

Background: Tyr-226 and Tyr-390 in the Φ29 DNA polymerase active site are implicated in the mechanism of translocation.
Results: Y226F and Y390F differ in their effects on translocation and on dNTP and pyrophosphate binding.
Conclusion: Mutations in the Φ29 DNA polymerase and exonuclease active sites perturb dNTP or pyrophosphate binding rates.
Significance: DNA polymerase architecture is finely tuned to integrate translocation and substrate binding.
Keywords: DNA Polymerase, Enzyme Kinetics, Enzyme Mechanisms, Molecular Motors, Single Molecule Biophysics, Translocation Mechanism
Abstract
The Φ29 DNA polymerase (DNAP) is a processive B-family replicative DNAP. Fluctuations between the pre-translocation and post-translocation states can be quantified from ionic current traces, when individual Φ29 DNAP-DNA complexes are held atop a nanopore in an electric field. Based upon crystal structures of the Φ29 DNAP-DNA binary complex and the Φ29 DNAP-DNA-dNTP ternary complex, residues Tyr-226 and Tyr-390 in the polymerase active site were implicated in the structural basis of translocation. Here, we have examined the dynamics of translocation and substrate binding in complexes formed with the Y226F and Y390F mutants. The Y226F mutation diminished the forward and reverse rates of translocation, increased the affinity for dNTP in the post-translocation state by decreasing the dNTP dissociation rate, and increased the affinity for pyrophosphate in the pre-translocation state. The Y390F mutation significantly decreased the affinity for dNTP in the post-translocation state by decreasing the association rate ∼2-fold and increasing the dissociation rate ∼10-fold, implicating this as a mechanism by which this mutation impedes DNA synthesis. The Y390F dissociation rate increase is suppressed when complexes are examined in the presence of Mn2+ rather than Mg2+. The same effects of the Y226F or Y390F mutations were observed in the background of the D12A/D66A mutations, located in the exonuclease active site, ∼30 Å from the polymerase active site. Although translocation rates were unaffected in the D12A/D66A mutant, these exonuclease site mutations caused a decrease in the dNTP dissociation rate, suggesting that they perturb Φ29 DNAP interdomain architecture.
Introduction
Replicative DNA polymerases (DNAPs)5 are molecular motors that translocate along their DNA substrates in single nucleotide increments as they catalyze template-directed DNA replication. The DNAP from the bacteriophage Φ29 is a B-family polymerase that catalyzes highly processive DNA synthesis (1–3), without the need for accessory proteins, such as sliding clamps or helicases, because it remains tightly associated with its DNA substrate and promotes downstream strand displacement during replication (1, 4, 5). In addition to its 5′–3′ polymerase active site, Φ29 DNAP has a 3′–5′ exonuclease active site, located in a separate domain of the protein, ∼30 Å from the polymerase active site (2–4, 6).
Crystal structures of the Φ29 DNAP binary complex with a primer-template DNA substrate bound in the polymerase active site (Fig. 1A) and of the Φ29 DNAP-DNA ternary complex with dNTP complementary to the templating base in the active site (Fig. 1B) have been determined (4). The architecture of the DNA polymerase domain is highly conserved and resembles a partially closed right hand. The palm subdomain contains residues that participate in the chemistry of catalysis, whereas the thumb subdomain positions the primer-template duplex in the active site. The fingers subdomain contains residues essential for binding incoming nucleotide substrates. In crystal structures of complexes containing complementary dNTP, the position of the fingers subdomain differs from its position in the binary complex structures; elements of this subdomain move in toward the active site cleft to achieve a tight steric fit with the nascent base pair (Fig. 1, A and B).
FIGURE 1.

Structural transitions in Φ29 DNAP-DNA complexes critical to the translocation step and to dNTP binding. Shown are crystal structure models for the Φ29 DNAP-DNA, post-translocation state binary complex in the fingers-open conformation (Protein Data Bank entry 2PZS) (A) and the Φ29 DNAP-DNA-dNTP, post-translocation state ternary complex in the fingers-closed conformation (Protein Data Bank entry 2PYJ) (B). C and D, close-up views of the polymerase active site from the structures shown in A and B, respectively. The structures are from Ref. 4 and were determined using the D12A/D66A mutant of Φ29 DNAP. In A and B, the protein backbone is rendered as a gray ribbon, with residues 359–395 in the fingers domain in red ribbon to highlight the conformation difference between the open binary complex and the closed ternary complex. The backbone positions of the Asp-12 and Asp-66 residues in the exonuclease domain are colored magenta. In A–D, the DNA primer strand is displayed in orange, the DNA template strand is yellow, and the templating base at n = 0 is in cyan. Residues Tyr-254, Tyr-226, and Tyr-390 are rendered in blue (space-filling in A and B, sticks in C and D). In B and D, the incoming dNTP is shown in green. In A and C, the side chains of Tyr-254 and Tyr-390 are stacked, obstructing the dNTP binding site; in B and D, both tyrosine side chains are rotated out of the stacking interaction, removing the steric impediment to the incoming dNTP. In C and D, the water molecule that mediates the interaction of the hydroxyl group of Tyr-390 with the −1 and −2 residues of the template strand of the duplex is shown as a red sphere. This water is part of an extensive network of water-mediated interactions with the minor groove of the active site-proximal duplex, a network that is precisely conserved between Φ29 DNAP and the B-family DNAP from bacteriophage RB69 (21). The black dashed lines indicate potential hydrogen bonding interactions for the hydroxyl groups of the Tyr-226 or Tyr-390 side chains, including the hydrogen bond between the two side chains (labeled 2.7 Å in D). In C, the dashed gray line between the hydroxyl groups of the Tyr-226 and Tyr-390 side chains in the binary complex illustrates the increased distance between the hydroxyl groups of Tyr-226 and Tyr-390 (>5 Å) when the fingers are in the open conformation.
In the fingers-open, post-translocation state binary complex, the side chains of Tyr-254 and Tyr-390 in the polymerase active site are stacked, in a conformation that sterically occludes dNTP binding (Fig. 1C). In the fingers-closed ternary complex, the side chains of Tyr-254 and Tyr-390 both rotate relative to their positions in the fingers-open binary complex, disrupting the stacking interaction between them and allowing the incoming dNTP to bind. The deoxyribose sugar of the dNTP stacks on Tyr-254, and the rotation of Tyr-390 brings its hydroxyl group into hydrogen bonding distance of the hydroxyl group of Tyr-226 (Fig. 1D).
These key structural differences between the open and closed complexes prompted the proposal of an elegant model for the structural mechanism of translocation (4), in which the spatial displacement of translocation is directly linked to the structural transition of fingers opening. In this view, the fingers-closed post-translocation state ternary complex serves as a model for the structure of the pre-translocation state complex, in which the nascent base pair between the templating base at n = 0 and the incoming complementary dNTP in the post-translocation state ternary complex occupies the site that would be occupied by the terminal base pair of the primer-template duplex in the pre-translocation state complex. The structure of the Φ29 DNAP binary complex in the fingers-open, post-translocation state indicates that the pre-translocation state in the fingers-open conformation is sterically precluded. Specifically, the orientation of Tyr-390 and Tyr-254 would clash with the terminal base pair of the duplex. Hence, fingers opening was proposed to compel the forward translocation (4).
Fluctuations between the pre-translocation and post-translocation states can be directly observed and quantified from ionic current time traces recorded when individual Φ29 DNAP-DNA complexes are held atop a nanoscale pore in an electric field (7–9). A single α-hemolysin (α-HL) nanopore is inserted into a lipid bilayer that separates two chambers (termed cis and trans) containing buffer solution (Fig. 2A). A patch clamp amplifier applies voltage across the bilayer and measures the ionic current that flows through the nanopore, which is carried by K+ and Cl− ions in the buffer. A typical ionic current trace that results when a binary complex between Φ29 DNAP and a DNA substrate (DNA1; Fig. 2B) is captured atop the nanopore at 180 mV applied potential is shown in Fig. 2C. The ionic current through the open pore (Fig. 2C, i) drops rapidly when a complex is captured (Fig. 2C, ii). The enzyme is too large to enter the nanopore. Thus, the Φ29 DNAP-DNA complex, with the enzyme bound at the primer-template junction of the DNA substrate, perches atop the pore. The DNA template strand of the captured complex is suspended through the nanopore lumen, which is just wide enough to accommodate a single strand of DNA (Fig. 2C, ii).
FIGURE 2.

Capture of Φ29 DNAP-DNA complexes on the α-HL nanopore. In the nanopore device (A), a single α-HL nanopore is inserted in a ∼25-μm diameter lipid bilayer separating two chambers (cis and trans) that contain buffer solution. A patch clamp amplifier applies voltage across the bilayer and measures ionic current, which is carried through the nanopore by K+ and Cl− ions. B, DNA1 is a hairpin, featuring a 14-base pair duplex and a single-stranded template region of 35 nucleotides. The primer strand is terminated with a 2′-H, 3′-H CMP residue, and the template strand contains a reporter group of five consecutive abasic (1′-H, 2′-H) residues spanning positions +8 to +12 (indicated as red letters X in the sequence). C, representative current trace for a binary complex formed between Φ29 DNAP and the DNA1 substrate, captured at 180 mV applied potential in buffer containing 10 mm K-Hepes, pH 8.0, 0.3 m KCl, 1 mm EDTA, 1 mm DTT, and 11 mm MgCl2. DNA and Φ29 DNAP were added to the nanopore cis chamber to final concentrations of 1 and 0.75 μm, respectively. Schematics diagrams above the current trace illustrate the sequence of events, which is described in the Introduction. In the schematic diagrams, the five consecutive abasic (1′, 2′-H) residues spanning positions +8 to +12 of the template strand, which serve as a reporter group, are shown as red circles. D, ionic current traces for Φ29 DNAP = DNA1 complexes, captured at 180 mV in the presence of 0 μm (i) or 40 μm (ii) dGTP. E, a three-state model in which translocation and dNTP binding are sequential: dNTP can bind to complexes (kon [dNTP]) only after the transition from the pre-translocation to the post-translocation state (r1); the transition from the post-translocation to the pre-translocation state (r2) cannot occur before the dissociation of dNTP (koff).
Captured Φ29 DNAP-DNA complexes reside atop the nanopore for several seconds, during which the measured ionic current fluctuates on the millisecond time scale between two amplitude levels (Fig. 2C, ii). Transition between the two amplitudes corresponds to movement of the DNA substrate relative to the enzyme and the nanopore; the distance of this displacement is ∼1 nucleotide (7, 10). Detection of the DNA displacement is achieved by the use of a reporter group comprising five consecutive abasic (1′-H, 2′-H) residues in the template strand (red letters X or red circles in Fig. 2, B and C (ii), respectively); a displacement of the reporter group in the nanopore lumen is manifested as a change in measured ionic current (7, 10). In the upper amplitude, the primer-template junction of the DNA substrate is bound in the polymerase active site, in the pre-translocation state. At 180 mV, the pre-translocation state amplitude is centered at ∼32 pA (Fig. 2, C (ii) and D (i)). In the lower amplitude, the primer-template junction of the DNA substrate resides in the polymerase active site, in the post-translocation state. The post-translocation state amplitude is centered at ∼26 pA at 180 mV (Fig. 2, C (ii) and D (i)). The amplitude fluctuations continue until complexes dissociate or are ejected, after which another complex can be captured. We have shown that the pre-translocation and post-translocation states are discrete kinetic states (8).
The primer strand of DNA1 bears a 2′-H, 3′-H terminus (Fig. 2B), and thus DNA1 supports the formation of Φ29 DNAP-DNA-dNTP ternary complexes but not the chemical step of phosphodiester bond formation. Binding of dGTP (complementary to the template dCMP residue at n = 0) to Φ29 DNAP-DNA1 complexes stabilizes the post-translocation state. In the absence of dNTP, complexes fluctuate rapidly between the two states (Fig. 2D, i); the addition of dGTP (40 μm; Fig. 2D, ii) causes the average dwell time in the lower amplitude, post-translocation state to increase, as a subpopulation emerges with longer dwell times. The kinetic mechanism of translocation and dNTP binding in individual Φ29 DNAP-DNA complexes (9) is described by a three-state model with four transition rates (Fig. 2E). In the three-state model, translocation and dNTP binding are sequential. dNTP can bind to complexes (kon[dNTP]) only after the transition from the pre-translocation to the post-translocation state (r1); the transition from the post-translocation to the pre-translocation state (r2) cannot occur before the dissociation of dNTP (koff) (Fig. 2E).
In the current study, we have examined the dynamics of translocation and substrate binding in individual complexes formed with the Y226F and Y390F mutants of Φ29 DNAP. Both Tyr-226 and Tyr-390 are highly conserved residues in B-family DNAPs (11, 12). Changing either Tyr-226 or Tyr-390 to phenylalanine disrupts the hydrogen bond between their hydroxyl groups, an interaction that may have a role in stabilizing the orientation of Tyr-390 in the fingers-closed, post-translocation ternary complex, or in the proposed fingers-closed, pre-translocation state complex (4). This hydrogen bond is the only structural interaction predicted to be directly affected by the Y226F mutation, although indirect effects on active site structure, including perturbations of other interactions in which Tyr-390 is a partner, cannot be excluded. The Y390F mutation directly disrupts the hydrogen bonding potential with Tyr-226 in the closed complex as well as the water-mediated interactions of Tyr-390 with the template strand in the DNA duplex in both the open and closed complexes. Earlier biochemical studies of the Y390F mutant have shown that it is severely impaired in DNA synthesis relative to the wild type Φ29 DNAP (11, 13, 14). In contrast to Y390F, biochemical studies of the Y226F mutant showed that although it is impaired in transfer of the primer strand from the polymerase to the exonuclease active site for DNA substrates bearing fully paired duplexes (12), it is not impaired in DNA synthesis ((12) (Fig. 3). Thus, the Y390F and Y226F mutations yield very different biochemical properties in Φ29 DNAP.
FIGURE 3.
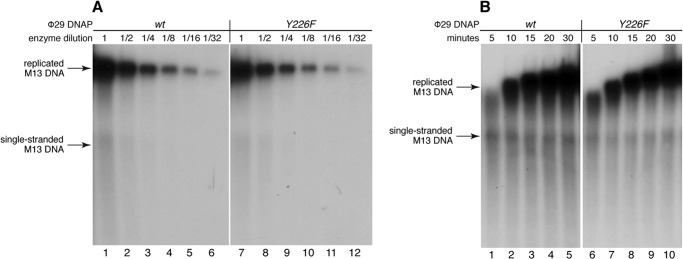
Processive DNA synthesis catalyzed by the Y226F mutant. A, DNA synthesis catalyzed by the wild type Φ29 DNAP (lanes 1–6) or the Y226F mutant (lanes 7–12) as a function of enzyme concentration. A series of 2-fold serial dilutions of each enzyme was tested, in which the highest concentration (lanes 1 and 7, indicated by enzyme dilution = 1) was 30 pm; the reactions were conducted at 30 °C for 30 min. B, DNA synthesis as a function of time for reactions catalyzed by the wild type Φ29 DNAP (lanes 1–5) or the Y226F mutant (lanes 6–10). Reactions were conducted using an enzyme concentration of 60 pm at 30 °C for the indicated times. In both A and B, the replication substrate was oligonucleotide-primed bacteriophage M13 single-stranded DNA (∼3.35 pm). The reaction products were resolved by electrophoresis in alkaline agarose gels.
EXPERIMENTAL PROCEDURES
DNA and Enzymes
DNA1 was synthesized at Stanford Protein and Nucleic Acid Facility and purified by denaturing PAGE. The DNA1 hairpin was annealed by heating at 90 °C for 4 min, followed by snap cooling in ice water.
Wild type Φ29 DNAP was obtained from Enzymatics (Beverly, MA). The D12A/D66A mutant was obtained from XPol Biotech (Madrid, Spain). The Y226F, Y390F, Y226F/D12A/D66A, and Y390F/D12A/D66A mutants were expressed in Escherichia coli BL21(DE3) cells and purified as described for the wild type Φ29 DNAP (15).
Nanopore Methods
Nanopore experiments were conducted as described (7, 9, 10, 16–18). Briefly, a single α-HL nanopore was inserted in an ∼25-μm diameter lipid bilayer that separates two chambers (cis and trans) containing buffer solution (10 mm K-Hepes, pH 8.0, 0.3 m KCl, and 1 mm EDTA). DTT was added to the nanopore cis chamber to a final concentration of 1 mm. MgCl2 and ddCTP were added to final concentrations of 11 mm and 400 μm, respectively, except in those experiments in Fig. 7, in which the effects of Mn2+ were assayed. In those experiments, MgCl2 and ddCTP were omitted, and MnCl2 was added to the cis chamber to a final concentration of 2 mm. DNA and Φ29 DNAP were added to the cis chamber to final concentrations of 1 and 0.75 μm, respectively, and dGTP or pyrophosphate were added as indicated in the figure legends. Ionic current was measured with an integrating patch clamp amplifier (Axopatch 200B, Molecular Devices) in voltage clamp mode. Data were sampled using an analog-to-digital converter (Digidata 1440A, Molecular Devices) at 100 kHz in whole-cell configuration and filtered at 5 kHz using a low pass Bessel filter.
FIGURE 7.
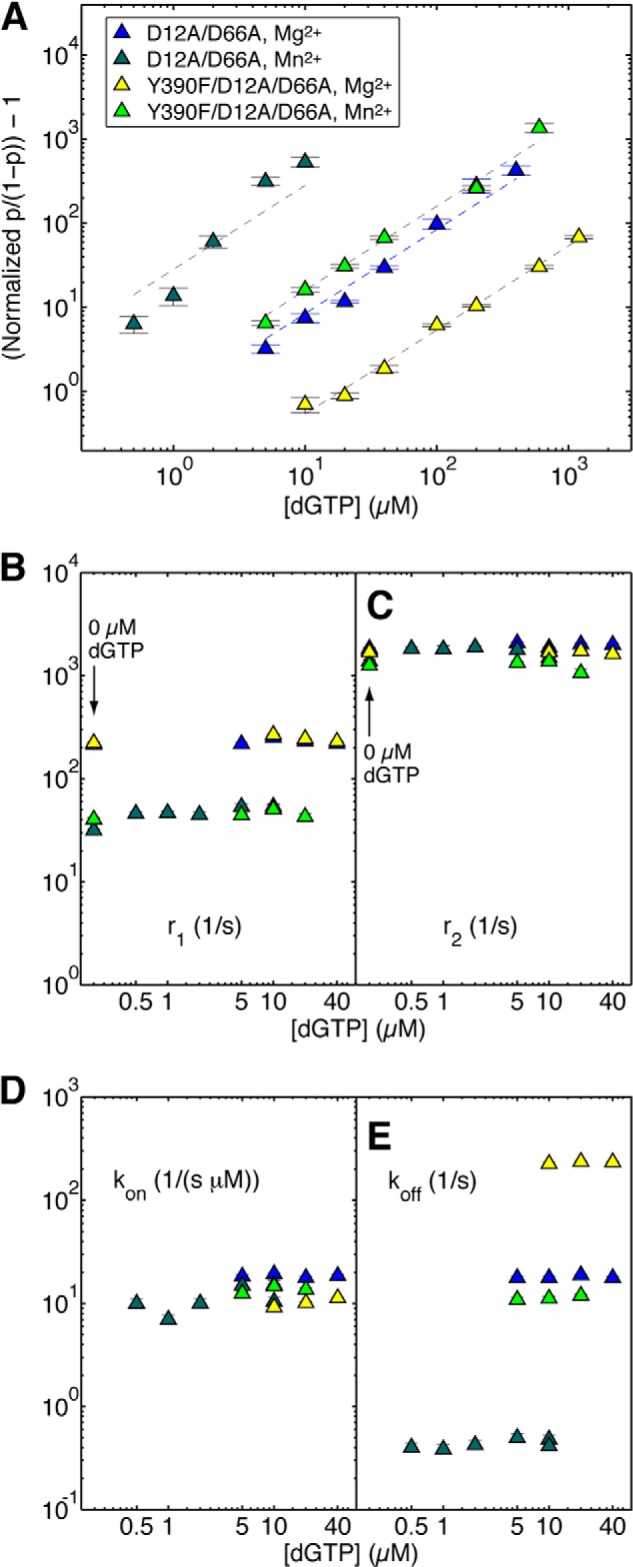
Effects of Mn2+ on dNTP binding to mutant Φ29 DNAP-DNA complexes. A, the (normalized p/(1 − p)) − 1 is plotted on a log scale as a function of dGTP concentration for complexes formed between DNA1 and the D12A/D66A mutant in the presence of Mg2+ (blue triangles) or Mn2+ (dark green triangles) and between DNA1 and the Y390F/D12A/D66A mutant in the presence of Mg2+ (yellow triangles) or Mn2+ (light green triangles). Complexes were captured at 180 mV. The data are plotted according to the concentration of dGTP added to the nanopore cis chamber. Because a fraction of the added dGTP is bound by complexes in the bulk phase (∼100-μl cis chamber volume, in which [DNA1] = 1 μm, and [enzyme] = 0.75 μm), the free [dGTP] is lower than the input [dGTP]. The difference is significant when the input [dGTP] is comparable with or lower than the concentration of complexes. This accounts for the difference between the data for D12A/D66A mutant in the presence of Mn2+ (A, dark green triangles) and the linear fitting because the linear relation is with respect to the free [dGTP]. Shown are plots of r1 versus [dGTP] (B) and r2 versus [dGTP] (C) for complexes formed between the D12A/D66A mutant or the Y390F/D12A/D66A mutant and DNA1, captured at 180 mV in the presence of Mg2+ or Mn2+. Because there is no zero value on the log scale plot, in B and C, the values of r1 and r2 for binary complexes of the two mutants, captured in the presence of Mg2+ or Mn2+ are placed on the plot at the position for 0.2 μm dGTP and are indicated by an arrow and label (0 μm dGTP). The binary complex translocation rates were determined using autocorrelation and the two-state model (8); the translocation rates in the presence of dGTP were determined using autocorrelation and the three-state model (Fig. 2E) (9). Shown are plots of kon versus [dGTP] (D) and koff versus [dGTP] (E) for complexes formed between the D12A/D66A mutant or the Y390F/D12A/D66A mutant and DNA1, captured at 180 mV in the presence of Mg2+ or Mn2+. The dNTP binding rates were determined using autocorrelation and the three-state model. Plot symbols in B–D are the same as in A. Each data point was determined from 15–30 ionic current time traces for individual captured complexes; each time trace had a duration of 5–10 s. Error bars, S.E.
Data Analysis
The value of p, the probability of the lower amplitude state, was determined from histograms of all sampled amplitude data points, generated with Clampfit software (Molecular Devices), at 0.2 pA bin width (7). Histograms were fit to a two-term Gaussian function using the Levenberg-Marquardt search algorithm provided in Clampfit.
In the methods for extracting translocation rates and dNTP binding rates, segments of ionic current time traces were exported from Clampfit to Matlab (MathWorks); the centers of the two amplitude clusters and their probabilities were accurately calculated using the maximum likelihood estimation on samples of measured amplitudes. For each ionic current time trace, the autocorrelation was calculated and fitted to its expected expression derived from the mathematical model. For experiments conducted in the absence of dNTP, a two-state kinetic model was used to describe the transitions between the two translocation states (8). In the presence of dNTP, a three-state kinetic model was employed to accommodate the additional dNTP-bound state (9). Kinetic rates were reconstructed by combining the results of maximum likelihood estimation on amplitude samples and the results of fitting the autocorrelation of the time trace to the corresponding model (8, 9). At each experimental condition, we used 15–30 time traces. For each kinetic rate, the reported value and S.E. were calculated based on values for these individual time traces.
Extraction of Dwell Time Samples
The dwell time samples in each of the two amplitude states used in the analyses in Fig. 8B and Table 3 were extracted using the single-channel detection function in Clampfit version 10 (Molecular Devices). This software uses a half-amplitude threshold method to assign transitions between two user-defined amplitude levels (19). Amplitude levels for each of the two states were determined for the single-channel searches from histograms of all sampled amplitude data points. Complexes were captured at 160 mV.
FIGURE 8.
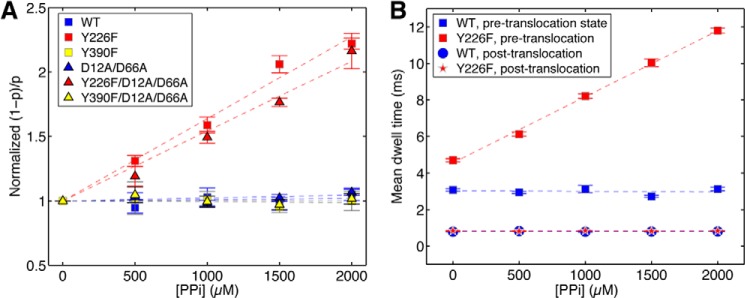
Pyrophosphate binding to mutant Φ29 DNAP-DNA complexes. A, the normalized (1 − p)/p (where (1 − p) is the probability of pre-translocation state occupancy, and the normalized (1 − p)/p is the value of (1 − p)/p in the presence of a given concentration of pyrophosphate, divided by the value for (1 − p)/p for the same Φ29 DNAP-DNA complex at 0 mm pyrophosphate) is plotted as a function of pyrophosphate concentration for complexes formed between wild type, Y226F, Y390F, D12A/D66A, Y226F/D12A/D66A, or Y390F/D12A/D66A Φ29 DNAP and DNA1. Plot symbols for each of the enzymes are given in the legend to Fig. 4. In B, the mean dwell times in the pre-translocation and post-translocation states for complexes formed between wild type Φ29 DNAP (blue squares, pre-translocation; blue circles, post-translocation) and the Y226F mutant (red squares, pre-translocation; red stars, post-translocation) are plotted as a function of pyrophosphate concentration. Complexes were captured at 160 mV. Each data point was determined from 15–30 ionic current time traces for individual captured complexes; each time trace had a duration of 5–10 s. Error bars, S.E.
TABLE 3.
Average dwell times in the pre-translocation and post-translocation states
All values are reported with the S.E.
| Φ29 DNAPa | [PPi] | Pre-translocation state mean dwell time | Post-translocation state mean dwell time |
|---|---|---|---|
| mm | ms | ms | |
| Wild type | 0 | 3.08 ± 0.07 | 0.79 ± 0.01 |
| 0.5 | 2.95 ± 0.05 | 0.82 ± 0.01 | |
| 1.0 | 3.13 ± 0.16 | 0.78 ± 0.01 | |
| 1.5 | 2.73 ± 0.04 | 0.79 ± 0.01 | |
| 2.0 | 3.13 ± 0.09 | 0.8 ± 0.01 | |
| Y226F | 0 | 4.7 ± 0.07 | 0.84 ± 0.02 |
| 0.5 | 6.13 ± 0.10 | 0.82 ± 0.01 | |
| 1.0 | 8.2 ± 0.11 | 0.85 ± 0.01 | |
| 1.5 | 10.04 ± 0.16 | 0.83 ± 0.01 | |
| 2.0 | 11.8 ± 0.12 | 0.84 ± 0.01 | |
| Y390F | 0 | 3.58 ± 0.04 | 0.97 ± 0.01 |
| 2.0 | 3.3 ± 0.04 | 0.94 ± 0.01 | |
| D12A/D66A | 0 | 3.89 ± 0.07 | 0.87 ± 0.01 |
| 2.0 | 3.65 ± 0.06 | 0.83 ± 0.01 | |
| Y226F/D12A/D66A | 0 | 5.92 ± 0.09 | 1.13 ± 0.02 |
| 2.0 | 14.96 ± 0.36 | 1.2 ± 0.03 | |
| Y390F/D12A/D66A | 0 | 3.58 ± 0.02 | 0.95 ± 0.01 |
| 2 | 3.52 ± 0.04 | 0.9 ± 0.01 |
a Complexes were formed between the indicated Φ29 DNAP enzymes and DNA1 and captured atop the nanopore at 160 mV applied potential.
Processive DNA Synthesis Assays
The incubation mixture contained 50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1 mm DTT, 4% (v/v) glycerol, 0.05% (v/v) Tween 20, 0.1 mg/ml BSA, 40 μm each dNTP and [α-32P]dATP (0.3 μCi), 100 ng of oligonucleotide-primed M13 ssDNA (∼3.35 pm in reaction mixture), and 30 ng of wild type or mutant Y226F Φ29 DNAP (dilution 1; 30 pm in reaction mixture), in a final volume of 15 μl. 2-Fold serial dilutions of the enzymes were carried out as indicated. After incubation for the indicated times at 30 °C, reactions were terminated by the addition of 30 mm EDTA, 0.5% SDS. The DNA was denatured by the addition of 0.4 m NaOH and subjected to alkaline electrophoresis in 0.7% agarose gels. After electrophoresis, the gels were dried and autoradiographed.
RESULTS AND DISCUSSION
Residues Tyr-226 and Tyr-390 in the polymerase active site of Φ29 DNAP are implicated in structural transitions essential to both the translocation step and to dNTP binding (4, 12–14). In this study, we examined the effects of introducing the Y226F or Y390F mutations on these DNAP functions in two contexts: 1) as single amino acid changes in the otherwise wild type Φ29 DNAP and 2) in combination with the D12A/D66A mutations in the exonuclease domain active site. We first determined the effects of the Y226F and Y390F mutations on the dynamics of the translocation step (Fig. 4). We used a robust method that employs autocorrelation and a two-state model (8) to extract the forward (r1; Fig. 4A) and reverse (r2; Fig. 4B) translocation rates from ionic current traces recorded when individual Φ29 DNAP-DNA binary complexes reside atop the nanopore.
FIGURE 4.
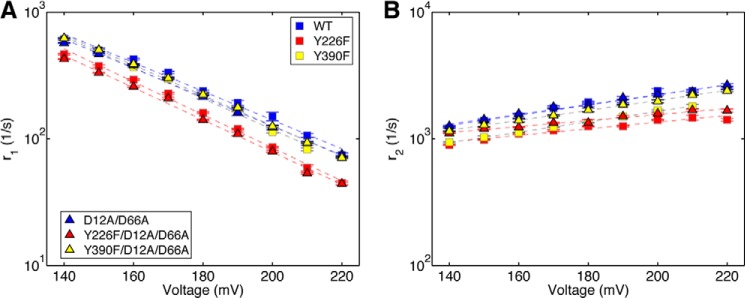
Transition rates of the Φ29 DNAP translocation step extracted from ionic current traces measured in absence of dNTP. Shown are plots of log(r1) versus voltage (A) and log(r2) versus voltage (B) for binary complexes formed between the wild type (blue squares), Y226F (red squares), Y390F (yellow squares), D12A/D66A (blue triangles), Y226F/D12A/D66A (red triangles), or Y390F/D12A/D66A (yellow triangles) Φ29 DNAP and DNA1. Each plotted point shows the mean ± S.E. In the absence of dNTP, the fluctuation rates between the pre-translocation and post-translocation states are fully described by a two-state model with two transition rates (8). Each plotted data point shows the means ± S.E., determined from 15–30 ionic current time traces for individual captured complexes; each time trace had a duration of 5–10 s.
We compared the translocation rates for wild type, Y226F, Y390F, D12A/D66A, Y226F/D12A/D66A, and Y390F/D12A/D66A Φ29 DNAP enzymes, as a function of applied force (voltage). The force opposes the forward translocation and thus decreases its rate (r1) while promoting the reverse translocation and increasing its rate (r2). The slope of log(r1) versus voltage is negative and proportional to the distance between the pre-translocation state and the transition state; the slope of log(r2) versus voltage is positive and proportional to the distance between the transition state and the post-translocation state. Plots of log(r1) versus voltage and log(r2) versus voltage show that the Y226F and Y390F mutations have modest effects on the translocation rates in the context of either the wild type or D12A/D66A backgrounds (Fig. 4); neither the vertical intercepts nor the slopes of log(rate) versus voltage exhibit large differences from those for the wild type enzyme, indicating that the mutations do not significantly change the energy landscape for the translocation step (8). The rates for complexes formed with each of the six enzymes and captured at 180 mV are given in Table 1.
TABLE 1.
Translocation rates for wild type Φ129 DNAP and mutants at 180 mV
All values are reported for data collected at 180 mV and are given with the S.E.
| Enzymea | r1b | r2c |
|---|---|---|
| s−1 | s−1 | |
| Wild type | 237.57 ± 3.88 | 1945 ± 21.5 |
| Y226F | 159.95 ± 2.06 | 1253.9 ± 9.7 |
| Y390F | 221.43 ± 2.33 | 1334.4 ± 9.0 |
| D12A/D66A | 214.36 ± 8.56 | 1830.7 ± 32.2 |
| D12A/D66A (in Mn2+) | 40.05 ± 0.88 | 1741.9 ± 23.8 |
| Y226F/D12A/D66A | 140.71 ± 1.81 | 1334.6 ± 77.3 |
| Y390F/D12A/D66A | 223.08 ± 4.6 | 1686.6 ± 58.8 |
| Y390F/D12A/D66A (in Mn2+) | 40.43 ± 1.74 | 1252.6 ± 27.6 |
a All enzymes were examined in Mg2+ unless otherwise specified.
b The forward translocation rate.
c The reverse translocation rate.
Across the range of voltages, the rates of the forward and reverse fluctuations across the translocation step for the D12A/D66A mutant are almost indistinguishable from those of the wild type enzyme (Fig. 4). The most notable effects on the translocation rates are caused by the Y226F mutation. Across the range of voltages, the forward translocation rates for the Y226F and Y226F/D12A/D66A mutants are ∼35% slower than those of the wild type and D12A/D66A enzymes, respectively (Fig. 4). The reverse translocation rates are also reduced by the Y226F mutation; r2 for the Y226F enzyme is ∼35% slower than it is for the wild type, and r2 for the Y226F/D12A/D66A is ∼15% slower than for the D12A/D66A enzyme (Fig. 4). The moderate effects on r1 and r2 of introducing the Y226F or Y390F mutations can be contrasted with the significant effects of active site proximal DNA substrate sequences on rates r1 and r2 in binary complexes formed with the wild type enzyme. For example, sequence changes at n = 0 of the template strand or in the −2 and −3 base pairs of the duplex can yield an ∼10-fold increase in r1, and an ∼15-fold decrease in r2, relative to DNA1 (8).
Complementary dNTP Binding to the Mutant Enzymes
We determined the effects of introducing the Y226F or Y390F mutations on complementary dNTP binding affinity in titration experiments, using complexes formed between each of the six Φ29 DNAP enzymes and DNA1, captured at 180 mV. To display these data, we plotted the normalized p/(1 − p), where p is the probability of post-translocation state occupancy, and the normalized p/(1 − p) is defined as the value of p/(1 − p) in the presence of a given concentration of dNTP, divided by the value of p/(1 − p) for the same Φ29 DNAP-DNA complex at 0 μm dNTP (7). The normalized p/(1 − p) is solely determined by the binding affinity of dNTP; it is independent of the transitions between the two translocation states in the absence of dNTP (and thus independent of any differences in the translocation rates among the six enzymes); the effect of these transitions is eliminated when p/(1 − p) is normalized by its value measured in the absence of dNTP. The normalized p/(1 − p) thus permits direct comparison of the post-translocation state dNTP binding affinities among the enzymes.
The Y226F and Y390F mutants differ dramatically in dNTP binding affinity. Relative to the wild type enzyme, the Y266F mutation increases the affinity for dNTP, whereas the Y390F mutation significantly decreases it (Fig. 5A). We also examined the dNTP binding affinities for Y226F and Y390F mutations in the D12A/D66A background. Fig. 5B shows the results of dGTP titration experiments for complexes formed with the wild type, D12A/D66A, Y226F, Y390F, Y226/D12A/D66A, and Y390F/D12A/D66A enzymes. Because of the wide range of dGTP concentrations used in the experiments and the differences in dNTP affinities among the enzymes, the normalized p/(1 − p) as a function of [dGTP] for the six enzymes is most clearly compared on a log scale plot of (normalized p/(1 − p)) − 1 (Fig. 5B). Interestingly, the D12A/D66A mutations themselves increase the affinity for dNTP relative to that of the wild type Φ29 DNAP (Fig. 5B). Residues Asp-12 and Asp-66 are located in the exonuclease domain, ∼30 Å from the polymerase active site (Fig. 1, A and B). Nonetheless, these mutations in the exonuclease active site have been shown to disrupt the ability of Φ29 DNAP to perform downstream strand displacement during DNA synthesis (5, 20). The effect of the D12A/D66A mutations on dNTP binding, taken together with the effects of these mutations on strand displacement, suggest that the exonuclease active site mutations perturb Φ29 DNAP interdomain architecture, yielding pleiotropic effects on enzyme function. When combined with either the Y226F or the Y390F mutations, the D12A/D66A mutations yield an increase in affinity over that of the polymerase site mutation alone (Fig. 5B).
FIGURE 5.
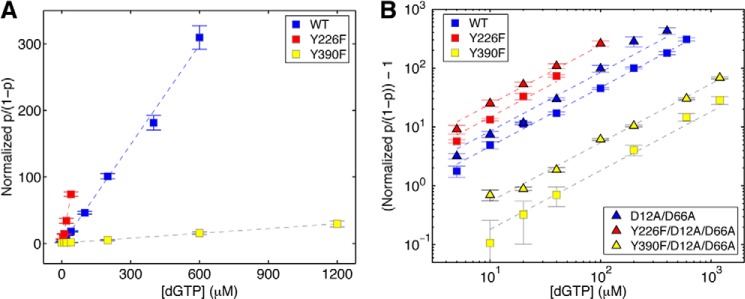
Complementary dNTP binding affinities of Φ29 DNAP mutants. The normalized p/(1 − p) (where p is the probability of post-translocation state occupancy, and the normalized p/(1 − p), defined as the value of p/(1 − p) in the presence of a given concentration of dNTP, divided by the value for p/(1 − p) for the same Φ29 DNAP-DNA complex at 0 μm dGTP (7)) is plotted (A) as a function of dGTP concentration for complexes formed between wild type, Y226F, and Y390F Φ29 DNAP. In B, (normalized p/(1 − p)) − 1 is plotted on a log scale as a function of dGTP concentration for complexes formed between the wild type, Y226F, Y390F, D12A/D66A, Y226F/D12A/D66A, or Y390F/D12A/D66A Φ29 DNAP and DNA1. Complexes were captured at 180 mV. Plot symbols for each of the enzymes are given in the legend to Fig. 4. Error bars, S.E. Each data point was determined from 15–30 ionic current time traces for individual captured complexes; each time trace had a duration of 5–10 s.
Dynamics of dNTP Binding to the Mutant Enzymes
To uncover the kinetic mechanisms by which the mutations in the Φ29 DNAP polymerase and exonuclease active sites perturb dNTP binding, we used autocorrelation and a three-state model (Fig. 2E) to simultaneously extract the forward (r1) and reverse (r2) translocation rates and the dNTP association (kon) and dissociation (koff) rates from ionic current traces for complexes captured in the presence of complementary dNTP (9). For each of the mutant enzymes, the plots of log(r1) versus voltage (Fig. 6A) and log(r2) versus voltage (Fig. 6B) obtained for complexes captured in the presence of dNTP are indistinguishable from those of the same enzyme in the absence of dNTP (Fig. 4, A and B). Thus, as we have shown for wild type Φ29 DNAP (9), the rates of fluctuation across the translocation step for the mutants are independent of [dNTP]; dNTP can only bind onto Φ29 DNAP complexes after they transition from the pre-translocation state to the post-translocation state and must dissociate from complexes prior to transition from the post-translocation state to the pre-translocation state.
FIGURE 6.

Translocation rates and dNTP association and dissociation rates determined simultaneously from ionic current traces measured in the presence of dNTP. Shown are plots of log(r1) versus voltage (A) and log(r2) versus voltage (B) for complexes formed between the wild type, Y226F, Y390F, D12A/D66A, Y226F/D12A/D66A, and Y390F/D12A/D66A Φ29 DNAP and DNA1, captured in the presence of dGTP. Also shown are plots of kon versus voltage (C) and koff versus voltage (D) for complexes formed between the wild type, Y226F, Y390F, D12A/D66A, Y226F/D12A/D66A, and Y390F/D12A/D66A Φ29 DNAP and DNA1, captured in the presence of dGTP. Rates were extracted from ionic current traces using the autocorrelation function and the three-state model shown in Fig. 2E. Plot symbols for each of the enzymes are given in the legend to Fig. 4. Error bars, S.E. Each data point was determined from 15–30 ionic current time traces for individual captured complexes; each time trace had a duration of 5–10 s. The data plotted are for complexes captured in the presence of the following dGTP concentrations: wild type, 10 μm; D12A/D66A, 10 μm; Y226F, 5 μm; Y390F, 20 μm; Y226F/D12A/D66A, 5 μm; and Y390F/D12A/D66A, 20 μm. Although we have shown that all four transition rates (r1, r2, kon, and koff) are independent of [dNTP] (9), the method of extracting the rates from the ionic current traces using autocorrelation and the three-state model (Fig. 2E) is most robust when using data collected under conditions where all three states are well sampled. For example, when the dNTP concentration is very low, the dGTP-bound state is not well sampled; when the dNTP concentration is very high, only the dGTP-bound state is well sampled. The dNTP concentrations optimal for the analysis vary with the dNTP binding affinity of each mutant.
We plotted the dGTP association rates (kon; Fig. 6C) and dissociation rates (koff; Fig. 6D) for complexes captured as a function of applied voltage. Neither kon nor koff for the mutant enzymes displays a systematic trend with the applied voltage (Fig. 6, C and D); as we have previously shown for the wild type enzyme (9), the dNTP binding rates are independent of applied force (Fig. 6, C and D). Therefore, to compare dNTP binding rates among the different enzymes, for each enzyme, we treat all data points for kon or koff as independent samples and calculate the mean and S.E. for each of the two rates (Table 2).
TABLE 2.
Complementary dNTP binding rates for wild type Φ29 DNAP and mutants
All values are reported with the S.E.
| Enzymea | konb | koffc | Kd |
|---|---|---|---|
| s−1 μm−1 | s−1 | μm | |
| Wild type | 18.88 ± 0.5 | 30.41 ± 0.59 | 1.621 ± 0.049 |
| Y226F | 13.94 ± 0.36 | 8.97 ± 0.21 | 0.654 ± 0.041 |
| Y390F | 8.33 ± 0.24 | 343.01 ± 5.80 | 41.61 ± 1.74 |
| D12A/D66A | 19.87 ± 0.27 | 18.02 ± 0.30 | 0.908 ± 0.023 |
| D12A/D66A (in Mn2+) | 11.45 ± 0.86 | 0.44 ± 0.02 | 0.040 ± 0.005 |
| Y226F/D12A/D66A | 17.84 ± 0.29 | 8.07 ± 0.18 | 0.453 ± 0.012 |
| Y390F/D12A/D66A | 10.42 ± 0.18 | 220.09 ± 2.63 | 21.23 ± 0.66 |
| Y390F/D12A/D66A (in Mn2+) | 13.64 ± 0.25 | 11.35 ± 0.25 | 0.835 ± 0.023 |
a All enzymes were examined in Mg2+ unless otherwise specified.
b The dGTP association rate constant.
c The dGTP dissociation rate.
d Kd values calculated from the ratio koff (s−1)/kon (s−1 μm−1).
The increase in dNTP binding affinity caused by the Y226F mutation (Fig. 5) is due to a significant decrease in koff (8.97 ± 0.21 s−1 for the Y226F mutant versus 30.41 ± 0.59 s−1 for the wild type enzyme; Table 2 and Fig. 6D). The Y226F mutation also causes a decrease in the dNTP association rate (kon = 13.94 ± 0.36 μm−1 s−1 for Y226F versus 18.88 ± 0.5 μm−1 s−1 for the wild type enzyme; Table 2 and Fig. 6C), but the effect of this decrease in kon on the binding equilibrium is more than offset by the decrease in the dissociation rate. For the Y390F mutant, kon is slower than it is for the wild type Φ29 DNAP (8.33 ± 0.24 μm−1 s−1 for Y390F versus 18.88 ± 0.5 μm−1 s−1 for the wild type enzyme; Table 2, Fig. 6C), and koff is >10 times faster than it is for wild type Φ29 DNAP (343.01 ± 5.80 s−1 for Y390F versus 30.41 ± 0.59 s−1 for the wild type; Table 2 and Fig. 6D). Therefore, changes in both dNTP binding rates contribute to the large decrease in dNTP binding affinity caused by the Y390F mutation.
The D12A/D66A mutations in the exonuclease active site have negligible effect on the dNTP association rate (Table 2 and Fig. 6C); they increase the dNTP binding affinity by decreasing koff (18.02 ± 0.30 s−1 for the D12A/D66A enzyme versus 30.41 ± 0.59 s−1 for the wild type enzyme; Table 2 and Fig. 6D). The increase in dNTP binding affinity of the Y226F/D12A/D66A mutant relative to the Y226F mutant (Fig. 5B) is attributable to the combination of a modest increase in the association rate (17.84 ± 0.29 μm−1 s−1 for the Y226F/D12A/D66A mutant versus 13.94 ± 0.36 μm−1 s−1 for the Y226F mutant; Table 2 and Fig. 6C) and a modest decrease in the dissociation rate (8.07 ± 0.18 s−1 for Y226F/D12A/D66A versus 8.97 ± 0.21 s−1 for Y226F; Table 2 and Fig. 6D). The increase in dNTP binding affinity for the Y390F/D12A/D66A mutant relative to the Y390F mutant (Fig. 5B) arises due to an increase in the association rate (10.42 ± 0.18 μm−1 s−1 for Y390F/D12A/D66A versus 8.33 ± 0.24 μm−1 s−1 for Y390F; Table 2 and Fig. 6C) and a more substantial decrease in the dissociation rate (220.09 ± 2.63 s−1 for Y390F/D12A/D66A versus 343.01 ± 5.80 s−1 for Y390F; Table 2 and Fig. 6D). Nonetheless, the dNTP dissociation rate for the Y390F/D12A/D66A mutant is still dramatically higher than it is for either the wild type enzyme (30.41 ± 0.59 s−1) or the D12A/D66A mutant (18.02 ± 0.30 s−1); in both backgrounds, the introduction of the Y390F mutation increases koff by >10-fold.
Rescue by Mn2+ of the Impairment in dNTP Binding Kinetics Caused by the Y390F Mutation
In the presence of Mg2+, the Y390F mutant is severely compromised in processive DNA synthesis relative to the wild type Φ29 DNAP, even at high concentrations of dNTPs (13). However, in the presence of Mn2+, the level of processive synthesis catalyzed by the Y390F mutant as a function of dNTP concentration is very similar to the level catalyzed by the wild type enzyme (13). We considered that the decrease in the dNTP association rate and the large increase in the dNTP dissociation rate caused by the Y390F mutation (Fig. 6D and Table 1) might contribute to the synthesis impairment observed in the presence of Mg2+ and whether the binding rates might be rescued in the presence of Mn2+. In particular, the large dissociation rate of Mg2+-dNTPs in the presence of the Y390F mutation could significantly decrease the probability of progressing to the chemical step upon dNTP binding. If so, the rescue of processive synthesis in the presence of Mn2+-dNTPs might be explained, at least in part, if the dissociation rate of Mn2+-dNTPs was slower than the rate for Mg2+-dNTPs. A decrease in the dissociation rate that brings the dwell time in the dNTP-bound state (1/koff) into a regime that is sufficient for chemistry could improve the function of the mutant enzyme in synthesis. (Note that it is not necessary that the dwell time in the dNTP-bound state be increased to the level of the wild type for this to obtain, only that it be increased sufficiently to increase the probability of chemistry.)
We compared the effects of the Y390F mutation on the translocation step and on dNTP binding in the presence of Mn2+ or Mg2+. To avoid bulk phase exonucleolytic degradation, we performed these experiments using the Y390F/D12A/D66A and D12A/D66A enzymes. In binary complexes, the identity of the divalent metal cation affects the equilibrium across the translocation step; for complexes formed in Mn2+, the probability of the post-translocation state is smaller than it is for complexes formed in Mg2+. When binary complexes formed between D12A/D66A and DNA1 are captured at 180 mV, p/(1 − p) in Mg2+ = 0.117 ± 0.002, and p/(1 − p) in Mn2+ = 0.023 ± 0.0004. For complexes formed with the Y390F/D12A/D66A enzyme and captured at 180 mV, p/(1 − p) in Mg2+ = 0.132 ± 0.001, and p/(1 − p) in Mn2+ = 0.032 ± 0.001. Plots of normalized p/(1 − p) for dGTP titration experiments show that complexes formed with both the D12A/D66A and Y390F/D12A/D66A mutants have a greater affinity for dNTP when they are captured in Mn2+ than when they are captured in Mg2+ (Fig. 7A). Nonetheless, when the two enzymes are compared in Mn2+, the Y390F/D12A/D66A mutant retains a significantly diminished affinity for dNTP relative to the D12A/D66A mutant (Fig. 7A).
To determine the kinetic mechanisms by which Mn2+ alters the translocation and dNTP binding, we compared the translocation fluctuation rates and the dNTP binding rates for complexes formed between the D12A/D66A and Y390F/D12A/D66A enzymes in Mg2+ or Mn2+. The shift in the translocation equilibrium toward the pre-translocation state caused by Mn2+ in binary complexes is primarily due to a significant decrease in the forward translocation rate relative to complexes formed in Mg2+ (Fig. 7, B and C, and Table 1). At 180 mV, for the D12A/D66A mutant, r1 in Mg2+ = 214.36 ± 8.56, and r1 in Mn2+ = 40.05 ± 0.88. For the Y390F/D12A/D66A mutant, r1 in Mg2+ = 223.08 ± 4.6, and in Mn2+, r1 = 40.43 ± 1.74. It is possible that this decrease in the forward translocation rate caused by Mn2+ may contribute to the generally decreased level of processive synthesis supported by Mn2+ relative to Mg2+ for both wild type and mutant enzymes (13). We will examine the effects of divalent metals on the dynamics of the Φ29 DNAP translocation step in detail in a separate study.
As we have shown for complexes captured in Mg2+ (Fig. 6, A and B) (9), when complexes are captured in Mn2+, neither r1 nor r2 is affected by [dNTP] (Fig. 7, B and C). The effect of Mn2+ on the dNTP association rate (relative to Mg2+) differed between the two enzymes (Table 2 and Fig. 7D). For the D12A/D66A mutant, kon was slower in Mn2+ (11.45 ± 0.86 μm−1 s−1) than in Mg2+ (19.87 ± 0.27 μm−1 s−1); for the Y390F/D12A/D66A mutant, kon increased very slightly in Mn2+ (kon in Mn2+ = 13.64 ± 0.25 μm−1 s−1; kon in Mg2+ = 10.42 ± 0.18 μm−1 s−1). For both enzymes, Mn2+ had a dramatic effect on the dNTP dissociation rates (Table 2 and Fig. 7E). For the D12A/D66A mutant, koff in Mg2+ = 18.02 ± 0.30 s−1, and koff in Mn2+ = 0.44 ± 0.02 s−1. For the Y390F/D12A/D66A mutant, koff in Mg2+ = 220.09 ± 2.63 s−1, and koff in Mn2+ = 11.35 ± 0.25 s−1. Thus, Mn2+ decreases the dNTP dissociation rate for the Y390F/D12A/D66A mutant to a value smaller than 30.41 ± 0.59 s−1, the dNTP dissociation rate for the wild type enzyme in Mg2+ (Table 2 and Fig. 6D). These data are consistent with the proposal that the rescue of processive DNA synthesis by the Y390F mutant in the presence of Mn2+ could result from a decrease in the dNTP dissociation rate for the mutant, relative to this rate for complexes formed in the presence of Mg2+.
The findings that both r1 and koff are significantly slower for complexes captured in Mn2+ than they are for complexes captured in Mg2+ suggests the possibility that Mn2+ may exert its effects on r1 and koff via a common mechanism; it may diminish the rate of fingers opening from the closed complex. The structural model proposed for the translocation step (4) predicts that decreasing the rate of fingers opening in the pre-translocation state would lead to a decrease in the rate of the forward translocation (r1). In the dNTP-bound post-translocation state, decreasing the rate of fingers opening could yield a decrease in the dissociation rate of dNTP (koff).
Pyrophosphate Binding to the Pre-translocation State in Mutant Enzyme Complexes
The strong (and opposing) effects of the Y226F and Y390F mutations on the kinetics of dNTP binding to post-translocation state complexes prompted us to examine whether these mutations affected the binding of pyrophosphate to Φ29 DNAP-DNA complexes. As a product of phosphodiester bond formation, pyrophosphate is bound to pre-translocation state complexes immediately following the chemical step. We compared the effects of pyrophosphate on complexes formed between each of the six Φ29 DNAP enzymes and DNA1 in pyrophosphate titration experiments (Fig. 8A). To highlight the effect of pyrophosphate on the pre-translocation state probability, we plotted the normalized (1 − p)/p as a function of pyrophosphate concentration, where p is the probability of the post-translocation state, and consequently (1 − p) is the probability of the pre-translocation state. The normalized (1 − p)/p is determined solely by the binding affinities of pyrophosphate. This quantity is independent of the transitions between the two translocation states in the absence of pyrophosphate (and thus independent of any differences in the translocation rates among the six enzymes; Fig. 4); the effect of these transitions is eliminated when (1 − p)/p is normalized by its value in the absence of pyrophosphate.
When pyrophosphate was titrated into the nanopore chamber in the presence of complexes formed between DNA1 and the wild type, Y226F, Y390F, D12A/D66A, Y226F/D12A/D66A, or Y390F/D12A/D66A enzymes, the equilibrium across the translocation step was shifted toward the pre-translocation state for the Y226F and Y226F/D12/D66A mutants, whereas the translocation equilibrium for the wild type, Y390F, D12A/D66A, and Y390F/D12A/D66A enzymes was unaffected (Fig. 8A). Thus, the introduction of the Y226F mutation specifically causes a concentration-dependent shift in the equilibrium across the Φ29 DNAP translocation step, toward the pre-translocation state. The enzymes bearing the Y226F mutation displayed a ∼2.5-fold increase in pre-translocation state probability at the highest concentration of pyrophosphate that could be tested without the risk of precipitation (Fig. 8A).
For each of the six enzymes, complexes captured in the presence of pyrophosphate resided atop the nanopore and fluctuated between the upper and lower amplitude states for tens of seconds, during which we rarely observed pyrophosphorolytic (or exonucleolytic) cleavage (which can be readily discerned by a change in ionic current amplitude (7, 10)). This is consistent with our prior finding that pyrophosphorolysis is extremely slow for complexes formed with DNA substrates bearing 3′-H termini (7).
To determine the dynamic mechanism by which pyrophosphate shifts the translocational equilibrium in complexes with enzymes bearing the Y226F mutation, we examined the effects of pyrophosphate on the average dwell time in the pre-translocation and post-translocation states. We used a half-amplitude threshold method (19) to extract dwell time samples from ionic current traces for complexes formed between each of the six enzymes and DNA1. Pyrophosphate causes a concentration-dependent, linear increase in the average pre-translocation state dwell time for complexes formed with the Y226F and Y226F/D12A/D66A mutants, whereas the average dwell time in the post-translocation state for these mutants is unaffected (Fig. 8B and Table 3). For complexes formed with the wild type, Y390F, D12A/D66A, or Y390F/D12A/D66A enzymes, neither the average dwell time in the pre-translocation state nor the average dwell time in the post-translocation state was affected by the presence of pyrophosphate (Fig. 8B and Table 3). Thus, the Y226F mutation specifically increases the affinity of the pre-translocation state for pyrophosphate. For the wild type, D12A/D66A, Y390F, and Y390F/D12A/D66A enzymes, the affinity for pyrophosphate is below the limit of detection of our assay.
The Y226F mutant has a slower forward translocation rate than the wild type enzyme (Fig. 4 and Table 1), and its forward translocation is further diminished in the presence of pyrophosphate (Fig. 8B). The immediate product of the chemical step in DNA synthesis is the fingers-closed, pre-translocation state complex with pyrophosphate bound, and the probability of fingers opening and the consequent pre-translocation to post-translocation transition may be lower when pyrophosphate is bound. Nonetheless, the Y226F mutant is not impaired in the rate or processivity of DNA synthesis when assayed in the bulk phase (Fig. 3). This suggests that the effects of the Y226F mutation on the forward translocation rate, revealed under opposing force, and on the affinity for pyrophosphate in the pre-translocation state do not reduce the forward translocation rate enough to impede the rate of DNA synthesis in the absence of an opposing force.
CONCLUSION
The Y390F and Y226F mutations in Φ29 DNAP both disrupt the hydrogen bond between the hydroxyl groups of these two conserved residues, an interaction that was proposed to stabilize the orientation of Tyr-390 in the fingers-closed, post-translocation ternary complex and in the fingers-closed, pre-translocation state complex (4). Nonetheless, biochemical studies revealed that the Y390F and Y226F mutations yield very different properties in Φ29 DNAP (11–14). In this study, we have shown that the Y390F and Y226F mutations differ in their effects on the dynamics of the translocation step and on the dynamics of substrate binding in both the pre-translocation and post-translocation states. Somewhat surprisingly, we found that neither the Y226F nor the Y390F mutation caused an increase in the forward translocation rate; this rate is decreased by the Y226F mutation and largely unaffected by the Y390F mutation. This indicates that the disruption of the hydrogen bond between the two residues does not destabilize the fingers-closed pre-translocation state, implying that the hydrogen bond makes a relatively small contribution to the free energy of this state.
Although the most dramatic consequences of introducing the Y390F or Y226F mutations are their opposing effects on dNTP binding rates and affinity, it is difficult to clearly attribute these effects to the destabilization of the fingers-closed, dNTP-bound, post-translocation state complex. The effects on the dNTP binding rates elicited by the Y390F mutation, particularly the significantly increased dNTP dissociation rate, could instead be due to a distortion of dNTP binding within the closed complex rather than destabilization of the closed complex itself, because the closed pre-translocation state does not appear to be destabilized by this mutation. Distortion of dNTP binding may arise as a consequence of the perturbation of the water-mediated interactions of Tyr-390 with the template strand in the DNA duplex.
The increased rate of dNTP dissociation observed with the Y390F mutation is not observed when the hydrogen bond is disrupted by the Y226F mutation; by contrast, this mutation leads to a dramatic decrease in the dNTP dissociation rate. The Y226F mutation also yields a higher affinity for pyrophosphate in the pre-translocation state, which may reflect a slower dissociation rate for this ligand. This suggests that the higher affinity for both dNTP and for pyrophosphate caused by the Y226F mutation may arise from a common mechanism. As with the Y390F mutation, it is possible that the effects of the Y226F mutation on ligand binding arise from an indirect effect on the water-mediated interactions of Tyr-390 with the template strand of the DNA duplex. In this scenario, the loss of the hydrogen bond between Tyr-390 and Tyr-226 results in a change in the distance or angle of the bonds formed between Tyr-390 and the template strand, leading to tighter binding of both dNTP in the post-translocation state and pyrophosphate in the pre-translocation state. Indeed, it cannot be ruled out that such a structural perturbation might also contribute to the effects of the Y226F mutation on the translocation rates.
It is difficult to unequivocally assign the effects of introducing the Y390F or Y226F mutations on the translocation kinetics or on substrate binding kinetics in the pre-translocation and post-translocation states directly to disruption of the hydrogen bonding potential between them in fingers-closed Φ29 DNAP-DNA complexes. These findings do not refute the proposed structural mechanism, which links the spatial displacement of translocation to the fingers opening transition (4), but they suggest that the hydrogen bond that forms between Tyr-390 and Tyr-226 in the fingers-closed state is not directly essential to the mechanism.
Acknowledgments
We are grateful to Ashley Cox for assistance in data analysis and to Robin Abu-Shumays and Mark Akeson for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health, NIGMS, Grant 1R01GM087484 (to K. R. L.). This work was also supported by United States National Science Foundation Grant DMS-0719361 (to H. W.) and by Spanish Ministry of Economy and Competitiveness Grant BFU2011-23645 (to M. S.).
- DNAP
- DNA polymerase
- α-HL
- α-hemolysin.
REFERENCES
- 1. Blanco L., Bernad A., Lázaro J. M., Martín G., Garmendia C., Salas M. (1989) Highly efficient DNA synthesis by the phage Φ29 DNA polymerase. Symmetrical mode of DNA replication. J. Biol. Chem. 264, 8935–8940 [PubMed] [Google Scholar]
- 2. Blanco L., Salas M. (1996) Relating structure to function in Φ29 DNA polymerase. J. Biol. Chem. 271, 8509–8512 [DOI] [PubMed] [Google Scholar]
- 3. Salas M., Blanco L., Lázaro J. M. M., de Vega M. (2008) The bacteriophage Φ29 DNA polymerase. IUBMB Life 60, 82–85 [DOI] [PubMed] [Google Scholar]
- 4. Berman A. J., Kamtekar S., Goodman J. L., Lázaro J. M., de Vega M., Blanco L., Salas M., Steitz T. A. (2007) Structures of Φ29 DNA polymerase complexed with substrate. The mechanism of translocation in B-family polymerases. EMBO J. 26, 3494–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morin J. A., Cao F. J., Lázaro J. M., Arias-Gonzalez J. R., Valpuesta J. M., Carrascosa J. L., Salas M., Ibarra B. (2012) Active DNA unwinding dynamics during processive DNA replication. Proc. Natl. Acad. Sci. U.S.A. 109, 8115–8120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kamtekar S., Berman A. J., Wang J., Lázaro J. E., de Vega M., Blanco L., Salas M., Steitz T. A. (2004) Insights into strand displacement and processivity from the crystal structure of the protein-primed DNA polymerase of bacteriophage Φ29. Mol. Cell 16, 609–618 [DOI] [PubMed] [Google Scholar]
- 7. Dahl J. M., Mai A. H., Cherf G. M., Jetha N. N., Garalde D. R., Marziali A., Akeson M., Wang H., Lieberman K. R. (2012) Direct observation of translocation in individual DNA polymerase complexes. J. Biol. Chem. 287, 13407–13421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lieberman K. R., Dahl J. M., Mai A. H., Akeson M., Wang H. (2012) Dynamics of the translocation step measured in individual DNA polymerase complexes. J. Am. Chem. Soc. 134, 18816–18823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lieberman K. R., Dahl J. M., Mai A. H., Cox A., Akeson M., Wang H. (2013) Kinetic mechanism of translocation and dNTP binding in individual DNA polymerase complexes. J. Am. Chem. Soc. 135, 9149–9155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lieberman K. R., Cherf G. M., Doody M. J., Olasagasti F., Kolodji Y., Akeson M. (2010) Processive replication of single DNA molecules in a nanopore catalyzed by Φ29 DNA polymerase. J. Am. Chem. Soc. 132, 17961–17972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blanco L., Bernad A., Blasco M. A., Salas M. (1991) A general structure for DNA-dependent DNA polymerases. Gene 100, 27–38 [DOI] [PubMed] [Google Scholar]
- 12. Truniger V., Lázaro J. M., Salas M., Blanco L. (1996) A DNA binding motif coordinating synthesis and degradation in proofreading DNA polymerases. EMBO J. 15, 3430–3441 [PMC free article] [PubMed] [Google Scholar]
- 13. Blasco M. A., Lázaro J. M., Bernad A., Blanco L., Salas M. (1992) Φ29 DNA polymerase active site. Mutants in conserved residues Tyr254 and Tyr390 are affected in dNTP binding. J. Biol. Chem. 267, 19427–19434 [PubMed] [Google Scholar]
- 14. Saturno J., Blanco L., Salas M., Esteban J. A. (1995) A novel kinetic analysis to calculate nucleotide affinity of proofreading DNA polymerases. Application to Φ29 DNA polymerase fidelity mutants. J. Biol. Chem. 270, 31235–31243 [DOI] [PubMed] [Google Scholar]
- 15. Lázaro J. M., Blanco L., Salas M. (1995) Purification of bacteriophage Φ29 DNA polymerase. Methods Enzymol. 262, 42–49 [DOI] [PubMed] [Google Scholar]
- 16. Benner S., Chen R. J., Wilson N. A., Abu-Shumays R., Hurt N., Lieberman K. R., Deamer D. W., Dunbar W. B., Akeson M. (2007) Sequence-specific detection of individual DNA polymerase complexes in real time using a nanopore. Nat. Nanotechnol. 2, 718–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Akeson M., Branton D., Kasianowicz J. J., Brandin E., Deamer D. W. (1999) Microsecond time-scale discrimination among polycytidylic acid, polyadenylic acid, and polyuridylic acid as homopolymers or as segments within single RNA molecules. Biophys. J. 77, 3227–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garalde D. R., Simon C. A., Dahl J. M., Wang H., Akeson M., Lieberman K. R. (2011) Distinct complexes of DNA polymerase I (Klenow fragment) for base and sugar discrimination during nucleotide substrate selection. J. Biol. Chem. 286, 14480–14492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Colquhoun D., Sigworth F. J. (1995) in Single-channel Recording (Sakmann B., Neher E., eds) pp. 483–587, Plenum Press, New York [Google Scholar]
- 20. Esteban J. A., Soengas M. S., Salas M., Blanco L. (1994) 3`→5` exonuclease active site of Φ29 DNA polymerase. Evidence favoring a metal ion-assisted reaction mechanism. J. Biol. Chem. 269, 31946–31954 [PubMed] [Google Scholar]
- 21. Wang M., Xia S., Blaha G., Steitz T. A., Konigsberg W. H., Wang J. (2011) Insights into base selectivity from the 1.8 Å resolution structure of an RB69 DNA polymerase ternary complex. Biochemistry 50, 581–590 [DOI] [PMC free article] [PubMed] [Google Scholar]