

Background: The potential mechanism of YHL-14 against cancer cells has not been explored.
Results: YHL-14 induces G2/M phase arrest and inhibition of cancer cell growth by up-regulation of p21 transcription and protein expression via modulation of Sp1 protein stability.
Conclusion: YHL-14 inhibits bladder and colon cancer cell growth through up-regulation of p21 expression.
Significance: This study identifies a novel mechanism underlying the anticancer effect of YHL-14.
Keywords: Anticancer Drug, Cell Growth, p38, MAPK, Protein Stability, Sp1, p21, Yuanhuacine
Abstract
Yuanhuacine (YHL-14), the major component of daphnane diterpene ester isolated from the flower buds of Daphne genkwa, has been reported to have activity against cell proliferation in various cancer cell lines. Nevertheless, the potential mechanism has not been explored yet. Here we demonstrate that YHL-14 inhibits bladder and colon cancer cell growth through up-regulation of p21 expression in an Sp1-dependent manner. We found that YHL-14 treatment resulted in up-regulation of p21 expression and a significant G2/M phase arrest in T24T and HCT116 cells without affecting p53 protein expression and activation. Further studies indicate that p21 induction by YHL-14 occurs at the transcriptional level via up-regulation of Sp1 protein expression. Moreover, our results show that p38 is essential for YHL-14-mediated Sp1 protein stabilization, G2/M growth arrest induction, and anchorage-independent growth inhibition of cancer cells. Taken together, our studies demonstrate a novel mechanism of YHL-14 against cancer cell growth in bladder and colon cancer cell lines, which provides valuable information for the design and synthesis of other new conformation-constrained derivatives on the basis of the structure of YHL-14 for cancer therapy.
Introduction
Cell cycle regulation involves a series of events to control cell division and duplication (1). This regulation process can be divided into two periods: interphase and mitotic (M) phase. During interphase, the cells accumulate the nutrients needed for mitosis and duplication of their DNA. During the mitotic (M) phase, the cell splits itself into two distinctly different cells. Interphase is subdivided into three phases, G1, S, and G2, whereas G0 means that cells have exited the cell cycle and have become quiescent (2). Abnormal cell cycle regulation is the hallmark of cancer (3). It is well known that disruption of the cell cycle can lead to growth arrest and, ultimately, apoptosis (4). The arrest of abnormally proliferating cancer cells in stages of the cell cycle could increase the sensitivity of proliferating cancer cells to treatment with other therapeutic approaches, such as radiation (5).
The flower buds of Daphne genkwa Siebold et Zucc. (Thymelaeaceae) have been used as a Chinese traditional medicine to treat various diseases, including cancer (6). For example, daphnane diterpene ester has been reported to have strong anticancer effects against multiple cancer cell lines, including L-1210 (mouse lymphocytic leukemia) cells, KB (epidermoid carcinoma of the mouth) cells (7), P-388 (mouse lymphocytic leukemia) cells, A-549 (human lung adenocarcinoma) cells, and human microvascular endothelial cells in vitro (8), as well as antileukemic effects in vivo (9). Yuanhuacine (YHL-14) is the major component of daphnane diterpene ester in the flower buds of D. genkwa. Recently it has been reported to be effective against cell proliferation in various cancer cell lines (8). However, the molecular mechanism underlying YHL-14 suppression of cancer cell growth has been barely elucidated.
Colon cancer has been the second most common cancer in women and the third most common in men on the basis of a United States epidemiology report in 2008 (11), with it being the fourth most common cause of cancer death after lung, stomach, and liver cancer (12). Although bladder cancer is the fourth most common type of cancer in men and the ninth most common cancer in women in the United States (13), invasive bladder cancers are responsible for 100% of deaths resulting from this disease. Thus, colon and bladder cancer are considered a serious threat to public health. Therefore, our objective in this study was to evaluate the anticancer ability of YHL-14 and elucidate the mechanism underlying YHL-14 inhibition of colon and bladder cancer cells.
EXPERIMENTAL PROCEDURES
Chemicals
Compound YHL-14 was isolated from the Daphne genera and provided by Dr. Shaojiang Song (Shenyang Pharmaceutical University, Shenyang, China). The purity was greater than 99%. The structure is shown in Fig. 1A. YHL-14 was dissolved in dimethyl sulfoxide to make a stock concentration at 80 mm. The stock YHL-14 was further diluted in DMEM with a final dimethyl sulfoxide concentration of 0.1% (v/v) for cell culture experiments. The same concentration (0.1% v/v) of dimethyl sulfoxide was used as a vehicle control in all experiments. The chemicals cycloheximide (CHX)3 was purchased from Calbiochem (San Diego, CA).
FIGURE 1.

YHL-14 inhibits cell monolayer growth and anchorage-independent growth in bladder and colon cancer cell lines. A, chemical structure of YHL-14. B, results of a coupled ATPase activity assay in the presence of varying concentrations of YHL-14 at 24 h. Incubation of cells with YHL-14 caused dose-dependent growth retardation of T24T, UMUC3, and HCT116 cells in vitro as observed in ATPase assays. Proliferation rates were determined in the indicated cells using a CellTiter-Glo luminescent cell viability assay kit. Results are presented as the mean ± S.D. of triplicate assays. *, significant inhibition of proliferation in comparison with vehicle control (p < 0.05). Error bars represent mean ± S.D. C and D, T24T cells were seeded in soft agar as described under “Experimental Procedures.” Representative images of colonies of T24T cells in a soft agar assay without or with YHL-14 (2 μm) were visualized under the microscope, and only colonies with over 32 cells were counted. Colonies are expressed as mean ± S.D. from five assays of three independent experiments. *, p < 0.05.
Antibodies
Antibodies specific against p21, p53, phospho-p53 at Ser-15, phospho-p90RSK at Ser-573, ERK1/2, phospho-ERK1/2 at Thr-202/Tyr-204, p38, phospho-p38 at Thr-180/Tyr-182, GAPDH, α-tubulin, β-actin, STAT3, and phospho-ATF2 at Thr-69/71 were purchased from Cell Signaling Technology (Danvers, MA). Anti-Sp1 antibody was bought from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-HA was purchased from Covance Antibody Service Inc. (Princeton, NJ).
Cell Culture
The human colon cancer cell line HCT116, p21 knockout (HCT116 p21−/−) cells, and p53 knockout (HCT116 p53−/−) cells were gifts from Dr. Bert Vogelstein (Howard Hughes Medical Institute, Sidney Kimmel Comprehensive Cancer Center, and the Johns Hopkins Medical Institutions) and were cultured in McCoy's 5A medium supplemented with 10% FBS (v/v) heat-inactivated FBS, 2 μm l-glutamine, and 25 μg/ml gentamycin. The human T24T bladder cancer cell line was a gift from Dr. Dan Theodorescu (14), and human UMUC3 was provided by Dr. Xue-Ru Wu (Departments of Urology and Pathology, New York University School of Medicine) (15). These cell lines were maintained in a 1:1 mixture of DMEM/Ham's F-12 medium and DMEM supplemented with 5% (v/v) or 10% (v/v) heat-inactivated FBS, 2 μm l-glutamine, and 25 μg/ml gentamycin, respectively, at 37 °C in a humidified atmosphere of 5% CO2.
Plasmids and Transfection
The dominant negative mutant of ERK1 (K71R) was a gift from Dr. Melanien H. Cobb and has been used in our previous studies (16, 17). The dominant negative mutant of p38 kinase (DN-p38) was a gift from Dr. Mercedes Rincon (Department of Medicine, University of Vermont, Burlington, VT) and has been used in our previous studies (18, 19, 20). Both the 2.4-kb and 200-bp lengths of the p21 promoter-driven luciferase reporter plasmids were provided by Dr. Jennifer A. Pietenpol (Vanderbilt Ingram Comprehensive Cancer Center, Vanderbilt University School of Medicine, Nashville, TN) (21). The sh-Sp1 construct was purchased from Fisher Scientific. The T24T (sh-Sp1), T24T (DN-ERK1 K71R), and T24T (DN-P38) transfectants were established by transfection of either DN-ERK1 or DN-p38, respectively, using PolyJetTM DNA in vitro transfection reagent (SigmaGen Laboratories, Gaithersburg, MD). The stable transfectants were established by selection with hygromycin for 4–6 weeks, and the surviving cells were pooled as stable mass transfectants.
Cell Proliferation Assay
Confluent monolayers of cells were trypsinized, and 1 × 103 viable cells suspended in 100 μl of medium were added to each well of 96-well plates. After adherence, cells were synchronized by replacement with 0.1% FBS culture medium and cultured for another 24 h. The proliferation of the cells was determined using the CellTiter-Glo luminescent cell viability assay kit (Promega, Madison, WI) with a luminometer (Wallac 1420 Victor2 multipliable counter system) as described in our previous publication (19).
Anchorage-independent Growth Assay in Soft Agar
An anchorage-independent growth assay in soft agar (soft agar assay) was carried out as described in our previous study (22). Briefly, 1 × 104 cells, with or without 2 μm of YHL-14 in 10% FBS basal medium Eagle containing 0.33% soft agar, were seeded over a bottom layer of 0.5% agar in 10% FBS BME in each well of 6-well plates. The plates were incubated in a 5% CO2 incubator at 37 °C for 3 weeks. Colonies were observed under the microscope, and only colonies with over 32 cells were counted. The results were normalized with a control.
Western Blot Analysis
Western blot analysis was generally carried out as described in our previous study (23). Briefly, cells were seeded in 6-well plates and cultured until 70–80% confluent in McCoy's 5A medium supplemented with 10% (v/v) heat-inactivated FBS or a 1:1 mixture of DMEM)/Ham's F-12 medium supplemented with 5% (v/v) heat-inactivated FBS. The culture medium was replaced with 0.1% FBS medium for 24 h. The cells were then treated with YHL-14 as indicated in the figure legends, and the cell extracts were subjected to Western blotting as described in our previous study (23). The protein bond recognized by specific antibodies was detected by an alkaline phosphatase-linked secondary antibody and an ECF Western blotting system (Amersham Biosciences, Piscataway, NJ).
RT-PCR
Total RNA was extracted with TRIzol reagent (Invitrogen), and cDNAs were synthesized with the Thermo-Script RT-PCR system (Invitrogen). The human β-actin cDNA used as an internal control was amplified by two specific primers: 5′-GCGAGAAGATGACCCAGATCA T-3′ (sense) and 5′-GCTCAGGAGGAGCAA TGATCTT-3′ (antisense). The human SP1 cDNA fragments were amplified by primers 5′-ATTAACCTCAGTGCATTGGGTA-3′ and 5′-AGGGCAGGCAAATTTCTTCTC-3′. The RT-PCR products were analyzed on 2% agarose gels, and, following staining with ethidium bromide, the images were scanned and visualized with a FluorChem SP imaging system (Alpha Innotech Inc., CA) as described previously (24).
Luciferase Reporter Assay
The different lengths of p21 promoter-driven luciferase reporter (2.4 kb and 200 bp) were transfected into T24T cells as described above. Stable transfectants suspended in normal cell culture medium were seeded into each well of 96-well plates and cultured in a 5% CO2 incubator at 37 °C until 70–80% confluence. The cells were treated with 2 μm of YHL-14 in 0.1% FBS medium for the time periods indicated in the figure legends. The cells were then extracted with a lysis buffer (25 mmol/liter Tris-phosphate (pH 7.8), 2 mm LEDTA, 1% Triton X-100, and 10% glycerol). The luciferase activity in the cell extracts was determined using a microplate luminometer (LB 96V, Berthold GmbH & Co. KG, Bad Wildbad, Germany) with a luciferase assay system (Promega) as described previously (25).
Flow Cytometry Assay
The cell cycle distributions were determined by flow cytometry as described previously (26). The cells were cultured in 6-well plates until they were 60% confluent. The cell culture medium was then replaced with 0.1% FBS medium and cultured for 24 h for synchronization. Following this, the cells were treated with YHL-14 for 12 h, collected with ice-cold PBS, and fixed with 5 ml of 75% ethanol at −20 °C overnight. The fixed cells were stained in buffer containing 0.1% Triton X-100, 0.2 mg/ml RNase A, and 50 μg/ml propidium iodide and then subjected to an EpicsXL flow cytometer (Beckman Coulter Inc., Miami, FL) for cell cycle analysis. The results were analyzed by histogram analysis software (Expo32 v. 1.2).
Cytosol and Nuclear Protein Extraction
Cytosol and nuclear proteins were prepared with the CelLytic-NuCLEAR extraction kit (Sigma) according to the protocols of the manufacturer and as described in our previous study (27).
Statistical Methods
Student's t test was applied for data analysis, and p < 0.05 was considered statistically significant.
RESULTS
YHL-14 Inhibits Cancer Cell Anchorage-independent Growth
YHL-14 is a diterpenoid compound with a molecular mass of 648 daltons, as shown in Fig. 1A. To evaluate the anticancer activity of YHL-14 against colon and bladder cancer cell lines, two bladder cancer cell lines, T24T and UMUC3, and a colon cancer cell line, HCT116, were treated with YHL-14 at different concentrations (2–16 μm) for 24 h. The cell proliferation of these cells was analyzed using an ATPase assay. As shown in Fig. 1B, although the HCT116 cell line was less sensitive to YHL-14 treatment, the cell growth rate was inhibited significantly in all three cancer cell lines in a dose-dependent manner. The IC50 of the T24T cell line was 1.83 ± 0.02, that of the UMUC3 cell line was 1.89 ± 0.02, and that of the HCT116 cell line was 14.28 ± 0.64. We then evaluated the potential inhibition of YHL-14 on anchorage-independent growth of T24T cells. The results showed that anchorage-independent growth of T24T cells in soft ager was impaired dramatically by YHL-14 treatment (Fig. 1, C and D).
YHL-14 Induces G2/M Phase Growth Arrest by Up-regulation of p21 Protein in a p53-independent Manner
To elucidate the molecular mechanisms underlying the anticancer effect of YHL-14, the potential effect of YHL-14 on the regulation of cell death and the cell cycle was determined by flow cytometry analysis in both T24T and HCT116 cancer cells. As shown in Fig. 2, A and B, treatment of cells with YHL-14 did not induce a marked effect on cell death, but it did induce a significant G2/M phase arrest in both T24T and HCT116 cells, suggesting that G2/M phase arrest induction might be associated with an anticancer effect of YHL-14. To elucidate the molecular mechanism potentially leading to G2/M growth arrest, we first evaluated the effect of YHL-14 treatment on major cell cycle regulators, and the results showed that YHL-14 treatment did not affect the expression of cell cycle-regulating proteins, including cyclin A, cyclin B1, cyclin D1, cyclin E, retinoblastoma, E2F1, and p27 (data not shown). Because p21 is a well known cell cycle inhibitor involved in cell cycle regulation, the effect of YHL-14 on p21 expression and expression of its upstream transcription factor, p53, was evaluated in both T24T and HCT116 cells. As shown in Fig. 3, A and B, following YHL-14 treatment for 12 h, p21 protein expression was significantly up-regulated in both T24T and HCT116 cells, whereas YHL-14 did not show any observable induction of p53 protein expression and p53 protein phosphorylation at Ser-15 under the same experimental conditions in both cell lines (Fig. 3, A–C). Interestingly, YHL-14 treatment also did not induce p53-dependent transcriptional activity in a p53-dependent luciferase reporter assay, whereas another control anticancer compound, F2, induced p21 expression, p53 protein expression, and p53-dependent transcriptional activation under the same experimental conditions (Fig. 3, D and E), suggesting that YHL-induced p21 expression was mediated via a p53-independent cascade. To completely rule out the possible role of p53 in p21 induction by YHL-14, HCT116 WT and HCT116 p53−/− cells were further employed to compare p21 protein induction following treatment of cells with YHL-14 and F2. As expected, YHL-14 treatment resulted in a similar level of p21 protein expression in both HCT116 WT and HCT116 p53−/− cells (Fig. 3C), whereas F2-induced p21 expression was completely blocked in HCT116 p53−/− cells compared with HCT116 WT cells (Fig. 3D). These results demonstrate that YHL-14 treatment leads to p21 protein induction in a p53-independent manner. To test the role of p21 induction by YHL-14 in the mediation of G2/M phase arrest, HCT116 p53−/−, HCT116 p21−/−, and HCT116 WT cells were employed (Fig. 3, C and F). As expected, YHL-14 treatment induced G2/M phase growth arrest in HCT116 WT cells and p53−/− cells at a similar level, whereas knockout of the p21 gene in HCT116 (HCT116 p21−/−) impaired the induction of G2/M phase growth arrest by YHL-14 (Fig. 3G). In contrast, p53 deletion dramatically inhibited G0/G1 phase growth arrest induced by the control anticancer compound F2 in HCT116 cells (Fig. 3H). These results demonstrate that YHL-14-induced G2/M phase arrest is mediated by up-regulation of p21 via a p53-independent cascade. Consistent with the induction of G2/M growth arrest, YHL-14 treatment also inhibited cancer cell proliferation and anchorage-independent cancer cell growth in both HCT116 WT and HCT116p53−/− cells but not in HCT116 p21−/− cells (Fig. 3, I and J). Our results demonstrate that p21 induction by YHL-14 is critical for G2/M growth arrest following YHL-14 treatment via a p53-independent axis.
FIGURE 2.

YHL-14 induces G2/M phase growth arrest in T24T and HCT116 cells. After synchronization, T24T and HCT116 cells were treated with YHL-14 as indicated for 12 h, and the cells were then fixed and subjected to cell cycle analysis by flow cytometry as described under “Experimental Procedures.” The results represent one of three independent experiments. AP, apoptosis; S, synthesis.
FIGURE 3.


p21 protein expression up-regulated by YHL-14 plays a key role in the induction of G2/M phase growth arrest in cancer cells. A and B, after synchronization, cells were treated with YHL-14 at the indicated concentrations for 12 h (A) or YHL-14 at 2 μm (T24T cells) or 16 μm (HCT116 cells) for the indicated time points (B). C and D, HCT116 WT, HCT116 p53−/−, and HCT116 p21−/− cells were treated with the indicated doses of YHL-14 (C) or F2 (D) for 12 h. The cell extracts were subjected to Western blotting with anti-p21, anti-p53, anti-phospho-p53 (Ser-15), or anti-GAPDH antibodies. E, HCT116 stable transfectant (8 × 103) that was stably transfected with the PG13-luciferase reporter was seeded into each well of a 96-well plate. After synchronization, cells were treated with the indicated concentrations of YHL-14 or F2 for 12 h, and the cells were then extracted for determination of luciferase activity. The results were presented as p53-dependent luciferase transactivity relative to vehicle control (relative p53 activity). Error bars show the mean ± S.D. from three independent experiments. F, HCT116 WT and HCT116 p21−/− cells were treated with YHL-14 at concentrations as indicated for 12 h, and the cell extracts were subjected to Western blotting with anti-p21 and anti-GAPDH antibodies. G and H, HCT116 WT, HCT116 p53−/−, and HCT116 p21−/− cells were treated with the indicated doses of YHL-14 (G) or F2 (H) for 12 h. The cells were then fixed and subjected to flow cytometry analysis. I, results of a coupled ATPase activity assay in the presence of varying concentrations of YHL-14 at 24 h. Incubation with YHL-14 caused dose-dependent growth effects of HCT116 wild-type, HCT116 p21−/−, and HCT116 p53−/− cells in vitro as observed in ATPase assays. Proliferation rates were determined in the indicated cells using a CellTiter-Glo luminescent cell viability assay kit. Results are presented as the mean ± S.D. of triplicate assays. J, HCT116 WT, p21−/−, and p53−/− cells were seeded in soft agar as described under “Experimental Procedures.” Representative images of colonies of these cells in a soft agar assay without or with YHL-14 (16 μm) were visualized under the microscope, and only colonies with more than 32 cells were counted. Colonies are expressed as mean ± S.D. from five assays of three independent experiments.
Sp1 Induction by YHL-14 Mediates p21 Gene Transcriptional Up-regulation
To understand the up-regulation of p21 protein upon YHL-14 treatment, the p21 mRNA level was first evaluated in YHL-14-treated cells. As shown in Fig. 4A, YHL-14 treatment led to an increase of p21 mRNA, suggesting that p21 protein expression was regulated at either transcriptional level or mRNA stability level. Consistent with mRNA induction, YHL-14 treatment also resulted in increases in p21 promoter-driven luciferase activity (Fig. 4B), indicating the transcriptional regulation of p21 by YHL-14. Moreover, bioinformation analysis suggested that the p21 gene promoter contains multiple transcription factor binding sites, including p53, Stat3, and Sp1 (Fig. 4C). We found that YHL-14 treatment resulted in Sp-1 protein induction (Fig. 4D), whereas it did not show an inducible effect on p53 expression (Fig. 3, A and B) or Stat3 expression (Fig. 4D), suggesting that Sp-1 might be the transcription factor that is responsible for p21 transcriptional induction following YHL-14 treatment. To test this notion, we used both full-length (2.4 kb) of the p21 promoter-driven luciferase reporter and a 200-bp length of the p21 promoter-drive luciferase reporter containing Sp1 binding sites only, as indicated in Fig. 4C, to transfect to T24T cells, respectively, and the stable transfectants were treated with YHL-14 for comparison of their promoter activities. The results showed that the p21 promoter reporter containing only Sp1 binding sites had a similar transcriptional activity to that observed in the full-length p21 promoter-driven luciferase reporter (2.4 kb) (Fig. 4E), suggesting that Sp1 is sufficient to mediate p21 promoter transcription following YHL-14 treatment. To further extend the role of Sp1 in mediating p21 induction following YHL-14 treatment, two Sp1-specific shRNA constructs (sh-Sp1) were used to transfect into T24T cells, and the two stable transfectants of T24T(sh-Sp1) were established as shown in Fig. 4F, left panel. Compared with the results obtained from T24T(vector), p21 expression was significantly alleviated in T24T(sh-Sp1) transfectants following YHL-14 treatment (Fig. 4F, right panel). Our results demonstrate that Sp1 plays an important role in YHL-14-induced p21 expression.
FIGURE 4.
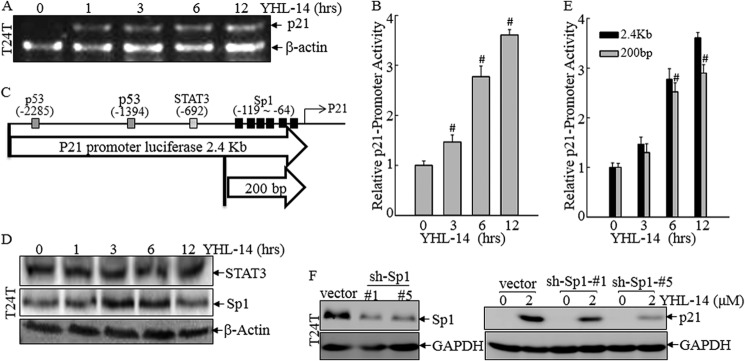
Sp1 induction by YHL-14 mediates p21 transcription. A, after synchronization, cells were treated with 2 μm YHL-14 for the indicated times. Total RNA was isolated, and RT-PCR was carried out to determine p21 mRNA levels. β-Actin mRNA levels were used as a loading control. B, T24T stable transfectant (8 × 103) that was stably transfected with the full-length (2.4 kb) of p21 promoter-driven luciferase reporter were seeded into each well of a 96-well plate. After synchronization, cells were treated with 2 μm YHL-14 at the indicated time, and the cells were then extracted for determination of luciferase activity. #, significant enhancement of p21 transcription compared with vehicle control (p < 0.05). Error bars represent mean ± S.D. C, schematic of various transcription factor binding sites in the p21 promoter-driven luciferase reporter. D, after synchronization, T24T cells were exposed to 2 μm YHL-14 at the indicated time, and then cells were extracted. The cell extracts were subjected to Western blotting with anti-STAT3, anti-SP1, and anti-β-actin antibodies. E, cells (8 × 103) stably transfected with either full-length (2.4 kb) or short (200 bp) p21 promoter-driven luciferase reporters were seeded into each well of a 96-well plate. After synchronization, cells were treated with YHL-14 at the indicated time, and the cells were extracted for determination of luciferase activity. #, significant induction of p21 promoter transcriptional activity (p < 0.05). Error bars represent mean ± S.D. F, T24T (vector), T24T (sh-Sp1) colon #1 and T24T (sh-sp1) colon #5 cells were exposed to the indicated doses of YHL-14 for 12 h. The cell extracts were subjected to Western blotting with anti-p21, anti-Sp1, or anti-GAPDH antibodies.
p38 Activation by YHL-14 Stabilizes Sp1 Protein
The results above show that Sp1 induction by YHL-14 plays a critical role in mediating p21 transcription and protein expression. Because Sp1 protein induction could be observed as early as 1 h following YHL-14 treatment, we anticipated that this Sp1 protein up-regulation could occur at either protein stabilization or translation. To exclude the possibility of YHL-14 regulation of Sp1 protein expression at the mRNA level, the effect of YHL-14 treatment on sp1 mRNA expression was evaluated. The results indicated that YHL-14 treatment did not show any effect on sp1 mRNA level for any of the time points tested (Fig. 5A). Moreover, we observed that Sp1 protein rapidly degraded in T24 cells following CHX treatment, whereas coincubation of T24T cells with YHL-14 and CHX significantly delayed Sp1 protein degradation (Fig. 5B). These results strongly suggest that YHL-14 regulates Sp1 protein degradation. Previous studies have reported that activation of ERKs and p38 could stabilize Sp1 protein (22, 23). Thus, we determined the potential activation of ERKs and p38 by YHL-14. Consistent with Sp1 protein expression, YHL-14 treatment did lead to activation of ERKs and p38 as early as 1 h (Fig. 5C). To determine the role of ERK and p38 in the induction of Sp1 and p21 expression, the domain-negative ERK1 K71R (DN-ERK1) or domain-negative p38 (DN-p38) constructs were transfected together with a 200-bp length of the p21 promoter-driven luciferase reporter into T24T cells. As shown in Fig. 5D, exogenous expression of DN-ERK1 was observed at the ERK protein level and verified by detection of its tagged HA expression. Ectopic expression of DN-ERK1 blocked endogenous ERK phosphorylation induced by YHL-14, whereas the basal level of ERK phosphorylation was elevated in comparison with that from T24T(vector) transfectant, as reported in our previous studies (Fig. 5D). However, blockage of ERK activation led to a slight increase in Sp1 and p21 protein induction by YHL-14, whereas it impaired phosphorylation of p90RSK, a well known ERK downstream substrate (Fig. 5D). Thus, ERK might not be involved in the up-regulation of Sp1 or p21 expression. On the other hand, overexpression of DN-p38 in T24T cells not only inhibited p38 phosphorylation and its well known substrate ATF-2 phosphorylation, but it also blocked Sp1 and p21 protein induction by YHL-14 (Fig. 5E). Consistent with p21 protein expression, YHL-14-induced p21 promoter transactivation was also impaired by the ectopic expression of DN-p38 and increased by DN-ERK1 overexpression (Fig. 5, F and G). Moreover, the inhibition of p38 activation by DN-p38 reversed the stabilization of Sp1 protein following YHL-14 treatment (Fig. 5H). Taken together, our results demonstrate that YHL-14 treatment can activate p38, which stabilizes Sp1 protein and, subsequently, leads to p21 transcription and protein expression.
FIGURE 5.

p38 activation by YHL-14 is crucial for Sp1 protein stabilization. A, total RNA was isolated and subjected to RT-PCR analysis in T24T cells treated with 2 μm YHL-14 for the indicated time. mRNA levels of sp1 were evaluated by RT-PCR. β-Actin mRNA levels were used as a loading control. B, after synchronization, T24T cells were exposed to 100 μg/ml CHX with or without 2 μm YHL-14 as indicated, and the cell extracts were subjected to Western blotting with anti-Sp1 or anti-β-actin antibodies. The blot intensity was quantitated by Quantity One software. Data were normalized with a control group. C, T24T Cells were treated with 2 μm of YHL-14 as indicated, and the cell extracts were then subjected to Western blotting with anti-p-ERK, anti-ERK, anti-p-p38, or anti-p38 antibodies. D and E, T24T (vector), T24T (DN-ERK1), and T24T (DN-p38) cells were treated with YHL-14 (2 μm) for 12 h or left untreated. The cell extracts were subjected to Western blotting with specific antibodies against HA, ERK, p-ERK, p-p90RSK Thr-573, SP1, p21, p-p38, p38, p-ATF2, α-tubulin, and β-actin. F and G, T24T (DN-ERK1), T24T (DN-p38), or T24T (vector) cells stably cotransfected with short (200 bp) p21 promoter-driven luciferase reporter plasmids were seeded into each well of a 96-well plate. After synchronization, cells were treated with YHL-14 at indicated time, and the cells were then extracted for determination of luciferase activity. #, significant increase of Sp1 transcription (p < 0.05); *, significant decrease (p < 0.05). Error bars represent mean ± S.D. H, after synchronization, the indicated cells were treated with 100 μg/ml CHX with or without 2 μm YHL-14 as indicated. The cell extracts were subjected to Western blotting with anti-SP1 or anti-β-actin antibodies. The band intensity was quantized by Quantity One software. Data were normalized with a control group.
p38 activation Is Crucial for YHL-14-induced G2/M Phase Growth Arrest and Required for YHL-14 Inhibition of Cancer Cell Anchorage-independent Growth
Our above results indicate an essential role of p38 activation in YHL-14 stabilization of Sp1 protein, and transcription of p21 gene. To further evaluate role of p38 in mediation of YHL-14 anti-cancer activity, we compared the effect of DN-p38 overexpression on YHL-induced cancer cell G2/M phase growth arrest and anchorage-independent growth. The results showed that G2/M phase growth arrest induced by YHL-14 could be reversed by DN-p38 overexpression in T24T cells (Fig. 6A). Consistently, the YHL-14 inhibition of T24T anchorage-independent growth was also partially reversely by the introduction of DN-p38 (Figs. 6B and 6C). Collectively, our results demonstrated that YHL-14-induced p38 activation played a crucial role in its mediation of G2/M phase growth arrest and inhibition of T24T cell anchorage-independent growth, further revealing that p38 activation mediates YHL-14 anti-cancer activity.
FIGURE 6.

p38 activation by YHL-14 mediates G2/M phase growth arrest and inhibition of T24T cell anchorage-independent growth. A, after synchronization, T24T (vector) and T24T (DN-p38) cells were treated with 2 μm YHL-14 for 12 h and then subjected to flow cytometry analysis. B and C, T24T (vector) and T24T (DN-p38) cells were subjected to an anchorage-independent growth assay in soft agar as described under “Experimental Procedures.” Representative images of colonies of T24T cells without or with 2 μm YHL-14 were captured under the microscope, and only colonies with more than 32 cells were counted. Colonies are expressed as mean ± S.D. from five assays of three independent experiments. *, significant inhibition compared with the vehicle control (p < 0.05).
DISCUSSION
The flower buds of D. genkwa Siebold et Zucc. (Thymelaeaceae), a medicinal plant distributed mainly in the mainland of China and Korea, has traditionally been used for treatment of cancer and is also valued for its antitussive, diuretic, and expectorant effects (28). Yuanhuacine (YHL-14), a compound of daphne diterpene ester, is the principal active component of the flowers of D. genkwa. YHL-14 has recently been reported to exhibit potent antiproliferative activity in various cancer cell lines (10, 29, 30), and this inhibition is associated with its targeting of topoisomerase-DNA complexes and blocking of the regulation step, thereby enhancing the formation of persistent DNA breaks and further leading to cell death. YHL-14 is, therefore, considered to be a DNA-damaging agent (31). This study demonstrates that YHL-14 treatment results in G2/M phase growth arrest and inhibits cancer cell monolayer growth and anchorage-independent growth in both human bladder cancer T24T and UMUC3 cells and/or the human colon cancer HCT116 cell line. Further studies showed that YHL-14 induced p21 up-regulation that was attributable to G2/M phase arrest 12 h following treatment in both T24T and HCT116 cells. p53 is thought to be responsible for p21 up-regulation (32). This study shows that YHL-14 treatment only induced p21 expression without affecting either p53 protein expression, p53 protein phosphorylation, or p53-dependent transcriptional activation, whereas another anticancer compound, F2, did show all of those effects in the same cell lines and under the same experimental conditions. Moreover, deletion of p53 in HCT116 cells did not affect p21 protein induction by YHL-14, whereas it blocked p21 protein expression following F2 treatment. Consistently, knockout of p21 expression in HCT116 cells impaired G2/M phase growth arrest, cell proliferation inhibition, and anchorage-independent growth inhibition following YHL-14 treatment, whereas deletion of p53 expression did not affect those biological effects on YHL treatment. Those results exhibited that anticancer effect of YHL-14 was mediated by its induction of p21 expression, which in turn induced G2/M phase growth arrest via the p53-independent pathway. Following the exclusion of p53, we found that rapid up-regulation of p21 by YHL-14 was mediated by transient up-regulation of nuclear transcription factor Sp1, which led to the enhancement of p21 gene transcription. The Sp1 regulation of p21 transcription following YHL-14 treatment was evident from the utilization of the p21 promoter-driven luciferase reporter and the Sp1 knockdown approach. We found that the transcription activity of ∼200-bp p21 promoter-luciferase reporter with only Sp1 binding sites was a comparable level with that of ∼2.4-kb p21 promoter-luciferase reporter with the transcription factors, including Sp1, p53, and Stat3. Knockdown of Sp1 expression in T24T cells blocked p21 induction because of YHL-14 treatment. Together with no effect of YHL-14 on Stat3 protein expression, p53 protein expression, and transcriptional activity, we concluded that Sp1 induction by YHL-14 mediated p21 expression. Our studies also found that the inhibition of p38 kinase activation by ectopic expression of DN-p38 not only resulted in the reduction of Sp1 protein stability and p21 protein expression, but it also counteracted the effects of YHL-14 on G2/M phase growth arrest and partially restored cancer cell anchorage-independent growth following YHL-14 treatment. Therefore, we demonstrated that p38 activation by YHL-14 mediated rapid and transient increased Sp1 protein stability and Sp1 protein expression, further leading to p21 protein induction and, in turn, resulting in cancer cell G2/M phase growth arrest and anchorage-independent growth inhibition.
p53, a sensitive DNA damage-responsive transcriptional factor, is always up-regulated in cellular response to DNA damage treatments (32, 33). p53 protein up-regulation could initiate p21-dependent cellular growth arrest and/or apoptotic responses (34). p21 is a protein that inhibits the activity of cyclin-CDK2 or cyclin-CDK1 complexes. Thus, p21/WAF1 functions as a regulator of cell cycle progression at G1 (35). It is well known that there are multiple p53 binding sites in the p21 promoter region. Thus, the p53-p21 axis is always regarded as a classic pathway for G0/G1 phase arrest induced by DNA-damaging agents (27). This notion was supported by our results obtained from an anticancer compound, F2, that induced p21 expression and G0/G1 growth arrest in a p53-dependent manner. However, by elucidating the anticancer effect of YHL-14, we disclosed a distinct mechanism underlying YHL-14 regulation of p21 as well as its function as a regulator of G2/M phase growth arrest rather than G0/G1 phase growth arrest. The explanation of this differential effect of p21 induction upon F2 and YHL-14 treatment might be due to multiple p53 downstream-targeted genes in addition to p21 following F2 treatment. The detailed molecular mechanisms for this differential effect are currently under investigation in our laboratory.
This study found that p21 was significantly transcriptionally up-regulated through an Sp1-dependent and p53-indpendent manner following YHL-14 treatment. As shown in Fig. 4C, there are six Sp1 binding sites in the p21 promoter region between −119 bp and the starting site of the human p21 gene (36, 37). Our results showed that YHL-14 treatment led to the induction of Sp1 protein expression and p21 promoter-driven luciferase activity. Moreover, the full-length (2.4 kb) p21 promoter luciferase reporter showed a similar transcriptional activity to that observed in a 200-bp p21 promoter luciferase reporter only containing Sp1 binding sites following YHL-14 treatment. Importantly, knockdown of Sp1 expression resulted in a dramatic reduction of p21 protein expression in T24T cells. Those results strongly indicate that Sp1 induction is essential for p21 transcription and protein induction upon YHL-14 treatment.
The p38 protein kinase is a member of the MAPK family that is activated in a cell response to environmental stress (UV light, oxidative stress, and ionizing radiation) and cytokines (e.g. TNFα) (44). MAPK activation by cytokines and receptor ligands generally leads to cell differentiation, whereas MAPK activation by environmental stress mediates growth arrest and apoptosis (45, 46). p38 activation has also been reported to mediate activation and expression of various downstream targets, including transcription factors (ATF2, Sp1, Mac, and MEF2), protein kinases (MAPKAPK2), and cell cycle control proteins (cyclin D1) (44, 46). Previous studies have reported that YHL-14 treatment inhibits the relaxation activity of Topo I toward DNA and induces DNA damage in 30 min (10). DNA damage could immediately activate p38 (47, 48). Unlike other MAPKs, p38 has been found to be distributed throughout the cytosol and nucleus because of its lack of a structure of nuclear localization signaling (18). It has also been reported that activation of p38 by a DNA-damaging agent could lead to its nuclear translocation (49). Our study found that p38 was activated by YHL-14 treatment and that YHL-14-induced phosphorylated p38 increased its nuclear translocation (Fig. 7A), suggesting that YHL-14-induced p38 activation might be associated with its DNA-damaging activity. Abnormal cell proliferation is necessary for cancer development (3). Induction of cell growth arrest and/or apoptosis are two of the major mechanisms that are responsible for the anticancer effect of most current antitumor agents. p38 activation has also been reported to play an important role in the mediation of full Sp1 protein phosphorylation at Thr-453 and Thr-739 and transactivation (38–40). In our study, Sp-1 up-regulation by YHL-14 was observed 3–6 h following YHL-14 treatment. The transient increase of Sp1 was due to YHL-14 inhibition of Sp1 protein degradation rather than due to the regulation of the mRNA level. Furthermore, YHL-14 not only induced p38 activation, but, also, this p38 activation was required for YHL-14 stabilization of Sp1 protein and induction of p21 expression, as demonstrated by the utilization of ectopic expression of DN-p38 in T24T cells. Thus, we demonstrated that YHL-14 treatment could stabilize Sp1 protein by induction of p38 activation. Our observation is somewhat unique because many recent studies show down-regulation of Sp1 and other Sp transcription factors by many naturally occurring anticancer agents (41–43). Taken together, our results demonstrate that the anticancer activity of YHL-14 is mediated by activated p38, which leads to Sp1 protein accumulation and transactivation, subsequently resulting in p21 gene transcription and protein expression as well as cancer cell G2/M phase growth arrest and anchorage-independent growth retardation, as shown in Fig. 7B. These results provide important new insights into the molecular basis underlying the anticancer activity of YHL-14 and contribute valuable information that is helpful for the design and synthesis of other new conformation-constrained derivatives for the treatment of cancers.
FIGURE 7.

YHL-14 induced phospho-p38 nuclear accumulation. A, cells (5 × 105) were seeded into 10-cm dishes. After synchronization, cells were treated with YHL-14 for 2 h, and the cells were extracted for isolation of cytoplasmic and nuclear fractions according to the protocol of the nuclear/cytosol fractionation kit (Biovision Inc.). The isolated protein fractions were subjected to Western blot analysis for determination of protein expression. B, proposed model for regulation of p21 and G2/M phase growth arrest by YHL-14.
Acknowledgments
We thank Dr. Jennifer A. Pietenpol (Vanderbilt-Ingram Comprehensive Cancer Center, Vanderbilt University School of Medicine) for the p21 promoter-driven luciferase reporters. We also thank Nedda Tichi for critical reading of the manuscript.
This work was supported, in whole or in part, by NCI, National Institutes of Health Grants CA177665, CA165980, and CA112557 and National Institutes of Health Grants NSFC81229002 and LR2012034.
- CHX
- cycloheximide.
REFERENCES
- 1. Sisodiya D., Dashora K., Pandey P. (2012) Cellular organization and cell reproduction. IJAPBC 1, 138–150 [Google Scholar]
- 2. De Souza C. P., Osmani S. A. (2007) Mitosis, not just open or closed. Eukaryot. Cell 6, 1521–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maton A., Hopkins Jean Johnson, David Susan LaHart, David Quon Warner, Wright Jill D. (1997) Cells. Building Blocks of Life, pp. 76–80, Pearson Prentice Hall, Upper Saddle River, NJ [Google Scholar]
- 4. Evan G. I., Vousden K. H. (2001) Proliferation, cell cycle and apoptosis in cancer. Nature 411, 342–348 [DOI] [PubMed] [Google Scholar]
- 5. Vinay K. M., Abulk A. M., Nelson F. (2004) Robbins and Cotran Pathologic Basis of Disease, pp. 331–340, Elsevier's Health Sciences, Philadelphia, PA [Google Scholar]
- 6. Zhou J. J., Xie G., Yan X. (2013) Encyclopedia of traditional Chinese Medicines, Vol. 5, pp. 227–229, Springer, Berlin [Google Scholar]
- 7. Hall I. H., Liou Y. F., Oswald C. B., Lee K. H. (1986) The effects of genkwadaphnin and gnidilatidin on the growth of P-388, L-1210 leukemia and KB carcinoma cells in vitro. Eur. J. Cancer Clin. Oncol. 22, 45–52 [DOI] [PubMed] [Google Scholar]
- 8. Zhan Z. J., Fan C. Q., Ding J., Yue J. M. (2005) Novel diterpenoids with potent inhibitory activity against endothelium cell HMEC and cytotoxic activities from a well known TCM plant Daphne genkwa. Bioorg. Med. Chem. 13, 645–655 [DOI] [PubMed] [Google Scholar]
- 9. Bhandurge P., Rajarajeshwari N., Ganapaty S., Pattanshetti S. (2013) The Gnidia genus: A review. Asian Journal of Biomedical and Pharmaceutical Sciences 3, 1–31 [Google Scholar]
- 10. Zhang S., Li X., Zhang F., Yang P., Gao X., Song Q. (2006) Preparation of yuanhuacine and relative Daphne diterpene esters from Daphne genkwa and structure-activity relationship of potent inhibitory activity against DNA topoisomerase I. Bioorg. Med. Chem. 14, 3888–3895 [DOI] [PubMed] [Google Scholar]
- 11. Jemal A., Bray F., Center M. M., Ferlay J., Ward E., Forman D. (2011) Global cancer statistics. CA-Cancer J. Clin. 61, 69–90 [DOI] [PubMed] [Google Scholar]
- 12. WHO Library Cataloguing-in-Publication Data (2010) World Health Statistics. World Health Organization, pp. 18–23, Geneva [Google Scholar]
- 13. WHO Library Cataloguing-in-Publication Data (2009) Global health risks: mortality and burden of disease attributable to selected major risks. World Health Organization, pp. 1–25, Geneva [Google Scholar]
- 14. Harding M. A., Theodorescu D. (2010) RhoGDI signaling provides targets for cancer therapy. Eur. J. Cancer 46, 1252–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cummins J. M., Kohli M., Rago C., Kinzler K. W., Vogelstein B., Bunz F. (2004) X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res. 64, 3006–3008 [DOI] [PubMed] [Google Scholar]
- 16. Robbins D. J., Zhen E., Owaki H., Vanderbilt C. A., Ebert D., Geppert T. D., Cobb M. H. (1993) Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J. Biol. Chem. 268, 5097–5106 [PubMed] [Google Scholar]
- 17. Huang C., Ma W. Y., Dong Z. (1999) The extracellular-signal-regulated protein kinases (Erks) are required for UV-induced AP-1 activation in JB6 cells. Oncogene 18, 2828–2835 [DOI] [PubMed] [Google Scholar]
- 18. Raingeaud J., Gupta S., Rogers J. S., Dickens M., Han J., Ulevitch R. J., Davis R. J. (1995) Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 270, 7420–7426 [DOI] [PubMed] [Google Scholar]
- 19. Zhang D., Song L., Li J., Wu K., Huang C. (2006) Coordination of JNK1 and JNK2 is critical for GADD45α induction and its mediated cell apoptosis in arsenite responses. J. Biol. Chem. 281, 34113–34123 [DOI] [PubMed] [Google Scholar]
- 20. Huang C., Ma W. Y., Maxiner A., Sun Y., Dong Z. (1999) p38 kinase mediates UV-induced phosphorylation of p53 protein at serine 389. J. Biol. Chem. 274, 12229–12235 [DOI] [PubMed] [Google Scholar]
- 21. Westfall M. D., M. D. J., Sniezek Joseph C., Pietenpol Jennifer A. (2003) The ΔNp63 phosphoprotein Binds the p21 and 14-3-3σ promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol. Cell. Biol. 23, 2264–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang D., Li J., Costa M., Gao J., Huang C. (2010) JNK1 mediates degradation HIF-1α by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res. 70, 813–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ding J., Li J., Xue C., Wu K., Ouyang W., Zhang D., Yan Y., Huang C. (2006) Cyclooxygenase-2 induction by arsenite is through a nuclear factor of activated T-cell-dependent pathway and plays an antiapoptotic role in Beas-2B cells. J. Biol. Chem. 281, 24405–24413 [DOI] [PubMed] [Google Scholar]
- 24. Cai T., Li X., Ding J., Luo W., Li J., Huang C. (2011) A cross-talk between NFAT and NF-κB pathways is crucial for nickel-induced COX-2 expression in Beas-2B cells. Curr. Cancer Drug Targets 11, 548–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu J., Zhang D., Mi X., Xia Q., Yu Y., Zuo Z., Guo W., Zhao X., Cao J., Yang Q., Zhu A., Yang W., Shi X., Li J., Huang C. (2010) p27 suppresses arsenite-induced Hsp27/Hsp70 expression through inhibiting JNK2/c-Jun- and HSF-1-dependent pathways. J. Biol. Chem. 285, 26058–26065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luo W., Liu J., Li J., Zhang D., Liu M., Addo J. K., Patil S., Zhang L., Yu J., Buolamwini J. K., Chen J., Huang C. (2008) Anti-cancer effects of JKA97 are associated with its induction of cell apoptosis via a Bax-dependent and p53-independent pathway. J. Biol. Chem. 283, 8624–8633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. el-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K. W., Vogelstein B. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 [DOI] [PubMed] [Google Scholar]
- 28. Hong J. Y., Chung H. J., Lee H. J., Park H. J., Lee S. K. (2011) Growth inhibition of human lung cancer cells via down-regulation of epidermal growth factor receptor signaling by yuanhuadine, a daphnane diterpene from Daphne genkwa. J. Nat. Prod. 74, 2102–2108 [DOI] [PubMed] [Google Scholar]
- 29. Hong J. Y., Nam J. W., Seo E. K., Lee S. K. (2010) Daphnane diterpene esters with anti-proliferative activities against human lung cancer cells from Daphne genkwa. Chem. Pharm. Bull. 58, 234–237 [DOI] [PubMed] [Google Scholar]
- 30. Park B. Y., Min B. S., Ahn K. S., Kwon O. K., Joung H., Bae K. H., Lee H. K., Oh S. R. (2007) Daphnane diterpene esters isolated from flower buds of Daphne genkwa induce apoptosis in human myelocytic HL-60 cells and suppress tumor growth in Lewis lung carcinoma (LLC)-inoculated mouse model. J. Ethnopharmacol. 111, 496–503 [DOI] [PubMed] [Google Scholar]
- 31. Zhang S. (2009) Evaluation of poly(D,L-lactide-co-glycolide) microspheres for the lung-targeting of yuanhuacine, a novel DNA topoisomerase I inhibitor. J. Drug Target. 17, 286–293 [DOI] [PubMed] [Google Scholar]
- 32. Lakin N. D., Jackson S. P. (1999) Regulation of p53 in response to DNA damage. Oncogene 18, 7644–7655 [DOI] [PubMed] [Google Scholar]
- 33. Gartner A. (2000) A conserved checkpoint pathway mediates DNA damage-induced apoptosis and cell cycle arrest in C. elegans. Mol. Cell 5, 435–443 [DOI] [PubMed] [Google Scholar]
- 34. Macleod K. F. (1995) p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 9, 935–944 [DOI] [PubMed] [Google Scholar]
- 35. Harper J. W. (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816 [DOI] [PubMed] [Google Scholar]
- 36. Gartel A. L. (2000) Sp1 and Sp3 activate p21 (WAF1/CIP1) gene transcription in the Caco-2 colon adenocarcinoma cell line. Oncogene 19, 5182–5188 [DOI] [PubMed] [Google Scholar]
- 37. Gartel A. L., Radhakrishnan S. K. (2005) Lost in transcription. p21 repression, mechanisms, and consequences. Cancer Res. 65, 3980–3985 [DOI] [PubMed] [Google Scholar]
- 38. Chuang J. Y. (2008) Phosphorylation by c-Jun NH2-terminal kinase 1 regulates the stability of transcription factor Sp1 during mitosis. Mol. Biol. Cell 19, 1139–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. D'Addario M., Arora P. D., McCulloch C. A. (2006) Role of p38 in stress activation of Sp1. Gene 379, 51–61 [DOI] [PubMed] [Google Scholar]
- 40. Tan N. Y., Khachigian L. M. (2009) Sp1 phosphorylation and its regulation of gene transcription. Mol. Cell. Biol. 29, 2483–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chintharlapalli S., Papineni S., Ramaiah S. K., Safe S. (2007) Betulinic acid inhibits prostate cancer growth through inhibition of specificity protein transcription factors. Cancer Res. 67, 2816–2823 [DOI] [PubMed] [Google Scholar]
- 42. Chadalapaka G., Jutooru I., Safe S. (2012) Celastrol decreases specificity proteins (Sp) and fibroblast growth factor receptor-3 (FGFR3) in bladder cancer cells. Carcinogenesis 33, 886–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jutooru I., Chadalapaka G., Lei P., Safe S. (2010) Inhibition of NFκB and pancreatic cancer cell and tumor growth by curcumin is dependent on specificity protein down-regulation. J. Biol. Chem. 285, 25332–25344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zarubin T., Han J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18 [DOI] [PubMed] [Google Scholar]
- 45. Mikhailov A., Shinohara M., Rieder C. L. (2005) The p38-mediated stress-activated checkpoint. A rapid response system for delaying progression through antephase and entry into mitosis. Cell Cycle 4, 57–62 [DOI] [PubMed] [Google Scholar]
- 46. Mikhailov A. (2007) The G2 p38-mediated stress-activated checkpoint pathway becomes attenuated in transformed cells. Curr. Biol. 17, 2162–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eom H. J., Choi J. (2010) p38 MAPK activation, DNA damage, cell cycle arrest and apoptosis as mechanisms of toxicity of silver nanoparticles in Jurkat T cells. Environ. Sci. Technol. 44, 8337–8342 [DOI] [PubMed] [Google Scholar]
- 48. Bulavin D. V. (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411, 102–107 [DOI] [PubMed] [Google Scholar]
- 49. Wood C. D. (2009) Nuclear localization of p38 MAPK in response to DNA damage. Int. J. Biol. Sci. 5, 428–437 [DOI] [PMC free article] [PubMed] [Google Scholar]