

Background: Vaccinia viral protein A27 associates with viral membrane protein A17 for anchoring to the viral membrane.
Results: A27 specifically interacts with two binding regions within the N-terminal domain of A17.
Conclusion: The A27-A17 interaction is mediated through a specific and cooperative binding mechanism.
Significance: As demonstrated, the F1 and F2 bindings are critical for A27 anchoring to the viral membrane and virion assembly.
Keywords: Pox Viruses, Protein Structure, Protein-Protein Interactions, Viral Protein, Virus Assembly
Abstract
The vaccinia viral protein A27 in mature viruses specifically interacts with heparan sulfate for cell surface attachment. In addition, A27 associates with the viral membrane protein A17 to anchor to the viral membrane; however, the specific interaction between A27 and A17 remains largely unclear. To uncover the active binding sites and the underlying binding mechanism, we expressed and purified the N-terminal (18–50 residues) and C-terminal (162–203 residues) fragments of A17, which are denoted A17-N and A17-C. Through surface plasmon resonance, the binding affinity of A27/A17-N (KA = 3.40 × 108 m−1) was determined to be approximately 3 orders of magnitude stronger than that of A27/A17-C (KA = 3.40 × 105 m−1), indicating that A27 prefers to interact with A17-N rather than A17-C. Despite the disordered nature of A17-N, the A27-A17 interaction is mediated by a specific and cooperative binding mechanism that includes two active binding sites, namely 32SFMPK36 (denoted as F1 binding) and 20LDKDLFTEEQ29 (F2). Further analysis showed that F1 has stronger binding affinity and is more resistant to acidic conditions than is F2. Furthermore, A27 mutant proteins that retained partial activity to interact with the F1 and F2 sites of the A17 protein were packaged into mature virus particles at a reduced level, demonstrating that the F1/F2 interaction plays a critical role in vivo. Using these results in combination with site-directed mutagenesis data, we established a computer model to explain the specific A27-A17 binding mechanism.
Introduction
Vaccinia virus is the prototype of the Orthopoxvirus genus of the Poxviridae family, the genome of which is ∼190 kb in size and encodes more than 200 viral proteins (1, 2). Vaccinia virus has a wide host range and replicates in the cytoplasm of the infected cells. Two forms of infectious virus particles are produced by the infected cells as follows: the mature virus (MV),3 which accounts for more than 90% of infectious particles, and the extracellular virus. The MV contains ∼80 viral proteins (3–5), of which at least 16 play a role in the virus entry process. The MV attaches to cell surface glycosaminoglycans and laminin through four envelope proteins: H3, D8, A27, and A26 (6–10). The viral L1 protein also mediates nonglycosaminoglycan binding to cells (11). Subsequently, the MV particles cluster at membrane lipid rafts (12), interact with integrin (13) and CD98 (14), and then undergo endocytosis (15–17). After uptake into cellular vesicles, the acidic environment triggers membrane fusion mediated by the viral entry fusion complex (18). Despite the fact that the vaccinia virus entry pathways differ depending on the virus strain (19) and cell type (17), recent studies have shown that three cell lines, HeLa, CHO, and L cells, exhibit the following phenomenon; the deletion of A25/A26 proteins in the wild-type WR strain induces a switch in virus entry from endocytosis to plasma membrane fusion (19, 20). These results support the model that in these three cell lines A25 and A26 proteins are acid-sensitive fusion suppressors of the MV (20). Neither the A25 nor the A26 protein contains a transmembrane region, and the incorporation of the A25 and A26 protein complex into the MV particles relies on the A27 protein (20–22), which, in turn, binds to an integral viral membrane A17 protein for packaging into MV particles (23). Thus, it is noteworthy that, in addition to binding to cell surface HS, the A27 protein functions as an adaptor between the A26 and A17 proteins and that the levels of A25 and A26 proteins in the MV would be minimal in the absence of A27 tethering to A17.
A27 consists of 110 amino acids encoding five functional domains (Fig. 1A), including an N-terminal heparin-binding sequence (HBS, 21–32 residues) essential for heparin binding to the cell surface (6), a flexible spacer (33–42 residues), an α-helical coiled-coil domain (CCD, 43–65 residues) critical for A27 oligomerization, a disulfide-linked domain (DLD, 66–84 residues) (24), and a C-terminal leucine zipper domain (LZD, 85–110 residues) responsible for anchoring to the viral membrane via interaction with A17 (23, 25). Previous studies have suggested that the N- and C-terminal domains are structurally distinct. For instance, the HBS is a flexible random coil, whereas the CCD, DLD, and LZD form rigid consecutive heptad repeated coiled coils (25, 26). Despite their structural differences, both the N-terminal HBS and the C-terminal coiled coils cooperatively provide the specificity required for the interaction of the A27 protein with HS on the cell surface (26). Moreover, a penta-amino acid segment, KKPEA, in the flexible HBS forms a turn-like structure critical for heparin binding in vitro and HS binding on cells (26). Finally, the crystal structure of a truncated A27 protein (21–84 residues) was recently resolved and, consistent with previous results, revealed that the A27 protein assembles as a trimer containing two parallel α-helices and one antiparallel α-helix (27). Based on the current knowledge of the A27 protein structure, in this study we aim to understand the molecular criteria required for the interaction between the LZD of the A27 protein and the A17 protein.
FIGURE 1.

Illustration of the functional domains in the vaccinia viral envelope protein A27 and the viral membrane protein A17. A, A27 protein used in this study is divided into five functional domains as follows: a flexible Lys/Arg-rich domain (21–32 residues) known as the heparin-binding site (HBS) (6), a flexible spacer (33–42 residues), an α-helical CCD (43–65 residues) critical for A27 oligomerization, DLD (66–84 residues), and LZD (85–110 residues) responsible for anchoring to the viral membrane via interaction with A17. Two adjacent Cys residues (CC) at positions 71 and 72 are labeled by triangles. The three Leu residues within the LZD and part of the DLD responsible for the A17 interaction are shown below (23). Eleven A27(21–110) mutants were used in this study, and the mutated residues are highlighted in the last panel. B, A17 (18–203 residues) consists of an N-terminal domain (18–60 residues), two transmembrane (TM) domains (61–98 and 113–161 residues), and a C-terminal domain (162–203 residues). In the infected cells, A17 is synthesized as a 23-kDa precursor of 203 amino acids and is subsequently cleaved at both the N and C termini during maturation (30), as indicated by arrows. The three A17 fragments used in this study are shown below: A17-N, A17-C, and A17-S, in which the A17-N sequence 24LFTEEQQQSFM34 was scrambled to 24QLESQFQTMEF34. These A17 fragments tagged with thioredoxin were expressed in E. coli, and the tag was removed by factor Xa cleavage (for details, see “Experimental Procedures”). Eight A17-N mutants, including five scrambled-sequence mutants and three single mutants, were synthesized using a solid phase synthesizer. The mutated or scrambled residue(s) are highlighted. To facilitate the HSQC analysis, two residues, Phe33 and Phe25, were specifically 15N isotope-labeled. The Coomassie Brilliant Blue staining of A27 and A17 recombinant proteins is shown in C.
The viral membrane protein A17 is conserved in Poxviridae and is required for MV assembly (28–30). When the A17 gene is repressed, the accumulation of large, electron-dense aggregates becomes surrounded by vesicular structures (28, 30). In infected cells, A17 is synthesized as a 23-kDa precursor of 203 amino acids and undergoes phosphorylation by viral F10 kinase (31–33) and subsequent cleavage at the “AGX” consensus motif present in both the N and C termini by viral protease I7 during maturation (Fig. 1B) (23, 31, 34). The processed A17 protein spans the membrane twice, and both ends of the protein are oriented toward the cytoplasm in the infected cells (35, 36). After being packaged into MV particles, the N terminus of A17 is exposed on the surface of the virions, whereas the C terminus appears embedded within the viral membranes (36, 37). A17 exists predominantly as dimers and forms stable complexes with A27 (23, 25, 38) and A14 (23, 31, 33).
To address the active binding sites and the underlying mechanism responsible for the specific interaction between A27 and A17, we produced N-terminal (18–50 residues) and C-terminal (162–203 residues) fragments of A17, denoted A17-N and A17-C, and we analyzed their interactions with the A27 protein through a variety of techniques, including NMR, CD, and SPR. Based on these in vitro results, a molecular model revealing a cooperative interaction between the A17 and A27 proteins during viral protein complex formation is proposed. Finally, we constructed recombinant vaccinia viruses expressing an A27 protein containing mutations within the A17-binding sites to validate our model.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis
The in vitro mutagenesis of A27 protein (21–110 residues) was performed (Yao-Hong Biotechnology Inc., Taiwan) to substitute the Val78, Phe80, Arg81, Asn84, Glu87, Thr88, Ile94, Ser95, Lys98, Lys99, or Val102 residues to Ala. The resulting mutant A27 ORFs were cloned into pET21a (Novagen) as described previously (6). All of the mutations in the A27 ORFs were confirmed by DNA sequencing.
Purification of Recombinant Proteins
The preparations of all of the recombinant A27 proteins were based on a previously described method (7), and these recombinant proteins contained a T7 tag at the N terminus and a hexahistidine tag at the C terminus, which did not interfere with the function of wild-type A27 (6). For heteronuclear NMR studies, the transformed bacteria were grown at 37 °C in M9 medium supplemented with [15N]ammonium chloride (1 g/liter) and [13C]glucose (1 g/liter) (Cambridge Isotope Laboratories, Inc., Andover, MA) as the sole nitrogen and carbon sources, respectively, to an absorbance at 600 nm of 0.8. The cells were then induced for 2 h at 37 °C with 1 mm isopropyl 1-thio-β-d-galactopyranoside and harvested; the proteins were purified on a nickel-nitrilotriacetic acid affinity column, as described previously (6).
To achieve better yields, the plasmids containing the A17 constructs, namely A17-N (18–50 residues of WT-A17), A17-C (162–203 residues of WT-A17), and A17-S (same as A17-N, but the 24–34 residues were scrambled), were modified through the attachment of a thioredoxin fusion protein, a hexahistidine tag, and a factor Xa cleavage site at the N terminus. These A17 fragments tagged with thioredoxin were expressed in Escherichia coli and purified by Ni2+-nitrilotriacetic acid-agarose beads (Novagen). For heteronuclear NMR analysis, A17-N, A17-C, and A17-S were expressed in M9 medium, as described above. The tag was then removed by incubation with factor Xa protease at 25 °C for 1 h in 0.1 mm factor Xa buffer (pH 8.0) followed by purification through HPLC. After purification, the protein samples were exchanged into NMR buffer (10 mm sodium phosphate buffer (pH 6.5), 150 mm sodium chloride, 50 mm dithiothreitol (DTT), and 10% D2O). The final samples were concentrated to 0.2 mm and transferred into Shigemi NMR tubes. The purification of these proteins was verified by SDS-PAGE, and the concentration was measured through the BCA method (Pierce).
33-mer A27-binding Peptide Synthesis
Eight 33-mer peptides corresponding to A17-N (GVLDKDLFTEEQQQSFMPKDGGMMQNDYGGMND) were synthesized using an automated, solid phase synthesizer 433A (Applied Biosystems, Stafford, TX) based on Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry and PyBOP activation. The peptides were purified by HPLC using a reverse phase C-18 column (SUPELCO, 250 × 10 mm, 5 μm) on an Agilent 1100 system. The purity and homogeneity were confirmed by MALDI-TOF mass spectrometry.
SPR
Real time SPR experiments were performed on a BIAcore Biosensor 3000 system (BIAcore, Uppsala, Sweden) as described previously (24). CM5 sensor chips were purchased from BIAcore. To analyze the A27 (21–110 residues, termed as A27 hereafter) binding affinity of A17, we covalently immobilized the A27 protein on the CM5 chip surface. The immobilization procedure included three steps in 10 mm acetate running buffer at pH 5.0 and a flow rate of 10 μl/min. First, the carboxyl groups of the dextran matrix on the surface of the CM5 sensor chip was activated by a mixture of 0.2 m 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide and N-hydroxysuccinimide to create active succinimide esters. In the following step, the A27 protein was passed over the CM5 surface, and the esters on the surface reacted spontaneously with the primary amine or nucleophilic groups of A27 to allow the A27 to covalently link to the dextran matrix. In the final step, the cells were washed with 1 m ethanolamine (pH 8.5, 10 μl/min) for 7 min to remove the unbound proteins. The A27-coated surfaces were created with an enhanced response unit value of 7000–8000. The recombinant proteins A17-N, A17-C, and A17-S containing thioredoxin at five different concentrations (10, 5, 2.5, 1.25, and 0.625 μm) were injected over the A27-coated surface for 300 s for association, and running buffer (10 mm HEPES (pH 7.0), containing 0.15 m NaCl and 0.005% Polysorbate 20) was then injected for 300 s for dissociation at a flow rate of 30 μl/min. In a control experiment, thioredoxin at five different concentrations (10, 5, 2.5, 1.25, and 0.625 μm) was solely injected over the same A27-coated surface to determine the noise level in the SPR response, which indicated that thioredoxin does not interact with immobilized A27. Parallel injections of analytes over a control surface were performed for background measurements. After subtraction of nonspecific binding curves, the association and dissociation rate constants were derived from either a two-state binding model (39) or a 1:1 Langmuir binding model (40). The coated surface was regenerated for subsequent injections by three successive injections of 30 μl of 1 m NaCl, 30 μl of 2 m MgCl2, and 30 μl of 100 mm imidazole (30 μl/min).
NMR Spectroscopy
The 1H NMR spectra were recorded on a Bruker DRX500 spectrometer operating at 500.13 MHz. The spectra were collected in H2O/D2O (90:10) at 25 °C and a repetition time of 1.5 s. The chemical shifts were referenced to 2,2-dimethyl-2-silapentane-5-sulfonate according to the water resonance calibrated at 25 °C. The NOESY spectra (mixing time of 300 ms) and TOCSY (mixing time of 80 ms) spectra were recorded over a spectrum bandwidth of 4.7 kHz using 512 t1 blocks of 80 scans (NOESY) or 72 scans (TOCSY), each consisting of 2048 t2 points. The two-dimensional data sets were processed using the 60° sine-squared bell window function in both dimensions and zero-filling to obtain 2048 × 1048 data points prior to Fourier transformation.
Heteronuclear correlation NMR measurements were performed on Bruker 600-MHz or 800-MHz spectrometers equipped with a 5-mm inverse triple resonance (1H/13C/15N) z axis gradient probe. For the backbone resonance assignments, 1H-15N HSQC (41), HNCA (42), HN(CO)CA (42), HN(CA)CB (43), and HN(COCA)CB (43) experiments were conducted at 25 °C. The detailed procedures for NMR measurement, data acquisition, and analysis have been described previously (44).
CD Spectroscopy
The CD spectra were recorded on a Jasco J-815 spectrometer equipped with a water bath for temperature control. All CD spectra were collected at 25 °C using a quartz cuvette with a 1-mm path length and a protein concentration of 50 μm. The step size was 0.2 nm with a 1.0-nm bandwidth at a scan speed of 50 nm/min. Each spectrum shown represents the average of three measurements. All spectra were collected in 50 mm potassium phosphate buffer with background buffer correction.
Cell Culture, Reagents, and Viruses
HeLa, L, BSC40, and BSC40 cells expressing the A27 protein (BSC40-A27) (27) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen). The Western Reserve (WR) strain of vaccinia virus and WR-ΔA27L were described previously (10, 27). The purification of vaccinia MV was performed through 36% sucrose cushion followed by CsCl gradient purification as described previously (45, 46). Anti-A27 and anti-H3 antibodies were described previously (8, 22). Anti-A17 antibodies were raised in New Zealand White rabbits against a synthetic peptide (AGVLDKDLFTEEQQQSFMPKD) containing A17 sequences.
Construction of WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A Viruses
In vitro mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene) to obtain three mutant A27L ORFs encoding the full-length WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A. Next, the wild-type and three mutant A27L ORFs were individually cloned into pMJ601 to produce pMJ601-A27-Rev, pMJ601-A27-E87A, pMJ601-A27-I94A, and pMJ601-A27L-E87A,I94A, all of which were confirmed by DNA sequencing. The resulting plasmids were transfected individually into 293T cells, and the cells were subsequently infected with WR-ΔA27L at an m.o.i. of 1 pfu/cell, cultured for 2–3 days, and harvested for recombinant virus isolation on BSC40-A27 cells, as described previously (27), with three rounds of plaque purification in 1% agar with 150 μg/ml of 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal). To obtain purified MV stocks, BSC40 cells were infected with WR, WR-ΔA27L, WR-A27-E87A, WR-A27-I94A, or WR-A27-E87A,I94A viruses at an m.o.i. of 1 pfu/cell and subsequently harvested at 2–3 days p.i. for CsCl gradient purification as described previously (45, 46).
Plaque Formation on BSC40 and BSC40-A27 Cells
BSC40 and BSC40-A27 cells were seeded in 6-well plates at 2.7 × 105 cells per well. The next day, the cells were infected with WR-A27-Rev, WR-ΔA27L, WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A viruses at 37 °C for 1 h and subsequently cultured in growth medium containing 1% agar. At 3 days p.i., the cells were fixed in 4% formaldehyde and stained with 1% crystal violet in 20% ethanol (Sigma) to visualize the plaques.
Coimmunoprecipitation
HeLa cells were either mock-infected or infected with each virus at an m.o.i. of 5 pfu/cell and harvested at 24 h p.i. The cells were lysed in lysis buffer (1% Triton X-100, 150 mm NaCl, 1 mm EDTA, and 50 mm Tris (pH 7.4)) supplemented with protease inhibitors, and the insoluble materials were removed by centrifugation (10,000 × g, 15 min, 4 °C). The cell lysates were further precleared by incubation with 50 μl of a 50% protein A-Sepharose bead slurry with gentle agitation for 1 h at 4 °C in a tube rotator. After brief centrifugation, the precleared supernatant was incubated with anti-A17 (1:100) or anti-A27 (1:100) antibodies and 50 μl of a 50% protein A-Sepharose bead slurry at 4 °C for 2 h. The immunoprecipitates were washed four times with lysis buffer and stored for immunoblot analyses.
Cell Fusion Assay (Fusion from Without)
Cell fusion assays were performed as described previously (27). In brief, L cells expressing GFP or RFP were mixed at a 1:1 ratio and seeded in 96-well plates (4 × 104 cells/well). The next day, the cells were pretreated with 40 μg/ml cordycepin (Sigma) for 60 min and subsequently infected with WT WR, WR-ΔA27L, WR-A27-Rev, WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A viruses at a m.o.i. of 50 pfu/cell; these infections were performed in duplicate. After infection at 37 °C for 30 min, the cells were incubated at 37 °C in the presence of 40 μg/ml cordycepin and photographed at 2 h p.i. using a Zeiss Axiovert fluorescence microscope. The percentage of cells containing both GFP and RFP fluorescence was quantified to assess the degree of cell-cell fusion using the Axio Vision Rel. 4.8 software.
A27-A17 Complex Molecular Modeling and Docking Analysis
To resolve the A27-A17 complex formation, we performed molecular modeling of the 18-mer 20LDKDLFTEEQQQSFMPKD37 and 22-mer 80FRLENHAETLRAAMISLAKKID101, which are representative of the active binding sites of A17-N and A27, respectively. The molecular structures of these two polypeptides were created using the Discover Studio 3.1 program and typed using the CHARMM Force Field software. The structure of the 18-mer was built by setting up the constraints with the NOE data. Appropriate counterions, such as sodium and chloride ions, were added to neutralize the total charge, and the three-dimensional structure was subjected to analysis using a standard dynamic cascade with an implicit solvent model of Generalized Born Switching (GBSW) for 1 ps. Twenty conformers were accumulated and optimized by energy minimization to a root mean square gradient (0.00001). The most adequate conformer was selected, and the conformation of A27 was created as a typical α-helix template. The protein-protein interaction between the 18-mer A17 peptide and the 22-mer A27 peptide was obtained using a manual procedure to determine the best fit. The electrostatic interactions were calculated using the Particle Mesh Ewald method.
RESULTS
A27 Binds to the N Terminus of A17, as Revealed by HSQC
To determine the A27 and A17 active binding sites, we expressed and purified the wild-type recombinant A27 protein (21–110 residues) in E. coli. Additionally, a series of 12 Ala mutations were introduced into the LZD domain of the A27 protein (Fig. 1A). Furthermore, we expressed three A17 protein fragments in E. coli; these contained either the N-terminal 18–50 amino acids (A17-N) or the C-terminal 162–203 amino acids (A17-C), or a sequence scramble of the N-terminal sequence (A17-S). In addition, eight mutant constructs of A17-N were also generated (Fig. 1B). All of the recombinant proteins were soluble and purified by nickel affinity chromatography as described under “Experimental Procedures” (Fig. 1C).
The crystal structure of a truncated A27 protein (21–84 residues) was recently solved (27). Because of the intrinsic flexibility of HBS and the spacer (21–42 residues), its electron density maps were not visible, whereas the remaining part (43–84 residues) forms three coiled-coil arrangements in the crystal structure, which provide the spatial stability of the A27 assembly (27). Thus, the two-dimensional HSQC spectrum of A27 at pH 6.5 (Fig. 2A) shows only the signals originating from the flexible random coil (21–42 residues), whereas any remaining signals arising from the structured domains, such as CCD, DLD, and LZD (i.e. the A17 binding domain), were absent, consistent with the truncated A27 crystal structure (27) and previous reports (24, 26, 44). In contrast, as shown in Fig. 2, B and C, the two-dimensional HSQC spectra of both A17-C and A17-N gave rise to a much better spectral resolution under the same experimental conditions because all of the cross-peaks were present with no missing signals. Therefore, compared with A17-N or A17-C, A27 was less suitable for the study of the active binding residues of the A27-A17 interaction by HSQC. Thus, we used A17-N and A17-C as the target molecules in an attempt to uncover the active A27-binding residues.
FIGURE 2.

HSQC analyses and in vitro binding assay. Two-dimensional 1H/15N HSQC spectra of A27 (A), A17-C (B), A17-N (C), and A17-S (D) at pH 6.5. As observed in A, the HSQC pattern revealed only one-third of the total residues, and the remaining two-thirds of the residues involved in protein self-assembly were absent. B–D, A17 fragments/A27 binding were analyzed by HSQC, and the cross-peaks are colored in red in the absence of A27 and in blue in the presence of A27. B and D, no chemical shift perturbations were found. As indicated in C, two sets of segmental cross-peaks were perturbed due to the presence of A27: 32SFMPK36 (denoted as the F1 segment) and 20LDKDLFTEEQ29 (F2). The sequential assignments of the A27 and A17 fragments were determined by three-dimensional heteronuclear correlation NMR experiments (47, 48) based on the inter- and intra-residue chemical shift correlations. The single-letter abbreviations for amino acids are used. The numbering is based on the sequences of WT-A17 and WT-A27. E, representative cross-peaks of the F2 residues (Leu20 and Gln27) and the F1 residues (Phe33 and Met34) reveal a linear diminution as a function of the A17-N-A27 molar ratio (1:0, 1:0.25, 1:0.5, 1:0.75, and 1:1) at pH 6.5 from top to bottom. Notice that the F1 residues decrease faster than the F2 residues, implying that F1 has a stronger binding affinity than F2. In contrast, the cross-peaks of those residues not involved in A27 binding, such as Asp50, remained unchanged in the presence of A27. F, two-dimensional HSQC spectra of the synthetic peptides (from left to right) as follows: A17-S1, -S2, -S3, -S4 (upper row), and -S5, -L24A, -Q29A, and -P35A (lower row). As observed, the cross-peaks of the A17-N peptides in the presence (in blue) and absence of A27 (in red) arising from both Phe25 and Phe33 remain basically unchanged, implying that all of the A17-N mutants lost binding specificity for A27. For comparison, the two spectra were shifted by 0.235 ppm along the horizontal 1H dimension. Only two residues, Phe25 and Phe33, have been specifically 15N isotope-labeled in these synthetic peptides, corresponding to the upfield and downfield cross-peak signals along the vertical 15N dimension, respectively. For details, see “Experimental Procedures.” G, selective 1H/15N HSQC residual intensity ratio determined from the A17-N fragment with uniform 15N isotope labeling mixed with the A27 mutants (natural abundance). A17-N was mixed with the single mutants A27-E87A and A27-I94A and the double mutant A27-E87A,I94A, respectively, as well as the parental A27 for reference (at a 1:1 molar ratio). The residual intensity ratios were calculated from the intensity ratio of two sets of cross-peaks in the presence and absence of the A27 mutant and calibrated with respect to unperturbed Asp50. The effect of mutation upon F1 and F2 binding represented by Phe33 for F1 (black bar) and by Phe25 for F2 (gray bar) revealed that A27-E87A disrupts F2 binding and partially disables F1 binding, whereas A27-I94A caused a substantial disruption of both F1 and F2 binding. In addition, the double mutation A27-E87A,I94A completely abolished the F1 and F2 binding activities.
To facilitate the determination of the binding residues, we applied three-dimensional heteronuclear correlation NMR experiments (47, 48) to the A17 fragments through uniform 13C/15N isotope labeling at pH 6.5. Based on the intra- and inter-chemical shift correlations, we established the 1HN, 15NH, 13Cα, 13Cβ, and 13CO chemical shifts of these A17 fragments without any ambiguity (see Tables 1–3 and Fig. 2, B and C).
TABLE 1.
1HN, 15NH, 13Cα, 13Cβ, and 13CO inter-residual chemical shift correlations of A17-N fragment in PBS buffer at pH 6.5
| Residue | 1HN | 15NH | 13Cαi | 13Cαi-1 | 13Cβi | 13Cβi-1 | 13COi | 13COi-1 |
|---|---|---|---|---|---|---|---|---|
| Leu20 | 8.456 | 123.7 | 55.0 | 63.0 | 42.2 | 32.4 | 177.1 | 175.8 |
| Asp21 | 8.100 | 121.0 | 54.2 | 55.0 | 41.0 | 42.2 | 176.3 | 177.1 |
| Lys22 | 8.139 | 120.9 | 56.6 | 54.2 | 32.8 | 41.0 | 176.3 | 176.3 |
| Asp23 | 8.307 | 120.6 | 54.5 | 56.6 | 41.0 | 32.8 | 176.1 | 176.3 |
| Leu24 | 7.937 | 121.6 | 55.2 | 54.5 | 42.2 | 41.0 | 177.1 | 176.1 |
| Phe25 | 8.166 | 120.3 | 57.6 | 55.2 | 39.2 | 42.2 | 176.1 | 177.1 |
| Thr26 | 8.025 | 115.5 | 61.7 | 57.6 | 70.0 | 39.2 | 174.6 | 176.1 |
| Glu27 | 8.439 | 122.8 | 57.2 | 61.7 | 29.9 | 70.0 | 177.0 | 174.6 |
| Glu28 | 8.448 | 121.3 | 57.2 | 57.2 | 29.9 | 29.9 | 177.1 | 177.0 |
| Gln29 | 8.280 | 120.8 | 56.1 | 57.2 | 29.3 | 29.9 | 176.3 | 177.1 |
| Gln30 | 8.348 | 120.9 | 56.1 | 56.1 | 29.3 | 29.3 | 176.3 | 176.3 |
| Gln31 | 8.348 | 120.9 | 56.0 | 56.1 | 29.3 | 29.3 | 176.1 | 176.3 |
| Ser32 | 8.199 | 116.3 | 58.4 | 56.0 | 63.8 | 29.3 | 173.9 | 176.1 |
| Phe33 | 8.122 | 121.7 | 57.5 | 58.3 | 39.3 | 63.9 | 175.1 | 173.9 |
| Met34 | 8.109 | 123.5 | 52.8 | 57.5 | 32.5 | 39.3 | 173.6 | 175.1 |
| Pro35 | 63.0 | 52.8 | 32.2 | 32.5 | 177.0 | 173.6 | ||
| Lys36 | 8.487 | 121.8 | 56.3 | 63.0 | 32.8 | 32.2 | 176.5 | 177.0 |
| Asp37 | 8.323 | 120.9 | 54.2 | 56.3 | 41.1 | 32.8 | 176.7 | 176.5 |
| Gly38 | 8.368 | 109.4 | 45.6 | 54.2 | 41.1 | 174.9 | 176.7 | |
| Gly39 | 8.342 | 108.6 | 45.2 | 45.6 | 174.2 | 174.9 | ||
| Met40 | 8.197 | 119.5 | 55.5 | 45.2 | 32.8 | 176.3 | 174.2 | |
| Met41 | 8.434 | 121.3 | 55.5 | 55.5 | 32.8 | 32.8 | 176.1 | 176.3 |
| Gln42 | 8.368 | 121.3 | 55.7 | 55.6 | 29.3 | 32.8 | 175.5 | 176.1 |
| Asn43 | 8.421 | 119.8 | 53.1 | 55.8 | 38.9 | 29.3 | 174.6 | 175.5 |
| Asp44 | 8.272 | 120.5 | 54.1 | 53.2 | 41.0 | 38.9 | 176.0 | 174.6 |
| Tyr45 | 8.157 | 120.6 | 58.0 | 54.1 | 38.4 | 41.0 | 176.6 | 176.0 |
| Gly46 | 8.373 | 110.5 | 45.5 | 58.0 | 38.4 | 174.6 | 176.6 | |
| Gly47 | 8.021 | 108.6 | 45.2 | 45.5 | 174.2 | 174.6 | ||
| Met48 | 8.241 | 119.4 | 55.6 | 45.2 | 32.6 | 176.0 | 174.2 | |
| Asn49 | 8.478 | 119.8 | 53.1 | 55.6 | 39.2 | 32.6 | 173.9 | 176.0 |
| Asp50 | 7.906 | 126.1 | 55.9 | 53.1 | 41.9 | 39.2 | 180.8 | 173.9 |
TABLE 2.
1HN, 15NH, 13Cα, 13Cβ, and 13CO inter-residual chemical shift correlations of A17-C fragment in PBS buffer at pH 6.5
| Residue | 1HN | 15NH | 13Cαi | 13Cαi-1 | 13Cβi | 13Cβi-1 | 13COi | 13COi-1 |
|---|---|---|---|---|---|---|---|---|
| Tyr164 | 8.412 | 122.2 | 57.8 | 53.8 | 38.1 | 41.2 | 175.7 | 175.7 |
| Arg165 | 8.100 | 121.0 | 54.2 | 57.8 | 40.8 | 38.1 | 176.4 | 175.7 |
| Arg166 | 8.217 | 122.0 | 55.6 | 54.2 | 30.7 | 40.8 | 176.1 | 176.4 |
| Ser167 | 8.305 | 117.2 | 57.8 | 55.6 | 63.5 | 30.7 | 174.2 | 176.1 |
| Ile168 | 8.097 | 121.7 | 60.8 | 57.8 | 38.5 | 63.5 | 175.6 | 174.2 |
| Asn169 | 8.444 | 122.3 | 52.5 | 60.8 | 38.5 | 38.5 | 174.8 | 175.6 |
| Val170 | 8.073 | 120.4 | 61.7 | 52.5 | 32.4 | 38.5 | 176.0 | 174.8 |
| Thr171 | 8.277 | 118.7 | 61.7 | 61.7 | 69.2 | 32.4 | 174.0 | 176.0 |
| Ile172 | 8.208 | 125.2 | 58.2 | 61.7 | 38.1 | 69.2 | 174.0 | 174.0 |
| Pro173 | 63.0 | 31.5 | 176.9 | 174.0 | ||||
| Glu174 | 8.481 | 120.8 | 56.5 | 63.0 | 29.7 | 31.5 | 176.5 | 176.9 |
| Gln175 | 8.322 | 120.4 | 55.6 | 56.5 | 28.9 | 29.8 | 175.6 | 176.5 |
| Tyr176 | 8.208 | 120.9 | 57.8 | 55.6 | 38.5 | 28.9 | 175.9 | 175.6 |
| Thr177 | 8.018 | 115.8 | 61.3 | 57.8 | 69.6 | 38.5 | 174.0 | 175.9 |
| Cys178 | 8.272 | 121.0 | 57.8 | 61.3 | 27.6 | 69.6 | 174.1 | 174.0 |
| Asn179 | 8.472 | 121.3 | 53.0 | 57.8 | 38.5 | 27.6 | 174.5 | 174.1 |
| Lys180 | 8.124 | 122.5 | 54.0 | 53.0 | 32.4 | 38.5 | 174.3 | 174.5 |
| Pro181 | 62.6 | 31.5 | 176.4 | 174.3 | ||||
| Tyr182 | 8.276 | 120.9 | 57.8 | 62.6 | 38.5 | 31.5 | 175.8 | 176.4 |
| Thr183 | 7.911 | 117.2 | 60.8 | 57.8 | 69.6 | 38.5 | 173.6 | 175.8 |
| Ala184 | 8.129 | 126.5 | 52.5 | 60.9 | 18.9 | 69.6 | 177.9 | 173.6 |
| Gly185 | 8.282 | 107.9 | 45.1 | 52.5 | 18.8 | 173.8 | 177.9 | |
| Asn186 | 8.235 | 118.6 | 53.0 | 45.1 | 38.5 | 174.9 | 173.8 | |
| Lys187 | 8.286 | 122.0 | 56.0 | 53.0 | 32.8 | 38.5 | 176.2 | 174.9 |
| Val188 | 8.148 | 121.3 | 61.7 | 56.0 | 32.4 | 32.4 | 175.6 | 176.2 |
| Asp189 | 8.379 | 124.2 | 53.8 | 61.7 | 40.7 | 32.4 | 175.9 | 175.6 |
| Val190 | 7.973 | 118.9 | 61.7 | 53.8 | 32.4 | 40.7 | 175.5 | 175.9 |
| Asp191 | 8.365 | 123.5 | 53.8 | 61.7 | 40.7 | 32.4 | 175.5 | 175.5 |
| Ile192 | 7.985 | 122.6 | 58.2 | 53.9 | 38.9 | 40.7 | 176.9 | 175.5 |
| Pro193 | 63.0 | 31.5 | 176.8 | 176.9 | ||||
| Thr194 | 8.087 | 113.9 | 61.3 | 63.0 | 69.6 | 31.5 | 174.3 | 176.8 |
| Phe195 | 8.213 | 122.4 | 57.3 | 61.3 | 39.4 | 69.6 | 175.2 | 174.3 |
| Asn196 | 8.369 | 120.8 | 52.5 | 57.3 | 38.5 | 39.5 | 174.8 | 175.2 |
| Ser197 | 8.208 | 116.5 | 58.2 | 52.5 | 63.5 | 38.5 | 174.3 | 174.8 |
| Leu198 | 8.198 | 123.4 | 55.1 | 58.2 | 42.0 | 63.4 | 176.9 | 174.3 |
| Asn199 | 8.366 | 119.5 | 52.5 | 55.1 | 38.5 | 42.0 | 175.3 | 176.9 |
| Thr200 | 8.138 | 114.0 | 61.3 | 52.5 | 69.2 | 38.5 | 174.3 | 175.3 |
| Asp201 | 8.282 | 122.0 | 54.3 | 61.3 | 40.7 | 69.2 | 175.6 | 174.3 |
| Asp202 | 8.106 | 120.4 | 53.8 | 54.3 | 40.7 | 40.7 | 174.7 | 175.6 |
| Tyr203 | 7.549 | 124.8 | 58.6 | 53.8 | 39.0 | 40.7 | 180.3 | 174.7 |
TABLE 3.
1HN, 15NH, 13Cα, 13Cβ, and 13CO inter-residual chemical shift correlations of A17-S fragment in PBS buffer at pH 6.5
| Residue | 1HN | 15Nα | 13Cαi | 13Cαi-1 | 13Cβi | 13Cβi-1 | 13COi | 13COi-1 |
|---|---|---|---|---|---|---|---|---|
| Leu20 | 8.433 | 123.8 | 54.9 | 62.9 | 42.0 | 32.3 | 177.1 | 176.4 |
| Asp21 | 8.120 | 121.1 | 54.2 | 54.9 | 40.8 | 41.9 | 176.4 | 177.1 |
| Lys22 | 8.246 | 121.1 | 57.0 | 54.2 | 32.3 | 40.8 | 176.7 | 176.4 |
| Asp23 | 8.340 | 120.2 | 54.9 | 57.0 | 40.8 | 32.3 | 176.6 | 176.7 |
| Gln24 | 8.135 | 119.9 | 56.1 | 54.9 | 28.9 | 40.8 | 176.3 | 176.6 |
| Leu25 | 8.198 | 122.5 | 55.9 | 56.1 | 42.1 | 29.0 | 177.9 | 176.4 |
| Glu26 | 8.384 | 120.8 | 57.0 | 55.9 | 29.9 | 42.1 | 177.0 | 177.9 |
| Ser27 | 8.203 | 115.9 | 58.7 | 57.0 | 63.5 | 29.9 | 174.9 | 177.0 |
| Gln28 | 8.276 | 121.6 | 56.1 | 58.7 | 28.8 | 63.5 | 175.8 | 174.9 |
| Phe29 | 8.081 | 120.1 | 57.7 | 56.1 | 39.2 | 28.8 | 175.7 | 175.8 |
| Gln30 | 8.184 | 121.4 | 55.7 | 57.7 | 29.4 | 39.2 | 175.9 | 175.7 |
| Thr31 | 8.154 | 115.3 | 61.8 | 55.7 | 69.6 | 29.4 | 174.4 | 175.9 |
| Met32 | 8.296 | 122.6 | 55.2 | 61.8 | 32.7 | 69.6 | 175.5 | 174.4 |
| Glu33 | 8.232 | 122.1 | 56.1 | 55.2 | 30.4 | 32.7 | 175.6 | 175.5 |
| Phe34 | 8.252 | 122.1 | 56.1 | 56.1 | 38.8 | 30.4 | 173.7 | 175.6 |
| Pro35 | 62.9 | 56.1 | 32.1 | 38.8 | 177.1 | 173.7 | ||
| Lys36 | 8.482 | 122.2 | 56.5 | 62.9 | 32.8 | 32.1 | 176.6 | 176.6 |
| Asp37 | 8.350 | 120.8 | 54.2 | 56.5 | 41.1 | 32.8 | 176.8 | 176.6 |
| Gly38 | 8.369 | 109.4 | 45.6 | 54.2 | 41.1 | 175.0 | 176.8 | |
| Gly39 | 8.350 | 108.6 | 45.3 | 45.6 | 174.2 | 175.0 | ||
| Met40 | 8.188 | 119.5 | 55.6 | 45.3 | 32.7 | 176.3 | 174.2 | |
| Met41 | 8.418 | 121.4 | 55.6 | 55.6 | 32.5 | 32.7 | 176.1 | 176.3 |
| Gln42 | 8.364 | 121.4 | 55.7 | 55.7 | 29.4 | 32.5 | 175.6 | 176.1 |
| Asn43 | 8.418 | 119.9 | 53.1 | 55.8 | 39.0 | 29.4 | 174.6 | 175.6 |
| Asp44 | 8.271 | 120.5 | 54.0 | 53.1 | 41.0 | 39.0 | 176.1 | 174.7 |
| Tyr45 | 8.159 | 120.6 | 57.8 | 54.0 | 38.1 | 41.0 | 176.6 | 176.0 |
| Gly46 | 8.374 | 110.6 | 45.4 | 57.8 | 38.1 | 174.7 | 176.6 | |
| Gly47 | 8.022 | 108.6 | 45.1 | 45.4 | 174.3 | 174.7 | ||
| Met48 | 8.242 | 119.5 | 55.6 | 45.1 | 32.5 | 176.0 | 174.3 | |
| Asn49 | 8.477 | 119.8 | 53.2 | 55.6 | 39.2 | 32.5 | 173.9 | 176.0 |
| Asp50 | 7.910 | 126.1 | 55.9 | 53.2 | 42.1 | 39.2 | 180.0 | 173.9 |
To determine whether A27 binds to the N or C terminus of A17, we conducted an A27 titration experiment using A17-N and A17-C, respectively. In this in vitro binding assay, A17-N and A17-C labeled with the 15N isotope were titrated with A27 systematically at five different molar ratios, namely 1:0, 1:0.25, 1:0.5, 1:0.75, and 1:1 (pH 6.5), and monitored by HSQC. A17-C titrated with A27 produced an HSQC pattern (indicated in blue) identical to that obtained with A17-C alone (indicated in red), showing no chemical shift perturbation (Fig. 2B). In contrast, when A17-N was titrated with A27, two sets of residues were differently perturbed in the HSQC (Fig. 2C and supplemental Fig. S1), 32SFMPK36 and 20LDKDLFTEEQ29 (denoted as F1- and F2-binding sites hereafter), the cross-peak intensities of which were linearly decreased at different rates as a function of the molar ratio. As illustrated (Fig. 2E), the F1 signals represented by Phe33 and Met34 retained only ∼10%, whereas the F2 signals represented by Leu20 and Glu27 remained ∼30–35% of their original intensities at a molar ratio of 1:1, revealing the distinct diminution rates between these two binding sites. Because the F1 signals decreased relatively faster than those of F2, we concluded that the F1 site has a stronger A27 binding affinity than the F2 site. The cross-peaks for other residues, such as Gly38–Asp50, remained unchanged (Fig. 2, C and E, and supplemental Fig. S1), indicating that these residues are not involved in A27 binding.
N Terminus of A17 Has a Higher A27 Binding Affinity than the C Terminus as Revealed by SPR
In addition to HSQC, we used SPR as another in vitro binding assay to measure the A27/A17 binding affinity. In this binding assay, A27 was immobilized on a CM5 sensor chip, and the analyte, either A17-N or A17-C with a thioredoxin tag, was then passed over the immobilized A27 protein. Fig. 3, A and B, shows the SPR sensorgrams obtained at five different protein concentrations of the A17 fragments (A17-N and A17-C) and immobilized A27. In the negative control experiment, we confirmed that thioredoxin alone does not interact with A27 (supplemental Fig. S2). In the curve fitting simulation, we used a one-to-one Langmuir binding model (49) to determine the kinetic binding constants for the A27/A17-C interaction, and a binding constant KA of 3.40 × 105 m−1 was deduced (Table 4). In the case of A27/A17-N binding, we chose to use a two-state binding model to extract the kinetic binding constants to reduce the standard deviation and obtain a better fit. Accordingly, kinetic binding constants, ka1, kd1, ka2, kd2, and KA of 9.78 × 102 m−1 s−1, 2.14 × 10−3 s−1, 2.92 × 10−3 s−1, 3.93 × 10−6 s−1, and 3.40 × 108 m−1, respectively, were determined (Table 4). The KA value for A27/A17-N binding was 3 orders of magnitude higher than that found for A27/A17-C binding, suggesting that A27 preferentially interacts with A17-N rather than with A17-C. In addition, the A27/A17-N interaction showed a characteristic slow association (ka2 = 2.92 × 10−3 s−1) and an even slower dissociation binding mechanism in the second state (kd2 = 3.93 × 10−6 s−1), implying that the local conformation of A17-N may play a critical role in the regulation of specific A27 binding.
FIGURE 3.
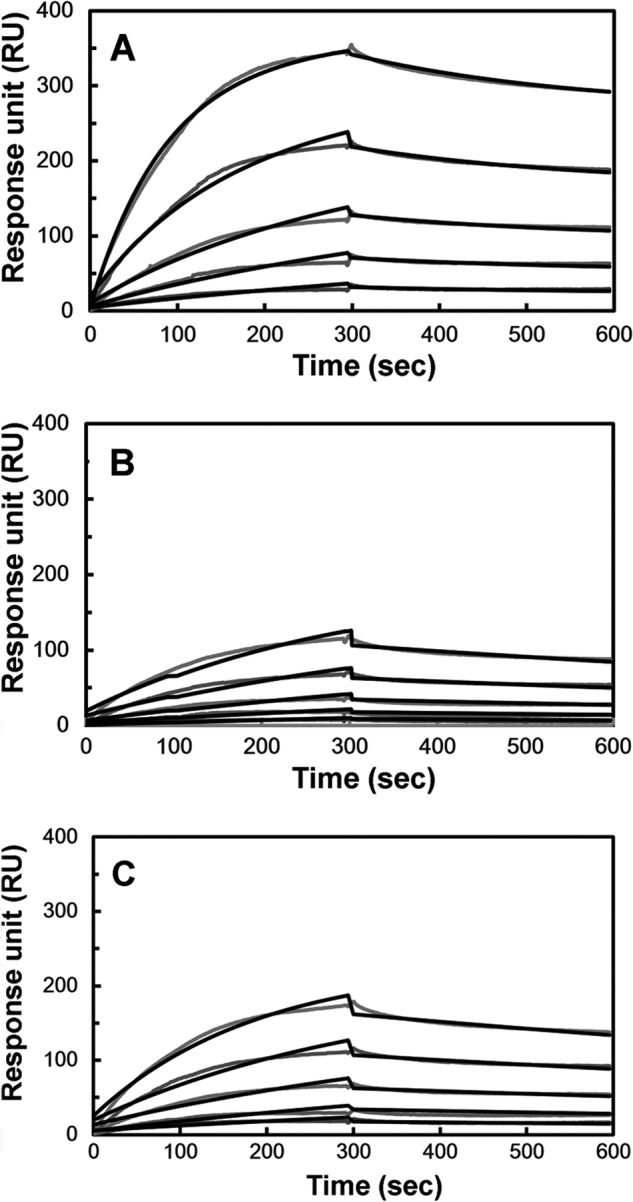
In vitro A27/A17 binding assay by SPR. The viral protein A27 was immobilized on a CM5 sensor flow chip. A17-N (A), A17-C (B), and A17-S (C) tagged with thioredoxin were injected onto the immobilized A27 at five different protein concentrations (10, 5, 2.5, 1.25, and 0.625 μm), and the association and dissociation kinetic interactions were monitored as a function of the protein concentration in real time. In the negative control, thioredoxin only gave no response when passing over a CM5 chip onto which A27 was immobilized (supplemental Fig. S2). The kinetic binding constants obtained from the sensorgrams of the A27/A17 fragments are shown (Table 4). For details, see text.
TABLE 4.
Binding constants (KA), association rate constant (ka), and dissociation rate constant (kd) of specific and nonspecific A27/A17 interactions
Dissociation binding constants (KA) were calculated from the ratio ka/kd. Data represent the means ± S.D. of at least four independent experiments at five different protein concentrations.

A27 Binding to the N Terminus of A17 Is Highly Specific
To verify whether the A27 and A17 interaction is specific, we generated a scrambled mutant of A17-N, denoted A17-S (Fig. 1B). In the A17-S construct, the A27-binding sequence 24LFTEEQQQSFM34 was scrambled to 24QLESQFQTMEF34. When A17-S was titrated with A27, the A17-S cross-peak pattern detected by HSQC remained intact, indicating that A17-S loses its binding specificity to A27 (Fig. 2D). Consistently, SPR detected only a weak binding constant KA of 5.46 × 105 m−1 (Fig. 3C), which is comparable with that observed from the nonspecific A27/A17-C interaction (Table 4). Because of the 3 orders of magnitude difference in the value of KA, the A27 and A17-N interaction appears to be highly specific, whereas the A27 and A17-S interaction was not.
Secondary Structural Analyses of A17-N Revealed a Random Coil Character
To analyze the secondary structure and local conformation, we applied circular dichroism (CD) and chemical shift index (CSI) analyses to the A17-N and A17-S fragments. In the CD approach, we analyzed the secondary structure of the A17-N and A17-S fragments and A27 through far-UV CD spectroscopy. The CD spectrum of A27 presented major ellipticity at 208 and 222 nm (Fig. 4A), suggesting a typical α-helical coiled-coil secondary structure. In contrast to the pattern produced by A27, both CD spectra of A17-N and A17-S (Fig. 4B) presented major ellipticity at 200 nm and a minor difference at 222 nm. Our CD data suggest a predominant random coil secondary structure for A17-N and A17-S.
FIGURE 4.

Secondary structure of A17 fragments based on CD and CSI analyses. Far-UV CD spectra of A27 (A), A17-N (dark line) and A17-S (gray line) (B). The A17-N spectrum indicated a superimposition of the major random coil and minor α-helix patterns, where the former exhibited ellipticity at 200 nm, and the latter exhibited ellipticity at 208 and 222 nm. Furthermore, the A17-S spectrum revealed a similar spectral pattern with the exception of a slightly lower value at ∼220 nm. In contrast to C, the A27 spectrum indicated a structural pattern distinct from that of a typical α-helix. 13Cα (C) and 13CO (D) chemical shift propensity index analysis of the A27 binding domain of A17-N and A17-S. The chemical shift index was determined as (δ − δrandom)/(δα − δrandom), where δ is the experimental chemical shift and δα and δrandom are the chemical shift values reported in the literature for a α-structure and random coil, respectively. The propensity index for the EEQQQ segment of A17-N is >0.3, suggesting a mild α-helical conformation. However, in the CSI analysis of A17-S, no such conformation is observed. The sequence numbering for A17 (single-letter code) is shown on the horizontal axis. C, respective F1- and F2-binding sites are indicated by an arrow bar.
In the CSI analyses of A17-N and A17-S fragments, we compared the 13Cα (13CO) chemical shifts (Tables 1 and 3) to the values reported for a random coil (50, 51), and a structural propensity index was deduced as (δ − δrandom)/(δα − δrandom), where δ is the experimental chemical shift and δα and δrandom are the chemical shift values reported in the literature for an α-helix and a random coil, respectively (50, 51). Ideally, a segment is considered to be an α-helix if the 13Cα and 13CO propensity indexes for three consecutive residues or more are equal to 1 and to be a random coil if these indexes are equal to 0. For indexes between 0 and 1, the propensity to form an α-helix is proportional to the propensity index. As resolved from the 13Cα and 13CO chemical shift propensity analyses (Fig. 4, C and D), the F2-binding site of A17-N is deduced to be a random coil, whereas the F1-binding site showed a slight β-strand propensity. Interestingly, both the 13CO and 13Cα CSI of the intersection (27EEQQQ31) were greater than 0.2, revealing a mild α-helical propensity. Because A17-S loses its specificity for binding to A27, we hypothesize that the local conformation of 27EEQQQ31 may play a critical role in the specific A27-A17 interaction.
To further explore the local conformations, we then performed two-dimensional NOESY and TOCSY analyses on a 33-mer peptide, corresponding to the A17-N fragment (for details see “Experimental Procedures”). Interestingly, the NOESY and TOCSY spectra of A17-N revealed consecutive short range NOE signals in the F1 and F2 sites only (Fig. 5, A and B). In the absence of any long range NOE signal, three (i, i + 2) NOE signals were found between 1HN of Thr26 and 1Hβ of Glu28, between 1Hβ of Ser32 and 1HN of Met34, and between 1Hβ of Met34 and 1HN of Lys36. We further noticed that these residues are localized close to or within the intersection with 27EEQQQ31 that had been revealed to have a mild α-helical character, as described above. Because these NOE signals may provide important conformation information for A17-N, we used the residues yielding NOE signals to derive distance constraints for a computer modeling analysis of the molecular conformation of the A17 and the A17-A27 complex. In contrast, the sequence-scrambled mutant A17-S showed only very low NOE signals in the respective TOCSY and NOESY spectra (see supplemental Fig. S3).
FIGURE 5.
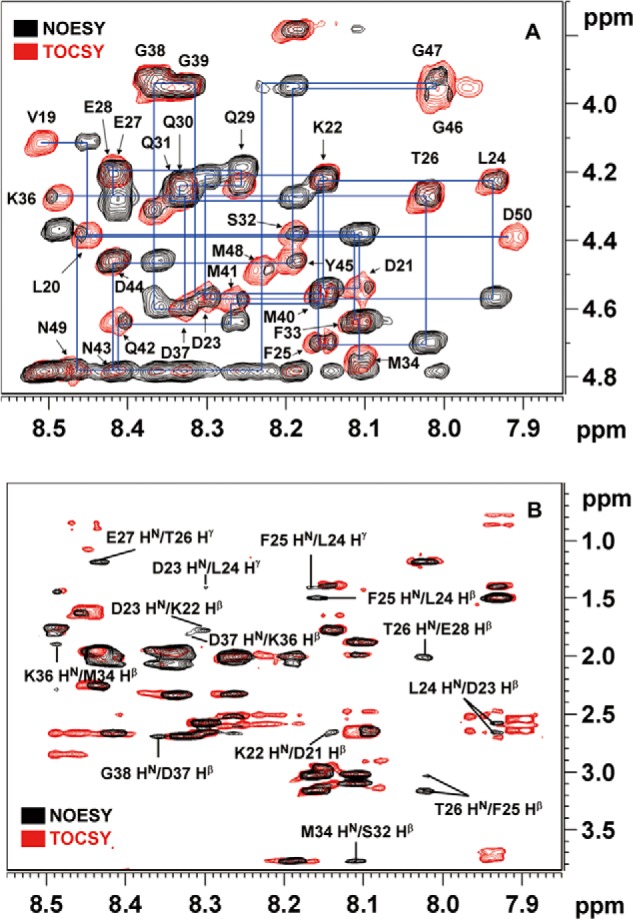
Chemical shift assignments and conformational analysis of the A17-N peptide by two-dimensional TOCSY and NOESY. A, sections of the 1HN-1Hα region of the NOESY and TOCSY spectra of the 33-mer A17-N peptide that binds to A27. The 1H chemical shift assignments were successfully determined using the inter- and intra-residual correlations in these spectra (supplemental Table S1). B, sections of 1HN-1Hβ and 1HN-1Hγ of the A17-N NOESY and TOCSY spectra. Consecutive inter-residual (i, i + 1) NOE signals were resolved as follows: between the 1Hβ of Asp21 and the 1HN of Lys22; between the 1Hβ of Lys22 and the 1HN of Asp23; between the 1HN of Asp23 and the 1Hγ of Leu24; between the 1Hβ of Asp23 and the 1HN of Leu24; between the 1Hβ of Leu24 and the 1HN of Phe25; between the 1Hγ of Leu24 and the 1HN of Phe25; between the 1Hβ of Phe25 and the 1HN of Thr26; between the 1Hγ of Thr26 and the 1HN of Glu27; between the 1Hβ of Gln30 and the 1HN of Gln31; and between the 1Hβ of Lys36 and the 1HN of Asp37. Additionally, (i, i + 2) NOE signals were found between the 1HN of Thr26 and the 1Hβ of Glu28, between the 1Hβ of Ser32 and the 1HN of Met34, and between the 1Hβ of Met34 and the 1HN of Lys36.
Site-directed Mutagenesis Studies Revealed a Cooperative Binding Mechanism
In this study, we proposed that the F1- and F2-binding sites of A17 specifically interact with A27 via a cooperative binding mechanism. In other words, both the F1 and F2 residues, as well those in between, appeared to be critical for the specific binding. To validate this hypothesis, we expressed and purified eight A17-N peptide mutants using a solid phase peptide synthesizer (for details see “Experimental Procedures”). These A17-N peptide mutants were divided into two series, namely scrambled and point mutation series (Fig. 1B). In the scrambled series, denoted A17-S1, -S2, and so on, five peptide mutants were created, the sequences of the associated residues of which were scrambled. In the point mutation series, three single mutations were made, A17-L24A, -Q29A, and -P35A. For the HSQC titration measurements, we specifically labeled two residues present in the F1- and F2-binding sites, Phe33 and Phe25, respectively, with 15N isotope labeling. If these residues are critical for the A27-A17 interaction, the scrambled sequence or point mutation would be expected to effectively deactivate the specific binding ability, and thus the HSQC signals of these mutants would remain the same after titration with or without A27. The HSQC spectra of the A17-S1, -S2, -S3, -S4, -S5, -L24A, -Q29A, and -P35A mutants titrated with A27 (Fig. 2F) confirmed this hypothesis, because the cross-peaks of the F1 and F2 residues remained unperturbed. Because the A27-A17 specific interaction is sensitive to each of these residues, we confirmed that the A27-A17 interaction is mediated through a cooperative binding mechanism.
In addition to the A17-N peptide mutants, we constructed a series of A27 mutants with mutations in the LZD (85–110 residues) in an attempt to further explore the cooperative binding mechanism (see Fig. 1A). The published crystal structure of A27 truncated protein (21–84 residues) (27) does not contain the LZD domain, which was predicted to be heptad repeats in which the hydrophobic residues at positions a and d contribute to stabilize the coiled-coil secondary structure (52–55). In fact, we found that site-directed mutations of the hydrophobic residues at the a and d positions of A27, such as M93A or L96A, deteriorate the α-helical structure by ∼20–30% compared with the parental A27 (data not shown). We rationalized that the residues at positions other than a and d may be appropriate targets for mutagenesis that would not interrupt the LZD secondary structure. Therefore, we chose the residues at the b, c, f, and g positions for Ala mutation to avoid disturbing the secondary structure of the A27 protein. A total of 12 A27 mutants were generated (Fig. 1A), and their A17 binding abilities with respect to the F1 and F2 sites were carefully examined by HSQC via interaction with 15N isotope-labeled A17-N. Using the parental A17-N without A27, the addition of the A27-F80A, -R81A, -N84A, and -E87A mutants produced less intense cross-peaks for the F1 residues and similarly intense cross-peaks for the F2 residues, suggesting a diminution at the F1 site and a disruption in the binding specificity at the F2 site (Table 5). The mutants in the other set, i.e. the A27-T88A, -I94A, -S95A, -K98A, and -K99A mutants, showed a disruption in binding to both the F1 and the F2 sites. Importantly, the disruption of the F1 binding of the A27 protein led to a loss in the binding specificity of the F2 site, underscoring the cooperative binding property of the A27-A17 interaction. Unlike A17 mutants that lose their A27 binding specificity through the loss of both their F1 and F2 specific binding activities (Fig. 2F), a disruption of the F2-binding residues in A27, such as A27-E87A, caused a partial disability of the F1 binding. A mutation of the F1 residues, such as A27-I94A, caused a disruption of both F1 and F2 binding, whereas mutations of F1 and F2 residues, i.e. the double mutant A27-E87A,I94A, completely abolished the F1 and F2 binding activities. In addition, the effects of site-directed mutagenesis on the F1 and F2 binding activities revealed by the HSQC residual intensity ratio of A17-N are summarized (see Fig. 2G). For the other mutants, such as A27-V78A and -V102A, the resulting HSQC spectra showed the same effect as that obtained for the parental A27, which suggests that these residues are outside the F1- and F2-binding sites. From our A27 mutation data (Table 5), the sequence-dependent binding activity was determined; Thr88–Lys99 corresponds to Ser32–Lys36 at the F1-binding site, and Phe80–Glu87 corresponds to Leu20–Gln29 at the F2-binding site. In this study, we concluded that the C terminus of the envelope protein A27 specifically interacts with the N terminus of the viral membrane protein A17 and that this interaction is mediated by a parallel, cooperative binding mechanism via the F1- and F2-binding sites in which the Thr88–Lys99 of A27 interacts with the Ser32–Lys36 of A17 at the F1-binding site and the Phe80–Glu87 of A27 binds to the Leu20–Gln29 of A17 at the F2-binding site.
TABLE 5.
A17 binding specificity of A27 mutants in PBS buffer at pH 6.5 by HSQC
The A27 mutants/A17 binding specificity were determined by HSQC spectroscopy, in which A17 fragment is uniformly 15N isotope-labeled, see text for details.
| A27 mutants | Mutation position | α-Helical contenta | F1 binding | F2 binding |
|---|---|---|---|---|
| A27-V78Ab | g | 93.0 | ++ | ++ |
| A27-F80A | b | 85.5 | + | + |
| A27-R81A | c | 89.0 | + | − |
| A27-N84A | f | 83.0 | + | − |
| A27-E87A | b | 95.8 | + | − |
| A27-T88A | c | 85.5 | − | − |
| A27-I94A | b | 89.3 | − | − |
| A27-S95A | c | 82.7 | − | − |
| A27-K98A | f | 91.9 | − | − |
| A27-K99A | g | 84.3 | − | − |
| A27-E87A,I94A | b | 89.7 | − | − |
| A27-V102Ab | c | 94.3 | ++ | ++ |
a The α-helical contents determined by CD spectroscopy were referred to in the α-helical content in parental A27(21–110) as 100% (Fig. 4A).
b In these A27 mutants, A27-V78A and -V102A, in which the mutation was beyond the A17 active binding sites (residues 80–100), were treated as negative controls.
Computer Modeling of A27-A17 Protein Complex
To better understand the cooperative binding mechanism of the A27-A17 interaction, we conducted a computer modeling analysis of the local conformation of the A17 fragment, which was represented by an 18-mer peptide (Leu20–Asp37) that was identified as the A27 binding domain, using the Discovery Studio software. In this computer simulation, we confined the 1H-1H inter-residual distance constraints to within 5 Å for those revealing NOE signals (Fig. 5B).
As shown, the backbone of the A17 fragment revealed a distorted, unstructured conformation, consistent with the observations from the CD and CSI analyses (Fig. 4, B–D). The F1-binding site presents as a twisted bended motif due to the presence of Pro35, whereas the F2-binding site has a zigzag backbone conformation; both the F1- and F2-binding sites are suitable for accommodating the α-helical motif. To reconstruct the A27-A17 protein complex, we conducted a molecular docking analysis of the α-helical A27 segment (Phe80–Asp101) and A17 fragment (Leu20–Asp37) in a parallel mode (Table 5). The observation of the number of inter- and intra-molecular interactions suggested a binding network and indicated a cooperative protein complex formation, particularly in the Arg90–Asp101 segment of A27 and the Gln29–Asp37 segment of its binding partner A17. As observed through the A27-A17 protein complex modeling (Fig. 6A), the aliphatic side chain of Ile94 (b) aligned in parallel with the side chain of Arg90 (e) such that these residues are somehow tilted nearly 90° from the adjacent side chain of Lys98 (f) in A27. This tridentate molecular setup of the Arg90, Ile94, and Lys98 residues of A27 is critical for the accommodation of the twist-bent A17 fragment, and the aromatic ring of the Phe33 residue of A17 is able to specifically interact with the amine group of the Lys98 residue of A27 through a cation-π interaction. At one end, the hydroxyl group of the Met34 residue of A17 forms a hydrogen bond with the hydrogen of the amino group in the side chain of the Lys98 residue of A27, whereas at the other end, the Gln29 hydrogen is able to bond to Arg90. Other residues are also involved in the specific A27-A17 protein interaction via intermolecular hydrogen bonding, namely the oxygen of the Ser32 residue of A17 with the hydroxyl group of the Ser95 residue of A27 and the guanidinium group of the Lys36 residue of A17 and the carboxyl group of the Asp101 residue of A27.
FIGURE 6.
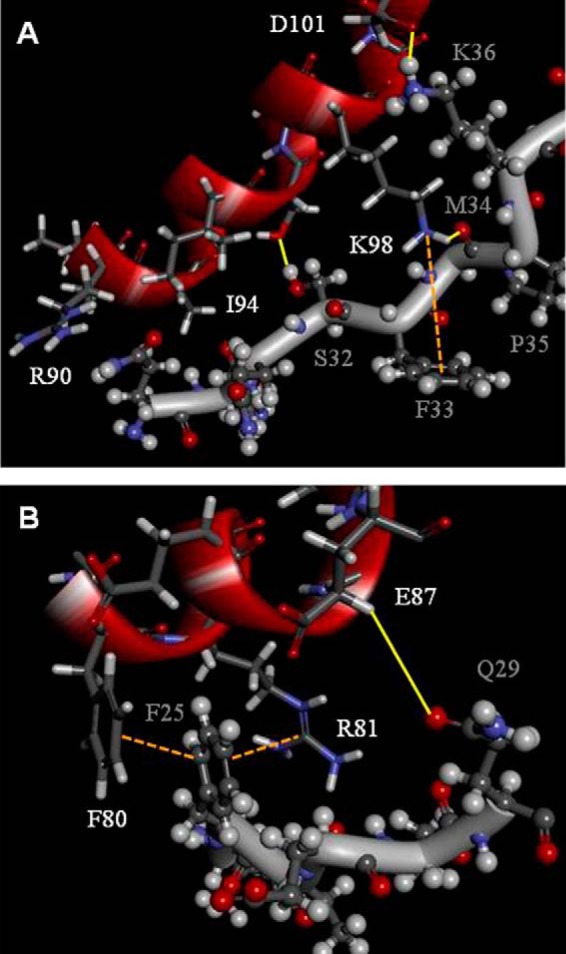
Computer modeling of A27 and A17 viral protein complex formation. A, representative of the F1-binding site, including the linker; the Gln29–Asp37 of the F1-binding site of A17 interacts with Arg90–Asp101 of A27. The amine group of Lys98 interacts with the aromatic ring of Phe33 to form a specific cation-π interaction. Intermolecular hydrogen bonds were found between the amine group of Lys98 and the hydroxyl group of Met34, between the hydroxyl group of Ser95 and the oxygen of Ser32, and between the guanidinium group of Lys36 and the carboxyl group of Asp101. B, representative F2-binding site. Leu20–Gln29 of A17 interact with Phe80–Glu87 of A27. Notably, A17 presents an α-helical zigzag backbone conformation. At one side, the aromatic ring of Phe25 interacts with the aromatic ring of Phe80 to form a π-π stacking interaction. At the opposite side, it interacts with the guanidinium group of Arg81 to form a cation-π interaction. In addition, the carboxyl group of Leu24 forms main chain interactions with the guanidinium side chain group of Arg81 to form a hydrogen bond. In this computer simulation, we confined the 1H-1H inter-residual distance constraints to within 5 Å for those revealing NOE signals (see Fig. 5B). The hydrogen bonds are highlighted by yellow solid lines and the cation-π interactions are labeled by orange dotted lines. The single-letter abbreviations of the amino acids of A27 and A17 are labeled in white and gray, respectively. For clarity, some of the side chains that are not involved in the A27-A17 interaction are not shown.
As indicated by our HSQC results (Fig. 2C), there are two residues, Gln30 and Gln31, between F1 and F2 that are not involved in the A27-A17 interaction. In addition, our CSI analysis suggests that the 27EEQQQ31 segment displays a zigzag distorted conformation. Interestingly, as revealed by our computer modeling, although Ile94 bypasses the Gln30 and Gln31 residues in the space between the F1 and F2 active binding sites and is not physically involved in the binding to A17, it has a pivotal role critical for the tridentate molecular alignment as described above and is therefore crucial for both F1 and F2 binding activities. In vitro binding assays revealed that the single mutant A27-I94A did substantially affect the A17 binding specificity (Table 5).
With respect to the F2-binding site of A17, the 24LFTEEQQQ31 segment displays a zigzag-distorted conformation, and the aromatic ring of Phe25 interacts with the side chains of the Phe80 and Arg81 residues in A27 (Fig. 6B). At one site, the aromatic ring of Phe25 interacts with the aromatic ring of Phe80 to form a π-π stacking interaction, whereas at the opposite side, it interacts with the guanidinium group of Arg81 through a cation-π interaction. Given the distorted conformation, hydrogen bonds are formed between the main chain carboxyl group of Leu24 and the guanidinium side chain of Arg81 (data not shown) and between the guanidinium group of Gln29 and the carboxyl group of Glu87. Notably, the side chains of Phe80, Arg81, Asn84, and Glu87 are delicately oriented such that any sequence disruption or single mutation may result in the destruction of the molecular alignment and thus the impairment of the binding specificity. This explains why the A27-R81A, -N84A, and -E87A mutants lose their F2 binding activity to A17 (Table 5). Based on this computer modeling, we predicted that the single mutations E87A and I94A and the double mutation E87A,I94A would disrupt the F1 and F2 cooperative binding accordingly.
F1- and F2-binding Sites of the A27 and A17 Protein Interactions Are Critical for A27 Packaging into Mature Virus
After the in vitro HSQC analyses with the A27-E87A, A27-I94A, and A27-E87A,I94A mutants, we extended these mutant protein analyses into recombinant vaccinia viruses to understand whether the Glu87 and Ile94 residues are critical for A27 and A17 protein interaction in vivo. We introduced the A27-E87A, A27-I94A, and A27-E87A,I94A mutations into the full-length WR-A27L ORF by in vitro mutagenesis. The mutant A27L ORFs were cloned into pMJ601 such that these proteins were expressed from a strong late promoter. The plasmids were individually transfected into cells infected with vaccinia A27 deletion virus, WR-ΔA27L, such that the A27-E87A, A27-I94A, or A27-E87A ORFs could be inserted into the nonessential thymidine kinase locus, and the resulting recombinant viruses were isolated as blue plaques using X-Gal (Fig. 7A). Because mutations of the A27 protein may cause a reduction in plaque size, which would make it difficult to isolate the recombinant viruses (56–58), we used a complementing cell line, BSC40-A27 (27), that expresses wild-type A27 protein in trans to ensure the successful isolation of these mutant A27 viruses. Indeed, all of the viruses were found as large plaques on BSC40-A27 cells (Fig. 7B). Interestingly, on BSC40 cells, only the wild-type (WR), WR-A27-Rev, and WR-A27-E87A mutant viruses formed large sized plaques. The WR-A27-I94A virus formed small but visible plaques, whereas WR-ΔA27 and the double mutant WR-E87A,I94A formed tiny plaques that were barely detectable (Fig. 7B). This finding suggests that the I94A mutation has a more severe effect than the E87A mutation on the role of A27 protein function in virus egress. This result also implies that E87A and I94A have some additive effects on the function of the A27 protein in plaque formation, although this was not revealed by the E87A single mutation. The levels of these A27 mutant proteins were analyzed by immunoblot analyses using cell lysates harvested at 24 h p.i. (Fig. 7C). As expected, the wild-type WR virus expressed the A27 protein, and the WR-ΔA27 did not. Furthermore, the WR-A27-Rev, WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A viruses expressed even more abundant levels of the A27 protein than the WR virus in the infected cells, suggesting that the A27 mutations did not have adverse effect on A27 protein stability and that the small plaque phenotype of the WR-A27-I94A and WR-A27-E87A,I94A viruses was not due to an insufficient A27 protein amount in the infected cells. The A26 protein level in all of the infected cells was comparable with the exception of the cells infected with WR-ΔA27 because the A26 protein became less stable. The levels of the other viral proteins, such as A17 (both precursor and mature forms) and H3, were comparable in the infected cells (Fig. 7C).
FIGURE 7.

Generation of recombinant vaccinia viruses expressing mutant A27 proteins in the infected cells. A, schematic representation of recombinant vaccinia virus genomes containing various A27L mutant ORFs. The wild-type WR virus is shown above each viral ORF, and the arrows indicate the direction of transcription. J2R represents the nonessential thymidine kinase locus. In all of the recombinant viruses except the WR virus, the viral A27L ORF is substituted with a dual-expression cassette, Luc-Gpt, which contains a luciferase (Luc) gene driven by a viral early promoter and the Eco-gpt (Gpt) gene driven by the viral p7.5k promoter. WR-A27-Rev, WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A also contain another expression cassette inserted in the J2R locus in which the WT-A27, A27-E87A, A27-I94A, or A27-E87A,I94A mutants are driven by a synthetic late promoter, and a lacZ gene is driven by the viral p11k promoter. The asterisks mark the mutant residues. B, morphology of plaques produced on BSC40 and BSC40-A27 cells by WR, WR-ΔA27L, WR-A27-Rev, WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A infections. BSC40 and BSC40-A27 cells were infected with each virus, fixed at 2 days p.i., stained with 1% crystal violet in 20% ethanol, and photographed. The tiny plaques are shown by triangles. C, immunoblot analysis of A27 protein in the infected cells. HeLa cells were infected with each virus at an m.o.i. of 5 pfu/cell, and the lysates were harvested at 24 h p.i. for immunoblot analyses with anti-A26 (1:1,000), anti-A27 (1:5,000), anti-A17 (1:1,000), and anti-H3 (1:1,000) antibodies.
To investigate whether A27 mutation affects its ability to bind to the A17 protein in vivo, HeLa cell lysates were harvested at 24 h p.i. and subjected to coimmunoprecipitation experiments using anti-A27 antibodies. As shown in Fig. 8A, the anti-A27 antibody brought down comparable levels of wild-type and mutant A27 protein. In addition, the wild-type WR A27, A27-Rev, and A27-E87A mutant proteins coimmunoprecipitated both precursor and mature A17 proteins from the infected cells; however, the A27-I94A and double mutant A27-E87A,I94A almost completely lost their ability to bind to the A17 protein. We then performed the reverse coimmunoprecipitation experiments using anti-A17 antibodies, which brought down both the precursor and mature forms of the A17 protein in the infected cells (Fig. 8B). Consistent with the above results, the anti-A27 antibody detected wild-type WR A27, A27-Rev, and A27-E87A proteins from the immunoprecipitates but not the A27-I94A and A27-E87A,I94A mutant proteins. Taken together, the coimmunoprecipitation results show that the F1-binding site on the A27 protein is more critical than the F2-binding site for interaction with the A17 protein in infected cells.
FIGURE 8.
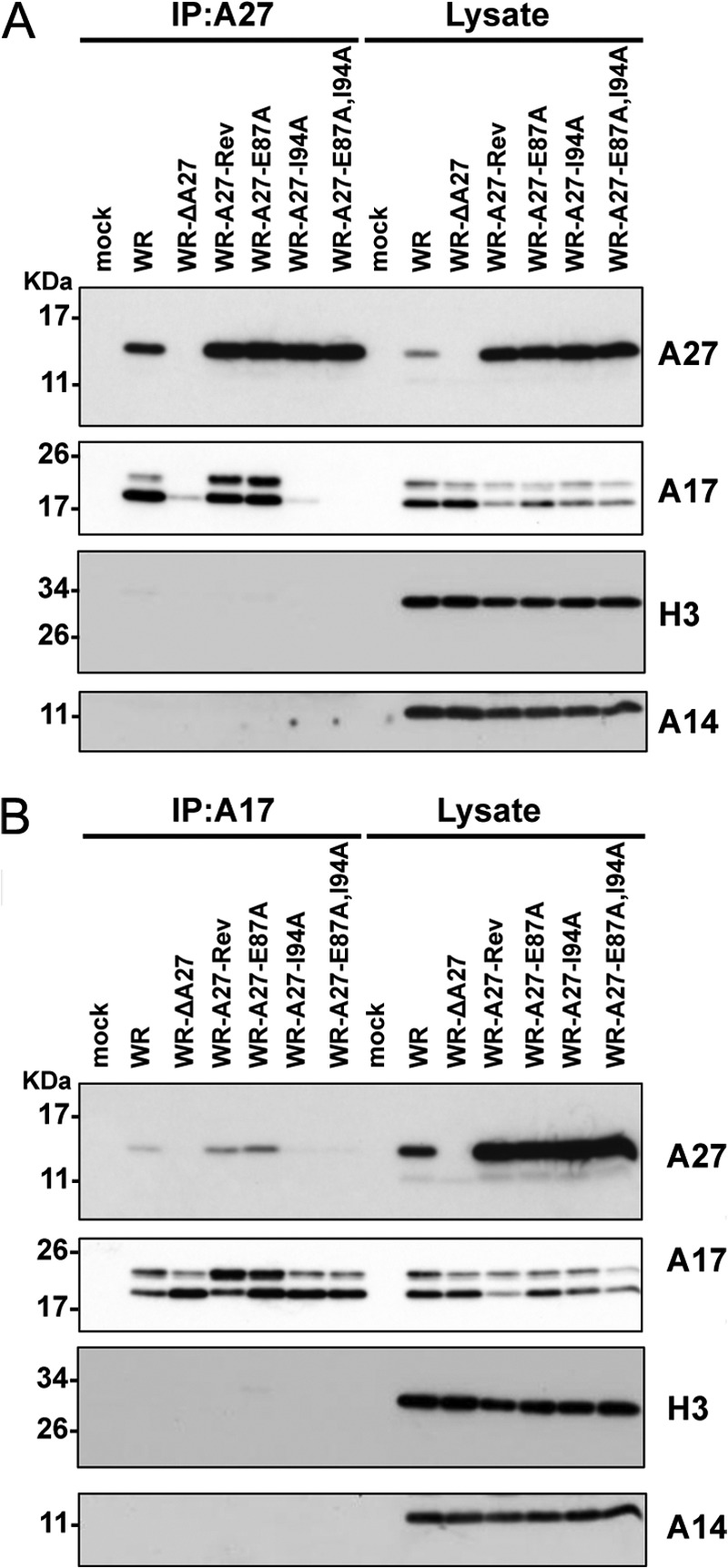
Coimmunoprecipitation analyses of the A27 and A17 interaction in virus-infected cells. HeLa cells were infected with WR, WR-ΔA27L, WR-A27-Rev, WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A at an m.o.i. of 5 pfu/cell and harvested 24 h p.i. The cell lysates were used for immunoprecipitation (IP) experiments using anti-A27 antibody (A) or anti-A17 antibody (B). The immunoprecipitates were subsequently separated through SDS-PAGE and analyzed by immunoblot analyses using anti-A27 (1:5,000), anti-A17 (1:1,000), anti-H3 (1:1,000), and anti-A14 (1:500) antibodies.
Because the association of the A27 protein with the A17 protein is thought to be critical for A27 protein assembly into mature virions, we asked whether the incorporation of these A27 mutant proteins into the MV particles was reduced. We therefore purified mature virus particles from BSC40 cells by CsCl gradient centrifugation as described under “Experimental Procedures.” One microgram of purified mature virions was separated on 10% SDS-PAGE, and silver staining revealed comparable purity (Fig. 9A). These virion proteins were transferred for immunoblot analyses (Fig. 9B), and similar amounts of the A17 proteins were detected in all of the purified mature viruses. The A27 and A26 proteins were abundant in the purified virus particles of WR and WR-A27-Rev; however, both proteins were found in lesser amounts in the mutant A27 virions in the following order: WR and WR-A27-Rev > WR-A27-E87A > WR-A27-I94A > WR-A27-E87A,I94A. Thus, although single F1- and F2-binding site mutations of A27 resulted in a partial reduction of the A27 incorporation into MV particles, double mutations of the F1- and F2-binding sites in the A27 protein also exhibited an additive effect to reduce the incorporation of A27 and A26 proteins into mature virus during the assembly stage.
FIGURE 9.

Structure and function analyses of A27 mutant viruses. A, silver staining of SDS-polyacrylamide gels containing 1 μg of purified mature virions of WR, WR-ΔA27L, WR-A27-Rev, WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A viruses. B, immunoblot analyses of WR, WR-ΔA27L, WR-A27-Rev, WR-A27-E87A, WR-A27-I94A, and WR-A27-E87A,I94A MV particles (1 μg) with anti-A27 (1:5000), anti-A17 (1:1000), and anti-A26 (1:1000) antibodies. The protein bands were scanned, and their relative intensity is shown at the bottom of each panel. C, cell-cell fusion at neutral pH induced by virus infections. L cells expressing GFP or RFP (1:1 mixture) were either mock-infected or infected with WR, WR-ΔA27L, WR-A27-Rev, WR-A27-E87A, WR-A27-I94A, or WR-A27-E87A,I94A viruses at an m.o.i. of 50 pfu/cell at 37 °C for 30 min and monitored for cell-to-cell fusion at neutral pH. The cell images were photographed at 2 h p.i. D, quantification of cell-cell fusion at neutral pH. The percentage of cells containing both GFP and RFP fluorescence was determined using Axio Vision Rel. 4.8 with a Zeiss Axiovert fluorescence microscope.
MV Particles Purified from Cells Infected with WR-A27-E87A,I94A Triggered Plasma Membrane Fusion at Neutral pH
Our previous results suggest that the vaccinia A26 protein on the MV controls the specificity of the viral entry pathway into HeLa, CHO, and L cells (20, 59). The wild-type WR strain MV particles contain the A26 protein and enter HeLa cells through an endocytosis pathway, whereas WRΔA26 MV particles enter cells through plasma membrane fusion, resulting in robust cell-cell fusion at neutral pH (20).
We thus investigated whether these recombinant A27 mutant viruses containing a reduced amount of A26 protein trigger plasma membrane fusion at neutral pH. L cells expressing either GFP or RFP were mixed at a 1:1 ratio, infected with each virus at an m.o.i. of 50 pfu/cell, and monitored for cell-cell fusion at neutral pH 2 h p.i. The mock cells and WR virus infection did not trigger cell-cell fusion, and the GFP- and RFP-expressing cells were well separated (Fig. 9C). In contrast, the WR-ΔA27L virus proceeded rapidly with plasma membrane fusion at neutral pH, resulting in gigantic fused cells that coexpressed GFP and RFP. As expected, the WR-A27-Rev virus behaved as the WR virus and did not trigger cell-cell fusion. WR-A27-E87A infection also did not induce cell-cell fusion, and WR-A27-I94A induced moderate cell-cell fusion, although the fusion level appeared not to be statistically significant. The double mutant WR-A27-E87A,I94A exhibited a clear fusion phenotype with multinucleate fused cells of smaller sizes than those observed with WR-ΔA27L. The quantitative cell-cell fusion data, which are presented in Fig. 9D, confirm that the A27-E87A,I94A double mutation reduced the A27 and A26 protein incorporation into MV and thereby resulted in an increase in cell-cell fusion. Taken together, the above in vivo recombinant virus studies reveal that the A17-A27 protein interaction through the F1- and F2-binding sites facilitates not only A27 incorporation but also A26 protein tethering into MV particles.
DISCUSSION
To study the A27 and A17 protein interaction, we first attempted to express an A17 truncated protein, A17-T1 (18–77 residues) with a transmembrane region (61–77 residues); however, the protein expression induced the formation of an inclusion body (data not shown). We therefore constructed only the A17 N-terminal and A17 C-terminal regions with no transmembrane domain to obtain soluble proteins. In this study, we report that the viral protein A27 anchors to the viral membrane through specific interactions with the F1- (32SFMPK36) and F2- (20LDKDLFTEEQ29) binding sites in A17 and that the F1 site provides higher affinity binding than the F2. To test whether either the F1 or F2 binding is sensitive to an acidic environment, we compared the HSQC pattern of A17-N acquired at pH 5.0 (supplemental Fig. S4) with that acquired at pH 6.5 (Fig. 2C) and found that some of the cross-peaks of the F2 residues, such as Phe25, Glu27, and Asn29, were shifted upfield in the range of 0.12–0.20 ppm in the 15N chemical shift in an acidic environment compared with the F1 residues (<0.05 ppm), which implies that the F2 residues are sensitive to acid treatment. To further verify whether the F2 binding is acid-dependent, we performed an HSQC A27 titration experiment on A17-N at pH 5.0. The F1 cross-peaks diminished at a similar rate as that obtained at pH 6.5, and the F2 cross-peaks diminished at a considerably slower rate (data not shown), suggesting that the F2 binding affinity at pH 5.0 is somehow weaker than that observed at pH 6.5. Thus, it is possible that A17 makes use of F2 binding to regulate the A27 binding affinity at low pH (5.0).
Additionally, we determined that the LZD (85–110 residues) of A27 is responsible for binding to A17-N. However, the LZD alone is insufficient for A27 binding because a synthesized LZD peptide of 26 amino acids does not specifically interact with A17-N (data not shown). The LZD of the A27 protein lacked the coiled-coil region (Asn43–Asn84), which contains a hydrophobic core region (Asn43–Glu55) within the α-helical coiled-coil region that has been implicated as responsible for inter-helical interactions (56–58). Moreover, three residues, Leu47, Leu51, and Leu54, positioned at a and d in the heptad repeat unit are critical for self-assembly and thus for the biological activities associated with binding to HS or heparin on the cell surface (56–58). In summary, we conclude that A27 requires the hydrophobic core region to stabilize the protein structure and to be capable of binding to the A17 protein.
The A27/A17 specific binding affinity was 3 orders of magnitude greater than its nonspecific binding affinity, suggesting that specific hydrogen bonding and cation-π interactions form a cooperative binding network such that scrambling the sequence or even introducing a single mutation to A17 may disrupt the side chain alignments and result in a loss of binding specificity (Fig. 2, D and F). Only the site-directed mutation of the A27 residues at the b, c, f, and g positions but not the a and d position was able to disrupt the A27-A17 side chain alignments without interrupting the α-helical backbone conformation. This allowed us to specifically identify the F1- and F2-binding residues. Furthermore, the specific A27-A17 interaction significantly decreased the molecular dynamics of unstructured A17, explaining why the HSQC cross-peaks of the A17-N titrated with A27 are diminished (Fig. 2C). This phenomenon resembles the missing cross-peaks observed in the HSQC spectrum of self-assembled rigid A27 (Fig. 2A).
It is plausible that the specific A27-A17 interaction may induce a conformational change in the A17-N fragment. To discern this, we examined the A27 titration HSQC spectra of A17-N with extra care. If this is indeed the case, based on the conformational change in the F1 and F2 residues, as well as associated neighboring residues, such as Gln30, Gln31, Asn37, and Gly38, their chemical shifts are thought to be perturbed accordingly. As evidenced (Fig. 2C and supplemental Fig. S1), however, none of these residues was perturbed, which implies that the abovementioned hypothesis is not the valid.
From the SPR results in combination with the HSQC and site-directed mutagenesis studies, we demonstrated that the A17-N-A27 complex forms through an unstructured/structured binding mechanism in which the unstructured random coil of the N-terminal domain in A17 specifically interacts with the structured C-terminal coiled-coil domain in A27. In our previous studies of A27, we presented a similar unstructured/structured molecular interaction for the A27-HS complex, in which the N-terminal random coil of A27 specifically interacts with an α-helical tetrasaccharide, HS, which was used as a mimic of the glycosaminoglycan molecules on the cell surface (52–55). In these two cases, the molecular interactions are driven by charge-charge interactions that require no specific conformation or local secondary structure for one of the binding partners. It is conceivable that a flexible unstructured motif is amenable to the binding of the structured binding site, and A27 appears to have evolved such an unstructured-structured binding mechanism for the mediation of its functions on the viral membrane.
In addition, A27 tends to form hexamers or higher degree oligomers that are critical for heparin or HS binding, and it is possible that the formation of oligomers facilitates the unstructured/structured interaction (24, 27). It has been reported that the A17 protein forms dimers embedded in the viral membrane and that its binding to A27 is important for A27 assembly into MV particles (23, 25, 38). Using recombinant vaccinia viruses expressing mutant A27 proteins, we showed that the interruption of the F1- or F2-binding site had differential effects on the A27-A17 protein interaction, whereas interruption of both F1- and F2-binding sites, i.e. A27-E87A,I94A, significantly impaired the incorporation of A27 and the tethering A26 proteins into MV particles. Thus, the vaccinia virus envelope protein A27 binding to A17 affects two important biological stages as follows: the virion assembly/egress stage (i.e. plaque size), and the infection pathways of the virus progeny (endocytosis versus plasma membrane fusion). In the future, more studies will be required to understand whether the F1/F2-binding sites are masked in the A17 precursor protein and only become accessible to A27 protein binding after the proteolytic processing of the A17 protein during virion assembly.
Supplementary Material
Acknowledgments
The NMR spectra were obtained at the High Field-Nuclear Magnetic Resonance Center supported by the National Research Program for Genomic Medicine of Taiwan and Core Facility for Protein Structural Analysis supported by National Core Facility Program for Biotechnology and Instrumentation Center at National Taiwan University.
This work was supported by Academia Sinica and the National Science Council Grants NSC 100-2113-M-001-016-MY2 (to D.-L. M. T.) and NSC 101-2320-B-001-023-MY3 (to W. C.).

This article contains supplemental Figs. S1–S4 and Table S1.
- MV
- mature virus
- HS
- heparan sulfate
- HBS
- heparin binding site
- SPR
- surface plasmon resonance
- HSQC
- heteronuclear single quantum coherence
- TOCSY
- total correlated spectroscopy
- RFP
- red fluorescent protein
- m.o.i.
- multiplicity of infection
- p.i.
- post-infection
- WR
- Western Reserve
- HBS
- heparin-binding sequence
- CCD
- coiled-coil domain
- DLD
- disulfide-linked domain
- LZD
- leucine zipper domain
- CSI
- chemical shift index.
REFERENCES
- 1. Goebel S. J., Johnson G. P., Perkus M. E., Davis S. W., Winslow J. P., Paoletti E. (1990) The complete DNA sequence of vaccinia virus. Virology 179, 247–266 [DOI] [PubMed] [Google Scholar]
- 2. Moss B. (1996) in Fields Virology (Fields B. N., Knipe D. M., Howley P. M., eds) 3rd Ed., pp. 2637–2671, Lippincott-Raven Publishers, Philadelphia [Google Scholar]
- 3. Chung C. S., Chen C. H., Ho M. Y., Huang C. Y., Liao C. L., Chang W. (2006) Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 80, 2127–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Resch W., Hixson K. K., Moore R. J., Lipton M. S., Moss B. (2007) Protein composition of the vaccinia virus mature virion. Virology 358, 233–247 [DOI] [PubMed] [Google Scholar]
- 5. Yoder J. D., Chen T. S., Gagnier C. R., Vemulapalli S., Maier C. S., Hruby D. E. (2006) Pox proteomics: mass spectrometry analysis and identification of vaccinia virion proteins. Virol. J. 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chung C. S., Hsiao J. C., Chang Y. S., Chang W. (1998) A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72, 1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hsiao J. C., Chung C. S., Chang W. (1998) Cell surface proteoglycans are necessary for A27L protein-mediated cell fusion: identification of the N-terminal region of A27L protein as the glycosaminoglycan-binding domain. J. Virol. 72, 8374–8379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsiao J. C., Chung C. S., Chang W. (1999) Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73, 8750–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin C. L., Chung C. S., Heine H. G., Chang W. (2000) Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 74, 3353–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chiu W. L., Lin C. L., Yang M. H., Tzou D. L., Chang W. (2007) Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J. Virol. 81, 2149–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Foo C. H., Lou H., Whitbeck J. C., Ponce-de-León M., Atanasiu D., Eisenberg R. J., Cohen G. H. (2009) Vaccinia virus L1 binds to cell surfaces and blocks virus entry independently of glycosaminoglycans. Virology 385, 368–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chung C. S., Huang C. Y., Chang W. (2005) Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J. Virol. 79, 1623–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Izmailyan R., Hsao J. C., Chung C. S., Chen C. H., Hsu P. W., Liao C. L., Chang W. (2012) Integrin β1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 86, 6677–6687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schroeder N., Chung C. S., Chen C. H., Liao C. L., Chang W. (2012) The lipid raft-associated protein CD98 is required for vaccinia virus endocytosis. J. Virol. 86, 4868–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Townsley A. C., Weisberg A. S., Wagenaar T. R., Moss B. (2006) Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J. Virol. 80, 8899–8908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang C. Y., Lu T. Y., Bair C. H., Chang Y. S., Jwo J. K., Chang W. (2008) A novel cellular protein, VPEF, facilitates vaccinia virus penetration into HeLa cells through fluid phase endocytosis. J. Virol. 82, 7988–7999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whitbeck J. C., Foo C. H., Ponce de Leon M., Eisenberg R. J., Cohen G. H. (2009) Vaccinia virus exhibits cell-type-dependent entry characteristics. Virology 385, 383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moss B. (2006) Poxvirus entry and membrane fusion. Virology 344, 48–54 [DOI] [PubMed] [Google Scholar]
- 19. Bengali Z., Satheshkumar P. S., Moss B. (2012) Orthopoxvirus species and strain differences in cell entry. Virology 433, 506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang S. J., Chang Y. X., Izmailyan R., Tang Y. L., Chang W. (2010) Vaccinia virus A25 and A26 proteins are fusion suppressors for mature virions and determine strain-specific virus entry pathways into HeLa, CHO-K1, and L cells. J. Virol. 84, 8422–8432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Howard A. R., Senkevich T. G., Moss B. (2008) Vaccinia virus A26 and A27 proteins form a stable complex tethered to mature virions by association with the A17 transmembrane protein. J. Virol. 82, 12384–12391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ching Y. C., Chung C. S., Huang C. Y., Hsia Y., Tang Y. L., Chang W. (2009) Disulfide bond formation at the C termini of vaccinia virus A26 and A27 proteins does not require viral redox enzymes and suppresses glycosaminoglycan-mediated cell fusion. J. Virol. 83, 6464–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodriguez D., Rodriguez J. R., Esteban M. (1993) The vaccinia virus 14-kilodalton fusion protein forms a stable complex with the processed protein encoded by the vaccinia virus A17L gene. J. Virol. 67, 3435–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ho Y., Hsiao J. C., Yang M. H., Chung C. S., Peng Y. C., Lin T. H., Chang W., Tzou D. L. (2005) The oligomeric structure of vaccinia viral envelope protein A27L is essential for binding to heparin and heparan sulfates on cell surfaces: a structural and functional approach using site-specific mutagenesis. J. Mol. Biol. 349, 1060–1071 [DOI] [PubMed] [Google Scholar]
- 25. Vázquez M. I., Rivas G., Cregut D., Serrano L., Esteban M. (1998) The vaccinia virus 14-kilodalton (A27L) fusion protein forms a triple coiled-coil structure and interacts with the 21-kilodalton (A17L) virus membrane protein through a C-terminal α-helix. J. Virol. 72, 10126–10137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shih P. C., Yang M. S., Lin S. C., Ho Y., Hsiao J. C., Wang D. R., Yu S. S., Chang W., Tzou D. L. (2009) A turn-like structure “KKPE” segment mediates the specific binding of viral protein A27 to heparin and heparan sulfate on cell surfaces. J. Biol. Chem. 284, 36535–36546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang T. H., Chang S. J., Hsieh F. L., Ko T. P., Lin C. T., Ho M. R., Wang I., Hsu S. T., Guo R. T., Chang W., Wang A. H. (2013) Crystal structure of vaccinia viral A27 protein reveals a novel structure critical for its function and complex formation with A26 protein. PLoS Pathog. 9, e1003563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodríguez D., Esteban M., Rodríguez J. R. (1995) Vaccinia virus A17L gene product is essential for an early step in virion morphogenesis. J. Virol. 69, 4640–4648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodríguez D., Risco C., Rodríguez J. R., Carrascosa J. L., Esteban M. (1996) Inducible expression of the vaccinia virus A17L gene provides a synchronized system to monitor sorting of viral proteins during morphogenesis. J. Virol. 70, 7641–7653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wolffe E. J., Moore D. M., Peters P. J., Moss B. (1996) Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70, 2797–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Betakova T., Wolffe E. J., Moss B. (1999) Regulation of vaccinia virus morphogenesis: phosphorylation of the A14L and A17L membrane proteins and C-terminal truncation of the A17L protein are dependent on the F10L kinase. J. Virol. 73, 3534–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Derrien M., Punjabi A., Khanna M., Grubisha O., Traktman P. (1999) Tyrosine phosphorylation of A17 during vaccinia virus infection: involvement of the H1 phosphatase and the F10 kinase. J. Virol. 73, 7287–7296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Unger B., Mercer J., Boyle K. A., Traktman P. (2013) Biogenesis of the vaccinia virus membrane: genetic and ultrastructural analysis of the contributions of the A14 and A17 proteins. J. Virol. 87, 1083–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Whitehead S. S., Hruby D. E. (1994) A transcriptionally controlled trans-processing assay: putative identification of a vaccinia virus-encoded proteinase which cleaves precursor protein P25K. J. Virol. 68, 7603–7608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krijnse-Locker J., Schleich S., Rodriguez D., Goud B., Snijder E. J., Griffiths G. (1996) The role of a 21-kDa viral membrane protein in the assembly of vaccinia virus from the intermediate compartment. J. Biol. Chem. 271, 14950–14958 [DOI] [PubMed] [Google Scholar]
- 36. Betakova T., Moss B. (2000) Disulfide bonds and membrane topology of the vaccinia virus A17L envelope protein. J. Virol. 74, 2438–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wallengren K., Risco C., Krijnse-Locker J., Esteban M., Rodriguez D. (2001) The A17L gene product of vaccinia virus is exposed on the surface of IMV. Virology 290, 143–152 [DOI] [PubMed] [Google Scholar]
- 38. Kochan G., Escors D., González J. M., Casasnovas J. M., Esteban M. (2008) Membrane cell fusion activity of the vaccinia virus A17-A27 protein complex. Cell Microbiol. 10, 149–164 [DOI] [PubMed] [Google Scholar]
- 39. Morton T. A., Myszka D. G., Chaiken I. M. (1995) Interpreting complex binding kinetics from optical biosensors: a comparison of analysis by linearization, the integrated rate equation, and numerical integration. Anal. Biochem. 227, 176–185 [DOI] [PubMed] [Google Scholar]
- 40. Karlsson R., Fält A. (1997) Experimental design for kinetic analysis of protein-protein interactions with surface plasmon resonance biosensors. J. Immunol. Methods 200, 121–133 [DOI] [PubMed] [Google Scholar]
- 41. Mori S., Abeygunawardana C., Johnson M. O., van Zijl P. C. (1995) Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108, 94–98 [DOI] [PubMed] [Google Scholar]
- 42. Grzesiek S., Döbeli H., Gentz R., Garotta G., Labhardt A. M., Bax A. (1992) 1H, 13C, and 15N NMR backbone assignments and secondary structure of human interferon-γ. Biochemistry 31, 8180–8190 [DOI] [PubMed] [Google Scholar]
- 43. Yamazaki T., Lee W., Arrowsmith C. H., Muhandiram D. R., Kay L. E. (1994) A suite of triple resonance NMR experiments for the backbone assignment of 15N, 13C, 2H labeled proteins with high sensitivity. J. Am. Chem. Soc. 116, 11655–11666 [Google Scholar]
- 44. Lin T. H., Chia C. M., Hsiao J. C., Chang W., Ku C. C., Hung S. C., Tzou D. L. (2002) Structural analysis of the extracellular domain of vaccinia virus envelope protein, A27L, by NMR and CD spectroscopy. J. Biol. Chem. 277, 20949–20959 [DOI] [PubMed] [Google Scholar]
- 45. Engelstad M., Smith G. L. (1993) The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 194, 627–637 [DOI] [PubMed] [Google Scholar]
- 46. Payne L. (1978) Polypeptide composition of extracellular enveloped vaccinia virus. J. Virol. 27, 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kay L. E. (1997) NMR methods for the study of protein structure and dynamics. Biochem. Cell Biol. 75, 1–15 [DOI] [PubMed] [Google Scholar]
- 48. Bax A., Vuister G. W., Grzesiek S., Delaglio F., Wang A. C., Tschudin R., Zhu G. (1994) Measurement of homo- and heteronuclear J couplings from quantitative J correlation. Methods Enzymol. 239, 79–105 [DOI] [PubMed] [Google Scholar]
- 49. Adamson A. W. (1960) Physical Chemistry of Surfaces, pp. 457–481, Interscience Publishers, Inc., New York [Google Scholar]
- 50. Wishart D. S., Sykes B. D. (1994) Chemical shifts as a tool for structure determination. Methods Enzymol. 239, 363–392 [DOI] [PubMed] [Google Scholar]
- 51. Wishart D. S., Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 52. Fass D., Harrison S. C., Kim P. S. (1996) Retrovirus envelope domain at 1.7 angstrom resolution. Nat. Struct. Biol. 3, 465–469 [DOI] [PubMed] [Google Scholar]
- 53. Akey D. L., Malashkevich V. N., Kim P. S. (2001) Buried polar residues in coiled-coil interfaces. Biochemistry 40, 6352–6360 [DOI] [PubMed] [Google Scholar]
- 54. Bowie J. U., Lüthy R., Eisenberg D. (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253, 164–170 [DOI] [PubMed] [Google Scholar]
- 55. Weissenhorn W., Dessen A., Harrison S. C., Skehel J. J., Wiley D. C. (1997) Atomic structure of the ectodomain from HIV-1 gp41. Nature 387, 426–430 [DOI] [PubMed] [Google Scholar]
- 56. Dallo S., Rodriguez J. F., Esteban M. (1987) A 14K envelope protein of vaccinia virus with an important role in virus-host cell interactions is altered during virus persistence and determines the plaque size phenotype of the virus. Virology 159, 423–432 [DOI] [PubMed] [Google Scholar]
- 57. Gong S. C., Lai C. F., Dallo S., Esteban M. (1989) A single point mutation of Ala-25 to Asp in the 14,000-Mr envelope protein of vaccinia virus induces a size change that leads to the small plaque size phenotype of the virus. J. Virol. 63, 4507–4514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rodriguez J. F., Smith G. L. (1990) IPTG-dependent vaccinia virus: identification of a virus protein enabling virion envelopment by Golgi membrane and egress. Nucleic Acids Res. 18, 5347–5351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chang S. J., Shih A. C., Tang Y. L., Chang W. (2012) Vaccinia mature virus fusion regulator A26 protein binds to A16 and G9 proteins of the viral entry fusion complex and dissociates from mature virions at low pH. J. Virol. 86, 3809–3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.