

Background: Ventral specification is regulated by Smad1-, Smad5-, and/or Smad9-mediated bone morphogenetic protein (BMP) signaling.
Results: Double knockdown instead of single knockdown of smad1 and smad9 induces dorsalization, which cannot be rescued by smad5 overexpression.
Conclusion: smad1 and smad9 act redundantly and downstream of smad5 to mediate ventral specification.
Significance: The regulation network and cooperative roles of BMP R-Smads in early development are clarified.
Keywords: Bone Morphogenetic Protein (BMP), Development, Gene Regulation, SMAD Transcription Factor, Zebrafish, Dorso-ventral Patterning, Myelopoiesis
Abstract
Bone morphogenetic proteins (BMPs) are multifunctional growth factors that play crucial roles during embryonic development and cell fate determination. Nuclear transduction of BMP signals requires the receptor type Smad proteins, Smad1, Smad5, and Smad9. However, how these Smad proteins cooperate in vivo to regulate various developmental processes is largely unknown. In zebrafish, it was widely believed that the maternally expressed smad5 is essential for dorso-ventral (DV) patterning, and the zygotically transcribed smad1 is not required for normal DV axis establishment. In the present study, we have identified zygotically expressed smad9, which cooperates with smad1 downstream of smad5, to mediate zebrafish early DV patterning in a functional redundant manner. Although knockdown of smad1 or smad9 alone does not lead to visible dorsalization, double knockdown strongly dorsalizes zebrafish embryos, which cannot be efficiently rescued by smad5 overexpression, whereas the dorsalization induced by smad5 knockdown can be fully rescued by overexpression of smad1 or smad9. We have further revealed that the transcription initiations of smad1 and smad9 are repressed by each other, that they are direct transcriptional targets of Smad5, and that smad9, like smad1, is required for myelopoiesis. In conclusion, our study uncovers that smad1 and smad9 act redundantly to each other downstream of smad5 to mediate ventral specification and to regulate embryonic myelopoiesis.
Introduction
The transforming growth factor β (TGF-β) family plays crucial roles in regulating diverse cellular and developmental processes, such as cell proliferation, differentiation, and migration and embryonic pattern formation. Signaling by members of the TGF-β superfamily is transduced by Smad proteins, which are classified into three subfamilies: the receptor-regulated Smads (R-Smads),2 the common mediator Smad (Co-Smad), and the inhibitory Smad (I-Smad) (1, 2). Among the R-Smad subfamily members, Smad2 and Smad3 mediate the TGF-β/activin pathways, whereas Smad1, Smad5, and Smad9 (also known as Smad8) transduce the bone morphogenetic protein (BMP) pathway (3, 4). BMPs consist of the largest subgroup within the TGF-β superfamily. In early development of vertebrates, BMPs are required for the dorso-ventral (DV) axis formation during gastrulation and embryonic hematopoiesis (5, 6).
The three BMP R-Smads share an extremely high degree of homology, and their cellular function is transducing BMP signaling. Consistent with this notion, much evidence shows that they have overlapping functions during development. In zebrafish and Xenopus, misexpression of either smad1 or smad5 in the embryo induces ventral fates (7–9). In chicken, the functions of Smad1 and Smad5 are largely interchangeable during spinal cord neurogenesis (10). In mammals, Smad1, Smad5, and Smad9 are believed to function redundantly in transducing the anti-Mullerian hormone signal and mediating regression of the Mullerian duct in males (11). In addition, Smad1 and Smad5 function redundantly in the apical ectodermal ridge and ventral ectoderm (12), in endochondral bone formation and in controlling tumor cell migration to distant locations (13, 14).
However, accumulating evidence suggests that BMP R-Smads are not always interchangeable in development. In zebrafish, smad1 and smad5 were believed to have distinct functions in regulating early DV patterning, and the previous results from loss-of-function and gain-of-function studies even led to controversial conclusions. For instance, smad5 mutants (such as sbn−/−) or morphants are strongly dorsalized (15–18), demonstrating that smad5 is essentially required for early DV patterning. In contrast, effective loss of function of smad1 does not cause any phenotype of dorsalization (19), although overexpression of smad1 strongly ventralizes zebrafish embryos as does smad5 overexpression (9). This implies that smad1 itself is not essential for zebrafish DV axis establishment. On the other hand, overexpression of smad1 rather than smad5 leads to complete rescue of zebrafish mutants of bmp2b (9), suggesting that smad1 might be a direct mediator for ventral specification in the bmp2b null condition. Ectopic overexpression of smad5 just after midblastula transition can rescue the sbn−/− dorsalized embryos, whereas no rescue effects could be observed if the exogenous smad5 is forced to express only during gastrulation (17). Besides, the expression of non-neuroectoderm-specific gata2 is mediated by Smad5 and completely independent of Smad1 before the gastrula stage, whereas at later stages, its expression is dependent upon Smad1 instead of Smad5 (20). These findings suggest that Smad5 acts before gastrulation and is not required during gastrulation in DV patterning. Nevertheless, how other non-Smad5 BMP R-Smads contribute to the patterning of DV axis in zebrafish remains largely unknown. Also, smad1 and smad5 are shown to play distinct roles in embryonic hematopoiesis in zebrafish (19).
In this study, we focus on how smad1, smad5, and smad9 are differentially regulated and how they cooperatively contribute to early development of zebrafish. We identify a zygotically expressed smad9 (previously named smad8) in the zebrafish genome. The tempo-spatial expression pattern of smad9 during embryogenesis is similar to that of smad1. Although the smad9 or smad1 knocked down embryos are not dorsalized, double knockdown of smad1 and smad9 leads to strong dorsalization, which cannot be rescued by smad5 overexpression. By contrast, overexpression of either smad1 or smad9 can fully rescue the dorsalized defects in smad5 morphants. Moreover, the transcriptional onsets of smad1 and smad9 are suppressed by each other, and Smad5 binds to the promoter regions and activates the promoter activities of smad1 and smad9. Altogether, our study reveals that smad1 and smad9 act redundantly and downstream of smad5 in regulating zebrafish DV patterning.
EXPERIMENTAL PROCEDURES
Animals
Zebrafish (Danio rerio) of the AB strain were raised in the China Zebrafish Resource Center (CZRC, Wuhan, China), maintained according to the Zebrafish Book (21), and staged as previously described (22). The experiments involving zebrafish were performed under the approval of the Institutional Animal Care and Use Committee of the Institute of Hydrobiology, Chinese Academy of Sciences.
Molecular Cloning and Construct Generation
According to the GenBankTM accession number NM_001004014, full-length zebrafish smad9 cDNA was amplified from the gastrula cDNA pool by reverse transcription PCR (RT-PCR), using a set of primers, 5′-GGAATTCGAAACAACCCGAATCTCCTG-3′ (P1) and 5′-CCGCTCGAGGTGTCTTGCGTGGCTATGAAG-3′ (P2). The PCR products were cloned into the EcoRI-XhoI sites of the pCS2+ expression vector and subjected to sequencing. To generate a Smad9-EGFP fusion protein expression construct, smad9 cDNA was amplified using P1 and 5′-GGGGTACCTTGGACACCGAGGAAATGGGG-3′ (P3) and cloned into the EcoRI-KpnI sites of the pEGFPN1 vector (Clontech). To generate Smad9-EGFP fusion expression construct that lacks the smad9 morpholino (MO) binding site, 5′-GGAATTCATGCACTCCTCTACCTCCATC-3′ (P4) and P3 were used for PCR amplification, and the PCR products were cloned into the pEGFPN1. To generate a smad5 expression construct in which the smad5 MO binding site is mutated, 5′-CGGGATCCATGACAAGTATGAGCTCCTTATTTTCCTTCACCAGCCCG-3′ and T3 primer (5′-ATTAACCCTCACTAAAGGGA-3′) were used to amplify mutated smad5 from pCS2+_zsmad5, and the PCR products were subcloned into the BamHI-EcoRI sites of pCS2+. The coding sequences of smad1, smad4, smad5, and smad9 were amplified and subcloned into pHAM, pCMVTag2B, and pCMVTag3C constructs to generate HA-, FLAG-, and Myc-tagged fusion constructs. The putative promoter region of smad1 was amplified from the zebrafish genomic DNA using the primers 5′-TTTTACCTTCAGAACTGCCTTAATCCATC-3′ (P5) and 5′-GAGCCATTCACAAACGTGTCAGTAGTAATCTCA-3′ (P6), and that of smad9 was amplified with 5′-GCAACTATTAAGAAAACATTGCACGGAT-3′ (P7) and 5′-AACTTACTTATGTGGTTGAAAACGCTCC-3′ (P8). The PCR products were cloned into pGL3-Basic (Promega) to generate constructs for the luciferase assay.
MO and mRNA Injections
Antisense MOs (Gene Tools, LLC) were designed to complement the translation start sites of smad9 (smad9 MO, 5′-TCGTGAGACGGGTTGATTTTAAATC-3′) and smad1 (smad1 MO, 5′-GGAAAAGAGTGAGGTGACATTCATT-3′). smad1 MO has only one base shift compared with the previously published smad1 MO (19). MO used for knockdown of smad5 was as described previously (16). To make synthetic capped RNA encoding smad1, smad5, smad9, bmp2b, bmp4, bmp7, and dominant negative BMP receptor 1a (DNBR1a), pCS2+-based constructs containing the corresponding cDNA were linearized by NotI, and mRNA was synthesized using the sp6 mMessage mMachine kit (Ambion). For microinjection, embryos were injected with mRNA and morpholinos of interest at the 1-cell stage as described previously (23).
RNA Isolation, Semiquantitative RT-PCR, and Quantitative RT-PCR
Total RNA was extracted from embryos and organs with TRIzol reagent following the manufacturer's manual (Invitrogen). 1 μg of total RNA was used to generate cDNA with first strand Moloney murine leukemia virus reverse transcriptase (Invitrogen) and oligo(dT)20 RT primer. The cDNA was then used in a polymerase chain reaction (PCR) accordingly: 95 °C for 5 min; 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 40 s for specific cycles; and 72 °C for 10 min. Primers and amplification cycles for each primer pair used were as follows: smad1, 5′-AGAGGTGTATGCCGAATGTTTG-3′ (forward) and 5′-CCATCTGGGTGAGGACTTTATC-3′ (reverse), 26 cycles; smad5, 5′-AAAACACCCGTCGCCACATC-3′ (forward) and 5′-AGCCCATCATTACGAGACAGAA-3′ (reverse), 22 cycles; and smad9, 5′-GAAGGCTCCAGGTGTCTCAT-3′ (forward) and 5′-GAAGCGGTTCTTGTTGTTTG-3′ (reverse), 26 cycles. An ef1a-specific primer pair (forward, 5′-TCACCCTGGGAGTGAAACAGC-3′; reverse, 5′-ACTTGCAGGCGATGTGAGCAG-3′) was used for PCR as an internal control (24). For quantitative PCR, the amplification was performed on a CFX96TM real-time PCR detection system (Bio-Rad) according to the manufacturer's instructions. Primers for smad5 were 5′-CTTTGAGGCCGTCTATGAGC-3′ (forward) and 5′-GGGTGCTTGTCACATCTTGT-3′ (reverse). A pair of β-actin primers (forward, 5′-CGAGCAGGAGATGGGAACC-3′; reverse, 5′-CAACGGAAACGCTCATTGC-3′) was used as the internal control (25).
Luciferase Reporter Assay
To analyze the putative promoter activities of smad1 and smad9 in different genetic background, the relative luciferase activities were determined by the Dual-Luciferase reporter assay system as described previously (26). Each embryo was coinjected with smad1 promoter-luc or smad9 promoter-luc (described previously) and a constitutively expressed TK-Renilla luciferase construct (Promega). These embryos were subsequently injected with the indicated RNA or morpholino samples. When the embryos developed to shield stage, sets of 30 embryos were lysed, the luciferase activity was measured with a Promega luminometer, and relative luciferase activity was calculated as described (27). Assays were performed in triplicates, and the average values and the standard variations were calculated. A p value less than 0.05 was considered statistically significant.
Cycloheximide Treatment
Embryos were cultured in fish medium containing 50 μg/ml protein synthesis inhibitor cycloheximide (CHX) from 2 h postfertilization (hpf) until the embryos were fixed for whole-mount in situ hybridization (28).
Whole-mount in Situ Hybridization
Whole-mount in situ hybridization (WISH) was performed using digoxigenin (DIG)-labeled antisense RNA probes and anti-DIG alkaline phosphatase-conjugated antibody as described (29). pCS2+ vectors containing smad1, smad5, and smad9 were digested with HindIII, and the antisense RNA probes were synthesized with T7 RNA polymerase (Promega). Molecular markers for analyzing DV patterning were cyp26a (30), foxi1 (31), sizzled (32), and ved (33). Antisense probes of l-plastin and mpx (34–36) were used to analyze the myelopoiesis in zebrafish development.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed with a ChIP assay kit (Upstate Biotechnology) as described with some modifications (37). Briefly, 2000 embryos were injected with 300 pg/embryo HA-smad5 mRNA or HA-sv40 mRNA. Immunoprecipitation was carried out using anti-HA antibody (Santa Cruz Biotechnology, Inc.). The relative amounts of smad1 or smad9 upstream region in immunoprecipitated chromatin and input control were measured using quantitative PCR described previously. The primers specific for the smad1 upstream region were 5′-AAACATGCACTCTAGCCTTCG-3′ (forward) and 5′-GTAGCCAACTCAATCTGGGAC-3′ (reverse). For the smad9 promoter region, primers were 5′-GAGTATTGTTACGTTCCCCTGCAG-3′ (forward) and 5′-CGCGGATGGATACACTTCGA-3′ (reverse). The exon of ribosomal protein rpl5b was served as a negative control, and the primers were 5′-GGGGATGAGTTCAATGTGGAG-3′ (forward) and 5′-CGAACACCTTATTGCCAGTAG-3′ (reverse) as described (38).
Cell Culture, Transfection, Immunoblotting, and Co-immunoprecipitation
HEK293T cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) plus 10% fetal bovine serum (Hyclone). DNA transfection into HEK293T cells was performed using Vigofect (Vigorous). Immunoprecipitation and immunoblotting were performed as described (39). The FLAG-tagged proteins were immunoprecipitated from cell lysates using agarose-conjugated anti-FLAG antibody (Santa Cruz Biotechnology). Antibodies against HA tag, FLAG tag, and Myc tag were purchased from Sigma-Aldrich and Santa Cruz Biotechnology.
RESULTS
smad1 Genetically Interacts with smad5 in Zebrafish DV Patterning
Previous studies have shown that, although ectopic expression of smad1 led to strong ventralization of zebrafish (9, 40), knockdown of smad1 did not give an obvious dorsalization phenotype (19). This is distinct from zebrafish smad5, whose gain of function ventralized the embryos and whose loss of function strongly dorsalized the embryos (17, 18). Because both Smad1 and Smad5 are widely believed to be the major transducers of the ventralizing BMP signals, we further asked whether smad1 carries ventralization activity intrinsically and whether smad1 genetically interacts with smad5 in DV patterning. First of all, we injected in vitro transcribed enhanced green fluorescent protein (egfp) mRNA into 1-cell stage zebrafish embryos. These embryos developed normally and showed normal expression of DV patterning-related genes, so we used uninjected embryos as controls for subsequent analysis. As shown in Fig. 1B, when smad1 mRNA was injected into zebrafish zygotes, the injected embryos were strongly ventralized, as characterized by an expansion of ventral posterior tissues at the expense of dorsal anterior tissues (41). According to the previous study (5), the ventralized embryos at 30 hpf were divided into three classes, V1–V3. Injection of 300 pg of smad1 mRNA per embryo caused ventralized phenotypes in 74% of the embryos, which were 47% V1 (showing slightly increased ventral tail fin and enlarged blood island), 20% V2 (showing reduced or absent notochord but remained head structures), and 7% V3 (showing absent notochord and anterior structures as well as expanded posterior). These results strongly suggest that zebrafish smad1 carries ventralization activity.
FIGURE 1.

smad1 genetically interacts with smad5 in DV patterning. A, an uninjected control embryo. B, a typical ventralized embryo that was injected with smad1 mRNA (300 pg/embryo). C, a typical smad1 morphant that was not dorsalized (2.4 ng/embryo). D, a typical dorsalized embryo that was injected with smad5 MO (1.6 ng/embryo). E, a typical weakly dorsalized embryo that was injected with 0.8 ng/embryo smad5 MO. F, a typical smad1 and smad5 double morphant (2 ng smad1 MO + 0.8 ng smad5 MO per embryo) that was strongly dorsalized. For each injection experiment, over 120 embryos were observed, and a representative phenotype is shown, and the number of embryos showing a corresponding phenotype is indicated in the top right corner of each panel. G–J, WISH analysis of smad5. smad5 is maternally expressed (G) and shows ubiquitous expression before gastrulation (H and I), and its transcripts are more prominently present in the ventral side of the embryo at 75% epiboly stage (J). K–N, WISH analysis of smad1. smad1 does not show maternal expression (K); its zygotic transcription does not begin at 30% epiboly (L), and it starts from 50% epiboly (M) until the gastrula stage in the ventral side of the embryo (N). Embryos in A–F are at 30 hpf, shown in a lateral view with anterior to the left; embryos in G, H, K, and L are shown in a lateral view with animal pole to the top; embryos in I, J, M, and N are shown in a lateral view with dorsal to the right. Arrow in B, expanded blood island; arrowhead in B, loss of head structure; arrowheads in C, thinner yolk and degenerative central neural system; arrow in C, slightly extended blood island due to increase in erythroid cells; arrow in F, loss of ventral tissues; arrows in J, M, and N, ventral location of signals. conc., concentration; epi, epiboly.
In order to further analyze the function of smad1 during DV patterning, translation-blocking MO was utilized for loss-of-function studies. Low dose injection of smad1 MO (1 ng/embryo) caused no obvious phenotype changes. When the injection dose was raised to 2.4 ng/embryo, the morphants showed degenerated anterior neurons and not well extended yolk stalk as well as slightly extended ventral blood island at 30 hpf (Fig. 1C), just like in a previous report (19) demonstrating that smad1 morphants have an increased number of erythroid cells at the ventral blood island, compared with the wild type embryos (Fig. 1A). Therefore, translational block of smad1 did not cause any dorsalization phenotype in zebrafish embryos, indicating that smad1 is not required by itself for DV axis establishment.
Then we were interested in the function of smad1 under conditions in which endogenous Smad5 activity is slightly lessened. We utilized different doses of smad5 MO to attenuate Smad5 activity at different levels (16). We observed the dorsalized embryos at 30 hpf with phenotype strength from C1 to C4 (42). Injection of smad5 MO at a dose of 1.6 ng/embryo produced 36% C4 (showing significant coiling of the tail as well as dorsalization within the anterior regions), 54% C3 (showing shortened and twisted tail), and 10% C2 dorsalization (showing normal tail length with absent ventral tail vein and fin) (Fig. 1D). When the injection dose of smad5 was titrated to 0.8 ng/embryo, however, the injected embryos showed a very mild dorsalization phenotype, with 94% C1-C2 (Fig. 1E) and 6% C3. Intriguingly, coinjection of low dose of smad1 MO (2 ng/embryo) strongly dorsalized the injected embryos, showing 44% C4, 53% C3, and 3% C2 dorsalized embryos (Fig. 1F), the phenotype for which was even stronger than with the high dose (1.6 ng/embryo) injection of smad5 MO alone (36% C4, 54% C3, and 10% C2). Therefore, smad1 knockdown could strongly amplify the dorsalization effects resulting from mild knockdown of smad5, although no DV defects are observed in smad1 knockdown. Taken together, these findings suggest that smad5 or its downstream factors could compensate for the activity of smad1 when smad1 itself is knocked down.
Because knockdown of smad1 or smad5 resulted in distinct phenotypes, we further comparatively analyzed their expression patterns during embryogenesis via WISH. As shown in Fig. 1, G–J, smad5 is maternally expressed, and its transcripts are ubiquitously displayed until midgastrulation, when there is transcriptional enrichment in the ventral side of the embryo. The expression pattern of smad1 is distinctly different (Fig. 1, K–N). smad1 is not maternally expressed and does not start its transcription until 50% epiboly, before the onset of gastrulation. During gastrulation, its expression is restricted to the ventral side of the embryo.
smad9 Genetically Interacts with smad5 in Zebrafish DV Patterning
The aforementioned results suggest that there might be other factors that are downstream of smad5 to compensate for the activity of Smad1 in vivo. This is also supported by the previous study that a maternal-zygotic smad5 mutant (sbn−/−) could be rescued by overexpression of bmp2b, indicating that ventral-promoting Smad activity exists in the smad5 mutants (17). We speculate that another BMP R-Smad, Smad9 (43), might be the factor besides Smad1 that is downstream of Smad5 to induce ventral fate. The full coding region of smad9 (previously named smad8; see the Zebrafish Model Organism Database) was PCR-amplified from a cDNA pool of zebrafish embryos before 24 hpf. The predicted amino acid sequence of Smad9 protein is shown in Fig. 2A. Zebrafish Smad9 is closely related to the Smad9 proteins of human, rat, mouse, and Xenopus, especially in the MH1 and MH2 domains. All of the three zebrafish BMP R-Smads (Smad1, Smad5, and Smad9) share striking sequence similarities (Fig. 2B). The phylogenetic tree of R-Smads of several vertebrates shows that the zebrafish Smad9 is clustered with Smad9 rather than Smad1 or Smad5 of other species (Fig. 2C). The early expression profile of smad9 was analyzed by WISH and RT-PCR. As shown in Fig. 3, A–D, smad9 starts its transcription prior to the onset of gastrulation in the ventral side of the embryo and continues its expression in the ventral gastrula, the same as smad1. From the results of RT-PCR (Fig. 3E), smad5 has maternal expression, and its expression level decreases during gastrulation. Both smad1 and smad9 initiate their transcription from the 50% epiboly stage, and their zygotic expression tends to be stronger throughout development.
FIGURE 2.

Zebrafish Smad9 is highly homologous with BMP R-Smads among different species. A, the predicted zebrafish Smad9 (zSmad9) sequence is aligned with those of human (h), mouse (m), rat (r), Xenopus (x), and Drosophila Mad (dMad). Humans have two isoforms of Smad9, indicated as isoform A and isoform B. The MH1 and MH2 domains are highly conserved, whereas the linker regions are more divergent. B, comparison of the predicated protein sequences of zebrafish Smad1, Smad5, and Smad9. C, a phylogenetic tree of BMP R-Smads of different spices. MH1, Mad homologous domain 1; MH2, Mad homologous domain 2. Sequence alignments were performed using ClustalW, and phylogenetic alignment was generated using MegAlign.
FIGURE 3.

smad9 genetically interacts with smad5 in DV patterning. A–D, WISH analysis of smad9 expression during early development. smad9 does not show maternal expression (A), does not show zygotic expression at 30% epiboly (B), and starts its transcription from the 50% epiboly stage in the ventral side of the embryo (C) and continues expressing in the ventral side in gastrula stage (D). E, semiquantitative RT-PCR analysis of smad1, smad5, and smad9 during early development. ef1a was used as internal control. Only smad5 has maternal expression, and its transcriptional levels drop from the gastrula stage. smad1 and smad9 start to transcribe from the 50% epiboly stage, and their expression levels tend to show an increase during later stages. F, a typical ventralized embryo that was injected with smad9 mRNA (500 pg/embryo). G, a typical smad9 morphant embryo that is not dorsalized (6.4 ng/embryo). H, a typical smad9 and smad5 double morphant (4 ng of smad9 MO + 0.8 ng of smad5 MO per embryo) that is strongly dorsalized. I–L, smad9 morpholino used in the present study is specific and effective. The strong expression of Smad9-EGFP fusion protein (I) could be effectively blocked by coinjection of smad9 MO (J). The expression of Smad9(MU)-EGFP fusion protein (K) could not be blocked by coinjection of smad9 MO (L). In the smad9(MU)-egfp construct, the binding site of smad9 MO is mutated. For each injection experiment, over 120 embryos were observed, and a representative phenotype is shown, and the number of embryos showing corresponding phenotype is indicated in the top right corner of each panel. Embryos in A and B are shown in a lateral view with animal pole to the top; embryos in C and D are shown in a lateral view with dorsal to the right; and embryos in F–H are at 30 hpf and shown in a lateral view with anterior to the left. Arrows in C and D, ventrally expressed smad9 signals; arrows in F, smaller eyes and enlarged blood island; arrow in G, degenerative central neural system; arrow in H, loss of ventral tissues.
The developmental role of zebrafish smad9 was studied through overexpression and knockdown experiments. First, smad9 mRNA was synthesized and injected into 1-cell embryos. 68% of those injected embryos were ventralized (Fig. 3F), with 46% V1, 18% V2, and 4% V3. This indicates that smad9 carries intrinsic ventralizing activity, mimicking smad1. To knock down Smad9 expression in vivo, a translation-blocking MO was employed. To prove the effectiveness and specificity of smad9 MO (44), we coinjected the MO with the constructs coding for fusion protein Smad9-EGFP with or without the MO binding site separately. In comparison with the strong EGFP signal in the smad9-egfp construct-injected embryos (Fig. 3I), the MO-coinjected embryos showed depletion of EGFP expression (Fig. 3J). In addition, if the MO binding site in smad9 was mutated, the Smad9(MU)-EGFP expression could not be blocked by coinjection of smad9 MO (Fig. 3, K and L). These results provided evidence for the effectiveness and specificity of smad9 MO. Just like the smad1 morphants, the smad9 morphants were not dorsalized, even if the injection dose was as high as 6.4 ng/embryo (Fig. 3G). Similarly, smad9 MO (4 ng/embryo) was coinjected into smad5 mild morphants (0.8 ng/embryos), and the double morphants were analyzed for DV patterning defects at 30 hpf. As shown in Fig. 3H, the double morphant embryos were strongly dorsalized, with C4 of 40%, C3 of 55%, and C2 of 5%. The results show that smad9 knockdown could strongly strengthen the dorsalization of smad5 mild morphants. Therefore, we conclude that smad9 genetically interacts with smad5 in DV pattering of zebrafish in vivo.
Double Knockdown of smad1 and smad9 Leads to Strong Dorsalization
Altogether, the expression patterns of smad1 and smad9 are fairly similar to each other, and so are their overexpression and knockdown effects. The above results reveal that, although smad1 or smad9 is not essential by itself for the early DV axis establishment, they could help to pattern the DV axis in a condition in which Smad5 activity is slightly lessened. We further asked whether and how smad1 and smad9 cooperatively act in zebrafish DV patterning. A reasonable hypothesis is that smad1 and smad9 may function redundantly to each other; i.e. if only one of the redundant partners is inhibited, the other partner can be stimulated and compensate for the loss-of-function effects, and the overall amount of partners will not be affected (45). Therefore, in our case, the DV patterning might not be disrupted if smad1 or smad9 is knocked down alone. To examine this hypothesis, smad1 MO (2 ng/embryo) and smad9 MO (4 ng/embryo) were coinjected into embryos to knock down smad1 and smad9 simultaneously. The double morphants were allowed to develop to 30 hpf for analysis of DV defects. Intriguingly, double knockdown of smad1 and smad9 strongly dorsalized the embryos with 100% penetration (Fig. 4C), showing 69% C4, 28% C3, and 3% C2. This phenotype has never been observed in the single knockdown experiment of smad1 or smad9, which strongly supports the hypothesis that smad1 and smad9 have functional redundancy in regulating zebrafish DV patterning.
FIGURE 4.

Double knockdown of smad1 and smad9 leads to strong dorsalization that cannot be rescued by smad5 overexpression. A, a typical uninjected control embryo. B, a typical smad5 morphant (1.6 ng/embryo) showing a strong dorsalized phenotype. C, a typical smad1 and smad9 double morphant (2 ng of smad1 MO + 4 ng of smad9 MO per embryo) that is strongly dorsalized. D, a typical rescued embryo resulting from coinjection of smad5 MO (1.6 ng/embryo) and smad1 mRNA (200 pg/embryo). E, a typical rescued embryo resulting from coinjection of smad5 MO (1.6 ng/embryo) and smad9 mRNA (300 pg/embryo). F, a typical dorsalized embryo that was coinjected with smad1 MO (2 ng/embryo) and smad9 MO (4 ng/embryo) and smad5 mRNA (300 ng/embryo). For each injection experiment, over 120 embryos were observed, and a representative phenotype is shown, and the number of embryos showing corresponding phenotype is indicated in the top right corner of each panel. G, WISH analysis of the embryos injected with different reagents. Embryos were uninjected control or injected with MOs and mRNAs indicated at the top of each column. a–g, cyp26a staining, which labels neuroectoderm; h–n, foxi1 staining, which labels epidermal ectoderm; o–u, RNA in situ labeling by szl, which is a BMP target. b, i, p, c, j, and q, in smad1 MO- or smad9 MO-injected embryos, the expression of DV patterning markers was the same as that in the control embryos (a, h, and o). d, k, and r, in smad5 MO-injected embryos, the expression level of the cyp26a is elevated, and the expression of foxi1 and szl is strongly reduced. e, l, and s, in smad5 morphants coinjected with smad1 or smad9 mRNA, the enlarged expression of cyp26a is rescued to normal and the reduced expression of foxi1 and szl is also rescued to wild type level; f, m, and t, in smad1 and smad9 double morphants, the expression of cyp26a is strongly increased, and the expression of foxi1 and szl is strongly reduced. g, n, and u, the alteration of the expression of these markers in smad1 and smad9 double morphants cannot be rescued by smad5 mRNA injection. Embryos in A–F are shown in a lateral view with anterior to the left, 30 hpf; embryos in G, row 1, are at the 70% epiboly stage, shown in a lateral view with dorsal to the right; embryos in G, row 2, are at 70% epiboly, shown in an animal pole view with dorsal to the right; and embryos in G, row 3, are at the 60% epiboly stage, shown in an animal pole view with dorsal to the right. The results shown in G for WISH analysis represent over 90% of the embryos assayed.
To further confirm the DV defects in different morphants, the expression of a set of molecular markers indicating DV patterning was analyzed. At the gastrula stage, cyp26a labels neuroectoderm territory (30), foxi1 labels epidermal ectoderm (31), and sizzled (szl) represents a BMP signaling target (32). As shown in Fig. 4G, the expression patterns of these markers were not changed in the smad1 morphants (b, i, and p) or in the smad9 morphants (c, j, and q) (the WISH embryos shown in the Fig. 5G represent over 90% of the embryos assayed). In smad1 and smad9 double morphants, however, the expression of cyp26a was remarkably expanded toward the ventral side of the ectoderm (Fig. 4G, f), whereas the expression of foxi1 and szl was significantly reduced (Fig. 4G, m and t). These results were in accordance with the phenotypic analysis. Similar results of another target gene of BMP signaling, ved, were also obtained (data not shown). Therefore, we conclude that smad1 and smad9 act redundantly to each other in regulating DV patterning of zebrafish embryos.
FIGURE 5.

smad1 and smad9 are positively regulated by smad5 and suppress each other. A, WISH analysis of smad1 (a–j) and smad9 (k–t) in different types of embryos injected with different reagents. Compared with wild type embryos (a), the transcription of smad1 is up-regulated by injection of smad5 mRNA (b), smad1 MO (d), or smad9 MO (f) and by coinjection of smad1 MO and smad9 MO (g) or by coinjection of smad1 MO, smad9 MO, and smad5 mRNA (j); the transcription of smad1 is down-regulated by injection of smad5 MO (c) or smad9 mRNA (e) and by coinjection of smad1 MO and smad5 MO (h) or by coinjection of smad5 and smad9 MO (i). Compared with wild type embryos (k), the transcription of smad9 is up-regulated by injection of smad5 mRNA (l), smad1 MO (o), or smad9 MO (p) and by coinjection of smad1 MO and smad9 MO (q) or by coinjection of smad1 MO, smad9 MO, and smad5 mRNA (t); the transcription of smad9 is down-regulated by injection of smad5 MO (m) or smad1 mRNA (n) and by coinjection of smad1 MO and smad5 MO (r) or by coinjection of smad5 MO and smad9 MO (s). B, luciferase assay of zebrafish smad1 promoter activity. The activity of the smad1 promoter was up-regulated to 2.72-fold by smad5 mRNA injection and down-regulated to 0.58-fold by smad9 mRNA injection, and it was down-regulated to 0.50-fold by smad5 MO injection and up-regulated to 1.63-fold by smad9 MO injection. C, luciferase assay of zebrafish smad9 promoter activity. The activity of the smad9 promoter was up-regulated to 1.99-fold by smad5 mRNA injection and down-regulated to 0.55-fold by smad1 mRNA injection, and it was down-regulated to 0.56-fold by smad5 MO injection and up-regulated to 1.49-fold by smad1 MO injection. In B and C, embryo lysates were prepared at 5.4 hpf, and the luciferase activities were normalized to Renilla luciferase from pRL-TK; results were expressed relative to luciferase activities with constructs alone; each bar presents the mean value and the corresponding S.D. (error bar) from triplicate analysis. A statistically significant difference (p < 0.05) is indicated by a different lowercase letter above the bar. D, relative expression of smad5 in 50% epiboly embryos injected with different reagents by quantitative PCR analysis. All of the injected samples have no significant difference compared with uninjected control (p > 0.1). E, WISH analysis of smad1 (a–e) and smad9 (f–j) in the embryos injected with different reagents. a, a typical control embryo showing normal expression of smad1. b, a typical embryo that was injected with bmp2b mRNA (20 pg/embryo) with elevated expression of smad1. c, a typical embryo that was injected with bmp2b mRNA (20 pg/embryo) and smad5 MO (1.6 ng/embryo), showing absent expression of smad1. d, a typical embryo that was injected with DNBR1a mRNA (100 pg/embryo), showing absent expression of smad1. e, a typical embryo that was injected with DNBR1a mRNA (100 pg/embryo) and smad5 mRNA (300 ng/embryo), showing strong expression of smad1. f, a typical control embryo showing normal expression of smad9. g, a typical embryo that was injected with bmp2b mRNA (20 pg/embryo), with elevated expression of smad9. h, a typical embryo that was injected with bmp2b mRNA (20 pg/embryo) and smad5 MO (1.6 ng/embryo), showing absent expression of smad9. i, a typical embryo that was injected with DNBR1a mRNA (100 pg/embryo), showing absent expression of smad9. j, a typical embryo that was injected with DNBR1a mRNA (100 pg/embryo) and smad5 mRNA (300 ng/embryo), showing strong expression of smad9. All embryos in E are at the 50% epiboly stage, shown in a lateral view with dorsal to the right.
Because the phenotype of smad1 and smad9 double morphants is similar to that of smad5 morphants, and it is suggested that smad1 might be a transcriptional target of smad5 (9), the relationship among all of these Smads was extensively analyzed. First, we asked whether the dorsalized phenotype of smad5 morphants could be rescued by overexpression of smad1 or smad9. As shown in Fig. 4, the dorsalization caused by injection of smad5 MO (36% C4, 54% C3, and 10% C2; Fig. 4B) could be rescued by coinjection of smad1 mRNA (22% C3, 28% C2, and 50% wild type-like; Fig. 4D) or smad9 mRNA (22% C3, 32% C2, and 46% wild type-like; Fig. 4E). Second, we asked whether the dorsalization phenotype resulting from smad1 and smad9 double knockdown could be rescued by smad5 overexpression. Surprisingly, the strong dorsalization of smad1 and smad9 double morphants (69% of C4, 28% of C3, and 3% of C2) could not be effectively rescued by smad5 mRNA injection at different doses (67% C4, 26% C3, and 7% C2; Fig. 4F). The phenotypic results were further confirmed by WISH analysis with a set of molecular markers representing DV patterning and BMP signaling activity. As shown in Fig. 4G, smad5 morphants showed typical dorsalization of the ectoderm, with enlarged cyp26a-labeled and decreased foxi1-labeled territories (Fig. 4G, d and k) and a strong reduction of BMP signaling activity (Fig. 4G, r). Whereas in the smad5 MO- and smad1 or smad9 mRNA-coinjected embryos, the neuroectoderm territories were restored to the normal region or even reduced (Fig. 4G, e), the non-neural ectoderm expressing foxi1 labeled a much wider region (Fig. 4G, l), and szl expression was restored to the normal level (Fig. 4G, s). By contrast, the dorsalized phenotype of smad1 and smad9 double morphants could not be efficiently rescued by overexpression of smad5 (Fig. 4G, g, n, and u). The inability of ectopic smad5 overexpression to efficiently rescue the DV patterning defects in smad1 and smad9 double morphants suggests that Smad5 has transcriptional and functional activities differing from Smad1 and Smad9.
smad1 and smad9 Are Downstream Targets of smad5
To further clarify the regulation network among smad1, smad5, and smad9, we analyzed the transcription of different BMP type smad genes in smad1, smad5, or smad9 knocked down or overexpressed embryos at 50% epiboly, a stage when smad1 and smad9 start their zygotic transcription. Just as reported previously (9), smad1 transcription was strongly induced by smad5 misexpression (Fig. 5A, b), and smad1 transcription was absent if smad5 was knocked down (Fig. 5A, c). Similarly, the transcription of smad9 strongly increased in the smad5 overexpressed embryos (Fig. 5A, l) and nearly disappeared in smad5 morphants (Fig. 5A, m).
To better understand the transcriptional regulation of these smad genes, we further cloned the upstream regulatory region of smad1 of 4290 bp and the upstream regulatory sequence of smad9 of 3456 bp and utilized them to generate two luciferase assay constructs. Relative luciferase assays revealed that the activity of the smad1 promoter was significantly elevated to 2.72-fold by overexpression of smad5 and significantly decreased to 0.50-fold by knockdown of smad5 in the embryos at the 50% epiboly stage (Fig. 5B). Likewise, smad9 promoter activity showed a significant increase to 1.99-fold by smad5 overexpression and a significant decrease of 0.56-fold by smad5 knockdown (Fig. 5C). These findings reveal that the transcription of both smad1 and smad9 is dependent on smad5 and is positively regulated by smad5.
To clearly elucidate the transcriptional regulation between smad1 and smad9, we conducted transcriptional analysis of smad1 or smad9 in the embryos with up- or down-regulation of smad1 or smad9. First, smad9 mRNA or smad9 MO was injected into zebrafish embryos, and the injected embryos at 50% epiboly were subjected to WISH analysis of smad1. Strikingly, the transcription initiation of smad1 was highly repressed by smad9 overexpression (Fig. 5A, e) and significantly enhanced by smad9 knockdown (Fig. 5A, f). Second, smad1 mRNA or smad1 MO was injected, and the embryos at 50% epiboly were subjected to WISH analysis of smad9. The labeling signals of smad9 nearly disappeared in the smad1-overexpressed embryos (Fig. 5A, n), and the signals dramatically increased in the smad1 knocked down embryos (Fig. 5A, o). This demonstrates that the transcriptional onset of smad1 and of smad9 are negatively regulated by each other. Third, this conclusion was further confirmed by promoter activity analysis by luciferase assays. As expected, in smad9-overexpressed embryos, the activity of the smad1 promoter was down-regulated to 0.58-fold, and in smad9 morphants, it was up-regulated to 1.63-fold (Fig. 5B). Similarly, the activity of smad9 promoter was down-regulated to 0.56-fold in smad1-overexpressed embryos and up-regulated to 1.49-fold in smad1 morphants (Fig. 5C). Taken together, these results reinforce the notion that smad1 and smad9 act redundantly to each other at the transcriptional level, and these also give a reasonable explanation why double knockdown instead of single knockdown of smad1 or smad9 induces visible dorsalization effects.
In addition, we analyzed the regulation effects of smad1 and smad9 on themselves and on smad5. In smad1 knocked down embryos, the transcription of smad1 was strongly elevated (Fig. 5A, d), suggesting that smad1 is negatively self-regulated. In smad1 MO- and smad9 MO-coinjected embryos, the expression level of smad1 was dramatically elevated (Fig. 5A, g), higher than that in smad1 MO- or smad9 MO-injected embryos at the 50% epiboly stage, whereas coinjection of neither smad1 MO nor smad9 MO with smad5 MO could restore the transcription of smad1 (Fig. 5A, h and i), confirming that smad1 is downstream of smad5. In smad1 MO-, smad9 MO-, and smad5 mRNA-coinjected embryos, the expression of smad1 was greatly enhanced, spreading to the whole blastomere (Fig. 5A, j). Likewise, smad9 was negatively self-regulated; as in smad9 MO-injected embryos, the labeling signals of smad9 by WISH were strongly elevated (Fig. 5A, p). Additionally, the signals of smad9 increased even more in smad1 and smad9 double morphants (Fig. 5A, q), whereas in the background of smad5 morphants, neither knockdown of smad1 nor knockdown of smad9 could rescue the disappearance of smad9 transcription (Fig. 5A, r and s), confirming that smad9 is downstream of smad5. The strongest signal of smad9 was observed in smad1 MO-, smad9 MO-, and smad5 mRNA-coinjected embryos (Fig. 5A, t). On the other hand, the potential transcriptional regulation of smad5 by smad1 and smad9 was analyzed by quantitative RT-PCR (Fig. 5D). The amount of smad5 transcripts in smad1 mRNA-, smad1 MO-, smad9 mRNA-, or smad9 MO-injected embryos showed no significant difference when compared with wild type at 50% epiboly as well as with the embryos coinjected with smad1 MO and smad9 MO. These results further demonstrate that both smad1 and smad9 locate downstream of smad5, and neither of them exert any effects on the transcriptional regulation of smad5.
To analyze the transcriptional regulation of smad1 and smad9 by smad5 under different BMP signaling backgrounds, BMP activity-elevated and -abolished embryos were used for further study. As expected, elevated smad1 transcription was observed in bmp2b-overexpressed embryos (Fig. 5E, b) (9). In order to deplete the overall BMP signaling, a truncated dominant negative BMP receptor 1a, DNBR1a (46), was overexpressed in the embryos. In the DNBR1a mRNA-injected embryos, the transcription of smad1 was completely abolished (Fig. 5E, c), further showing that smad1 itself is a transcriptional target of BMP signaling. In bmp2b-overexpressed embryos, an enlarged domain and higher level of the smad9 expression were also observed (Fig. 5E, g), and in DNBR1a mRNA-injected embryos, smad9 transcription was likewise completely inhibited (Fig. 5E, h). Therefore, the normal transcription of smad9 also relies on proper BMP signaling activity. However, even in the bmp2b-overexpressed embryos, knockdown of smad5 could still efficiently block the transcription of smad1 or smad9 (Fig. 5E, c and h), suggesting that smad5 is necessary to mediate BMP-induced smad1 and smad9 transcription. By contrast, smad5 overexpression still strongly activated the transcription of smad1 and smad9 in the BMP-abolished embryos (Fig. 5E, e and j). These suggest that smad1 and smad9 are the direct transcriptional targets of smad5, and their transcriptional activities directly rely on smad5.
smad1 and smad9 Are Direct Transcriptional Targets of smad5
In order to uncover whether there is a direct or indirect effect of smad5 on activating smad1 and smad9, we used the translation inhibitor CHX from 2 hpf onward to block translation of the earliest zygotic mRNAs but allow the translation of injected smad5 mRNA (47). Transcriptional induction of ntl (no tail) requires intermediate zygotic translation steps after midblastula transition (MBT); thus, the absence of ntl signal served as a control of efficiency of CHX treatment (38) (Fig. 6A, a and b). We injected smad5 MO into 1-cell stage embryos to block the translation of maternal supplied smad5 transcripts, and as a result, the transcription of smad1 and smad9 was absent in the smad5 MO-injected embryos (Fig. 6A, d and h). After coinjection of mutated smad5 mRNA whose smad5 MO binding site was mutated but coding protein was not altered, the signals of smad1 or smad9 were restored and elevated in smad5 morphants (Fig. 6A, e and i). When CHX was added at 2 hpf, expression of smad1 and smad9 could still be rescued (Fig. 6A, f and j), indicating that smad1 and smad9 are the direct targets of Smad5. To further prove this, we performed a ChIP assay with extracts from embryos at the 50% epiboly stage injected with mRNA encoding HA-tagged Smad5. The precipitated chromatin was then analyzed by quantitative PCR using primer pairs that could amplify segments of the smad1 promoter or smad9 promoter. A segment of the rpl5b exon amplified by a specific primer pair was used as control (38). As shown in Fig. 6B, we observed a significant enrichment for the promoter regions of smad1 and smad9 in the immunoprecipitate from the injected samples compared with the HA-sv40 mRNA-injected control. In contrast, there was no enrichment of the control genomic region rpl5b. Thus, these data demonstrate that Smad5 directly binds to the promoters of smad1 and smad9 to activate their transcription.
FIGURE 6.

smad1 and smad9 are direct transcriptional targets of Smad5. A, WISH analysis of 6 hpf embryos injected with different reagents and treated by CHX or not. a, a typical control embryo showing normal expression of ntl. b, a typical embryo treated by CHX from 2 hpf, showing absent expression of ntl. d and h, embryos injected with smad5 MO, showing absent expression of smad1 (d) and smad9 (h). e and i, embryos coinjected with smad5 MO and mutated smad5 mRNA, showing restored and elevated expression of smad1 (e) and smad9 (i). f and j, embryos coinjected with smad5 MO and mutated smad5 mRNA and treated by CHX from 2 hpf, showing elevated expression of smad1 (f) or smad9 (j). In smad5 (MU) mRNA, the smad5 MO binding site is mutated, but the protein sequence coded by the mutated mRNA is identical to that coded by the wild type mRNA. All embryos shown in A are in a lateral view with animal pole to the top or dorsal to the right. B, ChIP analysis of HA-smad5 mRNA- or HA-sv40 mRNA-injected embryos with anti-HA antibody. A schematic diagram depicts the fragments of the smad1, smad9, and rpl5b genes that were amplified. The positions of quantitative PCR primers that were used to amplify smad1 and smad9 promoter fragments and the rpl5b exon fragment are indicated by arrows. Chromatin from HA-smad5 mRNA-injected (black bars) and HA-sv40 mRNA-injected (gray bars) embryos was sonicated and immunoprecipitated with anti-HA antibody. DNA levels of smad1 and smad9 promoter region and control sequence (rpl5b exon) in precipitate and input were measured by quantitative PCR. The smad1 promoter region was 2.43-fold enriched, and the smad9 promoter region was 2.44-fold enriched in the HA-smad5 mRNA-injected samples in comparison with the HA-sv40 mRNA-injected samples, whereas rpl5b did not show enrichment. Each bar presents the mean value and the corresponding S.D. (error bar) from triplicate analysis. *, statistically significant difference (p < 0.05). C, Smad4 interacts with Smad1, Smad5, and Smad9. HEK293T cells were transfected with FLAG-smad4 and HA-smad1, HA-smad5, or HA smad9. FLAG-Smad4-binding proteins were immunoprecipitated and analyzed by immunoblotting. D, binding affinity of FLAG-Smad4 against HA-Smad5 is not affected by coexpression of an increasing amount of Myc-Smad1 (1.2 and 2.5 μg) or Myc-Smad9 (1.2 and 2.5 μg). IP, immunoprecipitation; TCL, total cell lysate; WB, Western blot.
In order to address the intrinsic differences of Smad5, Smad1, and Smad9 proteins, we conducted co-immunoprecipitation experiments to analyze and compare their binding ability to Smad4, which is the co-Smad of BMP signaling (48, 49). We first investigated the interaction between zebrafish Smad4 and Smad1/Smad5/Smad9. In HEK293T cells, all of the zebrafish Smad1, Smad5, and Smad9 could bind to Smad4 (Fig. 6C). Then we analyzed whether the presence of Smad1 and Smad9 could disturb the binding affinity between Smad5 and Smad4. As shown in Fig. 6D, when Smad1 and Smad9 were transfected at increasing doses, the interaction of Smad4 and Smad5 was not affected.
smad9, Like smad1, Is Required for Myelopoiesis
It has been reported that smad1 and smad5 differently regulate zebrafish embryonic hematopoiesis; smad1 but not smad5 is required for the differentiation of macrophages and granulocytes, both belonging to the myeloid lineage (19). Because smad9 and smad1 have shown similar expression patterns and regulatory network in zebrafish early development, we proposed that they may share similar functions during embryonic hematopoiesis. First, smad9 was knocked down or overexpressed in zebrafish embryos, and the injected embryos at 24 hpf were subjected to analysis with a set of myelopoiesis-related markers. As shown in Fig. 7 (D and L), macrophages labeled by l-plastin and granulocytes labeled by mpx were obviously diminished in smad9 morphants at 24 hpf, which just mimicked the smad1 morphants (Fig. 7, C and K). This was distinctly different from what was observed in smad5 morphants, which did not show obvious defects of l-plastin- and mpx-labeled cells (Fig. 7, B and J). This revealed that smad9, like smad1, is required for zebrafish myelopoiesis. Second, smad9 mRNA was injected into smad1 morphants to check whether smad9 is able to replace the function of smad1 in myelopoiesis and vice versa. Coinjection of smad9 mRNA was able to rescue the developmental defects of l-plastin-labeled macrophages in smad1 morphants in 61% of the embryos (Fig. 7F) and mpx-labeled granulocytes in 59% of the embryos (Fig. 7N), and coinjection of smad1 mRNA could also restore the myeloid development in the smad9 morphants (Fig. 7, G and O; 57% rescue for macrophages and 70% rescue for granulocytes). In contrast, coinjection of smad5 mRNA was not able to rescue the defects of macrophages and granulocytes in smad9 morphants (Fig. 7, H and P; 100%), just like in the previous report (19) in which smad5 overexpression did not rescue the myeloid defects in smad1 morphants. Therefore, our study reveals that smad1 and smad9 not only share functional redundancy in early DV patterning but also are functionally exchangeable in regulating the differentiation of macrophages and granulocytes, both of which belong to the myeloid lineage.
FIGURE 7.
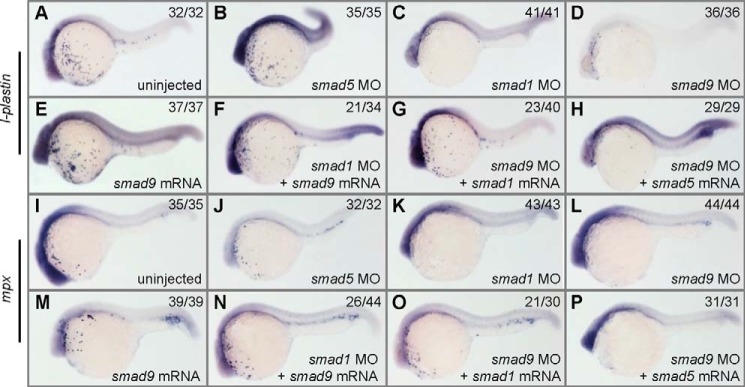
smad1 and smad9 regulate the differentiation of macrophages and granulocytes. Embryos were either uninjected control or injected with different mRNAs and MOs indicated in the images and processed at 24 hpf for WISH to identify l-plastin (A–H) and mpx (I–P) transcripts. l-plastin labels macrophages, and mpx labels granulocytes. The expression of l-plastin and mpx is normal in smad5 morphants (B and J) but is almost completely depleted in smad1 morphants (C and K), greatly reduced in smad9 morphants (D and L), and slightly increased in smad9 mRNA-injected embryos (E and M). Overexpression of smad1 or smad9 can rescue the absence of l-plastin- and mpx-labeled cells in the morphants of each other (F, G, N, and O). In contrast, overexpression of smad5 is unable to rescue the loss of these two types of cells in smad1 or smad9 morphants (H and P). For each assay, over 29 embryos were observed, and a representative phenotype is shown, and the number of embryos showing the corresponding phenotype is indicated in the top right corner of each panel. The injection doses of the mRNAs are as follows: smad1 mRNA, 150 pg/embryo; smad5 mRNA, 200 pg/embryo; smad9 mRNA, 250 pg/embryo. All embryos are shown in a lateral view with anterior to the left.
DISCUSSION
Smad1 and Smad5 are believed to be two major mediators transducing BMP signaling pathway, whereas BMP signals induce ventral fate in the early development of vertebrates (50). In zebrafish, the BMP ligands bmp2b and bmp7 are widely expressed soon after the MBT, and then their expression becomes restricted to the ventral half of the embryo by the onset of gastrulation (51). It was believed that Smad1 and Smad5 should both play critical roles in DV patterning of zebrafish. In accordance with this, overexpression of either smad1 or smad5 in zebrafish embryos leads to ventralized phenotypes (Figs. 1 and 2) (9), similar to the phenotypic effects of bmp2b or bmp4 overexpression (5). Overexpression of smad1 in a different genetic background suggests that smad1 is even more potent than smad5 in promoting ventral fate. For instance, overexpression of smad1 in a wild type background gives stronger ventralization than smad5, and smad1 overexpression in a smad5 mutant or bmp2b mutant leads to a rescue effect, whereas smad5 overexpression cannot rescue the bmp2b mutant (9). However, from loss-of-function studies, we may even reach an opposite conclusion. Large scale mutation screens in zebrafish have identified several smad5 mutants, such as pgy and sbn, which are strongly dorsalized (15), whereas no smad1 mutant has been identified; effective knockdown of smad1 expression by MO injection leads to no obvious defect on early DV patterning (Fig. 1C) (19). Here, we have revealed that a smad1-like gene, smad9, exists in the zebrafish genome, which could compensate for the Smad activity in the condition of smad1 loss of function. We further prove that zebrafish smad9 not only shows an expression pattern similar to that of smad1 but also acts redundantly with smad1 to mediate zebrafish DV patterning downstream of smad5.
It is proposed that the DV patterning in zebrafish can be divided into three distinct phases (17): an early Smad5- and Bmp2b-independent phase when an initial course of DV patterning is set up; an intermediate Smad5- and Bmp2b-dependent phase during which the initial DV patterning is refined and the morphogenetic Bmp2b gradient is established; and a later Smad5-independent phase after the onset of gastrulation when the Bmp2/4 gradient is interpreted. Direct evidence for this is the finding that the smad5 mutant, sbn−/−, can be rescued by smad5 overexpression just after MBT, but there is no rescue effect if the exogenous smad5 is strongly expressed during gastrulation and very weakly expressed at earlier stages (17). Our study suggests that the presence of Smad1 and Smad9 activities from 50% epiboly is the reason for the dispensability of Smad5 during gastrulation. The function of Smad5 during DV patterning is involved in setting up the putative morphogenetic BMP gradient along with other factors and, in addition, activates the expression of a cluster of genes, including smad1 and smad9. From 50% epiboly, these two Smads interpret the BMP gradient during gastrulation in a redundant manner, and Smad1 and Smad9 become the executors of BMP/Smad in promoting ventral specification. In regard to the functional redundancy of smad1 and smad9, if loss of function is utilized to study the relative roles of smad1 and smad9 in zebrafish DV patterning, we need to attenuate the activities of both of these two Smads in order to obtain informative results leading to precise conclusions.
The genetic regulation network and functional distinction of different BMP R-Smads in development are of great interest. It has been shown that Smad1 and Smad5 bind to different Smad-binding elements (SBEs) and activate different target genes (52–55). In different animal models, it is suggested that Smad1, Smad5, and Smad9 play different roles in various developmental processes. In chicken embryos, Smad9 is differently required during spinal cord neurogenesis compared with Smad1 and Smad5 (10). In Smad1 mutant mice, the allantois fails to fuse to the chorion, with the result that they fail to connect to the placenta and die at ∼10 days postcoitum (56, 57). Smad5 mutant mice also die at about 10 postcoitum but for different reasons, including angiogenic failure, mesenchymal apoptosis, and other defects (58, 59). However, Smad9 homozygous mutant mice develop normally and are viable and fertile (60). In the generation of dorsal spinal neural circuitry in mice, Smad1 is required to regulate dl1 axon outgrowth, whereas Smad5 is required for the generation of dorsal spinal neurons (61). In zebrafish, gene expression analysis revealed that smad1 morphants and smad5 morphants were deregulated for striking distinct genetic networks at the 1-somite stage (19). Our study reveals that smad1 and smad9 are two direct targets of smad5, that they start transcription prior the onset of gastrulation, and, more interestingly, that their transcription is negatively regulated by themselves and by each other (Fig. 5). The cross-regulated negative feedback loop between smad1 and smad9 and with themselves further supports the notion that smad1 and smad9 are redundant partners in regulating DV patterning because this phenomenon resembles the typical responsive circuit in the reciprocal regulation of redundant genes (45). In contrast, the upstream smad5 is not affected by up- or down-regulation of smad1 and smad9. Our results also suggest that the commonly known BMP targets, szl and ved, are probably the direct targets of smad1 and smad9 at the gastrula stage, instead of smad5, because their transcription is nearly abolished in smad5-overexpressed smad1/smad9 double morphants (Fig. 4) (data not shown). We could even predicate that most BMP/Smad targets at the gastrula stage are probably the targets of BMP/Smad1/9. The functional redundancy of smad1 and smad9 is also hinted at by a previous study (62). For instance, zebrafish protein phosphatase 4c (Ppp4c) utilizes Smad1 as an adaptor to bind to the endogenous id1 promoter, and depletion of both Smad1 and Smad9 activities could completely abolish this binding (62).
An interesting question is the mechanism of switching from Smad5 to Smad1 and Smad9 when gastrula begins. One possibility is that Smad1 and Smad9 can compete with Smad5 for interaction with Smad4, just as Smad3 competes with Smad2 for interacting with Smad4 and thus inhibits activin-induced goosecoid expression though Smad2/Smad4 (63). However, the ability of Smad5 to bind to Smad4 is not affected by the presence of an increasing amount of Smad1 or Smad9 (Fig. 6). Another possibility is that Smad1 and Smad9 rather than Smad5 are more potent activators for ventral promoting genes and BMP/Smad targets during gastrulation. The DNA element (GCAT) was previously characterized as the Smad1 SBE in the Xvent-2B (52) and Xretpos (53) promoters in Xenopus and in the vegf promoter in zebrafish (55). Several types of Smad5 SBEs were identified in different promoters, such as the DNA motif (GTCTAGAC) in the mouse smad7 promoter (54) and the DNA motif (TGTCTGAGAC) in the zebrafish vegf (55) promoter. We have searched the upstream region of a BMP target gene, szl, for the DNA element identical to Smad1 SBE or Smad5 SBEs. Interestingly, 11 DNA elements identical to the Smad1 SBE (GCAT) but no DNA elements identical to Smad5 SBEs (GTCTAGAC or TGTCTGAGAC) were found in a 2000-bp DNA fragment upstream of the szl coding sequence. This suggests that szl is probably the direct target of Smad1 or Smad9 rather than Smad5. The other possibility is that these three Smads may bind to different cofactors. Previous studies have shown that B-cell translocation gene 2 (Btg2), a primary P53 transcriptional target gene, shows strong interaction with Smad1 and Smad9 but not with Smad5 (64), and Smad nuclear interacting protein 1 (SNIP1) only interacts with Smad1 (65).
The embryonic hematopoiesis depends on BMP/Smad signaling (66), whereas Smad1 and Smad5 differentially regulate this process (19). A previous study has revealed that smad1 is required for the differentiation of the mature embryonic macrophages and granulocytes, whereas smad5 is required for the primitive erythropoiesis. Because smad1 and smad9 show similar expression patterns during development and redundant function in DV patterning, it is reasonable that smad9 is also involved in zebrafish hematopoiesis. In fact, myeloid defects are observed in embryos depleted for smad9, as in embryos depleted for smad1. Overexpression of smad9 efficiently rescues the defects in smad1 morphants and vice versa, and smad5 is not able to rescue the myeloid defects in smad1 or smad9 morphants. In other words, smad1 and smad9 are exchangeable in regulating myelopoiesis, whereas smad5 cannot replace them. Altogether, smad1 and smad9 not only share functional redundancy in regulating DV patterning but also have overlapping functions in myelopoiesis.
In conclusion, we have characterized the relative roles of smad1, smad5, and smad9 during zebrafish DV patterning, and we have further revealed that smad1 and smad9 act redundantly to each other and function downstream of smad5. Therefore, our study has clearly clarified the regulation network and the cooperative roles of BMP R-Smads in early development of zebrafish.
Acknowledgment
We thank Li Ming for fish husbandry.
This work was supported by China 973 Project Grants 2010CB126306 and 2012CB944504, the National Science Fund for Excellent Young Scholars of the National Natural Science Foundation of China (NSFC) Grant 31222052, NSFC Grant 30771100, and FEBL Grant 2011FBZ23 (to Y. H. S.).
- R-Smad
- Co-Smad, and I-Smad, receptor-regulated, common mediator, and inhibitory Smad, respectively
- BMP
- bone morphogenetic protein
- DV
- dorso-ventral
- MO
- morpholino
- CHX
- cycloheximide
- hpf
- hours postfertilization
- WISH
- whole-mount in situ hybridization
- DIG
- digoxigenin
- MBT
- midblastula transition
- SBE
- Smad-binding element.
REFERENCES
- 1. Massagué J., Wotton D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 19, 1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Attisano L., Wrana J. L. (2000) Smads as transcriptional co-modulators. Curr. Opin. Cell Biol. 12, 235–243 [DOI] [PubMed] [Google Scholar]
- 3. Massagué J. (1998) TGF-β signal transduction. Annu. Rev. Biochem. 67, 753–791 [DOI] [PubMed] [Google Scholar]
- 4. Derynck R., Feng X. H. (1997) TGF-β receptor signaling. Biochim. Biophys. Acta 1333, F105–F150 [DOI] [PubMed] [Google Scholar]
- 5. Kishimoto Y., Lee K. H., Zon L., Hammerschmidt M., Schulte-Merker S. (1997) The molecular nature of zebrafish swirl. BMP2 function is essential during early dorsoventral patterning. Development 124, 4457–4466 [DOI] [PubMed] [Google Scholar]
- 6. de Jong J. L., Zon L. I. (2005) Use of the zebrafish system to study primitive and definitive hematopoiesis. Annu. Rev. Genet. 39, 481–501 [DOI] [PubMed] [Google Scholar]
- 7. Graff J. M., Bansal A., Melton D. A. (1996) Xenopus Mad proteins transduce distinct subsets of signals for the TGF β superfamily. Cell 85, 479–487 [DOI] [PubMed] [Google Scholar]
- 8. Suzuki A., Chang C., Yingling J. M., Wang X. F., Hemmati-Brivanlou A. (1997) Smad5 induces ventral fates in Xenopus embryo. Dev. Biol. 184, 402–405 [DOI] [PubMed] [Google Scholar]
- 9. Dick A., Meier A., Hammerschmidt M. (1999) Smad1 and Smad5 have distinct roles during dorsoventral patterning of the zebrafish embryo. Dev. Dyn. 216, 285–298 [DOI] [PubMed] [Google Scholar]
- 10. Le Dréau G., Garcia-Campmany L., Rabadán M. A., Ferronha T., Tozer S., Briscoe J., Martí E. (2012) Canonical BMP7 activity is required for the generation of discrete neuronal populations in the dorsal spinal cord. Development 139, 259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Orvis G. D., Jamin S. P., Kwan K. M., Mishina Y., Kaartinen V. M., Huang S., Roberts A. B., Umans L., Huylebroeck D., Zwijsen A., Wang D., Martin J. F., Behringer R. R. (2008) Functional redundancy of TGF-β family type I receptors and receptor-Smads in mediating anti-Mullerian hormone-induced Mullerian duct regression in the mouse. Biol. Reprod. 78, 994–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wong Y. L., Behringer R. R., Kwan K. M. (2012) Smad1/Smad5 signaling in limb ectoderm functions redundantly and is required for interdigital programmed cell death. Dev. Biol. 363, 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Retting K. N., Song B., Yoon B. S., Lyons K. M. (2009) BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 136, 1093–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pangas S. A., Li X., Umans L., Zwijsen A., Huylebroeck D., Gutierrez C., Wang D., Martin J. F., Jamin S. P., Behringer R. R., Robertson E. J., Matzuk M. M. (2008) Conditional deletion of Smad1 and Smad5 in somatic cells of male and female gonads leads to metastatic tumor development in mice. Mol. Cell. Biol. 28, 248–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haffter P., Granato M., Brand M., Mullins M. C., Hammerschmidt M., Kane D. A., Odenthal J., van Eeden F. J., Jiang Y. J., Heisenberg C. P., Kelsh R. N., Furutani-Seiki M., Vogelsang E., Beuchle D., Schach U., Fabian C., Nüsslein-Volhard C. (1996) The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 123, 1–36 [DOI] [PubMed] [Google Scholar]
- 16. Lele Z., Bakkers J., Hammerschmidt M. (2001) Morpholino phenocopies of the swirl, snailhouse, somitabun, minifin, silberblick, and pipetail mutations. Genesis 30, 190–194 [DOI] [PubMed] [Google Scholar]
- 17. Hild M., Dick A., Rauch G. J., Meier A., Bouwmeester T., Haffter P., Hammerschmidt M. (1999) The smad5 mutation somitabun blocks Bmp2b signaling during early dorsoventral patterning of the zebrafish embryo. Development 126, 2149–2159 [DOI] [PubMed] [Google Scholar]
- 18. Kramer C., Mayr T., Nowak M., Schumacher J., Runke G., Bauer H., Wagner D. S., Schmid B., Imai Y., Talbot W. S., Mullins M. C., Hammerschmidt M. (2002) Maternally supplied Smad5 is required for ventral specification in zebrafish embryos prior to zygotic Bmp signaling. Dev. Biol. 250, 263–279 [PubMed] [Google Scholar]
- 19. McReynolds L. J., Gupta S., Figueroa M. E., Mullins M. C., Evans T. (2007) Smad1 and Smad5 differentially regulate embryonic hematopoiesis. Blood 110, 3881–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dee C. T., Gibson A., Rengifo A., Sun S. K., Patient R. K., Scotting P. J. (2007) A change in response to Bmp signalling precedes ectodermal fate choice. Int. J. Dev. Biol. 51, 79–84 [DOI] [PubMed] [Google Scholar]
- 21. Westerfield M. (2000) The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio), 4th Ed., University of Oregon Press, Eugene, OR [Google Scholar]
- 22. Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- 23. Chen C. H., Sun Y. H., Pei D. S., Zhu Z. Y. (2009) Comparative expression of zebrafish lats1 and lats2 and their implication in gastrulation movements. Dev. Dyn. 238, 2850–2859 [DOI] [PubMed] [Google Scholar]
- 24. Nordnes S., Krauss S., Johansen T. (1994) cDNA sequence of zebrafish (Brachydanio rerio) translation elongation factor-1 α. Molecular phylogeny of eukaryotes based on elongation factor-1 α protein sequences. Biochim. Biophys. Acta 1219, 529–532 [DOI] [PubMed] [Google Scholar]
- 25. Keegan B. R., Feldman J. L., Lee D. H., Koos D. S., Ho R. K., Stainier D. Y., Yelon D. (2002) The elongation factors Pandora/Spt6 and Foggy/Spt5 promote transcription in the zebrafish embryo. Development 129, 1623–1632 [DOI] [PubMed] [Google Scholar]
- 26. Alcaraz-Pérez F., Mulero V., Cayuela M. L. (2008) Application of the dual-luciferase reporter assay to the analysis of promoter activity in zebrafish embryos. BMC Biotechnol. 8, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y., Bourgeois C. F., Pang S., Kudla M., Dreumont N., Kister L., Sun Y. H., Stevenin J., Elliott D. J. (2009) The germ cell nuclear proteins hnRNP G-T and RBMY activate a testis-specific exon. PLoS Genet. 5, e1000707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poulain M., Lepage T. (2002) Mezzo, a paired-like homeobox protein is an immediate target of Nodal signalling and regulates endoderm specification in zebrafish. Development 129, 4901–4914 [DOI] [PubMed] [Google Scholar]
- 29. Thisse B., Heyer V., Lux A., Alunni V., Degrave A., Seiliez I., Kirchner J., Parkhill J. P., Thisse C. (2004) Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 77, 505–519 [DOI] [PubMed] [Google Scholar]
- 30. Kudoh T., Tsang M., Hukriede N. A., Chen X., Dedekian M., Clarke C. J., Kiang A., Schultz S., Epstein J. A., Toyama R., Dawid I. B. (2001) A gene expression screen in zebrafish embryogenesis. Genome Res. 11, 1979–1987 [DOI] [PubMed] [Google Scholar]
- 31. Rhinn M., Lun K., Luz M., Werner M., Brand M. (2005) Positioning of the midbrain-hindbrain boundary organizer through global posteriorization of the neuroectoderm mediated by Wnt8 signaling. Development 132, 1261–1272 [DOI] [PubMed] [Google Scholar]
- 32. Yabe T., Shimizu T., Muraoka O., Bae Y. K., Hirata T., Nojima H., Kawakami A., Hirano T., Hibi M. (2003) Ogon/Secreted Frizzled functions as a negative feedback regulator of Bmp signaling. Development 130, 2705–2716 [DOI] [PubMed] [Google Scholar]
- 33. Gilardelli C. N., Pozzoli O., Sordino P., Matassi G., Cotelli F. (2004) Functional and hierarchical interactions among zebrafish vox/vent homeobox genes. Dev. Dyn. 230, 494–508 [DOI] [PubMed] [Google Scholar]
- 34. Wang H., Long Q., Marty S. D., Sassa S., Lin S. (1998) A zebrafish model for hepatoerythropoietic porphyria. Nat. Genet. 20, 239–243 [DOI] [PubMed] [Google Scholar]
- 35. Herbomel P., Thisse B., Thisse C. (1999) Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development 126, 3735–3745 [DOI] [PubMed] [Google Scholar]
- 36. Bennett C. M., Kanki J. P., Rhodes J., Liu T. X., Paw B. H., Kieran M. W., Langenau D. M., Delahaye-Brown A., Zon L. I., Fleming M. D., Look A. T. (2001) Myelopoiesis in the zebrafish, Danio rerio. Blood 98, 643–651 [DOI] [PubMed] [Google Scholar]
- 37. Liu Z., Lin X., Cai Z., Zhang Z., Han C., Jia S., Meng A., Wang Q. (2011) Global identification of SMAD2 target genes reveals a role for multiple co-regulatory factors in zebrafish early gastrulas. J. Biol. Chem. 286, 28520–28532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Belting H. G., Wendik B., Lunde K., Leichsenring M., Mössner R., Driever W., Onichtchouk D. (2011) Pou5f1 contributes to dorsoventral patterning by positive regulation of vox and modulation of fgf8a expression. Dev. Biol. 356, 323–336 [DOI] [PubMed] [Google Scholar]
- 39. Wang L., Liu Y. T., Hao R., Chen L., Chang Z., Wang H. R., Wang Z. X., Wu J. W. (2011) Molecular mechanism of the negative regulation of Smad1/5 by CHIP. J. Biol. Chem. 286, 15883–15894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Müller F., Blader P., Rastegar S., Fischer N., Knöchel W., Strähle U. (1999) Characterization of zebrafish smad1, smad2 and smad5. The amino-terminus of smad1 and smad5 is required for specific function in the embryo. Mech. Dev. 88, 73–88 [DOI] [PubMed] [Google Scholar]
- 41. Hammerschmidt M., Serbedzija G. N., McMahon A. P. (1996) Genetic analysis of dorsoventral pattern formation in the zebrafish. Requirement of a BMP-like ventralizing activity and its dorsal repressor. Genes Dev. 10, 2452–2461 [DOI] [PubMed] [Google Scholar]
- 42. Mullins M. C., Hammerschmidt M., Kane D. A., Odenthal J., Brand M., van Eeden F. J., Furutani-Seiki M., Granato M., Haffter P., Heisenberg C. P., Jiang Y. J., Kelsh R. N., Nüsslein-Volhard C. (1996) Genes establishing dorsoventral pattern formation in the zebrafish embryo. The ventral specifying genes. Development 123, 81–93 [DOI] [PubMed] [Google Scholar]
- 43. Chen D., Zhao M., Mundy G. R. (2004) Bone morphogenetic proteins. Growth Factors 22, 233–241 [DOI] [PubMed] [Google Scholar]
- 44. Eisen J. S., Smith J. C. (2008) Controlling morpholino experiments. Don't stop making antisense. Development 135, 1735–1743 [DOI] [PubMed] [Google Scholar]
- 45. Kafri R., Springer M., Pilpel Y. (2009) Genetic redundancy. New tricks for old genes. Cell 136, 389–392 [DOI] [PubMed] [Google Scholar]
- 46. Maéno M., Ong R. C., Suzuki A., Ueno N., Kung H. F. (1994) A truncated bone morphogenetic protein 4 receptor alters the fate of ventral mesoderm to dorsal mesoderm. Roles of animal pole tissue in the development of ventral mesoderm. Proc. Natl. Acad. Sci. U.S.A. 91, 10260–10264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu C., Fan Z. P., Müller P., Fogley R., DiBiase A., Trompouki E., Unternaehrer J., Xiong F., Torregroza I., Evans T., Megason S. G., Daley G. Q., Schier A. F., Young R. A., Zon L. I. (2012) Nanog-like regulates endoderm formation through the Mxtx2-Nodal pathway. Dev. Cell 22, 625–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Whitman M., Raftery L. (2005) TGFβ signaling at the summit. Development 132, 4205–4210 [DOI] [PubMed] [Google Scholar]
- 49. Feng X. H., Derynck R. (2005) Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 [DOI] [PubMed] [Google Scholar]
- 50. von Bubnoff A., Cho K. W. (2001) Intracellular BMP signaling regulation in vertebrates. Pathway or network? Dev. Biol. 239, 1–14 [DOI] [PubMed] [Google Scholar]
- 51. Schier A. F., Talbot W. S. (2005) Molecular genetics of axis formation in zebrafish. Annu. Rev. Genet. 39, 561–613 [DOI] [PubMed] [Google Scholar]
- 52. Henningfeld K. A., Friedle H., Rastegar S., Knöchel W. (2002) Autoregulation of Xvent-2B. Direct interaction and functional cooperation of Xvent-2 and Smad1. J. Biol. Chem. 277, 2097–2103 [DOI] [PubMed] [Google Scholar]
- 53. Shima D. T., Kuroki M., Deutsch U., Ng Y. S., Adamis A. P., D'Amore P. A. (1996) The mouse gene for vascular endothelial growth factor. Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post-transcriptional regulatory sequences. J. Biol. Chem. 271, 3877–3883 [DOI] [PubMed] [Google Scholar]
- 54. Li W., Chen F., Nagarajan R. P., Liu X., Chen Y. (2001) Characterization of the DNA-binding property of Smad5. Biochem. Biophys. Res. Commun. 286, 1163–1169 [DOI] [PubMed] [Google Scholar]
- 55. He C., Chen X. (2005) Transcription regulation of the vegf gene by the BMP/Smad pathway in the angioblast of zebrafish embryos. Biochem. Biophys. Res. Commun. 329, 324–330 [DOI] [PubMed] [Google Scholar]
- 56. Tremblay K. D., Dunn N. R., Robertson E. J. (2001) Mouse embryos lacking Smad1 signals display defects in extra-embryonic tissues and germ cell formation. Development 128, 3609–3621 [DOI] [PubMed] [Google Scholar]
- 57. Lechleider R. J., Ryan J. L., Garrett L., Eng C., Deng C., Wynshaw-Boris A., Roberts A. B. (2001) Targeted mutagenesis of Smad1 reveals an essential role in chorioallantoic fusion. Dev. Biol. 240, 157–167 [DOI] [PubMed] [Google Scholar]
- 58. Chang H., Huylebroeck D., Verschueren K., Guo Q., Matzuk M. M., Zwijsen A. (1999) Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 126, 1631–1642 [DOI] [PubMed] [Google Scholar]
- 59. Yang X., Castilla L. H., Xu X., Li C., Gotay J., Weinstein M., Liu P. P., Deng C. X. (1999) Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development 126, 1571–1580 [DOI] [PubMed] [Google Scholar]
- 60. Arnold S. J., Maretto S., Islam A., Bikoff E. K., Robertson E. J. (2006) Dose-dependent Smad1, Smad5, and Smad8 signaling in the early mouse embryo. Dev. Biol. 296, 104–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hazen V. M., Andrews M. G., Umans L., Crenshaw E. B., 3rd, Zwijsen A., Butler S. J. (2012) BMP receptor-activated Smads confer diverse functions during the development of the dorsal spinal cord. Dev. Biol. 367, 216–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jia S., Dai F., Wu D., Lin X., Xing C., Xue Y., Wang Y., Xiao M., Wu W., Feng X. H., Meng A. (2012) Protein phosphatase 4 cooperates with Smads to promote BMP signaling in dorsoventral patterning of zebrafish embryos. Dev. Cell 22, 1065–1078 [DOI] [PubMed] [Google Scholar]
- 63. Labbé E., Silvestri C., Hoodless P. A., Wrana J. L., Attisano L. (1998) Smad2 and Smad3 positively and negatively regulate TGFβ-dependent transcription through the forkhead DNA-binding protein FAST2. Mol. Cell 2, 109–120 [DOI] [PubMed] [Google Scholar]
- 64. Park S., Lee Y. J., Lee H. J., Seki T., Hong K. H., Park J., Beppu H., Lim I. K., Yoon J. W., Li E., Kim S. J., Oh S. P. (2004) B-cell translocation gene 2 (Btg2) regulates vertebral patterning by modulating bone morphogenetic protein/smad signaling. Mol. Cell Biol. 24, 10256–10262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim R. H., Wang D., Tsang M., Martin J., Huff C., de Caestecker M. P., Parks W. T., Meng X., Lechleider R. J., Wang T., Roberts A. B. (2000) A novel smad nuclear interacting protein, SNIP1, suppresses p300-dependent TGF-β signal transduction. Genes Dev. 14, 1605–1616 [PMC free article] [PubMed] [Google Scholar]
- 66. Schmerer M., Evans T. (2003) Primitive erythropoiesis is regulated by Smad-dependent signaling in postgastrulation mesoderm. Blood 102, 3196–3205 [DOI] [PubMed] [Google Scholar]