

Background: Fn14 is a highly inducible member of the TNF receptor family.
Results: Large-magnitude mechanical stress induced Fn14 expression via JNK in osteoblasts.
Conclusion: Expression of Fn14 regulates mechanical stress-induced apoptosis in osteoblasts.
Significance: This is the first elucidation of the mechanism of excessive mechanical stress-induced apoptosis mediated by Fn14.
Keywords: Apoptosis, Jun N-terminal Kinase (JNK), Mechanotransduction, Osteoblasts, p38 MAPK, Fn14
Abstract
Bone mass is maintained by the balance between the activities of bone-forming osteoblasts and bone-resorbing osteoclasts. It is well known that adequate mechanical stress is essential for the maintenance of bone mass, whereas excess mechanical stress induces bone resorption. However, it has not been clarified how osteoblasts respond to different magnitudes of mechanical stress. Here we report that large-magnitude (12%) cyclic stretch induced Ca2+ influx, which activated reactive oxygen species generation in MC3T3-E1 osteoblasts. Reactive oxygen species then activated the ASK1-JNK/p38 pathways. The activated JNK led to transiently enhanced expression of FGF-inducible 14 (Fn14, a member of the TNF receptor superfamily) gene. Cells with enhanced expression of Fn14 subsequently acquired sensitivity to the ligand of Fn14, TNF-related weak inducer of apoptosis, and underwent apoptosis. On the other hand, the ASK1-p38 pathway induced expression of the monocyte chemoattractant protein 3 (MCP-3) gene, which promoted chemotaxis of preosteoclasts. In contrast, the ERK pathway was activated by small-magnitude stretching (1%) and induced expression of two osteogenic genes, collagen Ia (Col1a) and osteopontin (OPN). Moreover, activated JNK suppressed Col1a and OPN induction in large-magnitude mechanical stretch-loaded cells. The enhanced expression of Fn14 and MCP-3 by 12% stretch and the enhanced expression of Col1a and OPN by 1% stretch were also observed in mouse primary osteoblasts. These results suggest that differences in the response of osteoblasts to varying magnitudes of mechanical stress play a key role in switching the mode of bone metabolism between formation and resorption.
Introduction
Bone mass is maintained by the balance between the activities of bone-forming osteoblasts and bone-resorbing osteoclasts. The activity of bone cells is regulated in response to the changes of hormonal, neuronal, immunological, and mechanical environments (1, 2). It is well known that adequate mechanical stress is essential for the maintenance of bone mass (3, 4). On the other hand, bone resorption induced by excessive mechanical stress is observed locally in cases such as pseudoarthrosis with insufficient fixation, during orthodontic tooth movement, around dental implants, and beneath denture bases (5–8). Recent studies showed that receptor activator of nuclear factor κB ligand (RANKL)3 expression by osteocytes, the majority of bone cells, controls bone remodeling in adults as well as unloading-induced bone loss (9, 10). It has also been reported that the osteocyte network plays a significant role in unloading-induced bone loss (11, 12) or bone resorption during orthodontic tooth movement (13). On the other hand, the behavior of osteoblasts in response to excessive mechanical stress has been poorly understood. A previous study showed that the activation of caspase 3 and apoptosis has been observed in compression-loaded osteoblasts (14). However, the precise cues and mechanotransduction mechanisms that induce osteoblast apoptosis in response to large-magnitude mechanical stress have yet to be elucidated.
The MAPK pathway is a three-step sequential phosphorylation cascade that is composed of MAPK kinase kinase (MAP3K), MAPKK, and MAPK. Three major MAPKs, ERK, JNK, and p38 MAPK, are activated by various biological and physicochemical stressors and are pivotal in stress responses, including differentiation, cytokine production, and apoptosis (15). ERK and JNK2 act as positive regulators of osteoblast differentiation through the activation of runt-related transcription factor 2 (Runx2) by ERK (16) and induction of activating transcription factor 4 expression by JNK2 (17). The p38 MAPK pathway is essential for skeletogenesis through the activation of Runx2 by phosphorylation at several serine residues (18). However, we reported recently that transforming growth factor β-activated protein kinase 1 (TAK1), a mechanical stretch-activated MAP3K, induces IL-6 expression via downstream JNK, p38, and NF-κB pathways in both MC3T3-E1 cells and mouse primary osteoblasts (19). IL-6 has been shown to exacerbate rheumatoid arthritis through the expression of RANKL (1, 20), suggesting that the large magnitudes of mechanical stress that activate JNK and p38 in osteoblasts also suppress bone formation. However, it is not known whether JNK or p38 are involved in mechanical stress-induced apoptosis of osteoblasts.
In this study, we showed that the apoptosis signal-regulating kinase 1 (ASK1)-JNK1/2 pathway, which was activated by large-magnitude mechanical stretch, induced expression of the fibroblast growth factor-inducible 14 (Fn14, a member of the TNF receptor superfamily) gene in osteoblasts. TNF-related weak inducer of apoptosis (TWEAK)-Fn14 signaling contributed to osteoblast apoptosis evoked by large-magnitude stretch. On the other hand, the large-magnitude mechanical stretch-activated ASK1-p38 pathway induced expression of the monocyte chemoattractant protein 3 (MCP-3) gene. The first seven amino acids were required for shedding-mediated release of MCP-3, and the released MCP-3 induced chemotaxis of preosteoclasts. We also observed that ERK was preferentially activated by small-magnitude stretch and led to the expression of two osteogenic genes, collagen Ia (Col1a) and osteopontin (OPN). These observations shed light on the molecular mechanisms underlying the different effects of small- and large-magnitude mechanical stress on the behavior of osteoblasts.
EXPERIMENTAL PROCEDURES
Cell Culture
MC3T3-E1 cells were cultured in α-minimum essential medium containing 10% fetal bovine serum in 5% (v/v) CO2 at 37 °C. RAW264.7 cells were cultured in DMEM containing 10% fetal bovine serum in 5% (v/v) CO2 at 37 °C. The preparation of mouse primary osteoblasts has already been described (17).
Mechanical Stretch Application
Cyclic mechanical stretch experiments were performed using the ST-140 cell stretcher system (Strex). Before cell seeding, elastic cell culture chambers made of polydimethylsiloxane were coated with soluble swine type I collagen (Nitta gelatin) according to the protocol of the manufacturer. The cells were stretched for the indicated periods at a frequency of 10 cycles/min, each cycle consisting of 2 s of stretch and 2 s of relaxation.
Reagents and Antibodies
N-Acetyl-l-cysteine (NAC) was purchased from Wako. SP600125, SB203580, and U0126 were purchased from Calbiochem. CHX and MG132 were obtained from Sigma-Aldrich. Antibodies against phospho-JNK, phospho-p38, phospho-ERK, JNK2, p38, ERK, phospho-MKK3/6, phospho-MKK4, MKK6, phospho-TAK1 (P-Thr-187), Fn14, active caspase 3, actin, and FLAG were purchased from Cell Signaling Technology. Phospho-ASK1 antibody has been described previously (21). Antibodies to MKK4 (catalog no. 554105) and caspase 3 (catalog no. 611048) were obtained from BD Biosciences. Antibodies to TAK1 (catalog no. N579) and TWEAK (Ser-20) were purchased from Santa Cruz Biotechnology. JNK (catalog no. 06-749) and ASK1 (catalog no. C2) antibodies were purchased from Upstate Biotechnology and Anaspec, respectively. Antibody to MCP-3 (catalog no. AF-456-NA) was purchased from R&D Systems.
Immunoblot Analysis
Cells were lysed in lysis buffer containing 20 mm HEPES-KOH (pH 7.5), 250 mm NaCl, 1% (v/v) Triton X-100, 2 mm EGTA, 12.5 mm β-glycerophosphate, 1.5 mm MgCl2, 10 mm NaF, 1 mm Na3VO4, 1 mm phenylmethylsulfonylfluoride, and 10 μg/ml leupeptin. The cell extracts were clarified by centrifugation, and then the supernatants were boiled in SDS sample buffer. Cell lysates were subjected to SDS-PAGE and electroblotted onto a polyvinylidene difluoride membrane. After blocking with 2% (v/v) skim milk in TBST (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 0.1% (v/v) Tween 20), the membranes were incubated with the indicated antibodies. Antibody-antigen complexes were detected using an ECL system (GE Healthcare).
RNA Interference
Stealth siRNAs for mouse JNK1 (catalog no. MSS218561), JNK2 (catalog no. MSS218565), p38α (catalog no. MSS240943), p38β (catalog no. MSS207968), ASK1 (#1, catalog no. MSS218536; #2, catalog no. MSS218535), TWEAK (#1, NM_011614_stealth_501 and #2, NM_011614_stealth_722), and Stealth RNAi negative control were purchased from Invitrogen. MC3T3-E1 cells were transfected with 40 pmol/well of the indicated siRNA oligo using Lipofectamine RNAiMAX (Invitrogen). Knockdown was analyzed by immunoblotting with antibody to each protein.
Reactive Oxygen Species (ROS) Imaging
For detection of ROS, cells were pretreated with 20 μm of hydroxyphenyl fluorescein (Daiichi Pure Chemicals) for 20 min before mechanical stretching. The fluorescent images were obtained using an LSM 5 confocal laser-scanning unit coupled to an Axiovert 200 m inverted microscope with a Plan-Neofluar 40 × 0.75 objective lens (Carl Zeiss). The excitation wavelength was 488 nm, and the emission was filtered using a 505- to 530-nm barrier filter.
Quantitative RT-PCR Analysis
A detailed description of our quantitative PCR methodology has been provided in our previous study (19). Primer sequences were as follows. Mouse CCR1, 5′-GGTGATGCCATGTGCAAGC-3′ and 5′-GGGCCCTCAGGGCAAACAC-3′; CCR2, 5′-GACCAGAAGAGGGCATTGG-3′ and 5′-GCCGTGGSTGSSCTGSGG-3′; mouse Col1a, 5′-CACCCTCAAGAGCCTGAGTC-3′ and 5′-GCTTCTTTTCCTTGGGGTTC-3′; mouse Fn14, 5′-CGACAAGTGCATGGACTGCG-3′ and 5′-CCAGGACCAGACTAAGAGCGC-3′; mouse GAPDH, 5′-GCACAGTCAAGGCCGAGAATGG-3′ and 5′-GGTGAAGACACCAGTAGAC-3′; mouse MCP-3, 5′-CATCCACATGCTGCTGCTATGTC-3′ and 5′-CCACTTCTGATGGGCTTCAG-3′; mouse OPN, 5′-TACGACCATGAGATTGGCAGTGA-3′ and 5-TATAGGATCTGGGTGCAGGCTGTAA-3′; and mouse TWEAK, 5′-GGCCTCGAAATGGTGTCCTG-3′ and 5′-GCCACTCACTGTCCCATCC-3′.
Immunofluorescent Staining and TUNEL Staining
Cells were fixed with 4% (v/v) paraformaldehyde for 20 min at room temperature and permeabilized with 0.1% (v/v) Triton X-100. After blocking in 2% (w/v) BSA, cells were incubated with antibody to active caspase 3 at 4 °C. After washing with PBS, cells were probed with Alexa Fluor 555-conjugated anti-rabbit IgG antibody diluted 1:600 for 1 h at room temperature. TUNEL staining was performed using an in situ cell death detection kit (Roche) according to the protocol of the manufacturer. Fluorescent images were acquired with an LSM 5 microscope.
Plasmids and Transfection
Complementary DNA encoding Fn14WT was cloned using cDNA from MC3T3-E1 cells and inserted into a pCR-TOPO vector. cDNAs encoding Fn14D45A, Fn14K48R, and Fn14K109R were generated by site-directed mutagenesis using Fn14WT as a template. These Fn14 mutants were subcloned into pcDNA 3.0 with a C-terminal FLAG tag. Transfection of these expression plasmids was performed using Lipofectamine 2000 (Invitrogen) according to the protocol of the manufacturer.
CHX Chase Assay
Cells were stretched (Fig. 6A) or transfected with the indicated plasmids (Fig. 6C). Then the cells were treated with CHX (5 μg/ml) for the indicated periods. The cell lysates were analyzed by immunoblotting with the indicated antibodies.
FIGURE 6.

Fn14 was degraded by the ubiquitin-proteasome system. A, MC3T3-E1 cells were stimulated with cyclic mechanical stretch (12%) for 6 h. After stretch stimulation, cells were incubated with CHX (5 μg/ml) in the presence of dimethyl sulfoxide (DMSO) or MG132 (10 μm) for the indicated periods. The cell lysates were subjected to immunoblotting (IB) using the indicated antibodies. B, schematic of mouse Fn14WT, Fn14K48R, and Fn14K109R. All expression plasmids were FLAG-tagged at their C termini. C, CHX chase analysis of Fn14 mutants. MC3T3-E1 cells were transfected with the indicated expression plasmids. After 24 h, cells were treated with CHX for the indicated periods. Then, the cell lysates were subjected to immunoblotting using the indicated antibodies. O/N indicates overnight. D, in vivo ubiquitination (Ub) assay of Fn14WT and Fn14K109R. See “Experimental Procedures” for details. IP, immunoprecipitation. E, MC3T3-E1 cells were transfected with the indicated expression plasmids. After 24 h, cells were lysed and analyzed by immunoblotting with the indicated antibodies. A, C, D, and E, the data represent one of at least three experiments.
In Vivo Ubiquitination Assay
Cells were transfected with the indicated Fn14-FLAG expression plasmids and HA-ubiquitin. After 12 h, cells were incubated with fresh medium containing 0.5 μm MG132 for 16 h. Cells were then lysed in IP buffer containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 12 mm β-glycerophosphate, 1% (v/v) Triton X-100, 5 mm EGTA, 10 mm NaF, and 1 mm Na3VO4. The cell extracts were immunoprecipitated with FLAG antibody (M2, Sigma), and then the beads were washed with buffer A (20 mm Tris-HCl (pH 7.5), 500 mm NaCl, 1% Triton X-100, and 2 mm EGTA) and buffer B (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 2 mm EGTA). Subsequently, the beads were boiled and denatured in buffer B additionally containing 1% (w/v) SDS to disrupt non-covalent protein-protein interaction. The remaining bead-containing solutions were resuspended in a 50× volume of IP buffer to dilute the SDS, reimmunoprecipitated with FLAG M2 antibody, and analyzed by SDS-PAGE and immunoblotting.
Chemotaxis Assay
Cells were transfected with expression plasmids of FLAG-MCP-3WT or FLAG-MCP-3Δ7NT. After 24 h, conditioned medium was collected. Chemotaxis assay of RAW264.7 cells was performed using a transwell unit with 5-μm pore size (Corning). Briefly, the collected conditioned medium was added to the lower part of the transwell unit. Then, RAW264.7 cells (1.5 × 105 in 100 μl) were loaded into the upper part of the transwell unit. After 3 h, the transwell membrane was methanol-fixed and stained with eosin. Then, the cells on the top side of the membrane were wiped off, and cells trapped on the bottom side of the membrane were counted with a microscope.
RESULTS
JNK and p38 Activated by a Large-magnitude Mechanical Stretch Negatively Regulate Col1a and OPN Expression in Osteoblasts
To explore the role of MAPK pathways in the mechanical stress response, we first analyzed the effect of mechanical stretch on the activities of ERK, JNK, and p38. Upon mechanical stretch loading, ERK was strongly activated by a small-magnitude stretch (1%) (Fig. 1A). On the other hand, a large-magnitude stretch (12%) was required for clear activation of JNK and p38. An earlier study reported that mechanical stress-induced ERK activation contributed to Col1a expression through Runx2 activation (22). Our own quantitative RT-PCR analysis showed that expression of both Col1a and OPN was consistently induced by 1% stretch (Fig. 1, B and C). The expression of Col1a and OPN was suppressed significantly by treatment with U0126, a MEK1/2 inhibitor. On the other hand, Col1a and OPN expression was not enhanced upon 12% stretch (Fig. 1, D and E). Intriguingly, Col1a and OPN expression upon 12% stretch was recovered by treatment with SP600125 or SB203580, inhibitors of JNK or p38, respectively, suggesting that the activities of JNK and p38 suppressed the mechanical stress-induced expression of Col1a and OPN (Fig. 1, D and E).
FIGURE 1.

Mechanical stretch induces activation of ERK1/2, JNK1/2, and p38α via Ca2+ influx in MC3T3-E1 cells. A, activation of MAPKs in response to different magnitudes of mechanical stretch. MC3T3-E1 cells were stimulated with the indicated durations of cyclic stretch for 20 min. The kinase activity of MAPKs was detected by immunoblotting (IB) using P-ERK, P-JNK, and P-p38 antibodies. B and C, MC3T3-E1 cells were pretreated with or without U0126 (10 μm) for 60 min. The cells were then loaded with or without cyclic stretch (1%) for 6 h. Quantitative PCR analysis was then performed as under “Experimental Procedures.” Relative expression levels were normalized on the basis of the expression of GAPDH mRNA. Error bars indicate mean ± S.E. *, p < 0.05, Student's t test. D and E, MC3T3-E1 cells were pretreated with or without SP600125 (10 μm) or SB203580 (10 μm) for 60 min. The cells were then loaded with or without cyclic stretch (1%) for 6 h. The expression levels of Col1a mRNA were measured by quantitative RT-PCR. Relative expression levels were normalized on the basis of the expression of GAPDH mRNA. Error bars indicate mean ± S.E. *, p < 0.05, Student's t test. F and G, mouse calvarium-derived primary osteoblasts were prepared and subjected to quantitative PCR analysis as in B and C. H and I, mouse calvarium-derived primary osteoblasts were prepared and subjected to quantitative PCR analysis as in D and E. J, identification of MAPK isoforms activated by mechanical stretch. MC3T3-E1 cells were transfected with single or combination siRNA as indicated. After 72 h, the cells were subjected to cyclic stretch loading (12%) for 30 min. The lysates were immunoblotted with the indicated antibodies. C indicates control. K, MC3T3-E1 cells were subjected to cyclic stretch for the indicated periods in the presence or absence of 5 mm EGTA. The lysates were immunoblotted with the indicated antibodies. The data in A, J, and K represent one of at least three experiments.
We have reported previously that a large-magnitude cyclic stretch induced activation of JNK and p38 not only in MC3T3-E1 cells but also in mouse primary osteoblasts (19). Therefore, we asked whether enhanced expression of Col1a and OPN via ERK, which was activated by a small-magnitude cyclic stretch, was also observed in mouse primary osteoblasts. As shown in Fig. 1, F–I, the results obtained with MC3T3-E1 cells were essentially confirmed in the experiments using primary osteoblasts. These results also suggest that activation of JNK and p38 in the context of mechanical overloading acts as a negative regulator of bone formation.
Identification of JNK and p38 Isoforms Activated by Mechanical Stretch
To determine the stretch-activated isoforms of JNK and p38 in MC3T3-E1 cells, we performed an RNAi analysis using siRNA against JNK1, JNK2, p38α, and p38β (Fig. 1J). An immunoblot analysis using anti-phospho-JNK (P-JNK) antibody revealed that both 46-kDa and 54-kDa isoforms (46 kDa, p46; 54 kDa, p54) of JNK were activated upon mechanical stretch. Of these isoforms, the lower and upper halves of P-JNK (p46) were attenuated by single knockdown of JNK1 and JNK2, respectively. On the other hand, P-JNK (p54) was attenuated by knockdown of JNK2 but not JNK1, and the effect was equivalent to double knockdown of JNK1 and JNK2. These results indicate that the stretch-activated endogenous JNK isoforms are JNK1 (p46) and JNK2 (p46 and p54).
On the other hand, phosphorylated p38 detected using anti-phospho-p38 (P-p38) was attenuated by single knockdown of p38α and double knockdown of p38α and p38β. However, single knockdown of p38β had little effect on P-p38. Knockdown of p38α but not p38β markedly decreased the amount of p38 protein, suggesting that the observed minimal effect of p38β knockdown on P-p38 might be due to the very low expression of p38β in osteoblasts. Taken together, these results indicate that p38α is the major isoform activated by mechanical stretch. We next examined whether extracellular Ca2+ influx induced the activation of JNK and p38 (Fig. 1K). The activity of all endogenous JNK and p38 isoforms were strongly suppressed in the presence of EGTA, indicating that extracellular Ca2+ influx is critical for the mechanical stretch-induced activation of JNK and p38. Similar results were obtained for the activation of MAPKKs.
ROS Mediate JNK and p38 Activation in Mechanical Stretch
A recent report demonstrated the correlation between a decrease in loading-induced bone formation and the accumulation of ROS in osteoblasts (23). In addition, ROS have been reported to contribute to the regulation of the osteoblast and osteocyte life span by modulating apoptosis (24). To investigate the involvement of mechanical stretch-induced ROS generation, we examined whether mechanical stretch caused ROS generation using the fluorescent probe hydroxyphenyl fluorescein (Fig. 2A). We detected stretch-induced ROS generation that was abolished in the presence of EGTA, suggesting that Ca2+ influx is required for mechanical stretch-induced ROS generation.
FIGURE 2.
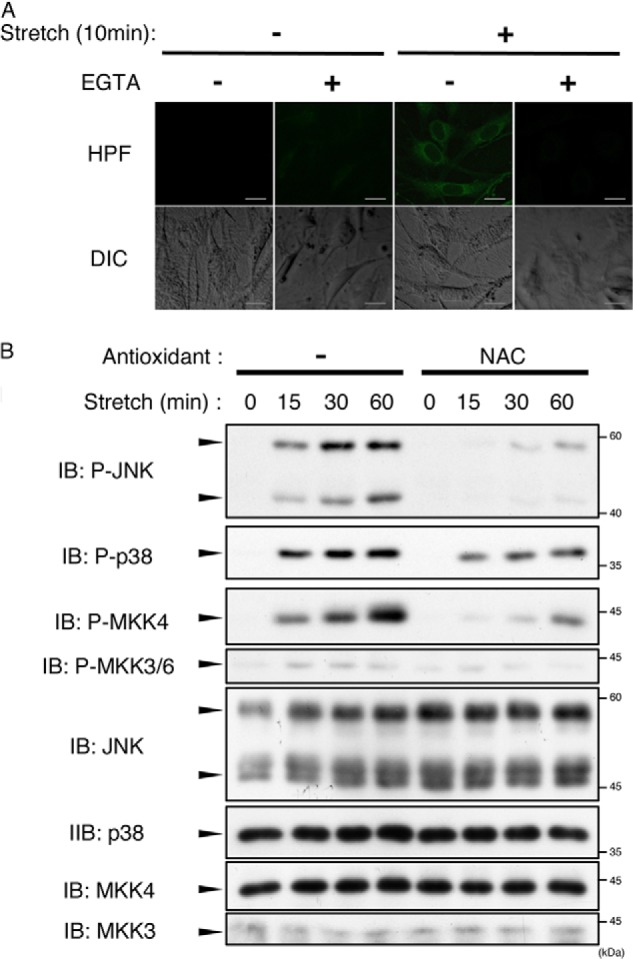
Mechanical stretch-induced ROS generation was involved in activation of JNK and p38. A, MC3T3-E1 cells were pretreated with hydroxyphenyl fluorescein (HPF) (20 μm) for 20 min, and then cyclic stretch (12%) was loaded for 10 min in the presence or absence of 5 mm EGTA. Fluorescent images and images of Nomarski differential interference contrast (DIC) are shown. B, MC3T3-E1 cells were pretreated with or without NAC (10 mm) for 60 min and then subjected to cyclic stretch loading (12%) for the indicated periods. The cell lysates were analyzed by immunoblotting (IB) with the indicated antibodies. The data represent one of at least three experiments.
We then investigated whether stretch-induced ROS were required for JNK and p38 activation (Fig. 2B). Treatment of cells with the antioxidant NAC substantially suppressed JNK and p38 activation as well as activation of MAPKKs. In our previous study, we demonstrated that 12% cyclic stretch-induced Ca2+ influx activated TAK1 (a MAP3K), which, in turn, enhanced the phosphorylation of JNK and p38. However, the mechanical stretch-induced activation of TAK1 was insensitive to NAC (see Fig. 3D). These results suggest that ROS-independent as well as ROS-dependent MAP3K was activated downstream of mechanical stretch-evoked Ca2+ influx.
FIGURE 3.

Mechanical stretch induced activation of ASK1 and TAK1 in a ROS-dependent and ROS-independent manner, respectively. A, mechanical stretch induced activation of ASK1. MC3T3-E1 cells were stimulated with cyclic mechanical stretch (12%) for the indicated periods. The lysates were analyzed by immunoblotting (IB) using anti-phospho-ASK1 (P-ASK1) antibody, which monitors the active state of ASK1 rendered by threonine 845 phosphorylation in the activation loop, and total ASK1 antibody. B, MC3T3-E1 cells were transfected with siRNA as a negative control or with siRNA for ASK1 (ASK1 #1 and ASK1 #2). After 72 h, the cells were subjected to cyclic stretch stimulation (12%) for the indicated periods and then lysed and analyzed with immunoblotting using the indicated antibodies. C, MC3T3-E1 cells were stimulated with cyclic stretch (12%) for 15 min in the presence or absence of EGTA (5 mm). The cell lysates were then subjected to immunoblotting using the indicated antibodies. D, MC3T3-E1 cells were pretreated with NAC (10 mm) for 60 min, and then the cells were stimulated with cyclic stretch (12%) for 15 min. The cells were then lysed and analyzed by immunoblotting using the indicated antibodies. The data represents one of at least three experiments.
Mechanical Stretch Activates ASK1 and TAK1 in ROS-dependent and ROS-independent Manners, Respectively
We attempted to identify the ROS-dependent MAP3K. Immunoblot analysis using anti-phospho-ASK1 (P-ASK1) antibody, which detects activating phosphorylation of threonine within the activation loop, revealed that ASK1 MAP3K was activated in response to mechanical stretch (Fig. 3A). RNAi of ASK1 substantially reduced JNK and p38 activation, indicating that ASK1 serves as the upstream MAP3K of both JNK and p38 in mechanical stretch (Fig. 3B). Furthermore, similar to TAK1, this activation was eliminated by EGTA treatment (Fig. 3C). However, antioxidant treatment selectively inhibited ASK1 activation without any effect on TAK1 activity (Fig. 3D). Consistent with this, activation of JNK and p38 was completely inhibited by EGTA treatment but only partially by NAC, whereas the same concentration of NAC completely inhibited ASK1 activation (Figs. 1K, 2B, and 3D). These results indicated that, subsequent to stretch-evoked Ca2+ influx, ASK1 and TAK1 were activated in ROS-dependent and ROS-independent manners, respectively.
Mechanical Stretch Induces Expression of Fn14 and MCP-3 via JNK and p38, Respectively
To explore the biological roles of ASK1-JNK/p38 pathway activation in mechanical stretch-loaded osteoblasts, we next analyzed the genes whose expressions were enhanced by mechanical stretch. RT-PCR analysis was performed with MC3T3E-1 cells subjected to mechanical stretch for 6 h. However, no known bone remodeling-related gene was identified as a downstream target of the ASK1-JNK/p38 pathway. Therefore, we performed microarray screening and RT-PCR analysis. This study identified two genes, Fn14 (encoded by Tnfrsf12a, a member of the TNF receptor family) and MCP-3 (encoded by Ccl7, a member of C-C motif chemokine), whose mRNA expression was up-regulated by mechanical stretch. Furthermore, expression of Fn14 and MCP-3 was diminished by knockdown of ASK1 (Fig. 4, A and B) or NAC treatment (data not shown), indicating that Fn14 and MCP-3 are downstream target genes of the ROS-ASK1 signaling pathway.
FIGURE 4.

Mechanical stretch enhanced Fn14 expression via the ASK1-JNK pathway. A and B, MC3T3-E1 cells were transfected with siRNA as a negative control or with siRNA for ASK1 (ASK1 #1). After 72 h, the cells were subjected to cyclic stretch stimulation (12%) for 6 h. The expression levels of Fn14 mRNA were measured by quantitative RT-PCR. Relative expression levels were normalized on the basis of the expression of GAPDH mRNA. Error bars indicate mean ± S.E. *, p < 0.05, Student's t test. C and D, MC3T3-E1 cells were pretreated with dimethyl sulfoxide, SP600125 (10 μm), or SB203580 (10 μm) for 60 min and then stimulated with cyclic stretch (12%) for 6 h. The expression levels of Fn14 mRNA were measured by quantitative RT-PCR. Relative expression levels were normalized on the basis of the expression of GAPDH mRNA. Error bars indicate mean ± S.E. *, p < 0.05, Student's t test. E and F, mouse calvarium-derived primary osteoblasts were prepared and subjected to quantitative PCR analysis as in C and D. G, MC3T3-E1 cells were pretreated with dimethyl sulfoxide (DMSO), SP600125 (10 μm), or SB203580 (10 μm) for 60 min and then stimulated with cyclic stretch (12%) for the indicated periods. The cells were then lysed and subjected to immunoblotting (IB) using the indicated antibodies. H, MC3T3-E1 cells were transfected with single siRNAs or a combination of siRNAs as indicated. After 72 h, the cells were subjected to cyclic stretch loading (12%) for 6 h. The cells were then lysed and analyzed by immunoblotting using the indicated antibodies. C indicates control. G and H, the data represent one of at least three experiments.
To assess the involvement of JNK and p38 in the expression of Fn14 and MCP-3, we next tested the effects of SP600125 and SB203580 on the mechanical stretch-induced expression of Fn14 and MCP-3 (Fig. 4, C and D). Interestingly, Fn14 expression was attenuated by SP600125 but not by SB203580. In contrast, expression of MCP-3 was suppressed by SB203580 but not by SP600125. Similar results were obtained in mouse primary osteoblast culture (Fig. 4, E and F). These expression patterns were also confirmed at the protein level by immunoblot analysis (Fig. 4G).
Moreover, RNAi analysis revealed that the expression of Fn14 was decreased slightly by single knockdown of JNK1 or JNK2 and strongly by double knockdown of JNK1 and JNK2 (Fig. 4H). Knockdown of p38α/β did not suppress Fn14 expression. On the basis of these observations, we concluded that the ROS-ASK1 signaling pathway was activated by mechanical stretch and regulated the expression of Fn14 through activation of JNK1/2.
Fn14 Is Involved in Mechanical Stretch-induced Cell Death
We next investigated the role of Fn14 in mechanical stretch loading of osteoblasts. Previous studies have shown that TWEAK is the ligand of Fn14 and that TWEAK-Fn14 signaling promotes apoptosis in several cell lines, such as HT-29 cells or HeLa cells (25). Therefore, we tested the possibility that mechanical stretch induces apoptosis via the TWEAK-Fn14 signaling pathway in osteoblasts. Cells were first stimulated with mechanical stretch for 6 h and then left untreated to prevent further stretch-induced gene expression. An immunoblot analysis showed that active caspase 3 was observed 12 h after stimulation (Fig. 5A), suggesting that mechanical stretch induces apoptosis of osteoblasts. To assess the potency of 12% stretch to activate caspase 3, we compared the level of stretch-activated caspase 3 with those of serial concentrations of H2O2-activated caspase 3 in MC3T3-E1 cells. The results indicated that the level of stretch-activated caspase 3 was between those of 0.05 mm H2O2- and 0.1 mm H2O2-activated caspase 3 (Fig. 5B).
FIGURE 5.

TWEAK-Fn14 signaling is required for mechanical stretch-induced cell death. A, MC3T3-E1 cells were stimulated with cyclic mechanical stretch (12%) for 6 h. After stretch stimulation, cells were incubated for the indicated periods. The cells were then lysed and analyzed by immunoblotting (IB) using the indicated antibodies. B, MC3T3-E1 cells were stimulated with cyclic mechanical stretch (12%) for 6 h. After stretch stimulation, cells were incubated for 12 h in the presence or absence of H2O2 with the indicated concentrations. The cells were then lysed and analyzed by immunoblotting using the indicated antibodies. C, MC3T3-E1 cells were transfected with the indicated expression plasmids. After 24 h, cells were lysed and analyzed by immunoblotting using the indicated antibodies. D, MC3T3-E1 cells were transfected with siRNA as a negative control or with siRNA for TWEAK (TWEAK #1 and TWEAK1 #2). After 72 h, the cells were subjected to cyclic stretch stimulation (12%) for 6 h, and then the cells were incubated for 12 h. The cell lysates were subjected to immunoblotting using the indicated antibodies. E, quantitative RT-PCR of TWEAK under the same conditions as in D. Error bars indicate mean ± S.E. *, p < 0.05, Student's t test. F, MC3T3-E1 cells were stimulated with cyclic mechanical stretch (12%) for 6 h. After stretch stimulation, cells were incubated for 12 h. The cells were then subjected to immunostaining with anti-active caspase 3 and TUNEL staining. A–D and F, the data represent one of at least three experiments. DIC, differential interference contrast.
We next examined the possibility that the TWEAK-Fn14 signaling pathway was involved in the apoptosis process using RNAi. Experiments with knockdown of Fn14 were not effective because of the fact that Fn14 was highly inducible by mechanical stress in MC3T3-E1 cells (data not shown). However, it has been reported that TWEAK and Fn14 have a one-on-one ligand-receptor relationship that and TWEAK does not cross-react with other TNFR family members (26). In addition, we observed that overexpression of Fn14WT but not Fn14D45A, a mutant that does not possess binding affinity for TWEAK (27), induced caspase 3 activation (Fig. 5C). These observations suggest that endogenous TWEAK is required for caspase 3 activation in the Fn14-expressing condition. Therefore, we examined the effect of RNAi for TWEAK on the mechanical stress-induced activation of caspase 3. As shown in Fig. 5D, activation of caspase 3 observed 12 h after stimulation was suppressed substantially by siRNA for TWEAK.
The expression level of TWEAK mRNA did not change significantly throughout the experimental period (Fig. 5E). However, the protein level of TWEAK was reduced after 12-h incubation (Fig. 5D), suggesting that the level of TWEAK is regulated posttranscriptionally. These observations also suggest that synchronized coexistence of Fn14 and TWEAK protein is essential for activation of caspase 3.
Fluorescent microscopic analysis revealed that these active caspase 3-positive cells were also TUNEL-positive, indicating that the double-positive cells were led to apoptosis (Fig. 5F). One of the unique characters of apoptotic cells is their rounded shape. It was observed that TUNEL and active caspase 3 double-positive cells had a unique rounded shape compared with those of TUNEL- and active caspase 3-negative cells (Fig. 5F, fourth and fifth rows). Moreover, the double-positive cells disappeared following RNAi against TWEAK (Fig. 5F). These observations indicate that TWEAK-Fn14 signaling, which is evoked by mechanical stretch, plays a critical role in stretch-induced cell death.
Immunoblot analysis showed that the Fn14 expression level decreased rapidly following the termination of stretch loading (Fig. 5, A and D), raising the possibility that Fn14-induced apoptosis is negatively regulated by degradation of Fn14 itself. A CHX-based protein chase assay showed that the decay of Fn14 protein following the termination of stretch loading was prevented by treatment of the cells with MG132, a proteasome inhibitor (Fig. 6A). These results suggest that ubiquitination-dependent proteasomal degradation was involved in the decay of Fn14 protein.
To identify the lysine residue(s) that is responsible for proteasomal degradation of Fn14, we prepared two Fn14 mutants, Fn14K48R and Fn14K109R, in which Lys-48 and Lys-109 were substituted by arginine, respectively (Fig. 6B). A CHX chase assay revealed that Fn14K109R, but not Fn14K48R, acquired resistance to degradation (Fig. 6C). Indeed, polyubiquitination of Fn14K109R was attenuated markedly compared with that of Fn14WT (Fig. 6D), indicating that Lys-109 is the critical site of polyubiquitination required for the proteasomal degradation of Fn14. Furthermore, overexpression of Fn14K109R activated caspase 3 more potently than Fn14WT (Fig. 6E). These observations indicate that the timing and duration of Fn14 expression is strictly controlled not only by transcriptional regulation but also by stability regulation, allowing cells to exhibit transient sensitivity to its ligand, TWEAK.
Cleavage and Secretion of MCP-3 Requires Seven N-terminal Amino Acids
We next investigated the mechanism of maturation of MCP-3, a downstream target gene of the ASK1-p38 pathway. It has been reported that MCP-3 has a signal peptide at its N terminus and that mature protein is produced after cleavage at Ala-23 (28). Bioinformatic analysis using SignalP 3.0 raised the possibility that cleavage at Ala-23 requires the sequence of the first seven amino acids. Therefore, we generated expression constructs of N-terminal, FLAG-tagged, wild-type MCP-3 (FLAG-MCP-3WT) and mutant MCP-3, which lacks the N-terminal seven amino acids (FLAG-MCP-3Δ7NT) (Fig. 7A). Cell lysates and conditioned medium from cells transfected with each constructs were subjected to immunoblotting, and MCP-3 was detected using anti-FLAG or anti-MCP-3 antibody (Fig. 7B). MCP-3 protein from cells transfected with FLAG-MCP-3WT was detected from both cell lysates and conditioned medium using anti-MCP-3 antibody. Secreted MCP-3 isolated from medium had a lower molecular weight than full-length MCP-3 isolated from cell lysates. In addition, this secreted form of MCP-3 was not detected by anti-FLAG antibody. In contrast, FLAG-MCP-3Δ7NT was detected in the cell lysates by both anti-FLAG and anti-MCP-3 antibodies but not in the conditioned medium. These results suggest that cleavage and secretion of MCP-3 require the seven N-terminal amino acids. Moreover, full-length MCP-3 and the cleaved form of MCP-3 were not detected in conditioned medium and cell lysates, respectively, suggesting that proteolytic secretion is performed on the surface of the plasma membrane, which is termed shedding.
FIGURE 7.
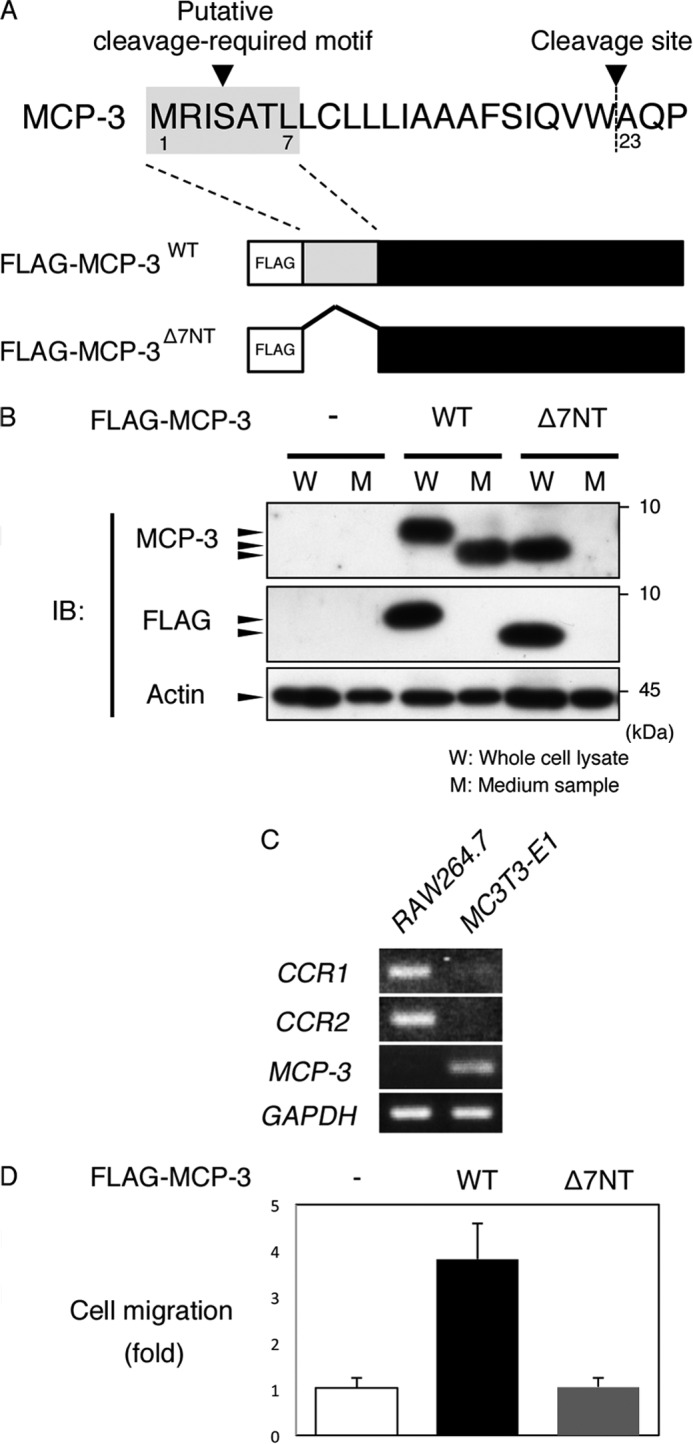
N-terminal seven amino acid-mediated cleavage and secretion is required for the function of MCP-3. A, amino acid sequence of the N-terminal region of mouse MCP-3 and schematic of FLAG-MCP-3WT and FLAG-MCP-3Δ7NT. B, MC3T3-E1 cells were transfected with the indicated expression plasmids. After 12 h, the medium was replaced with serum-free fresh medium and incubated for 12 h. Then, the conditioned medium and cell lysate were analyzed by immunoblotting (IB) using the indicated antibodies. C, RT-PCR analysis of CCR1, CCR2, MCP-3, and GAPDH in RAW264.7 preosteoclasts and MC3T3-E1 osteoblasts. D, MC3T3-E1 cells were transfected with the indicated expression plasmids. After 12 h, the medium was replaced with serum-free fresh medium and incubated for 12 h. A RAW264.7 chemotaxis assay was performed using a Corning transwell unit. See “Experimental Procedures” for details.
We next compared the chemotaxis-promoting activity of the conditioned medium from FLAG-MCP-3WT-expressing cells with that of FLAG-MCP-3Δ7NT-expressing cells. Major receptors of MCP-3, C-C chemokine receptor (CCR) 1 and 2, were strongly expressed in RAW264.7 preosteoclasts but were expressed very little in MC3T3-E1 osteoblasts (Fig. 7C). A chemotaxis assay using a transwell unit showed that conditioned medium from FLAG-MCP-3WT-transfected cells induced chemotaxis of RAW264.7 preosteoclasts 3.8-fold above that of control cells (Fig. 7D). This chemotaxis-inducing activity was abolished in the conditioned medium from FLAG-MCP-3Δ7NT-transfected cells. These results indicate that seven N-terminal amino acid-mediated cleavage and secretion is required for the activity of MCP-3.
DISCUSSION
It has been established that osteoblasts play a key role in adequate mechanical stress-induced osteogenesis (2, 4, 12). However, it has been largely unknown how osteoblasts behave in excessive mechanical stress-induced bone loss. In this study, loading with small-magnitude mechanical stretch (1% cyclic stretch) induced expression of Col1a and OPN via the ERK pathway in MC3T3-E1cells and mouse primary osteoblasts (Fig. 1). In contrast, not only ERK but also JNK and p38 were activated by large-magnitude mechanical stretch (12% cyclic stretch). However, Col1a and OPN expression was not enhanced in this case (Fig. 1). Furthermore, inhibiting the kinase activity of JNK and p38 resulted in the recovery of Col1a and OPN expression. These results suggest that ERK is responsible for the induction of osteoblast differentiation when mechanical stretch is loaded on the cells and that JNK and p38 activated by large-magnitude mechanical stress act as gatekeepers that prohibit bone formation in mechanical stress-overloaded cells.
We identified ASK1 as the MAPK3K responsible for the activation of the JNK and p38 pathways in cells loaded with large-magnitude stretch and revealed that ASK1 was activated by mechanical stretch-induced Ca2+ influx and subsequent ROS generation (Figs. 2 and 3). We have reported previously that JNK and p38 were also activated via the Ca2+-CaMKII-TAK1 pathway in mechanical stretch-loaded cells (19). In this study, we observed that TAK1 activation was ROS-independent (Fig. 3). These observations indicate that both ROS-dependent and ROS-independent activation of JNK and p38 takes place in cells loaded with mechanical stretch.
We observed de novo expression of Fn14 via mechanical stretch-activated ASK1-JNK1/2 (Fig. 4). Fn14 is an immediate early response gene and has putative binding sites for not only AP-1 (−1812TGACGTCA−1819) but also NF-κB (−2199AGGGAGTCCC−2208) in its promoter region. We have already reported that NF-κB is also activated by the mechanical stress-activated TAK1 signaling pathway (19). These results raise the possibility that activation of both the ASK1 and TAK1 signaling pathways contributes to inducing the expression of Fn14 by mechanical stretch.
Mechanical stretch-induced Fn14 expression was suppressed by RNAi or kinase activity inhibition of JNK (Fig. 4). Conversely, inhibition of the kinase activity of p38 or expression of p38 by RNAi enhanced the expression of Fn14. Because JNK activity was enhanced by RNAi against p38 (Fig. 1) or SB203580 (data not shown), we suggest that p38 suppresses JNK activity and that release of this suppression leads to enhanced expression of Fn14. Therefore, the expression levels of Fn14 in stretch-loaded cells may be controlled, in part, by the balance of activities between JNK and p38.
Fn14 binds to its ligand, TWEAK, and regulates proliferation, differentiation, and apoptosis (25). Fn14 signaling in osteoblasts has been reported to provide antiosteogenic functions such as suppression of differentiation, enhanced RANKL expression, and sclerostin expression (29, 30). We observed that large-magnitude mechanical stretch induced caspase 3 activation (Fig. 5). In addition, although overexpression of Fn14WT induced caspase 3 activation, this was not the case for Fn14D45A, a mutant that does not possess a binding affinity for TWEAK (27) (Fig. 5). These findings suggest that apoptosis induction by endogenous TWEAK is dependent on the expression of Fn14 protein. Indeed, an analysis using RNAi showed that knockdown of TWEAK resulted in a decrease in active caspase 3 and TUNEL double-positive cells (Fig. 5). These results indicate that TWEAK/Fn14 signaling is required for large-magnitude mechanical stretch-induced apoptosis.
We observed that Fn14 protein levels rapidly decreased when stretch application was terminated following continuous loading for 6 h and that this protein decay was due to proteasomal degradation caused by polyubiquitination at Lys-109 (Fig. 6). These results suggest that the expression of Fn14 protein is induced and maintained while the cells are exposed to strong mechanical stress. We propose that this system of regulating Fn14 expression enables cells to acquire a sensitivity to TWEAK only while damaging mechanical stress is applied and that this, subsequently, leads to cell apoptosis. Apoptosis of osteoblasts plays an important role during skeletal development and bone remodeling (24, 31, 32). Bone morphogenetic protein has been reported to mediate apoptosis independently of its ability to induce differentiation (33). Intriguingly, we found that bone morphogenetic protein as well as the inflammatory cytokine TNF-α induced Fn14 expression, which is suppressed by treatment with the JNK inhibitor (data not shown). These results suggest that the JNK-Fn14 axis, which is activated not only by large-magnitude mechanical stress but also by other Fn14-inducing factors, works as a stress-monitoring signal cassette and eliminates deeply damaged cells by apoptosis.
We observed that the large-magnitude cyclic stretch-activated ASK1-p38 pathway induced expression of the MCP-3 gene (Fig. 4). MCP-3 has been reported to promote chemotactic recruitment of preosteoclasts (34), which was confirmed in this study (Fig. 7). CCR1, CCR2, CCR3, and CCR5 are identified as receptors for MCP-3 (28). Of these genes, CCR2−/− mice have been shown recently to exhibit increased bone mass because of reduced osteoclast numbers derived from decreased expression of RANK on the surface of CCR2−/− preosteoclasts (35). It has been reported that CCR1−/− preosteoclasts exhibit disorder in multinucleation and bone-resorbing activity (36). We also confirmed that CCR1 and CCR2 expression was observed strongly in RAW264.7 cells but little in MC3T3-E1 cells (Fig. 7). These observations suggest that large mechanical stress-induced MCP-3 expression by osteoblasts is involved in local recruitment of preosteoclasts and subsequent osteoclastogenesis.
It has been hypothesized in a mechanostat theory that bone has a mechanosensor that is responsible for shifting the balance of formation and resorption of bone to an altered equilibrium state in response to mechanical stress (37). Cell matrix stretch, fluid shear, and intercellular adhesion have been thought to be important factors of mechanosensing in vivo (38, 39). However, because it seemed difficult to set up an in vitro experimental system which fulfills cooperation of all these factors at the same time, we used cyclic stretch as a model system for matrix stretch in our experiments. We employed 12% stretch as a model for large-magnitude mechanical stress. However, we are not able to rule out the possibility that synergistic cooperation of these factors amplifies small-magnitude mechanical stress by each factor into strong intracellular signaling cascades in vivo.
Moderate mechanical stress is required to maintain a metabolic balance in osteogenic-dominant bone remodeling. From clinical findings, it has been generally known that excessive mechanical stress induces bone resorption. However, the molecular index that distinguishes between moderate and excessive mechanical stress has not been defined experimentally. In this study, we showed that ERK activated by small-magnitude mechanical stress contributes to osteoblast differentiation. On the other hand, JNK activated by large-magnitude mechanical stress not only suppresses differentiation but also drives strongly damaged cells to undergo apoptosis. In addition, p38 that was activated by large-magnitude mechanical stress induced production of MCP-3, an osteoclastic chemokine (Fig. 8). These findings suggest that the magnitudes and modes of mechanical stress that are required for osteogenic-dominant bone remodeling lie between those that activate ERK and do not activate JNK and p38.
FIGURE 8.
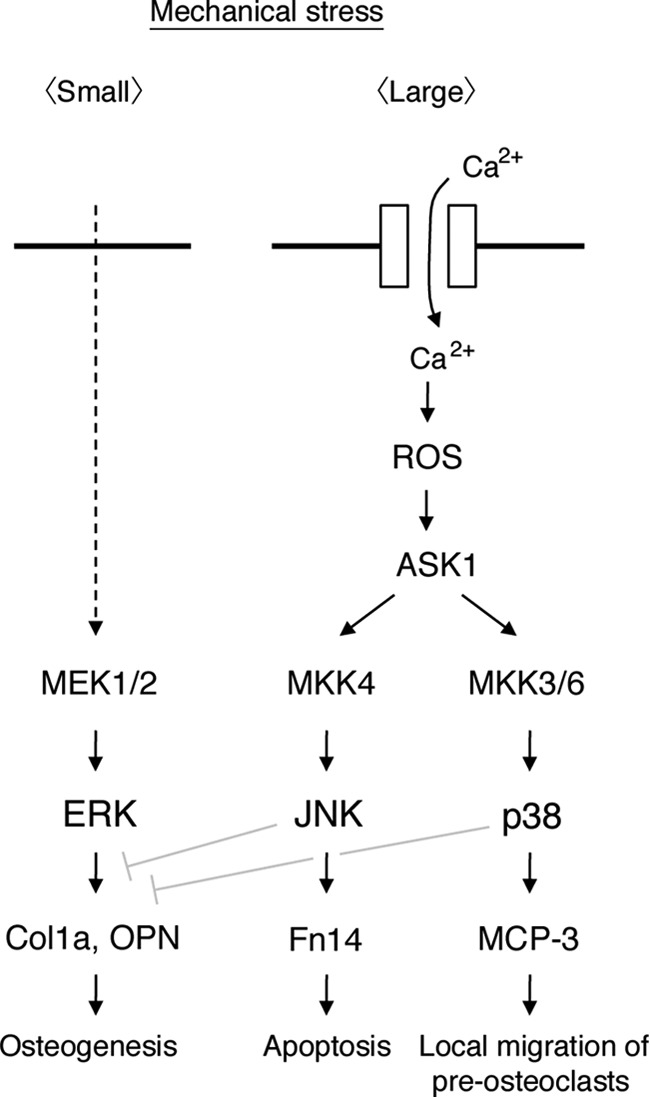
Proposed signaling model for mechanical stretch-induced MAPK activation. See “Discussion” for details.
Acknowledgments
We thank the members of the Department of Biochemistry, Institute of Development, Aging and Cancer, and Advanced Prosthetic Dentistry, Graduate School of Dentistry, Tohoku University, for discussions. We also thank Dr. Hisanori Horiuchi, Department of Molecular and Cellular Biology, Institute of Development, Aging and Cancer, Tohoku University, for generous support during the course of this study and Naomi Hoshi, Mami Shoji, Junko Hagawa, and Yuki Sato for office administrative work.
This work was supported by a grant-in-aid for Scientific Research from the Japan Society for Promotion of Science and Ministry of Education, Culture, Sports, Science and Technology of Japan.
- RANKL
- receptor activator of nuclear factor κB ligand
- MAP3K
- MAPK kinase kinase
- NF-κB
- nuclear factor κB
- TWEAK
- TNF-related weak inducer of apoptosis
- NAC
- N-acetyl-l-cysteine
- CHX
- cycloheximide
- OPN
- osteopontin
- CCR
- C-C chemokine receptor.
REFERENCES
- 1. Takayanagi H. (2007) Osteoimmunology. Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 7, 292–304 [DOI] [PubMed] [Google Scholar]
- 2. Zaidi M. (2007) Skeletal remodeling in health and disease. Nat. Med. 13, 791–801 [DOI] [PubMed] [Google Scholar]
- 3. Ehrlich P. J., Lanyon L. E. (2002) Mechanical strain and bone cell function. A review. Osteoporos. Int. 13, 688–700 [DOI] [PubMed] [Google Scholar]
- 4. Morey E. R., Baylink D. J. (1978) Inhibition of bone formation during space flight. Science 201, 1138–1141 [DOI] [PubMed] [Google Scholar]
- 5. Fu J. H., Hsu Y. T., Wang H. L. (2012) Identifying occlusal overload and how to deal with it to avoid marginal bone loss around implants. Eur. J. Oral Implantol. 5, S91–103 [PubMed] [Google Scholar]
- 6. Loboa E. G., Beaupré G. S., Carter D. R. (2001) Mechanobiology of initial pseudarthrosis formation with oblique fractures. J. Orthop. Res. 19, 1067–1072 [DOI] [PubMed] [Google Scholar]
- 7. Menini M., Signori A., Tealdo T., Bevilacqua M., Pera F., Ravera G., Pera P. (2012) Tilted implants in the immediate loading rehabilitation of the maxilla. A systematic review. J. Dent. Res. 91, 821–827 [DOI] [PubMed] [Google Scholar]
- 8. Suenaga H., Yokoyama M., Yamaguchi K., Sasaki K. (2012) Time course of bone metabolism at the residual ridge beneath dentures observed using (18)F-fluoride positron emission computerized-tomography/computed tomography (PET/CT). Ann. Nucl. Med. 26, 817–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-Hora M., Feng J. Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F., Penninger J. M., Takayanagi H. (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 [DOI] [PubMed] [Google Scholar]
- 10. Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., O'Brien C. A. (2011) Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moriishi T., Fukuyama R., Ito M., Miyazaki T., Maeno T., Kawai Y., Komori H., Komori T. (2012) Osteocyte network. A negative regulatory system for bone mass augmented by the induction of Rankl in osteoblasts and Sost in osteocytes at unloading. PloS ONE 7, e40143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tatsumi S., Ishii K., Amizuka N., Li M., Kobayashi T., Kohno K., Ito M., Takeshita S., Ikeda K. (2007) Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 5, 464–475 [DOI] [PubMed] [Google Scholar]
- 13. Matsumoto T., Iimura T., Ogura K., Moriyama K., Yamaguchi A. (2013) The role of osteocytes in bone resorption during orthodontic tooth movement. J. Dent. Res. 92, 340–345 [DOI] [PubMed] [Google Scholar]
- 14. Goga Y., Chiba M., Shimizu Y., Mitani H. (2006) Compressive force induces osteoblast apoptosis via caspase-8. J. Dent. Res. 85, 240–244 [DOI] [PubMed] [Google Scholar]
- 15. Takeda K., Naguro I., Nishitoh H., Matsuzawa A., Ichijo H. (2011) Apoptosis signaling kinases. From stress response to health outcomes. Antioxid. Redox Signal. 15, 719–761 [DOI] [PubMed] [Google Scholar]
- 16. Kanno T., Takahashi T., Tsujisawa T., Ariyoshi W., Nishihara T. (2007) Mechanical stress-mediated Runx2 activation is dependent on Ras/ERK1/2 MAPK signaling in osteoblasts. J. Cell. Biochem. 101, 1266–1277 [DOI] [PubMed] [Google Scholar]
- 17. Matsuguchi T., Chiba N., Bandow K., Kakimoto K., Masuda A., Ohnishi T. (2009) JNK activity is essential for Atf4 expression and late-stage osteoblast differentiation. J. Bone Miner. Res. 24, 398–410 [DOI] [PubMed] [Google Scholar]
- 18. Greenblatt M. B., Shim J. H., Zou W., Sitara D., Schweitzer M., Hu D., Lotinun S., Sano Y., Baron R., Park J. M., Arthur S., Xie M., Schneider M. D., Zhai B., Gygi S., Davis R., Glimcher L. H. (2010) The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Invest. 120, 2457–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fukuno N., Matsui H., Kanda Y., Suzuki O., Matsumoto K., Sasaki K., Kobayashi T., Tamura S. (2011) TGF-β-activated kinase 1 mediates mechanical stress-induced IL-6 expression in osteoblasts. Biochem. Biophys. Res. Commun. 408, 202–207 [DOI] [PubMed] [Google Scholar]
- 20. Kapoor M., Martel-Pelletier J., Lajeunesse D., Pelletier J. P., Fahmi H. (2011) Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7, 33–42 [DOI] [PubMed] [Google Scholar]
- 21. Tobiume K., Saitoh M., Ichijo H. (2002) Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J. Cell. Physiol. 191, 95–104 [DOI] [PubMed] [Google Scholar]
- 22. Jeon Y. M., Kook S. H., Son Y. O., Kim E. M., Park S. S., Kim J. G., Lee J. C. (2009) Role of MAPK in mechanical force-induced up-regulation of type I collagen and osteopontin in human gingival fibroblasts. Mol. Cell. Biochem. 320, 45–52 [DOI] [PubMed] [Google Scholar]
- 23. Zhou H., Newnum A. B., Martin J. R., Li P., Nelson M. T., Moh A., Fu X. Y., Yokota H., Li J. (2011) Osteoblast/osteocyte-specific inactivation of Stat3 decreases load-driven bone formation and accumulates reactive oxygen species. Bone 49, 404–411 [DOI] [PubMed] [Google Scholar]
- 24. Ambrogini E., Almeida M., Martin-Millan M., Paik J. H., Depinho R. A., Han L., Goellner J., Weinstein R. S., Jilka R. L., O'Brien C. A., Manolagas S. C. (2010) FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 11, 136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winkles J. A. (2008) The TWEAK-Fn14 cytokine-receptor axis. Discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 7, 411–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bossen C., Ingold K., Tardivel A., Bodmer J. L., Gaide O., Hertig S., Ambrose C., Tschopp J., Schneider P. (2006) Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J. Biol. Chem. 281, 13964–13971 [DOI] [PubMed] [Google Scholar]
- 27. Brown S. A., Hanscom H. N., Vu H., Brew S. A., Winkles J. A. (2006) TWEAK binding to the Fn14 cysteine-rich domain depends on charged residues located in both the A1 and D2 modules. Biochem. J. 397, 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Menten P., Wuyts A., Van Damme J. (2001) Monocyte chemotactic protein-3. Eur. Cytokine Netw. 12, 554–560 [PubMed] [Google Scholar]
- 29. Ando T., Ichikawa J., Wako M., Hatsushika K., Watanabe Y., Sakuma M., Tasaka K., Ogawa H., Hamada Y., Yagita H., Nakao A. (2006) TWEAK/Fn14 interaction regulates RANTES production, BMP-2-induced differentiation, and RANKL expression in mouse osteoblastic MC3T3-E1 cells. Arthritis Res. Ther. 8, R146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vincent C., Findlay D. M., Welldon K. J., Wijenayaka A. R., Zheng T. S., Haynes D. R., Fazzalari N. L., Evdokiou A., Atkins G. J. (2009) Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFα induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J. Bone Miner. Res. 24, 1434–1449 [DOI] [PubMed] [Google Scholar]
- 31. Jilka R. L., Weinstein R. S., Parfitt A. M., Manolagas S. C. (2007) Quantifying osteoblast and osteocyte apoptosis. Challenges and rewards. J. Bone Miner. Res. 22, 1492–1501 [DOI] [PubMed] [Google Scholar]
- 32. Miura M., Chen X. D., Allen M. R., Bi Y., Gronthos S., Seo B. M., Lakhani S., Flavell R. A., Feng X. H., Robey P. G., Young M., Shi S. (2004) A crucial role of caspase-3 in osteogenic differentiation of bone marrow stromal stem cells. J. Clin. Invest. 114, 1704–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haÿ E., Lemonnier J., Fromigué O., Guénou H., Marie P. J. (2004) Bone morphogenetic protein receptor IB signaling mediates apoptosis independently of differentiation in osteoblastic cells. J. Biol. Chem. 279, 1650–1658 [DOI] [PubMed] [Google Scholar]
- 34. Yu X., Huang Y., Collin-Osdoby P., Osdoby P. (2004) CCR1 chemokines promote the chemotactic recruitment, RANKL development, and motility of osteoclasts and are induced by inflammatory cytokines in osteoblasts. J. Bone Miner. Res. 19, 2065–2077 [DOI] [PubMed] [Google Scholar]
- 35. Binder N. B., Niederreiter B., Hoffmann O., Stange R., Pap T., Stulnig T. M., Mack M., Erben R. G., Smolen J. S., Redlich K. (2009) Estrogen-dependent and C-C chemokine receptor-2-dependent pathways determine osteoclast behavior in osteoporosis. Nat. Med. 15, 417–424 [DOI] [PubMed] [Google Scholar]
- 36. Hoshino A., Iimura T., Ueha S., Hanada S., Maruoka Y., Mayahara M., Suzuki K., Imai T., Ito M., Manome Y., Yasuhara M., Kirino T., Yamaguchi A., Matsushima K., Yamamoto K. (2010) Deficiency of chemokine receptor CCR1 causes osteopenia due to impaired functions of osteoclasts and osteoblasts. J. Biol. Chem. 285, 28826–28837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frost H. M. (1987) Bone “mass” and the “mechanostat.” A proposal. Anat. Rec. 219, 1–9 [DOI] [PubMed] [Google Scholar]
- 38. Heisenberg C. P., Bellaïche Y. (2013) Forces in tissue morphogenesis and patterning. Cell 153, 948–962 [DOI] [PubMed] [Google Scholar]
- 39. Ross T. D., Coon B. G., Yun S., Baeyens N., Tanaka K., Ouyang M., Schwartz M. A. (2013) Integrins in mechanotransduction. Curr. Opin. Cell Biol. 25, 613–618 [DOI] [PMC free article] [PubMed] [Google Scholar]