 9.1 (9.3) |
| Rpim (%) | 6.7 (33.7) | 9.7 (42.0) | 4.4 (35.7) |
| Rmeas (%) | 20.2 (103.4) | 29.1 (128.0) | 13.4 (109.1) |
| Mean I/σ(I) | 10.0 (2.7) | 8.7 (2.5) | 14.2 (2.4) |
| Mosiacity | 0.54° | 0.60° | 0.32° |
| Refinement | |||
| Non-hydrogen atoms | 3011 | ||
| Protein | 2838 | ||
| Water | 159 | ||
| Resolution, Å | 90.0–2.2 | ||
| Rfactor, % | 14.0 | ||
| Rfree, % | 16.0 | ||
| r.m.s.d. from ideality | |||
| Bond lengths, Å | 0.02 | ||
| Bond angles, Å | 1.53 | ||
| Chirality | 0.09° | ||
| Planarity | 0.007° | ||
| Dihedrals | 14.4° | ||
| Ramachandran plot (%) | |||
| Preferred regions | 96.4 | ||
| Allowed regions | 2.8 | ||
| Disallowed regions | 0.8 | ||
| B factors, Å2 | |||
| Average main chain | 55.037 | ||
| Average side chain and water | 59.137 | ||
| Average all atoms | 57.122 | ||
Structure Determination and Refinement
SeMet-SabA formed small rod crystals under similar precipitation conditions but using lower protein concentration (3 mg/ml). These cryo-protected crystals belong to the same space group and have similar unit cell parameters to the native SabA crystals. A fluorescence scan was carried out with a SeMet-SabA crystal around the selenium absorption edge to determine the wavelengths corresponding to the peak (0.9690 Å) and infection point (0.9795 Å). Anomalous diffraction data sets were collected with the SeMet-SabA crystals at the peak (2.90 Å) and infection point (3.3 Å) wavelengths (Table 1).
Automated experimental phase calculations were carried out using Auto-Rickshaw (14, 15) by the two-wavelength anomalous diffraction method. A solution was identified in the space group P3121 with one SabA molecule per asymmetric unit (solvent content of 80%). The calculations identified seven selenium sites using SHELXD (16). The sites were refined using MLPHARE/PHASER/BP3 (17–19). The resulting phase were subjected to phase extension and density modification using PIRATE (13) and the automated model building was performed using the program ARP/wARP (20) at 2.9 Å. The initial ARP/wARP model was fragmented and had wrong sequence assignment in the electron density map. Using the calculated positions of selenium and density-modified electron density map, the SabA structure model was built manually, and alternated with refinement cycles using Phenix (21) or REFMAC5 (22). Using the native data set, the final SabA structure was refined to 2.2 Å with Rfactor and Rfree of 14.0 and 16.0%, respectively (Table 1).
Surface Plasmon Resonance Analysis of Interaction of SabA with Lewis Antigens
Surface plasmon resonance (SPR) experiments were performed using a Biacore T100 biosensor system (GE Healthcare) at 25 °C as described previously (23) with the following modifications. All analyses of purified His-tagged SabA N-terminal adhesin domain were performed in PBS at pH 6.8 at a flow rate of 30 μl per min. SabA was diluted to 0.1 mg/ml and loaded on Flow Cell 2 of a nickel-nitrilotriacetic acid sensor chip with 10 min of contact time. Flow cell 1 was loaded with purified heat denatured (99 °C for 5 min) SabA diluted to 0.1 mg/ml as reference. Serial dilutions of Lewis antigens (Elicityl SA, Crolles, France) were prepared to 0.00625, 0.0125, 0.025, 0.05, and 0.1 mm and run using single-cycle kinetics as follows. After the injection of five glycan dilutions, the chip was regenerated with EDTA. Subsequently, the chip was reloaded with Ni2+ and SabA before the injection of the next Lewis antigen to be tested. A 10-min dissociation time was allowed after the addition of each analyte. SPR signals were analyzed using the Biacore Evaluation software to determine KD.
RESULTS
Structure of SabA Adhesin Domain
The mature SabA adhesin protein (residues 1–460, lacking the trans-membrane domain) was expressed in E. coli, refolded from inclusion bodies, and used in crystallization experiments. The crystal structure of the SabA adhesin was solved to 2.2 Å and revealed a predominantly α-helical molecule shaped similar to a golf putter (Fig. 1B) and comprising a “handle” and a “head” region. All residues were visible except for 26 residues from the N terminus and 61 residues from the C terminus. Residues 114 and 182–184, which are located at the tips of two highly mobile loops (as estimated through b-value analysis), were also not visible in electron density.
The handle domain (Fig. 1B, in blue) forms a rigid shaft that comprises the N- and C-terminal helices interacting to form an anti-parallel two-helix coiled-coil bundle. The C terminus helix and the disordered linker region (residues 400–460) connect to the predicted β-barrel trans-membrane domain (Fig. 1B).
The central head domain essentially comprises a bundle of α-helices, juxtaposed at approximately right angles to the handle domain. The core region of the head domain is a four-helix antiparallel coiled-coil bundle (Helix-1–4; a tetratricopeptide repeat repeat; Fig. 1B in magenta). Except for Helix-1, which is interrupted in the middle by a short loop (Fig. 1B, marked with an asterisk), the other three α-helices are continuous and are ∼30 residues in length. The four-helix bundle is a common structural scaffold motif found in a variety of proteins and, accordingly, DALI (24) and 3-D BLAST (25) searches reveal that the head domain is distantly homologous to a number of different proteins, e.g. hydrolases (Protein Data Bank code 1OR0; Z-score, 8.3) (26), pectin methylesterase inhibitor (Protein Data Bank code 1×8Z; Z-score, 7.6) (27), and part of the BRO-1 domain (3um2; Z-score, 5.7) (28). Fig. 2 shows an example of the superposition between SabA and a pectin methylesterase inhibitor.
FIGURE 2.
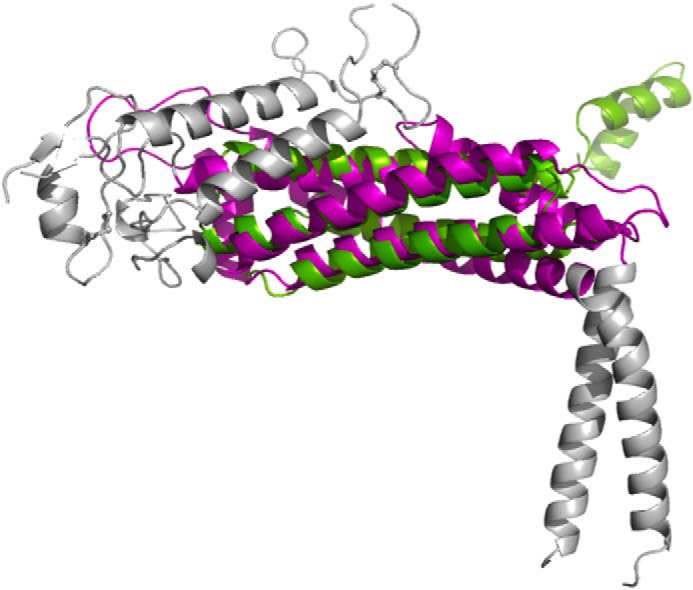
Structural superposition of SabA (magenta and gray) and pectin methylesterase inhibitor (green) around the tetratricopeptide repeat portion as calculated by DALI.
The connecting region between Helix-1 and -2 comprises 140 residues (Fig. 1B, in gold) and is made up of short α-helices, loops, and a small β-sheet (residues 110–112 and 117–119). Predominant features within the Helix-1/2 connecting region include a pair of interacting ∼20 residue α-helices (residues 144–163 of Helix-1a and 191–209 of Helix-1b) that sits on top of Helix-1 and -2 of the core four-helix bundle. The loops joining Helix-1a/Helix-1b and Helix-1b/Helix-2 are both constrained by disulfide bonds, each of which is formed by a pair of closely spaced cysteines (four to five amino acids apart; Cys-135/Cys-141 and Cys-173/Cys-178). Sequence alignments (Fig. 3) of SabA from different Helicobacter strains reveal both disulfide bonds are highly conserved in the SabA branch of the family. Interestingly, however, Cys-141 is absent in the BabA branch of the family (Fig. 3). Instead, in BabA, we note a conserved cysteine at position 108 (SabA numbering; Fig. 3, marked with a #); from a structural perspective, we suggest this residue would be appropriately positioned to form a disulfide bond with the conserved Cys-135.
FIGURE 3.

Primary sequence alignment of SabA and BabA N-terminal adhesion regions. The alignment was generated with Clustal Omega using 33 SabA and 41 BabA sequences from various H. pylori strains (KEGG database). The figure shows a representative of the complete alignment (five sequences from each protein). Residues in yellow are identical in all SabA and BabA, red are identical surface residues in the predicted binding pocket, cyan are cysteine residues involved in disulfide bonds in SabA. The position of the alternative conserved cysteine residue in BabA is marked (#). Ser-80 and Pro-81 (@) map to the interruption in Helix-1, and Gly-357 (§) causes a slight kink in Helix-4. Tyr-148, Lys-152, Gln-159, and Gln-162 were mutated to alanine in this study are marked with an asterisk.
Sequence/Structure Insights Suggest Functionally Conserved Portions of the SabA Structure
We also analyzed the structure of the SabA adhesin domain with respect to sequence conservation. The primary sequences of BabA (41 sequences) and SabA (33 sequences) from multiple H. pylori strains were aligned to identify identical residues that might be important for the function of these adhesins (Fig. 3). We further used the web-based automatic ligand-binding-site prediction program POCASA (29) to predict pockets and cavities in the SabA head domain, with the rationale that these might represent potential ligand binding sites (Fig. 4, A and B). Three adjacent pockets stretching from the non-structured tip of the head domain to the grove formed between Helix-1 and -4 were present. Interestingly, many of the identical SabA/BabA surface residues (Fig. 3, highlighted in red, and Fig. 4, C and D, cyan sticks) lined along the POCASA predicted pockets, suggesting that these cavities may play a role in the binding of host cell ligands. In particular, we noted a deep (∼9 Å) positively charged cavity (Fig. 4E), lined at the bottom by Trp-97 and at one side Helix-1a with identical residues Lys-152, Gln-159, and Gln-162 (Fig. 4, C and D). In our structure, a glycerol molecule (cryo-protectant) is found in this cavity (Fig. 4E), forming hydrogen bond interactions with the side chain of Lys-152 and Gln-159. We further note three other conserved residues, Ser-80 and Pro-81 map to the interruption in Helix-1 and Gly-357 causes a slight kink in Helix-4 (Fig. 4, C and D).
FIGURE 4.

Ligand-binding pockets calculated by an automatic ligand-binding-site prediction program (POCASA). A and B, the binding pockets (dark gray surface) are predicted mostly toward to the tip of the SabA head domain. C and D, view from the top toward the predicted ligand binding pockets (removed in D for clarity). A number of highly conserved residues (cyan sticks), in particular Ser-80, Pro-81, Trp-97, Tyr-148, Lys-152, Gln-159, and Gln-162, line the surface of these pockets. Tyr-148, Lys-152, Gln-159, and Gln-162 were mutated to alanine in this study are marked with an asterisk. A glycerol molecule (pink sticks) is observed in the crystal structure and forms hydrogen bond interactions with Lys-152 and Gln-159. E, electrostatic potential surface representation of the SabA head domain showing the predicted pockets (gray dots) generated using CCP4 mg (30). A glycerol molecule (pink sticks) binds in a positively charged cavity.
The Interaction of SabA Adhesin Domain with Binding Partners
The SabA crystal structure was solved in the absence of any ligand molecule. Previous experimental studies suggested that the SabA ligands include sialic acid or sialyl-LewisX (5, 6). Accordingly, we attempted to co-crystallize or soak-in sialic acid or sialyl-LewisX; however, these experiments were unsuccessful. We then performed SPR experiments based on small molecule single cycle kinetics to examine the interaction of SabA with a variety of sugars. SabA bound sialyl-LewisX with a KD of 19.9 ± 2.7 μm (Table 2 and supplemental Fig. 3A). It also bound to LewisX but at a 2.5-fold lower affinity with a KD of 50.4 ± 8.2 μm (Table 2 and supplemental Fig. 3B). No binding of SabA to other Lewis antigens, including LewisA, LewisB, and LewisY, was detected at ligand concentrations up to 1 mm, indicating that the observed binding of SabA to sialyl-LewisX and LewisX was highly specific (Table 2).
TABLE 2.
Affinity of SabA for Lewis antigens determined by surface plasmon resonance
|
KD determineda,b |
||
|---|---|---|
| Sialyl-LewisX | LewisX | |
| SabA wild-type | 19.9 μm ± 2.7 | 50.4 μm ± 8.2 |
| SabA Y148A | 21.1 μm ± 5.6 | 151.7 μm ± 30.5 |
| SabA K152A | 24.5 μm ± 2.1 | 62.8 μm ± 13.6 |
| SabA Q159A | NCDIc | NCDIc |
| SabA Q162A | 31.7 μm ± 7.1 | NCDIc |
a No interaction was observed between SabA and SabA mutants with LewisA, LewisB, and LewisY.
b Data shown are mean KD values determined from four independent experiments.
c NCDI indicates no concentration-dependent interaction was detected (KD > 1 mm).
To test whether the aforementioned glycerol-bound positively charged cavity of SabA functions as a glycan-binding site, we generated SabA mutants with point substitution of Tyr-148, Lys-152, Gln-159, or Gln-162 by alanine. These residues are predicted to play key roles in the interaction of SabA with ligands for the following reasons: (i) these four amino acids line along the surface of Helix-1a, with their side chains pointing toward the glycerol binding cavity (Fig. 4, C and D); (ii) Lys-152 and Gln-159 form hydrogen bonds with the bound glycerol in the crystal structure (Fig. 4, D and E); (iii) all four amino acids are highly conserved. Lys-152, Gln-159, and Gln-162 are conserved in both SabA and BabA (Fig. 3), whereas Tyr-148 is identical among all SabA orthologs but is replaced by the hydrophobic residue isoleucine in BabA.
The results of SPR analysis show that substitution of Gln-159 by alanine (Q159A) dramatically weakened the binding of SabA to sialyl-LewisX and LewisX by >50- and 20-fold, respectively (Table 2 and supplemental Fig. 3). In contrast, the substitution of Tyr-148, Lys-152, or Gln-162 by alanine (Y148A, K152A, and Q162A, respectively) did not significantly alter the affinity of SabA for sialyl-LewisX, although they reduced the binding of SabA to LewisX 3–20-fold (Table 2). Despite the obvious effects of the mutations on ligand binding to SabA, the mutations did not cause any global structural perturbation of SabA: gel filtration profiles (supplemental Fig. 1) and circular dichroism spectra (supplemental Fig. 2) of the SabA mutants are highly similar to that of their wild-type counterpart. Thus, we conclude that Gln-159 is of critical importance for the binding of SabA to sialyl-LewisX and LewisX. Furthermore, our data suggest that residues Tyr-148 and Gln-162 are essential only for the binding of SabA to LewisX but not to sialyl-LewisX. The charged side chain of Lys-152, which forms a hydrogen bond with the bound glycerol in the crystal structure, surprisingly does not seem to be required for the binding of SabA to either sialyl-LewisX or LewisX.
DISCUSSION
In this study, we present the structure of the SabA adhesin domain. These data reveal a “club-shaped” molecule that, on first inspection, presents a number of features consistent with a proposed function in interaction with host-cell surface glycoproteins. For example, we note the presence of a highly conserved cavity in the SabA head domain that would provide an attractive binding site for a potential ligand. Although, to date, we have been unable to crystallize the SabA adhesin domain in the presence of a bound carbohydrate ligand, we were able to detect the binding of SabA protein to sialyl-LewisX (KD of 19.9 ± 2.7 μm) by small molecule single cycle kinetics SPR, which is a highly sensitive method for analyzing molecular interactions. Surprisingly, we also detected an interaction between SabA and LewisX, which occurred with weak affinity (KD of 50.4 ± 8.2 μm). In contrast, binding to SabA was not detected using LewisA, LewisB, and LewisY as the analyte. These results are in agreement with previous reports that the sialic acid moiety is required for optimal binding of SabA to sialyl-LewisX and that SabA binding is highly specific to LewisX glycans (3, 5, 6). Additionally, this study shows for the first time that the adhesion domain of SabA in isolation is fully functional and sufficient for selective binding to sialyl-LewisX or LewisX.
To understand the molecular basis of SabA ligand binding, we mutated four highly conserved residues selected based on the analysis of the primary and tertiary structures of SabA and the structure of the predicted ligand-binding cavities. Of the four mutations tested, Q159A has the most significant effect, abolishing SabA binding to both sialyl-LewisX and LewisX. These data suggest that Gln-159 plays a critical role in ligand binding. In contrast, although Lys-152 is conserved, the K152A mutation does not cause significant changes in the binding affinity to sialyl-LewisX or LewisX.
Substituting Tyr-148 and Gln-162 with alanine significantly reduced LewisX-binding (3- to >20-fold) but had little or no effect on sialyl-LewisX-binding. Tyr-148 and Gln-162 are found on either end of the putative ligand-binding pocket and form part of the surface of the cavity. The side chains of these residues also form multiple Van der Waals interaction with other parts of the protein (Tyr-148 with the loop region; Gln-162 with residues in Helix-1 and Helix-4). It is plausible that changing these residues to the smaller alanine may alter the shape of the binding pocket in such a way that binding to the smaller LewisX trisaccharide becomes less favorable compared with the binding of the larger sialyl-LewisX tetrasacchaide. This remains to be ascertained by further mutagenesis of SabA and structural studies on SabA-LewisX and SabA-sialyl LewisX complexes.
In summary, our study shows that the N-terminal domain of SabA functions as a sugar-binding adhesion domain, in which a cavity lined by conserved amino acids likely serves as a highly selective ligand-binding site. Notably, this region consists of amino acid residues that are not only conserved among SabA orthologs but also between SabA and BabA. Thus, the findings in this study have significant implications for understanding the structure-function relationship of not only SabA but also that of BabA. It would be of interest to test whether mutating the corresponding residues in BabA will influence the specificity or affinity of LewisB-binding to BabA.
Previous studies suggested that SabA-mediated binding of H. pylori to sialyl glycoconjugates and sialyl gangliosides requires NeuAcα2–3Gal disaccharide as the minimal binding epitope (5), whereas the results in this study indicate that the adhesion domain of SabA binds to not only sialyl-LewisX but also LewisX. This difference in glycan-binding specificity between the purified SabA adhesion domain and SabA expressed on intact bacteria could be due to differences in steric hindrance, hydrophobicity and/or charges of the local environments of SabA. Alternatively, other domains/regions of SabA may play a role in fine-tuning the glycan-binding specificity. Future investigations are required to further understand the molecular basis of the ligand-binding specificity of SabA and hence its role for pathogenesis in the complex gastric environment during chronic H. pylori infection. Such knowledge will be instrumental for the development of new drugs against H. pylori-induced chronic gastritis and gastric cancer.
Supplementary Material
Acknowledgments
We thank the Australian synchrotron (MX-2 Beamline) and staff for beamline support.
The atomic coordinates and structure factors (code 4O5J) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Table 1 and Figs. 1–3.
- SabA
- sialic acid binding adhesin
- BabA
- LewisB antigen-binding adhesin
- SetMet-SabA
- selenomethionine-substituted SabA
- SPR
- surface plasmon resonance
- KD
- dissociation constant.
REFERENCES
- 1. Kuipers E. J., Michetti P. (2005) Bacteria and mucosal inflammation of the gut: Lessons from Helicobocter pylori. Helicobacter 10, 66–70 [DOI] [PubMed] [Google Scholar]
- 2. Magalhães A., Reis C. A. (2010) Helicobacter pylori adhesion to gastric epithelial cells is mediated by glycan receptors. Braz. J. Med. Biol. Res. 43, 611–618 [DOI] [PubMed] [Google Scholar]
- 3. Mahdavi J., Sondén B., Hurtig M., Olfat F. O., Forsberg L., Roche N., Angstrom J., Larsson T., Teneberg S., Karlsson K. A., Altraja S., Wadström T., Kersulyte D., Berg D. E., Dubois A., Petersson C., Magnusson K. E., Norberg T., Lindh F., Lundskog B. B., Arnqvist A., Hammarström L., Borén T. (2002) Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297, 573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sheu B. S., Sheu S. M., Yang H. B., Huang A. H., Wu J. J. (2003) Host gastric Lewis expression determines the bacterial density of Helicobacter pylori in babA2 genopositive infection. Gut 52, 927–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aspholm M., Olfat F. O., Nordén J., Sondén B., Lundberg C., Sjöström R., Altraja S., Odenbreit S., Haas R., Wadström T., Engstrand L., Semino-Mora C., Liu H., Dubois A., Teneberg S., Arnqvist A., Borén T. (2006) SabA is the H. pylori hemagglutinin and is polymorphic in binding to sialylated glycans. PLoS Pathog. 2, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walz A., Odenbreit S., Mahdavi J., Borén T., Ruhl S. (2005) Identification and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays. Glycobiology 15, 700–708 [DOI] [PubMed] [Google Scholar]
- 7. Aspholm M., Kalia A., Ruhl S., Schedin S., Arnqvist A., Linden S., Sjostrom R., Gerhard M., Semino-Mora C., Dubois A., Unemo M., Danielsson D., Teneberg S., Lee W. K., Berg D. E., Boren T. (2006) Helicobacter pylori adhesion to carbohydrates. Methods Enzymol. 417, 293–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Unemo M., Aspholm-Hurtig M., Ilver D., Bergström J., Borén T., Danielsson D., Teneberg S. (2005) The sialic acid binding SabA adhesin of Helicobacter pylori is essential for nonopsonic activation of human neutrophils. J. Biol. Chem. 280, 15390–15397 [DOI] [PubMed] [Google Scholar]
- 9. Alm R. A., Bina J., Andrews B. M., Doig P., Hancock R. E., Trust T. J. (2000) Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect. Immun. 68, 4155–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode in Methods in Enzymology (Charles W., Carter J., eds) pp. 307–326, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 11. Leslie A. G., Powell H. R. (2007) Processing diffraction data with MOSFLM. Evolving Methods for Macromolecular Crystallography 245, 41–51 [Google Scholar]
- 12. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 13. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Panjikar S., Parthasarathy V., Lamzin V. S., Weiss M. S., Tucker P. A. (2005) Auto-rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an x-ray diffraction experiment. Acta Crystallogr. D Biol. Crystallogr. 61, 449–457 [DOI] [PubMed] [Google Scholar]
- 15. Panjikar S., Parthasarathy V., Lamzin V. S., Weiss M. S., Tucker P. A. (2009) On the combination of molecular replacement and single-wavelength anomalous diffraction phasing for automated structure determination. Acta Crystallogr. D Biol. Crystallogr. 65, 1089–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schneider T. R., Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 17. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 19. Pannu N. S., McCoy A. J., Read R. J. (2003) Application of the complex multivariate normal distribution to crystallographic methods with insights into multiple isomorphous replacement phasing. Acta Crystallogr. D Biol. Crystallogr. 59, 1801–1808 [DOI] [PubMed] [Google Scholar]
- 20. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huergo L. F., Rahman H., Ibrahimovic A., Day C. J., Korolik V. (2013) Campylobacter jejuni Dps protein binds DNA in the presence of iron or hydrogen peroxide. J. Bacteriol. 195, 1970–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tung C. H., Huang J. W., Yang J. M. (2007) κ-α plot derived structural alphabet and BLOSUM-like substitution matrix for rapid search of protein structure database. Genome Biol. 8, R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim J. K., Yang I. S., Rhee S., Dauter Z., Lee Y. S., Park S. S., Kim K. H. (2003) Crystal structures of glutaryl 7-aminocephalosporanic acid acylase: insight into autoproteolytic activation. Biochemistry 42, 4084–4093 [DOI] [PubMed] [Google Scholar]
- 27. Hothorn M., Wolf S., Aloy P., Greiner S., Scheffzek K. (2004) Structural insights into the target specificity of plant invertase and pectin methylesterase inhibitory proteins. Plant Cell 16, 3437–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mu R., Dussupt V., Jiang J., Sette P., Rudd V., Chuenchor W., Bello N. F., Bouamr F., Xiao T. S. (2012) Two distinct binding modes define the interaction of Brox with the C-terminal tails of CHMP5 and CHMP4B. Structure 20, 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu J., Zhou Y., Tanaka I., Yao M. (2010) Roll: a new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 26, 46–52 [DOI] [PubMed] [Google Scholar]
- 30. McNicholas S., Potterton E., Wilson K. S., Noble M. E. (2011) Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 67, 386–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.