

Background: STIM1 gates TRPC channels, but the interacting domains are unknown.
Results: The TRPC N and C terminus coiled coil domains interact to restrict access of STIM1. Their dissociation by cell stimulation promotes STIM1 interaction.
Conclusion: The STIM1 Orai1-activating region (SOAR) domain interacts with the TRPC C terminus CCD to open the channels.
Significance: The findings reveal how STIM1 opens the TRPC channels to control receptor-stimulated Ca2+ influx.
Keywords: Calcium, Calcium Signaling, Gating, Signaling, TRP Channels, Gating Mechanism, STIM1
Abstract
Transient receptor potential canonical (TRPC) channels mediate a critical part of the receptor-evoked Ca2+ influx. TRPCs are gated open by the endoplasmic reticulum Ca2+ sensor STIM1. Here we asked which stromal interaction molecule 1 (STIM1) and TRPC domains mediate the interaction between them and how this interaction is used to open the channels. We report that the STIM1 Orai1-activating region domain of STIM1 interacts with the TRPC channel coiled coil domains (CCDs) and that this interaction is essential for opening the channels by STIM1. Thus, disruption of the N-terminal (NT) CCDs by triple mutations eliminated TRPC surface localization and reduced binding of STIM1 to TRPC1 and TRPC5 while increasing binding to TRPC3 and TRPC6. Single mutations in TRPC1 NT or C-terminal (CT) CCDs reduced interaction and activation of TRPC1 by STIM1. Remarkably, single mutations in the TRPC3 NT CCD enhanced interaction and regulation by STIM1. Disruption in the TRPC3 CT CCD eliminated regulation by STIM1 and the enhanced interaction caused by NT CCD mutations. The NT CCD mutations converted TRPC3 from a TRPC1-dependent to a TRPC1-independent, STIM1-regulated channel. TRPC1 reduced the FRET between BFP-TRPC3 and TRPC3-YFP and between CFP-TRPC3-YFP upon stimulation. Accordingly, knockdown of TRPC1 made TRPC3 STIM1-independent. STIM1 dependence of TRPC3 was reconstituted by the TRPC1 CT CCD alone. Knockout of Trpc1 and Trpc3 similarly inhibited Ca2+ influx, and inhibition of Trpc3 had no further effect on Ca2+ influx in Trpc1−/− cells. Cell stimulation enhanced the formation of Trpc1-Stim1-Trpc3 complexes. These findings support a model in which the TRPC3 NT and CT CCDs interact to shield the CT CCD from interaction with STIM1. The TRPC1 CT CCD dissociates this interaction to allow the STIM1 Orai1-activating region within STIM1 access to the TRPC3 CT CCD and regulation of TRPC3 by STIM1. These studies provide evidence that the TRPC channel CCDs participate in channel gating.
Introduction
The receptor-evoked Ca2+ signal entails Ca2+ release from internal stores, primarily the ER,3 which is followed by activation of the store-operated Ca2+ influx channels (SOCs) at the plasma membrane. The two established receptor-stimulated SOCs are the Orai (1–3) and transient receptor potential subfamily C channels (TRPCs) (4, 5). Both channels are gated by the ER Ca2+ sensor STIM1. STIM1 has several domains that participate in gating of Orai and TRPC channels. The N-terminal, ER-resident portion of STIM1 has an EF hand Ca2+ binding domain and a SAM domain. Ca2+ binding to the EF hand prevents STIM1 clustering and access to Orai1 (2, 6, 7) and TRPC channels (8). The SAM domain participates in clustering of STIM1 upon depletion of ER Ca2+ (9, 10). The cytoplasmic portion of STIM1 begins with the first coiled coil domain (CC1), an inhibitory helix (11, 12) that is followed by the STIM1 Orai- activating region (SOAR) (13) (also known as CAD (14) and CCb9 (15)); a C-terminal inhibitory domain (16); a serine/proline region; and a polybasic, lysine-rich domain (K-domain) (17).
Gating of Orai1 by STIM1 is only partially understood. SOAR is the only domain needed for activation of Orai1 (13–15) and the other Orai channels (18). SOAR folds as a coiled coil domain (12) and functions as a dimer (12, 13). The activation of Orai channels by SOAR requires the interaction of SOAR with the C-terminal coiled coil regions of Orai1 (18, 19), Orai2, and Orai3 (20, 21). Little information is available on the interaction and regulation of the TRPC channels by STIM1. STIM1 appears to directly interact with TRPC1, TRPC4, and TRPC5, but not with TRPC3, TRPC6, and TRPC7 (8). However, STIM1 mediates the heteromultimerization of TRPC3 with TRPC1. Thereby, TRPC1 confers STIM1 dependence to TRPC3 by an unknown mechanism (22). STIM1 gates TRPC channels by electrostatic interaction of the last two STIM1 lysines with two conserved aspartates/glutamates in the C terminus of the TRPC channels (23). However, the K-domain of the endoplasmic reticulum-resident STIM1 does not mediate and is not required for the interaction of STIM1 with TRPC channels (8), although a recent study reported a role of the K-domain in the interaction of plasma membrane-resident STIM1 with TRPC1 and the regulation of TRPC1-mediated Ca2+ influx by external Ca2+ (24). How STIM1 physically interacts with TRPC channels, the functional significance of the interaction, and how TRPC1 confers STIM1 dependence to TRPC3 is not known. This study attempts to address these topics.
Most TRPC channels have N-terminal (NT) and C-terminal (CT) coiled coil domains (CCDs). We reasoned that, because the SOAR-Orai1 interaction is mediated by their CCDs, CCDs may also mediate TRPC-STIM1 interaction. Indeed, we found that SOAR mediates STIM1 interaction with the TRPC CCDs. The intact TRPC1 NT CCD and CT CCD appear to be required for interaction with and activation by STIM1. Remarkably, disruption of the TRPC3 NT CCD unmasked the interaction of TRPC3 with STIM1 and converted TRPC3 from a STIM1-regulated, TRPC1-dependent channel to a STIM1-regulated, TRPC1-independent channel. Disruption of the TRPC3 CT CCD eliminated all forms of interaction and regulation of TRPC3 by STIM1. Interestingly, upon receptor stimulation, TRPC1 reduced the FRET between BFP-TRPC3 and TRPC3-YFP and between CFP and YFP in CFP-TRPC3-YFP. STIM1 dependence of TRPC3 is conferred by the TRPC1 C terminus. Our findings lead to a model in which the TRPC3 NT and CT CCDs interact to shield the CT CCD from interaction with STIM1. The TRPC1 CT CCD dissociates this interaction to allow SOAR within STIM1 access to the TRPC3 CT CCD and regulation of TRPC3 by STIM1. These findings provide a molecular mechanism by which STIM1 interacts with and regulates TRPC channels.
EXPERIMENTAL PROCEDURES
Plasmid Construction Solutions
The BFP-TRPC3 clone (25) was provided by Dr. Barbara Miller (Pennsylvania State University, Hershey, PA). Dr. Craig Montell (University of California Santa Barbara) provided the polyclonal antibody recognizing TRPC3. Dr. Leonidas Tsiokas (University of Oklahoma, Oklahoma City, OK) provided the monoclonal antibody recognizing TRPC1. Dr. Yizheng Wang (Shanghai Institutes of Biological Sciences, Shanghai, China) provided the monoclonal antibody recognizing TRPC3. Dr. Lutz Birnbaumer (NIEHS, National Institutes of Health) provided the Trpc1−/− and Trpc3−/− mice. Dr. Klaus Groschner (Medical University of Graz, Graz, Austria) provided the pyrazole 10. The HA-TRPC1, FLAG-TRPC1, FLAG-TRPC3, FLAG-TRPC5, FLAG-TRPC6, FLAG-TRPC7, YFP-STIM1 (wild-type and ΔERM), and FLAG-mCherry-Orai1(R91W) clones have been described previously (8, 13). To prepare CFP-TRPC3-YFP, CFP was PCR-cloned into the KpnI site in-frame in the TRPC3-YFP plasmid. CFP was amplified from the pAMCFPN1 vector from Clontech, digested with KpnI enzyme, and ligated with the TRPC3-YFP plasmid that was also digested with KpnI. The entire reading frame was sequenced to confirm the direction of insertion of CFP. All TRPC1 and TRPC3 point mutations were made using site-directed mutagenesis (Agilent Technologies) in their respective vectors: N-terminal HA-tagged pRK5 and FLAG-tagged p3XFLAG-CMV 7.1. The human STIM1 siRNA sequence is 5′-GGCUCUGGAUACAGUGCUC-3′. The various TRPC1 and TRPC3 N- and C-terminal fragments were generated by PCR and cloned into the p3XFLAG-CMV 7.1 vector (Sigma-Aldrich) using NotII (5′) and SalI (3′). Some of these fragments also had an HA tag inserted between the FLAG and cDNA using the NotI (5′ and 3′) site. The antibodies used were monoclonal antibody recognizing STIM1 (BD Biosciences), polyclonal antibody recognizing GFP (Invitrogen), monoclonal HRP-conjugated antibody recognizing HA (Santa Cruz Biotechnology) and monoclonal HRP-conjugated antibody recognizing FLAG (Sigma-Aldrich).4
Mice and Preparation of Salivary Gland Cells
All experiments with animals have been approved by the National Institutes of Health Animal Use Committee and adhered to National Institutes of Health guidelines. Generation of Trpc1−/− mice (26) and Trpc3−/− mice (27) has been described elsewhere. Dispersed acinar and duct fragments were prepared from mouse submandibular glands as detailed before (28). In brief, mice were sacrificed by inhalation of CO2, and the glands were quickly removed into solution containing 140 mm NaCl, 5 mm KCl, 1 mm CaCl2, 1 mm MaCl2, 10 mm HEPES (pH 7.4) (with NaOH), 10 mm glucose, 10 mm pyruvate, 0.02% trypsin inhibitor, and 0.1% bovine serum albumin. The glands were minced finely and digested in the same solution that also contained 0.5 mg/ml collagenase P for 10 min at 37 °C. After replacement with fresh digestion medium, the digestion continued for an additional 7–10 min until liberation of the acini and duct fragments. The digest was washed three times, and the acini and ducts were suspended with the same solution without the digestive enzymes. The cells were loaded with Fura-2 by incubation with 5 μm Fura-2/AM for 20 min at room temperature, washed, and used to measure Ca2+ as described (27).
Cell Culture and Transfection
HEK 293 cells and HeLa cells were grown according to the recommendation of the supplier. The cells were plated on poly-l-lysine-coated 6-well plates (day 0) so that they would be 80–90% confluent the next day (day 1). The cells were then transiently transfected for 6 h with the desired constructs using Lipofectamine 2000 (Invitrogen), replaced with regular HEK growth medium (DMEM + 10% FBS + 1% penicillin-streptomycin), and, 24 h later, were harvested and lysed (day 2) and used for surface and coimmunoprecipitation analysis.
Harvesting Total Protein Extract and Coimmunoprecipitation
Transfected cells were collected and lysed using 500 μl of binding buffer (1× PBS buffer containing 1 mm NaVO3, 10 mm sodium pyrophosphate, 50 mm NaF (pH 7.4), and 1% Triton X-100). The cell extracts were sonicated, and insoluble material was spun down at 30,000 × g at 4 °C for 20 min. For the coimmunoprecipitation experiments, antibody recognizing STIM1 or antibody recognizing GFP (1 μg) was added to cell extract (100 μl) and incubated for 2 h at 4 °C. Then, 50 μl of a 1:1 slurry of protein A-Sepharose 4B beads was added to the antibody:extract mixture and incubated for an additional 1 h at 4 °C. Beads were washed three times for 10 min with binding buffer, and proteins were released from the beads with 50 μl of SDS loading buffer. Protein (25 μl) was loaded onto 8 or 10% tris-glycine SDS-PAGE gels, which were transferred onto a PVDF membrane for Western blot analysis.
Surface Biotinylation
Transfected cells still adherent to the plate were washed once with 1× PBS on ice. EZ-Link Sulfo-NHS-SS-Biotin (0.5 mg/ml, Pierce) was added to the cells for 30 min on ice. Afterward, the biotin was quenched with 50 mm glycine on ice for 2 × 5 min. The cells were then processed as described above to make cell extracts. A 1:1 slurry of immobilized avidin beads (50 μl, Pierce) was added to 100 μl of the cell extract and incubated for 2 h at 4 °C. Beads were washed three times for 10 min with binding buffer, and proteins were released from the beads with SDS loading buffer (50 μl). Protein (25 μl) was loaded onto 8% tris-glycine SDS-PAGE gels, which were transferred onto a PVDF membrane for Western blot analysis.
Fluorescence Measurements
Fura-2 fluorescence was measured by placing Fura-2-loaded salivary gland cells in an open perfusion chamber and perfusing continuously with prewarmed (37 °C) solution containing 140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 10 mm Hepes (pH 7.4 with NaOH), 10 mm glucose and either 1.5 mm CaCl2 or 0.2 mm EGTA (Ca2+-free solution) as specified. Fluorescence was recorded with a Photon Technology International imaging system at excitation wavelengths of 340 and 360 nm, and the light emitted at wavelengths higher than 530 nm. Images where then analyzed to obtain the 340/380 ratios from ducts, and the ratios were normalized to the initial fluorescence and averaged. The signals were plotted as the mean ± S.E. For FRET measurements, COS-7 cells were transfected with BFP-TRPC3 and TRPC3-eGFP and with an empty vector or HA-TRPC1. The cells were perfused with a solution containing 1 mm Ca2+ and switched to Ca2+-free solution during the stimulation with 100 μm ATP. Images were captured with a confocal system (FV1000, Olympus) equipped with a UplanSApo ×60 oil immersion objective (numerical aperture 1.35, Olympus) at ×3 magnification. The cells were excited by a 440-nm laser, and the emission was recorded at 535 nm. For time-lapse imaging, images were captured every 11s. Photobleaching was minimized by using 0.1% laser power. Images were processed with Photoshop CS3 to obtain the CFP:eGFP ratio and determine FRET efficiency. For controls, BFP-TRPC3 was expressed with the eGFP plasmid. No FRET signal was observed under control conditions.
Current Measurements
Electrophysiological recordings were performed 24h after transfection of the full-length channel or the mutants in untreated cells or cells treated with scrambled or STIM1 or TRPC1 siRNA. The siRNA sequences and transfection conditions were as described previously (29). Transfected cells were identified by YFP, CFP, or BFP fluorescence. Current was measured in pipette solution containing 140 mm CsCl, 2 mm MgCl2, 1 mm ATP, 5 mm EGTA, and 1.5 mm CaCl2 to clamp free Ca2+ at 70 nm and 10 mm HEPES (pH 7.2) with CsOH. This solution eliminates the K+ current and prevents inhibition of the TRPC channel by cytoplasmic Ca2+. The bath solution contained 140 mm NaCl, 5 mm KCl, 0.5 mm EGTA or 2 mm CaCl2, and 10 mm HEPES (pH 7.4) (with NaOH). HEK 293 cells were transfected with 1.5 μg/well of total cDNA mixture including TRPC channels and the M3 muscarinic receptor and with or without STIM1, Orai1(R91W), and TRPC coiled coil fragments. Current was always stimulated with 100 μm of the M3 receptor ligand carbachol. The current was recorded by 400-ms rapid alterations of membrane potential from −100 to +100 mV from a holding potential of 0 mV. The current recorded at +80 mV was used to calculate current density as picoampere/picofarad, and the current recorded in multiple experiments was used to obtain the mean ± S.E. and calculate significance.
Prediction of TRPC Channel Coiled Coil Domains
To map putative CCDs on TRPC channels, we used Marcoil 1.0 software. Marcoil 1.0 lists the coiled coil probability in percent and the heptad phase with the highest probability. The predicted heptad phases are shown in the figures for each of the TRPC channels. Candidate CCDs that were analyzed had a typical coiled coil probability of 70–90%, meaning that each amino acid in this region had a 70–90% chance of being in a coiled coil. Selective and strategic mutations were made within this putative coiled coil domain to reduce the coiled coil probability to close to 0% both in silico and in the channels. Most of the mutations that drastically reduced the coiled coil probability were made on hydrophobic residues in position a or position d of the heptad repeat. Next, the region of this heptad repeat was put through an online helical wheel analysis program to plot the location of the anticipated cluster of hydrophobic residues on one side of the coiled coil. These hydrophobic residues form the fundamental units of coiled coil-mediated interaction between two proteins. The effect of triple mutations in the predicted wheels on total and surface expression of the channels and their interaction with STIM1 are shown in the figures.
Statistics
All results are given as mean ± S.E., and significance was analyzed by Student's t test or by analysis of variance.
RESULTS
SOAR Interacts with the TRPC Channel Coiled Coil Domains
In a previous study, we showed that full-length STIM1 interacts with TRPC1, TRPC4, and TRPC5 but not with TRPC3, TRPC6, and TRPC7 (8). In Fig. 1a we show that SOAR mediates the interaction of STIM1 with TRPC channels. As was found with STIM1, SOAR interacts with TRPC1, TRPC4, and TRPC5 but minimally with TRPC3 and TRPC6 and not with TRPC7. The minimal interaction with TRPC3 and TRPC6 may be due to the presence of a basal low level of TRPC1-TRPC3 and TRPC4-TRPC6 complexes, where TRPC1 and TRPC4 facilitate the interaction of TRPC3 and TRPC6, respectively, with STIM1 (see Ref. 22).
FIGURE 1.

Interaction of TRPC channels with SOAR and effect of N-terminal triple point mutations on the interaction and on TRPC channel surface expression. a, SOAR interacts with TRPC1, TRPC4, and TRPC5 but not with TRPC3, TRPC6, and TRPC7. IP, immunoprecipitation. b and c, mutations of three residues in the NT coiled coil domains of the indicated TRPC channels eliminate their surface expression and reduce the interaction of TRPC1, TRPC4, and TRPC5 with SOAR but enhance the interaction of TRPC3 and TRPC6 with SOAR. The mutants used were as follows: TRPC1, Y224S/L227S/L238S; TRPC3, L241S/L245S/L248S; TRPC5, Y241S/L244S/L255S; and TRPC6, L300S/L304S/L307S. The blots in a–c are one of three similar experiments. d, the predicted N-terminal and C-terminal CCDs of TRPC1 and TRPC3, the N-terminal CCDs of TRPC5 and TRPC6, and the tested mutations in the hydrophobic region that make up the coiled coil-mediated interactions.
SOAR interacts with the Orais CT CCD (13, 19, 21). To examine the role of TRPC channel CCDs in their interaction with STIM1, the predicted fold and mutations that disrupt them were generated using a helical wheel plotting program. The results are shown in Fig. 1d for the TRPC1 and TRPC3 NT and CT coiled-coil domains and for the TRPC5 and TRPC6 NT CCDs. The program predicted that maximal disruption of the CCDs requires mutation of three residues, as shown in Fig. 1d. The mutations decrease the interaction of STIM1 with TRPC1 and TRPC5 (Fig. 1, b and c). However, unexpectedly, the mutations markedly increase the interaction of STIM1 with TRPC3 and TRPC6 (Fig. 1c). In addition, the mutations eliminated the surface expression of all TRPC channels examined (Fig. 1, b and c). These findings indicate that the TRPCs NT CCDs are involved either in folding or trafficking of TRPC channels. A role of the TRPC3 CT CCD in trafficking has been suggested previously (30). More important for our discussion, the TRPC channel CCDs participate in isoform-specific interaction with STIM1. To further explore the latter, we selected to focus on TRPC1 and TRPC3 because they form heteromultimers (22, 31, 32) that are mediated by STIM1 and in which TRPC1 confers STIM1-dependent regulation to TRPC3 (22).
Role of TRPC1 CCDs in Channel Regulation by STIM1
Next, we tested the effect of single mutations in the CCDs of the TRPC channels on the interaction with STIM1 to avoid the effect of the triple mutations on surface localization. Fig. 2 depicts the effect of single mutations in the NT and CT CCDs on surface TRPC1 and their interaction and regulation by STIM1. Single mutations in both TRPC1 NT and CT CCDs reduce the interaction with STIM1 without affecting surface TRPC1 (Fig. 2, a and b). Fig. 2c shows an example of TRPC1 current measurement when activated by the M3 muscarinic receptors. Orai1(R91W) is channel-dead but binds STIM1 similar to wild-type Orai1 (19) and, thus, can be used as a STIM1 scavenger (33). The summary in Fig. 1d shows that the current is inhibited by scavenging STIM1 with the Orai1(R91W) mutant and that the current is fully restored by overexpressing STIM1 (Fig. 2d, black columns). Significantly, the TRPC1 NT and CT CCD mutations that reduce STIM1 interaction also inhibit activation of TRPC1 current by receptor stimulation, and the current can be rescued by overexpression of STIM1. Hence, although the TRPC1 CT CCD is the STIM1 binding site (see below), in the case of full-length TRPC1 (and perhaps TRPC4 and TRPC5), the NT CCD appears to be required for stabilization or promotion of TRPC1 binding to the TRPC1 CT CCD.
FIGURE 2.

Role of N- and C-terminal single point mutations in TRPC1 interaction with and regulation by STIM1. HEK cells were transfected with STIM1 and the TRPC1 wild type or mutants (a–d) and the M3 receptor (c and d). a and b show the effect of mutations in the N- or C-terminal coiled coil domains of TRPC1, respectively, on its interaction with STIM1. Anti-HA-HRP (TRPC1) and anti-GFP (STIM1) were used for detection of TRPC1 and STIM1. The blots are representative of three experiments. IP, immunoprecipitation. c and d, example of activated TRPC1 current trace and current/voltage relationship. Here and in all experiments, the channels were activated by 100 μm M3 receptor agonist carbachol. The current/voltage relationship was taken at the time marked by ■ and after subtraction of the leak current measured in the presence of NMDG. pA, picoampere; pF, picofarad. d, TRPC1-independent current was taken as the current measured with the channel-dead mutant TRPC1(F562A) (see Ref. 29). *, p < 0.05 or better with respect to TRPC1 current; #, p < 0.01 with respect to the current measured in the absence of STIM1. The results are the mean ± S.E. of the number of cells analyzed, which are listed in the columns.
Role of TRPC3 CCDs in Channel Regulation by STIM1
Fig. 3a shows that several single mutations in the TRPC3 NT CCD reproduce the dramatic increase in interaction of TRPC3 with SOAR. Importantly, they did so with minimal or no effect on surface TRPC3. Fig. 3b shows that the single mutations similarly increased the interaction of TRPC3 with full-length STIM1. For further analysis, we selected the TRPC3(L241S) and TRPC3(L245S) mutants. We reasoned that the increased STIM1-TRPC3 interaction by mutation of the NT CCD may have been due to exposure of the TRPC3 STIM1 binding site that is hidden by the NT CCD. This site could be the TRPC3 CT CCD. To examine this possibility, we generated the single TRPC3(I807S) and the double TRPC3(L241S/I807S) and TRPC3(L245S/I807S) mutants. Fig. 3c shows that, although TRPC3(I807S) has no apparent effect on STIM1 interaction, it abolished the enhanced interaction of STIM1 with TRPC3(L241S) and TRPC3(L245S). These single and double mutants had no effect on surface TRPC3 (Fig. 3c), so their effect on channel activity could be studied.
FIGURE 3.

N- and C-terminal single point mutants increase TRPC3 interaction with SOAR and STIM1. HEK cells were transfected with the indicated TRPC3 mutants and SOAR (a) or STIM1 (b and c). Interaction was evaluated by coimmunoprecipitation (IP) (a–c) and surface localization by biotinylation. The blot in a is one of three experiments, and the blot in b is one of two experiments. c, effect of the C-terminal CCD TRPC3(I807S) mutation on the interaction of the wild type, TRPC3(L241S), and TRPC3(L245S) with STIM1. The blots are representative of four experiments. TRPC3 was detected with antibodies recognizing FLAG and STIM1 and recognizing GFP. The minor high molecular weight band in a may represent TRPC3 multimers, and the lower band in b is due to some STIM1 degradation.
The enhanced interaction of TRPC3(L245S) with STIM1 predicts that more extensive inhibition of STIM1 should be required to inhibit its function compared with wild-type TRPC3. Two independent protocols were used to test this prediction. Fig. 4a shows the effect of different concentrations of STIM1 siRNA on expression of native STIM1. Fig. 4a shows that treatment with a higher concentration of siRNA is required to inhibit current by TRPC3(L245S) compared with TRPC3. Fig. 4, c and d, shows that about three times more Orai1(R91W) scavenger is required to inhibit TRPC3(L245S) compared with TRPC3 currents. The TRPC3 current could be restored by overexpression of STIM1 using 0.5 μg of STIM1 cDNA. STIM1 at 0.3 μg only partially restored the TRPC3(L245S) current. We could not use a higher amount of STIM1 because the use of high STIM1 inhibited both the TRPC3 and TRPC3(L245S) currents.
FIGURE 4.

Effect of scavenging STIM1 by various amounts of Orai1(R91W) on current density of TRPC3, TRPC3(L245S), TRPC3(I807S), and TRPC3(I807S/L245S). a, HEK cells were treated with different concentrations of STIM1 siRNA for 72 h prior to evaluation of STIM1 abundance by Western blot analyses using anti-STIM1 antibodies in two separate experiments (IB). HEK cells were treated with scrambled (SCR) or the indicated concentrations of STIM1 siRNA. After 48 h, the cells were transfected with TRPC3 or TRPC3(L245S) and the M3 receptor, and, 24 h later, they were used to measure TRPC3 current. Each point is the mean ± S.E. of six to eight experiments. b–d, HEK cells were transfected with wild-type TRPC3 or TRPC3(L245S) and different amounts of Orai1(R91W). b, example current/voltage relationship in medium containing 2 mm Ca2+. The columns in c and d show the effect of Orai1(R91W) and Orai1(R91W)+STIM1 on current density. pA, picoampere; pF, picofarad. e, HEK cells were transfected with TRPC3(I807S) or TRPC3(L245S/I807S), the M3 receptor, and the indicated concentrations of Orai1(R91W). The results in c–e are the mean ± S.E. of the number of cells analyzed, which are listed in the columns. *, p < 0.05; #, p < 0.01 compared with the respective controls.
The predicted effect of the I807S mutation alone and when combined with TRPC3(L245S) is that it should eliminate the STIM1 dependence of both TRPC3 and TRPC3(L245S). This prediction is tested in Fig. 4e. Indeed, TRPC3(I807S) is resistant to scavenging of STIM1. Moreover, the function of TRPC3(L245S/I807S) is also resistant to scavenging of STIM1. Hence, the I807S mutation that eliminates STIM1 binding to TRPC3 makes TRPC3 function as a STIM1-independent channel.
The TRPC1 C-terminal CCD Mediates STIM1 Dependence of TRPC3
TRPC3 functions as a STIM1-dependent channel only in the presence of TRPC1 (Ref. 22 and below). The enhanced interaction of TRPC3(L245S) with STIM1 raises the possibility that TRPC3(L245S) may function as a STIM1-dependent channel independently of TRPC1. To test this, we measured the effect of STIM1 scavenging on the function of TRPC3(L245S) expressed in cells treated with scrambled or TRPC1 siRNA. Fig. 5, a and b, shows that the TRPC3(L245S) current is inhibited by scavenging STIM1 with Orai1(R91W) in the presence and absence of TRPC1. Hence, TRPC3(L245S) appears to be directly regulated by STIM1 and does not require TRPC1 to access STIM1.
FIGURE 5.

Effect of TRPC1 C-terminal coiled coil domain on regulation of TRPC3 by STIM1. a and b, example current/voltage relationship in Ca2+-free medium and summary (b) examining the effect of scavenging STIM1 by 0.5 μg Orai1(R91W) on TRPC3(L245S) function in cells treated with scrambled (Scr) and TRPC1 siRNA. The results in b are the mean ± S.E. of nine cells. pA, picoampere; pF, picofarad; Con, control. c, the BFP-TRPC3 channel tagged at the N terminus with BFP and TRPC3-YFP tagged at the C terminus with YFP were transfected alone or together with AH-tagged TRPC1 in HeLa cells. After 24 h, the FRET between BFP and YFP was measured before and after stimulation with 100 μm ATP. d, the CFP-TRPC3-YFP channel tagged at both the N terminus with CFP and at the C terminus with YFP was transfected alone or together with AH-tagged TRPC1 in HeLa cells. After 24 h, the FRET between CFP and YFP was measured before and after stimulation with 100 μm ATP. The traces in c and d are the mean ± S.E. of the indicated number of analyzed cells. e, effect of TRPC1 C and N terminus CCDs on the interaction of TRPC3 and mutants with STIM1 and their interaction with STIM1 as analyzed by coimmunoprecipitation (IP). The blots are representative of three similar experiments. f, effect of the isolated TRPC1 CT CCD and NT CCD on the regulation of TRPC3 by STIM1 in cells treated with TRPC1 siRNA. The average carbachol-activated current densities are given as mean ± S.E. of the number of cells analyzed as listed in the columns.
The requirement of TRPC1 for TRPC3 to function as a STIM1-regulated channel suggests that TRPC1 exposes the TRPC3 CT CCD, which is the TRPC3 STIM1-binding site. To test this prediction, we first measured the effect of TRPC1 on the FRET signal between TRPC3 tagged with BFP on the N terminus (BFP-TRPC3) and TRPC3 tagged with YFP on the C terminus (TRPC3-YFP). Fig. 5c shows that TRPC1 had no significant effect on the basal FRET signal between BFP-TRPC3 and TRPC3-eGFP. Notably, cell stimulation with 100 μm ATP to activate the native P2Y2 receptors in HeLa cells resulted in a reduction in the FRET signal only in the presence of TRPC1. This suggests that cell stimulation enhances the formation of the TRPC1-TRPC3 complexes. To determine the effect of TRPC1 on the interaction between the N and C termini of TRPC3, we prepared CFP-TRPC3-YFP tagged at both the C and N termini that is fully active (data not shown). Fig. 5d shows that the FRET signal observed between the CFP and YFP was nearly constant during cell stimulation in the absence of TRPC1. Notably, TRPC1 markedly reduced the CFP-TRPC3-YFP FRET upon cell stimulation, suggesting that TRPC1 dissociates the TRPC3 N and C termini. Next, to identify the TRPC1 domain mediating the STIM1 dependence of TRPC3, we prepared TRPC1 NT and CT CCD constructs and tested their interaction with STIM1 and their effect of TRPC3 interaction with STIM1. Fig. 5e shows that the TRPC1 CT CCD, but not the TRPC1 NT CCD, interacts with STIM1. Moreover, the TRPC1 CT CCD, but not the NT CCD, enhances the interaction of TRPC3 with STIM1. Finally, binding of STIM1 to the TRPC1 CT CCD has no apparent effect on the interaction of STIM1 with TRPC3(L241S) and TRPC3(L245S), suggesting that their enhanced binding with STIM1 is already saturated and independent of TRPC1.
To determine whether the TRPC1 CT CCD mediates the STIM1 dependence of TRPC3, we tested the ability of the TRPC1 NT and CT CCDs to reconstitute STIM1 dependence to TRPC3 in cells treated with TRPC1 siRNA. Fig. 5f shows that knockdown of TRPC1 makes TRPC3 activity resistant to inhibition of STIM1 by the dominant negative STIM1(ΔERM) (8) or to scavenging STIM1 with Orai1(R91W). Thus, in the absence of TRPC1, TRPC3 function becomes STIM1-independent. That is, in the absence of TRPC1, TRPC3 is set in a mode where it can still be opened by receptor stimulation but is no longer gated by STIM1. To test which TRPC1 domain confers STIM1 dependence to TRPC3, we determined the effect of the TRPC1 NT CCD and CT CCD on TRPC3 gating by STIM1. Fig. 5f shows that expression of the TRPC1 CT CCD alone was sufficient to fully restore STIM1 dependence to TRPC3.
The results with siTRPC1 in Fig. 5f imply that the TRPC1-TRPC3 regulatory interaction takes place between the native TRPCs. To test this, we measured the interaction of the native Trpc1, Trpc3, and Stim1 in salivary gland cells and the role of Trpc1 and Trpc3 on Ca2+ influx activated by the Gq-coupled M3 receptors, for which Stim1 is obligatory. Fig. 6a shows that knockout of Trpc1 and Trpc3 in mice similarly reduced receptor-stimulated Ca2+ influx by about 50%. The relatively selective TRPC3 inhibitor pyrazole 10 (34) also inhibited Ca2+ influx by about 50% (Fig. 6b). Significantly, Fig. 6c shows that the inhibition of Trpc3 by pyrazole 10 in Trpc1−/− cells had no effect on the influx, suggesting that the native Trpc1 and Trpc3 in salivary gland cells cooperate to mediate the same component of the receptor-mediated Ca2+ influx. Finally, cell stimulation assembles the Trpc1-Stim1-Trpc3 complex, as revealed by their enhanced mutual coimmunoprecipitation in stimulated cells (Fig. 6d).
FIGURE 6.

Role and interaction of endogenous Trpc1 and Trpc3 in salivary gland ducts. a, Ca2+ was measured in Fura-2-loaded salivary gland ducts isolated from wild-type, Trpc1−/−, and Trpc3−/− mice. The ducts were perfused with solutions containing 1.5 mm Ca2+ or 0.2 mm EGTA (Ca2+ free conditions) as indicated by the bars. b and c, Ca2+ was measured in salivary gland ducts from wild-type (b) and Trpc1−/− mice (c) treated with 3 μm Trpc3 inhibitor pyrazole 10 (Pyr 10). The traces are the mean ± S.E. of at least five duct fragments analyzed in each experiment obtained in three (a) or two (b and c) experiments in ducts isolated from three (a) or two (b and c) mice of each phenotype. d, mutual coimmunoprecipitation (IP) of Trpc1/Stim1/Trpc3 in resting and stimulated salivary gland cells. Coimmunoprecipitation was tested in resting cells (R) and cells stimulated with the M3 receptor ligand carbachol (100 μm) and treated with the SERCA pump inhibitor CPA 25 μm (S). In the inputs, the heavy bands are not specific, and the bands representing the probed proteins are marked by arrows and identified by expression of the proteins in HEK cells as controls. The antibodies recognizing Trpc1 did not recognize the protein without enrichment to show the Trpc1 input. Combinations of monoclonal and polyclonal antibodies were used to immunoprecipitate Stim1 that coimmunoprecipitated Trpc1 and Trpc3 and to immunoprecipitate Trpc1 that coimmunoprecipitated Trpc3 and Stim1. The experiments are separate duplicates of resting and stimulated cells and are one of three similar experiments. IB, immunoblot.
DISCUSSION
This study identifies the domains for STIM1-TRPCs interaction and their role in activation of TRPC channels by STIM1. The biochemical and functional findings indicate that the STIM1 SOAR domain mediates the interaction with the TRPC channels, as was found for the interaction of STIM1 with the Orai channels (13–15). Disruption of the TRPC channel coiled coil domains reveals their role in channel trafficking and their central role in TRPC channel interaction with STIM1. Mutation of three residues in the NT CCD of all TRPCs examined resulted in complete inhibition of their surface expression (Fig. 1). At present, it is not known whether this is due to misfolding of the channels or due to a specific role of the NT CCD in trafficking.
Disruption of the TRPC1 CCDs reduced their interaction with STIM1. The findings in Fig. 2 lead to several conclusions. The NT and CT TRPC1 CCDs participate in interaction of TRPC1 with STIM1, TRPC1 is regulated by STIM1, and interaction of STIM1 with the CCDs is required for this regulation. The single mutations in the CCDs weakened, but did not eliminate, the interaction of TRPC1 with STIM1. Consequently, activation of the TRPC1 mutants by receptor stimulation can be restored by overexpression of STIM1. The basis of the involvement of the two TRPC1 CCDs in STIM1 binding is not clear at present because the isolated CT, but not the NT, of TRPC1 binds to STIM1 (Fig. 5). One possible explanation is that, in the full-length TRPC1, the NT CCD interacts with and stabilizes the CT CCD structure rather than hiding it from STIM1. Note that TRPC1 interacts with STIM1 with no need to expose the STIM1 binding site, suggesting that its STIM1 binding site may not be hidden and that receptor stimulation only enhances the interaction between TRPC1 and STIM1. In a previous study, we showed that TRPC1 is gated by electrostatic interaction between the last two STIM1 lysines and two conserved C-terminal aspartates in TRPC1 (23). On the basis of the previous and current findings, we propose that the SOAR interacts with the CT CCD of TRPC1 to present STIM1(Lys684/Lys685) to TRPC1(Asp639/Asp640) to gate the channel.
In sharp contrast with the findings with TRPC1 and TRPC5, disruption of the TRPC3 and TRPC6 NT CCD markedly increased their interaction with STIM1. To understand this unexpected finding and reveal the role of the CCDs in STIM1 regulation of TRPCs, we focused on TRPC3, its interaction with STIM1, and how TRPC1 confers STIM1 dependence to TRPC3. A FRET analysis with N- and C-terminally tagged TRPC3 (Fig. 5c) suggests that TRPC3 can form homomultimers, with the N and C termini sufficiently close to generate a FRET signal. Interestingly, TRPC1 had no apparent effect on the basal FRET signal, suggesting that TRPC1 and TRPC3 do not form stable heteromultimers prior to cell stimulation. This is further supported by the in vivo findings of an increased Trpc1/Stim1/Trpc3 coimmunoprecipitation upon cell stimulation (Fig. 6d). Analysis of the CFP-TRPC3-YFP FRET suggested that, in the heteromultimers, the TRPC1 C terminus facilitates the dissociation of the TRPC3 N- and C-terminal CCDs to promote interaction of TRPC3 with STIM1. That dissociation of the TRPC3 NT CCD/CT CCD by mutations in the NT CCD was sufficient to markedly increase the interaction of TRPC3 with STIM1 in the absence of TRPC1 indicates that TRPC3 does have a STIM1 binding site, that the STIM1 binding site is at the TRPC3 CT CCD, and that the STIM1 binding site is shielded by the NT CCD.
Together, these findings are compatible with the model depicted in Fig. 7. In the inactive state, the TRPC1 and TRPC3 NT and CT CCDs interact with each other to shield the STIM1 binding site and prevent regulation of TRPC1 and TRPC3 by STIM1. TRPC1 interacts with STIM1 and can be directly gated by STIM1 (8), and both its N and C terminus CCDs participate in STIM1 interaction (Fig. 2). Hence, when TRPC1 is not in a complex with TRPC3 (and perhaps other TRPC channels like TRPC4 and TRPC5), cell stimulation facilitates the interaction of the STIM1 SOAR with the TRPC1 CCDs to mediate the gating of TRPC1 by STIM1. However, when TRPC1 is in complex with TRPC3, cell stimulation facilitates the formation and enhances the interaction between the TRPC1-TRPC3 heteromultimers and dissociates between the TRPC1 NT and CT CCDs. In the heteromultimers, the free TRPC1 CT CCD interacts with the TRPC3 CCDs to dissociate them, making the TRPC3 CT CCD available for interaction with STIM1 and, thereby, allowing the activation of TRPC3 by STIM1.
FIGURE 7.
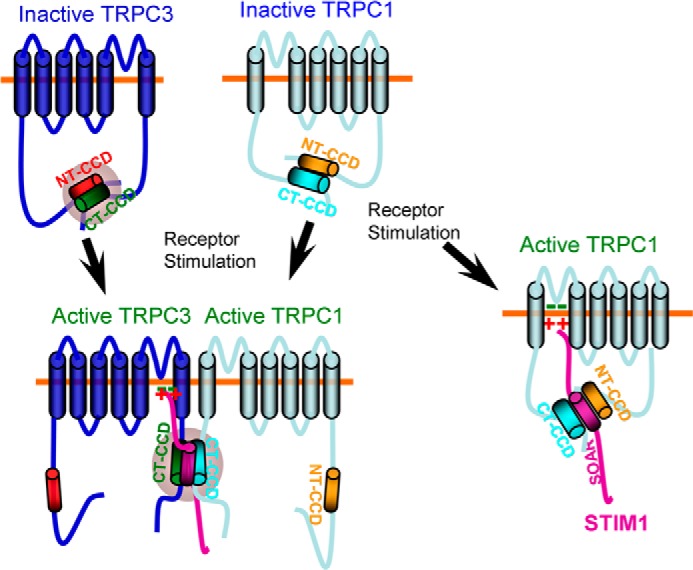
A model depicting gating of TRPC3 by STIM1. In the inactive state, the TRPC1 and TRPC3 N- and C-terminal CCDs interact to shield the STIM1 C-terminal CCD in both channels. TRPC1 can be directly activated by STIM1 when STIM1 interacts with a domain formed by the interacting TRPC1 NT and CT CCDs (right). Another conformation of TRPC1 is formed by receptor stimulation when TRPC1 is in complex with TRPC3 and serves to gate TRPC3. When in complex with TRPC3, receptor stimulation dissociates the TRPC1 CCDs, allowing the TRPC1 CT CCD to access the TRPC3 CCDs to dissociate them and expose the TRPC3 CT CCD so that it can interact with the STIM1 SOAR domain and present the polybasic domain to the conserved positive charges, which then open the channel.
In the in vivo situation, this mechanism provides an additional guard for activation of TRPC3 during receptor stimulation. Perhaps more significantly, this mechanism provides the cells with a mean of determining the mode of TRPC3 function. As described here and in a previous study (33), TRPC3 can function both as a STIM1-dependent and STIM1-independent channel, depending on the presence of TRPC3 and TRPC1 in the same cell. When TRPC3 operates as STIM1-gated channel, its function becomes coupled to Ca2+ content in the ER and is subject to all regulatory modalities of STIM1. The single-channel conductance of TRPC3 is much higher than that of the canonical SOC Orai1. Perhaps recruitment of TRPC3 to the SOC serves as a means of regulating the extent of SOC activity on the basis of demand, which includes sustaining the Ca2+ signal and reloading the stores, especially when the stores become overdepleted in pathological situations.
Acknowledgments
We thank Dr. Barbara A. Miller (Pennsylvania State University, Hershey, PA) for the BFP-TRPC3 clone, Dr. Craig Montell (University of California Santa Barbara) for the polyclonal antibody recognizing TRPC3, Dr. Leonidas Tsiokas (University of Oklahoma, Oklahoma City, OK) for the monoclonal antibody recognizing TRPC1, Dr. Yizheng Wang (Shanghai Institutes of Biological Sciences, Shanghai, China) for the monoclonal antibody recognizing TRPC3, Dr. Lutz Birnbaumer (NIEHS, National Institutes of Health) for the Trpc1−/− and Trpc3−/− mice, and Dr. Indu Ambudkar (NIDR/National Institutes of Health) for the Trpc1−/− mice, Dr. Klaus Groschner (Medical University of Graz, Graz, Austria) for pyrazole 10. We also thank Dr. Ambudkar for discussions.
This work was funded, in whole or in part, by intramural National Institute of Health grant Z1A-DE000735 (to S. M.); National Institutes of Health Grant Z01-ES101684 (to L. B.); and National Institutes of Health Grant 5R00HL093297 (to J. P. Y.). This work was also supported by National Research Foundation of Korea (NRF) grants (MSIP) NRF-2013R1A1A1010783 (to K. P. L.) and 2007-0056092 and 2012R1A2A1A01003487 (to D. M. S.) funded by the Korean government.
Plasmids are available upon request but require an MTA approved by NIDCR, National Institutes of Health.
- ER
- endoplasmic reticulum
- SOC
- store-operated Ca2+ influx channel
- TRPC
- transient receptor potential canonical
- STIM
- stromal interaction molecule
- SOAR
- stromal interaction molecule 1 Orai1-activating region
- NT
- N-terminal
- CT
- C-terminal
- CCD
- coiled coil domain.
REFERENCES
- 1. Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S. H., Tanasa B., Hogan P. G., Lewis R. S., Daly M., Rao A. (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185 [DOI] [PubMed] [Google Scholar]
- 2. Zhang S. L., Yeromin A. V., Zhang X. H., Yu Y., Safrina O., Penna A., Roos J., Stauderman K. A., Cahalan M. D. (2006) Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. U.S.A. 103, 9357–9362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vig M., Peinelt C., Beck A., Koomoa D. L., Rabah D., Koblan-Huberson M., Kraft S., Turner H., Fleig A., Penner R., Kinet J. P. (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Birnbaumer L. (2009) The TRPC class of ion channels. A critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. Annu. Rev. Pharmacol. Toxicol. 49, 395–426 [DOI] [PubMed] [Google Scholar]
- 5. Worley P. F., Zeng W., Huang G. N., Yuan J. P., Kim J. Y., Lee M. G., Muallem S. (2007) TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 42, 205–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roos J., DiGregorio P. J., Yeromin A. V., Ohlsen K., Lioudyno M., Zhang S., Safrina O., Kozak J. A., Wagner S. L., Cahalan M. D., Veliçelebi G., Stauderman K. A. (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E., Jr., Meyer T. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang G. N., Zeng W., Kim J. Y., Yuan J. P., Han L., Muallem S., Worley P. F. (2006) STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 8, 1003–1010 [DOI] [PubMed] [Google Scholar]
- 9. Stathopulos P. B., Li G. Y., Plevin M. J., Ames J. B., Ikura M. (2006) Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region. An initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 281, 35855–35862 [DOI] [PubMed] [Google Scholar]
- 10. Stathopulos P. B., Zheng L., Li G. Y., Plevin M. J., Ikura M. (2008) Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 135, 110–122 [DOI] [PubMed] [Google Scholar]
- 11. Muik M., Fahrner M., Schindl R., Stathopulos P., Frischauf I., Derler I., Plenk P., Lackner B., Groschner K., Ikura M., Romanin C. (2011) STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 30, 1678–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang X., Jin H., Cai X., Li S., Shen Y. (2012) Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. U.S.A. 109, 5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yuan J. P., Zeng W., Dorwart M. R., Choi Y. J., Worley P. F., Muallem S. (2009) SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., Walz T., Garcia K. C., Dolmetsch R. E., Lewis R. S. (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawasaki T., Lange I., Feske S. (2009) A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 385, 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jha A., Ahuja M., Maléth J., Moreno C. M., Yuan J. P., Kim M. S., Muallem S. (2013) The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell Biol. 202, 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liou J., Fivaz M., Inoue T., Meyer T. (2007) Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. U.S.A. 104, 9301–9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee K. P., Yuan J. P., Zeng W., So I., Worley P. F., Muallem S. (2009) Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc. Natl. Acad. Sci. U.S.A. 106, 14687–14692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muik M., Frischauf I., Derler I., Fahrner M., Bergsmann J., Eder P., Schindl R., Hesch C., Polzinger B., Fritsch R., Kahr H., Madl J., Gruber H., Groschner K., Romanin C. (2008) Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem. 283, 8014–8022 [DOI] [PubMed] [Google Scholar]
- 20. Derler I., Schindl R., Fritsch R., Romanin C. (2012) Gating and permeation of Orai channels. Front Biosci. 17, 1304–1322 [DOI] [PubMed] [Google Scholar]
- 21. Frischauf I., Muik M., Derler I., Bergsmann J., Fahrner M., Schindl R., Groschner K., Romanin C. (2009) Molecular determinants of the coupling between STIM1 and Orai channels. Differential activation of Orai1–3 channels by a STIM1 coiled-coil mutant. J. Biol. Chem. 284, 21696–21706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan J. P., Zeng W., Huang G. N., Worley P. F., Muallem S. (2007) STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 9, 636–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zeng W., Yuan J. P., Kim M. S., Choi Y. J., Huang G. N., Worley P. F., Muallem S. (2008) STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 32, 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jardin I., Dionisio N., Frischauf I., Berna-Erro A., Woodard G. E., López J. J., Salido G. M., Rosado J. A. (2013) The polybasic lysine-rich domain of plasma membrane-resident STIM1 is essential for the modulation of store-operated divalent cation entry by extracellular calcium. Cell. Signal. 25, 1328–1337 [DOI] [PubMed] [Google Scholar]
- 25. Tong Q., Hirschler-Laszkiewicz I., Zhang W., Conrad K., Neagley D. W., Barber D. L., Cheung J. Y., Miller B. A. (2008) TRPC3 is the erythropoietin-regulated calcium channel in human erythroid cells. J. Biol. Chem. 283, 10385–10395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu X., Cheng K. T., Bandyopadhyay B. C., Pani B., Dietrich A., Paria B. C., Swaim W. D., Beech D., Yildrim E., Singh B. B., Birnbaumer L., Ambudkar I. S. (2007) Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1−/− mice. Proc. Natl. Acad. Sci. U.S.A. 104, 17542–17547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim M. S., Hong J. H., Li Q., Shin D. M., Abramowitz J., Birnbaumer L., Muallem S. (2009) Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee M. G., Schultheis P. J., Yan M., Shull G. E., Bookstein C., Chang E., Tse M., Donowitz M., Park K., Muallem S. (1998) Membrane-limited expression and regulation of Na+-H+ exchanger isoforms by P2 receptors in the rat submandibular gland duct. J. Physiol. 513, 341–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim M. S., Zeng W., Yuan J. P., Shin D. M., Worley P. F., Muallem S. (2009) Native store-operated Ca2+ influx requires the channel function of Orai1 and TRPC1. J. Biol. Chem. 284, 9733–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wedel B. J., Vazquez G., McKay R. R., St. J. Bird G., Putney J. W., Jr. (2003) A calmodulin/inositol 1,4,5-trisphosphate (IP3) receptor-binding region targets TRPC3 to the plasma membrane in a calmodulin/IP3 receptor-independent process. J. Biol. Chem. 278, 25758–25765 [DOI] [PubMed] [Google Scholar]
- 31. Liu X., Bandyopadhyay B. C., Singh B. B., Groschner K., Ambudkar I. S. (2005) Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J. Biol. Chem. 280, 21600–21606 [DOI] [PubMed] [Google Scholar]
- 32. Cheung K. K., Yeung S. S., Au S. W., Lam L. S., Dai Z. Q., Li Y. H., Yeung E. W. (2011) Expression and association of TRPC1 with TRPC3 during skeletal myogenesis. Muscle Nerve 44, 358–365 [DOI] [PubMed] [Google Scholar]
- 33. Lee K. P., Yuan J. P., So I., Worley P. F., Muallem S. (2010) STIM1-dependent and STIM1-independent function of transient receptor potential canonical (TRPC) channels tunes their store-operated mode. J. Biol. Chem. 285, 38666–38673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schleifer H., Doleschal B., Lichtenegger M., Oppenrieder R., Derler I., Frischauf I., Glasnov T. N., Kappe C. O., Romanin C., Groschner K. (2012) Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca2+ entry pathways. Br. J. Pharmacol. 167, 1712–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]