

Background: Huntington disease is a fatal neuropsychiatric disorder caused by aberrant protein folding and interactions.
Results: An interaction network composed of primary and secondary huntingtin-interacting proteins is significantly enriched for pathways implicated in HD, including RhoGTPases.
Conclusion: Huntingtin interacts with members of the Rho GTPase signaling pathways and regulates filipodial dynamics.
Significance: This protein interaction network provides a resource for HD target discovery.
Keywords: Cell Death, Cell Motility, Computational Biology, Huntington Disease, Neurodegeneration, Neurodegenerative Diseases, Protein-Protein Interactions, Proteomics, Rho GTPases
Abstract
Huntington disease (HD) is an inherited neurodegenerative disease caused by a CAG expansion in the HTT gene. Using yeast two-hybrid methods, we identified a large set of proteins that interact with huntingtin (HTT)-interacting proteins. This network, composed of HTT-interacting proteins (HIPs) and proteins interacting with these primary nodes, contains 3235 interactions among 2141 highly interconnected proteins. Analysis of functional annotations of these proteins indicates that primary and secondary HIPs are enriched in pathways implicated in HD, including mammalian target of rapamycin, Rho GTPase signaling, and oxidative stress response. To validate roles for HIPs in mutant HTT toxicity, we show that the Rho GTPase signaling components, BAIAP2, EZR, PIK3R1, PAK2, and RAC1, are modifiers of mutant HTT toxicity. We also demonstrate that Htt co-localizes with BAIAP2 in filopodia and that mutant HTT interferes with filopodial dynamics. These data indicate that HTT is involved directly in membrane dynamics, cell attachment, and motility. Furthermore, they implicate dysregulation in these pathways as pathological mechanisms in HD.
Introduction
Huntington disease is an autosomal dominant neurodegenerative disease caused by a CAG repeat expansion in the first exon of the HTT gene that encodes the protein huntingtin (HTT) (1). HD3 manifests with progressive motor and psychiatric impairments caused by neuronal dysfunction and loss in the cortex and striatum (2, 3). Huntingtin is involved in a variety of cellular functions, including vesicle transport, transcription, and energy metabolism (4). HD pathogenesis is generally thought to result from a combination of a gain of toxic properties by mutant HTT as well as a loss of normal huntingtin function (5). Huntingtin is an ∼350-kDa protein containing a polyglutamine (polyQ) region, a proline-rich region (PRR), HEAT (Huntingtin, elongation factor 3, protein phosphatase 2A, target of rapamycin 1) repeats, and a number of caspase cleavage sites (4, 6). Several studies have emphasized a critical role of misfolded N-terminal fragments of mutant HTT (7, 8) that are natural products of HTT processing (9). HTT is known to have a large number of interacting proteins involved in a diverse range of biological processes. Numerous studies have shown that polyQ expansion in HTT may alter biological processes that are essential for cellular homeostasis and neuronal survival through impairment of its protein binding activities (10–13).
A number of large scale screens aimed to elucidate new pathways involved in HD pathogenesis by defining HTT partners using yeast two-hybrid (Y2H) and affinity purification approaches (14–17). Analyses of binary interactions or complexes identify the first level of HTT-interacting proteins. We have previously described a protein interaction network derived from a comprehensive Y2H screen using HTT as a bait. That study reported 102 high confidence HTT-interacting proteins, and many of these were shown to be modifiers of mutant HTT toxicity in a Drosophila model of HD (16). In this study, we report Y2H screening results for these primary HTT-interacting partners derived from a genome-scale interaction map (18, 19). Using the 102 HTT primary partners identified in our first screen, we identified a secondary interactome of 2038 known partners to build an expanded huntingtin protein interaction network (HDNet). This network includes HTT-primary, primary-primary, and primary-secondary interacting proteins. We analyzed the connectivity properties of these proteins at the two levels, showing significantly high interconnectivity between HDNet members compared with random proteins in a global curated interaction network from Human Protein Resource Database (HPRD).
Integration of data from different “omics” approaches has been shown to improve functional annotations and to help to formulate biological hypotheses (20). Combination of biological annotations with integration of gene expression data from post-mortem HD brain highlighted the role of Rho family GTPase signaling proteins in HD pathology. Using cell models, we showed that components of this pathway are modifiers of expanded polyQ-induced toxicity. We also showed that mutant HTT interferes with BAIAP2-induced filopodia formation, validating the role of Rho signaling in mutant HTT toxicity. Our study provides a comprehensive resource of binary protein interactions that define novel pathways contributing to Huntington disease pathology.
EXPERIMENTAL PROCEDURES
Y2H Screen
Y2H screens were performed as described previously (21). The screen for primary HTT partners has been described in Kaltenbach et al. (16). Interactions of primary partners have been extracted from a genome-wide Y2H screen as described previously (18). Y2H screens were performed in 96-well plates by mating in each well 5 × 106 cells of a yeast clone expressing a single bait with 5 × 106 clonally diverse cells from a prey library. After mating overnight, the matings were plated onto medium that was selected simultaneously for the mating event, the expression of the ORF selection markers, and the activity of the metabolic reporter genes ADE2 and HIS3. Yeast that grew on this selection medium (“positives”) were counted and transferred into liquid medium in a 96-well format. Cloned inserts were amplified from plasmid PCR. Liquid cultures grown from positive yeast colonies were used as templates in PCRs that amplified either both bait and prey cDNA inserts or prey inserts only in screens where the baits had been sequenced before the matings.
To determine identity of the coding genes from which the primary and secondary sequence fragments were derived, we first aligned cDNA fragments to the set of human RNA sequences downloaded from NCBI (ncbi.nih.gov). All Y2H search data and DNA sequences used to determine interaction pairs reported in this study are included in supplemental Table S2.
Network Topology Analysis
HTT primary and secondary proteins were anchored to the PPI network defined by HPRD (Release 9). The shortest distances between any two nodes in the PPI network were computed through the Matlab package MATLAB BGL. The largest subgraph contains 9219 proteins, and 64 primary and 1167 secondary proteins could be found in this subgraph, respectively. All network metrics were based on this largest subgraph. To approximate the distribution of the shortest distance observed in HPRD, we randomly sampled 1 million networks with each having 64 nodes as controls to the primary proteins, and 100,000 networks with each having 1167 nodes as controls to the secondary proteins.
Functional Annotation of the Network
Total data set and the subgroups of primary and secondary partners of HTT were analyzed using Ingenuity Pathway Knowledge Base (Ingenuity System, Mountain View, CA). The three groups were subjected to IPA Core analysis. Canonical pathways were considered as significantly enriched with a false discovery rate corrected p value of <0.01. Enriched canonical pathways were ranked based on the corrected p value obtained for the whole HDNet dataset and were represented using the IPA comparison analysis tool. The most significantly enriched lists were visualized in Cytoscape (22), using a union of HDNet and HPRD networks (23). Proteins were represented using node color for HD dysregulation data (24, 25) based on significance level (corrected p value of <0.01) and node shape for level of interaction with HTT (primary/secondary). Edges were represented as lines and dashed lines based on the source of interaction, HDNet and HPRD respectively. Only connected nodes are represented in Figs. 4 and 5 for better visibility; information of all nodes is found in supplemental Table S4.
FIGURE 4.

Modules identified by functional and gene expression enrichment analysis. Functional groups identified using ingenuity pathway and enriched gene expression dysregulation analyses are represented as modules based on a combination of HDNet and HPRD interactions. Node shape indicates the interaction type (diamond indicates primary and hexagon indicates secondary) with HTT from HDNet data. Node color indicates dysregulation in gene expression (log2 fold-change is indicated; yellow indicates increased and blue indicates decreased). Solid lines indicate interaction from HDNet. Dashed lines indicate interactions from HPRD.
FIGURE 5.

Additional functional modules identified by ingenuity pathway analysis. Proteins identified using IPA canonical pathways, but not significantly enriched in gene expression dysregulation, are represented as modules based on a combination of HDNet and HPRD interactions. Node shape indicates the interaction type (diamond indicates primary and hexagon indicates secondary) with HTT from HDNet data. Node color indicates dysregulation in gene expression (log2 fold-change is indicated; yellow indicates increased and blue indicates decreased). Solid lines indicate interaction from HDNet. Dashed lines indicate interactions from HPRD.
Domain Identification
The mapped protein segments were searched against NCBI conserved domain database (version 3.05) to obtain domain information, with settings of E-value cutoff of 0.01 and filtering low complexity regions. Whenever multiple domains were found on the same protein segments, only the most specific hits were kept. By definitions from the conserved domain database, the specificity of hits decreases in the order of specific domain, superfamily, and multiple domains.
Plasmids, siRNA, and Lentiviruses
BAIAP2 cDNA was obtained from Open Biosystems (MHS1011-59012), PCR-amplified with Pfu Turbo (Agilent) as recommended by manufacturer, with the following primers: forward primer, 5′-CACCATGGCTCTGTCTCGCTC-3′, and reverse primer, 5′-TGGCCATCTGCTGAGGAG-3′. cDNA was next cloned into the Gateway pENTR-D-TOPO vector; C-terminal V5-tagged BAIAP2 was generated by LR reaction with pDEST40 following the manufacturer's protocol (Invitrogen). Clones were verified with sequencing at all the steps. pDEST40-LacZ was used as a negative control. Entry clones containing HTT N-terminal fragments 1–558 (with 23Q or 135Q) fused to a C-terminal GFP tag have been described previously (26). LR reaction was performed with pLenti CMV Puro DEST vector (w118-1, Addgene); integrity of clones and length of CAG repeats were verified by sequencing at all steps. Lentiviral production and infection were performed in HEK293FT as described previously (27). Full-length HTT plasmids were described previously (28). Dharmacon siGENOME SMARTpools (Thermo Scientific) were used for siRNA disruption of CASP3 (M-043042), BAIAP2 (M-046696), EZR (M-046568), PI3KR1 (M-041079), PIK3R2 (M-041085), PIK3R3 (M-041300), PAK2 (M-040615), WAVE (M-061823), and RAC1 (M-041170) using nontargeting pool (D-001206) as a control.
Cell Culture and Transfection
Mouse striatal cell lines expressing full-length HTT with either wild-type (7Q/7Q) or mutant (111Q/111Q) polyglutamines were handled as described previously (29), maintained at 33 °C in a humidified atmosphere of 95% air and 5% CO2 in DMEM containing 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were nucleofected using kit L (Amaxa) with 3 μg of siRNA for two million cells, as described previously (28). Mouse NIH-3T3 and human HEK293T cells were maintained at 37 °C in a humidified atmosphere of 95% air and 5% CO2 in DMEM containing 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Infection of HEK293T cells was performed as described previously (27). Cells were selected with puromycin (Invitrogen) at a concentration of 1 μg/ml culture media 48 h after infection. NIH-3T3 and HEK293T cells were transfected using Lipofectamine 2000 reagent following the manufacturer's protocol (Invitrogen), with full-length HTT-17Q and HTT-138Q, C-terminal V5-tagged BAIAP2, or Dharmacon smart pool BAIAP2 siRNA.
Caspase Assays
Cell death assays were performed as described previously (28). Briefly, transfected cells were plated on collagen-treated 96-well plates and incubated for 48 h. After 24 h of serum starvation, activation of caspase 3/7 was measured in cells using Apo 3/7 HTS high throughput screen assay kit (Cell Technology), using Fusion Alpha-FP HT (PerkinElmer Life Sciences) microplate reader with excitation and emission wavelengths of 420 and 520 nm, respectively. Toxicity is represented as caspase 3/7 activity in change in relative fluorescence units/min/mg of protein, relative to STHdh111Q/111Q treated with nontargeting siRNA pool. Tests were performed in triplicates; t test was used for statistical analysis (GraphPad Prism).
Western Blotting and Co-immunoprecipitation
Cells were seeded on 6-well plates, harvested, and lysed with M-PER (Thermo Scientific) 72 h after transfection for Western blotting. Primary antibodies anti-BAIAP2, PIK3R1 (Genscript), Ezrin, PAK2 (Cell Signaling), PIK3R2 and PIK3R3 (R&D Systems), RAC1 (Sigma), WASF1 (Upstate), HTT (Millipore), and β-actin (Sigma) were used for protein level quantification and normalization following the manufacturers' protocols. Cells were seeded on 6-well plates, harvested, and lysed with RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1% sodium deoxycholic acid, 0.1% SDS, protease and phosphatase inhibitors) 48 h after transfection for co-immunoprecipitation. For V5-tagged protein immunoprecipitation, lysates were incubated with anti-V5-agarose affinity gel for 90 min and washed three times in RIPA buffer following the manufacturer's protocol (Sigma). For GFP-tagged precipitation, lysates were pre-cleared with protein G-Sepharose beads (GE Healthcare) for 1 h, incubated overnight with GFP antibody (mouse, Covance), and incubated with protein G-Sepharose beads for 1 h before three washes with RIPA buffer. All steps were performed at 4 °C. Antibodies used for detection of tagged proteins were anti-V5 (Invitrogen), anti-GFP (Cell Signaling).
Immunofluorescence and Filopodia Counting
NIH-3T3 were plated on poly-l-lysine eight-chamber slides (BD Biosciences). 48 h after transfection, cells were rinsed once with 1× PBS and fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.2% Triton X-100 for 2 min. After 30 min of incubation in blocking solution (5% normal donkey serum in PBS), cells were incubated overnight at 4 °C with anti-HTT (Millipore) and anti-V5 (invitrogen) primary antibodies. After incubation with respective secondary antibodies coupled with Alexa Fluor 488 and 647 (Invitrogen) for 1 h at room temperature, cells were incubated with rhodamine-phalloidin coupled with Alexa Fluor 555 (Invitrogen) for 20 min at room temperature to detect filamentous actin. Cells were washed three times with PBS and mounted using Prolong Gold with DAPI (Invitrogen). 15 cells per experiment were imaged using the confocal microscope LSM 780 (Zeiss). Three replicates per condition were performed. Images were analyzed with Zeiss Zen (2011) software; V5- and HTT-positive cells were digitalized using ImageJ process “find edges”; the number of actin-positive filopodia per cell was counted manually for each condition. Number of filopodia per cell was expressed as relative value compared with average filopodia number in control cells. Filopodia were double counted by a blinded experimenter. t test was used for statistical analysis (GraphPad Prism). Co-localization experiments were performed using the same methods and materials.
RESULTS
Network Properties of Huntingtin-interacting Proteins in HDNet
We had previously performed two large scale screens to identify novel HTT-interacting proteins using Y2H and mass spectrometry-based methods (16). For the Y2H screen, polyQ containing HTT fragment baits of amino acids 1–90, 1–450, or 1–740 were screened in both wild-type (23Q) and mutant (>45Q) forms against multiple prey libraries generated from human cDNA. Stringent criteria have been used to define high confidence core dataset composed of 102 direct HIPs. We then extracted interaction data for these 102 HIPs to include secondary interacting proteins. This resulted in a network containing 2141 nodes as follows: HTT, 102 primary and 2038 secondary interacting proteins connected through 3235 edges. These edges include 102 interactions of primaries with HTT, 51 primary-primary interactions, and 3082 primary-secondary interactions, representing data obtained from 9423 individual Y2H-positive yeast colonies (Fig. 1 and supplemental Tables S1 and S2). We refer to this network as HDNet.
FIGURE 1.

HDNet is an HTT interactome composed of primary and secondary interacting proteins. A, network is composed of HTT (red), 102 primary partners, and 2038 secondary partners connected though 3235 edges. B, first level of HTT-interacting proteins shows HTT-primary and primary-primary interactions. Node colors represent gene expression dysregulation in HD human caudate sample.
HDNet is centered on HTT with two levels of partners and includes interactions between primary nodes (Fig. 1B). Because we do not have information about interactions between secondary nodes, the network does not have a scale-free topology. To characterize the connectivity properties of HDNet members in the context of a scale-free network, we mapped HTT primary and secondary partners in the curated large scale HPRD protein interaction network (HPRD Release 9) (23). We then calculated the shortest path among them, using the literature-curated interactions described in HPRD. With nodes defined according to Entrez Gene IDs, 79 out of 102 primary and 1408 out of 2038 secondary interacting proteins were present in the largest sub-network in HPRD, which consists of 8783 of the total 9019 proteins (Fig. 2A).
FIGURE 2.
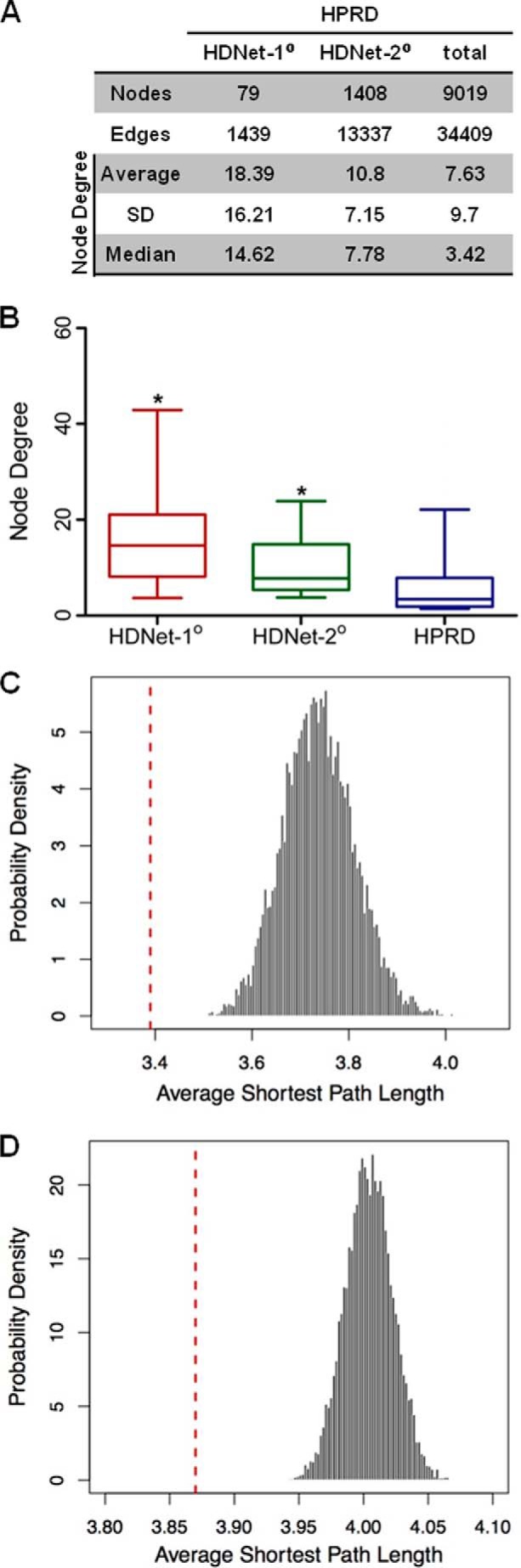
Connectivity properties of HDNet. A, distribution of node degrees of HDNet partners in the HPRD network. B, average node degrees of primary and secondary partners compared HPRD nodes in HPRD network (with average, S.D., and median, p value <0.001). *, p < 0.05. C, distribution of average shortest path lengths from HPRD subnetworks with the same node degree distribution as primary partners (red line indicates average shortest path length between primary nodes). D, distribution of average shortest path lengths from HPRD subnetworks with the same node degree distribution as secondary partners (red line indicates average shortest path length between secondary nodes).
This analysis showed that both primary and secondary proteins present in HDNet have significantly higher average node degrees as compared with the average for HPRD as a whole (18.25 and 10.80 versus 7.63, both p value < 0.001). This is true for median node degrees as well, indicating that the high connectivity of HDNet proteins in HPRD is not due to the presence of highly connected outliers (Fig. 2, A and B). The average shortest path lengths among both primary (3.39) and secondary HIPs (3.87) are significantly shorter than that observed in HPRD as a whole (4.25; Fig. 2, C and D; both p value <2.2e-16; t test). The significantly higher connectivity observed among HDNet primary and secondary nodes within the HPRD background suggests that these groups of proteins are enriched for shared functions and/or membership in common biological pathways (30). However, high node degrees could also contribute to short path lengths between nodes. We thus analyzed the properties of randomly generated networks to determine whether this was the case. We first tested if the number of proteins in each group influenced the respective connectivity. We selected random cohorts of proteins with population sizes of HDNet primary (79) or secondary (1408) interactors from HPRD and calculated the average shortest path lengths for these. This process was repeated 106 times for the primary cohort simulation and 105 times for the secondary cohort simulation. In both cases, the shortest path length distributions were very similar to the HPRD as a whole, ∼4.25 in both cases (data not shown). Given that nodes in HDNet have a higher average node degree as compared with HPRD, we wanted to determine how this property might contribute to the shorter path lengths observed between primary and secondary HIPs in HPRD. To do this, we repeated the simulation with randomly selected cohorts as described above but preserved the node degree distributions of the primary and secondary nodes. These simulations were performed 104 times in each case. We observed that sub-networks simulated this way are less interconnected than either primaries or secondaries (Fig. 2, C and D, respectively).
Analyses of the network properties of primary and secondary nodes in HPRD indicate that HIPs identified in our Y2H screen tend to have higher node degrees and tend to be more highly interconnected. In both cases, these differences are statistically significant. The high degree of interconnectedness of HDNet proteins in the independently derived and curated HPRD protein interaction network indicates that these proteins have shared properties and are likely to participate in shared biological functions. The increased node degree distribution observed among HIPs also indicates that the HTT network is enriched for highly connected proteins.
Functional Analysis of HDNet
Functional analysis of HDNet was performed using the IPA tool (Ingenuity System Inc.). We found that 51 canonical pathways were significantly enriched (B–H, p value = or <0.01) among HDNet proteins (supplemental Table S3). The 20 most significant canonical pathways enriched in HDNet are shown in Fig. 3A. Analyses were also performed separately with either primary or secondary nodes alone. This was done to determine the relative contribution of each category of partners (direct and indirect) to pathway enrichments, as well as to uncover potential bias due to an overlap between our dataset and HTT-interacting proteins curated in the IPA knowledge base. Notably, the fourth most significantly enriched canonical pathway in HDNet is Huntington disease signaling. Some of the proteins in this pathway have been reported as HIPs and have been validated in multiple sources (16). Interestingly, HD signaling is also significantly enriched in secondary interaction partners. These results further validate the biological significance of HDNet.
FIGURE 3.
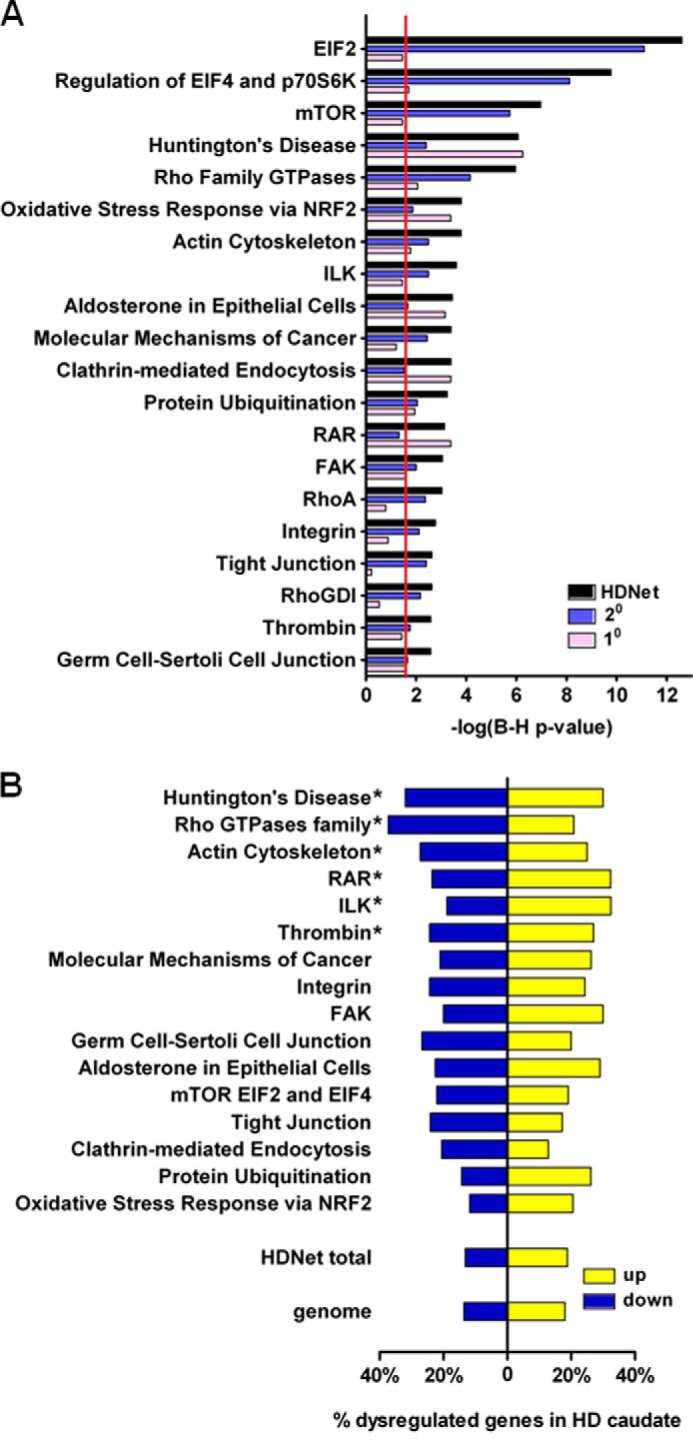
Functional characterization of primary and secondary HTT interaction partners using IPA. A, ingenuity pathway analysis of HDNet members and primary and secondary separately. The 20 most enriched canonical pathways are represented based on whole dataset ranking. Red line indicates threshold for significance (B-H corrected, p value = 0.05). B, enrichment analysis of IPA functional subgroups for gene expression dysregulation from human HD caudate (log2 fold-change is indicated; *, B-H corrected, p value <0.01).
The top three most significantly enriched pathways revealed by IPA analysis are related to protein synthesis and protein homeostasis as follows: EIF2, EIF4, p70S6K, and mammalian target of rapamycin signaling pathways (Fig. 3A). Primary HTT partners like PIK3R1, -2, and -3 (phosphoinositide 3-kinase regulatory) subunits and EIF3A (eukaryotic elongation factor 3 member A), as well as secondary partners like EIF and ribosomal proteins S and L family members are represented in all these canonical pathways (supplemental Table S3). Notably, PIK3R family members are present in a number of enriched pathways. Dysregulation of the PI3K pathway is associated with numerous cancers (31), inflammatory diseases (32), and metabolic diseases like diabetes (33). This pathway has also been shown to play a critical role in neurodegenerative diseases such as HD (34, 35).
Four other enriched canonical pathways are determined mainly by primary interacting partners as follows: NRF2-mediated oxidative stress response; aldosterone signaling in epithelial cells; clathrin-mediated endocytosis signaling, and retinoic acid receptor activation. NRF2, aldosterone, and retinoic acid receptor signaling pathway findings highlight the transcriptional role of HTT and its dysregulation in HD (36). The clathrin-mediated endocytosis pathway is known to be affected by mutant HTT (37, 38). Our data from primary and secondary interacting partners indicate that multiple components of these pathways interact with mutant HTT.
Enriched pathways driven by the secondary partners provide a broader view of global signaling pathways potentially impacted by HTT. These include the following: Rho family GTPase, actin cytoskeleton, and integrin-linked kinase signaling as well as molecular mechanisms of cancer. Interestingly, Rho family GTPase signaling is enriched in the secondary partner set, with Rho family, RHOA, and ARHGDI (RHOGDI), sharing components with actin signaling and integrin-linked kinase pathways. These canonical pathways are all involved in cell migration, adhesion, proliferation, and signal transduction (39, 40). Members of ARHGEF (anti-Rho guanine nucleotide-exchange factor), Rho GTPases (ROCK1 and RAC1), myosins and Wiskott-Aldrich syndrome protein families are found as secondary HTT partners. Several studies have showed that Rho GTPases, in particular ROCK1, have an effect on aggregation and degradation of mutant HTT (41, 42). As other components of these pathways have been highlighted independently with our Y2H screen, we suggest that this could also support the known role of HTT in vesicle trafficking as well as novel functions and pathological effects on cell adhesion and cell migration.
Correlation of HDNet with Transcriptional Dysregulation in HD
Integration of data from different types of genome-scale approaches has been shown to facilitate functional annotation and to help to formulate biological hypotheses (20, 43). To prioritize the exploration of enriched pathways and associated protein networks, we next integrated HDNet pathway cohorts with HD gene expression data. We focused on gene expression dysregulation data in grade 2 HD patient caudate nucleus (24, 25). Because some canonical pathways had significant overlap in gene members, highly similar pathways were combined into composite lists prior to analysis. EIF2, EIF4/p70S6K, and mammalian target of rapamycin signaling (Fig. 3A) were grouped into a pathway referred to as “mammalian target of rapamycin EIF2 and EIF4” (Fig. 3B). Similarly, Rho family GTPase, RhoA, and RhoGDI (Fig. 3A) signalings were grouped into a pathway referred to as “Rho GTPase family” (Fig. 3B). We then tested whether canonical pathways identified with ingenuity analysis and grouped pathways were enriched in dysregulated genes identified in HD caudate. Six canonical pathways were found enriched in dysregulated genes as compared with genome as well as genes represented in the entire HDNet dataset (Fig. 3B; hypergeometric test, B–H corrected p value < 0.01 and supplemental Table 4). Subnetworks of dysregulated proteins from enriched pathways are shown in Fig. 4. These subnetworks show interconnections based on a combination of HDNet and HPRD data. Subnetworks from canonical pathways without significant enrichment in genes dysregulated in HD caudate are shown Fig. 5. Notably, in this analysis to identify HIPs with dysregulated expression, the Huntington disease signaling pathway showed the highest enrichment of dysregulated genes. This analysis highlighted also the involvement of Rho GTPase family signaling, with a group of proteins found in more than one enriched canonical pathway. These include EZR (Ezrin), BAIAP2 (brain-specific angiogenesis inhibitor 1-associated protein 2), PI3K family members, cAMP-response element-binding protein, and cAMP-response element-binding protein-binding protein (supplemental Table S4 and Fig. 4)
Based on a combination of pathway analysis combined with gene expression data from HD brain, we focused on the Rho GTPase family module. Rho GTPases are involved in numerous cell processes acting downstream of growth factor receptor activation. Exchange of GDP for GTP by guanine exchange factors activates small GTPases and results in binding to their downstream effectors. GTP hydrolysis by GTPase-activating proteins inhibits their function. RhoGDIs (Rho guanine nucleotide-dissociation inhibitors) regulate Rho GTPase activity by disassociating them from plasma membrane (44). Rho GTPases such as RAC1 or CDC42 activate WAVE complexes (nucleation-promoting factors, including WAS, WASL (N-WASP), and WAVE1–3 (WASF1)), which in turn activate Arp2/3 complex, leading to induction of actin polymerization and remodeling of cytoskeletal interactions. Formins, like mDia, protect barbed ends of actin filaments from capping, thereby promoting elongation. Activated RAC1 binds to BAIAP2 to form a protein complex with WAVE to induce membrane ruffling and actin polymerization through Arp2/3 complex (45). Rac also activates PAK proteins, responsible for the delivery of WAVE protein to the plasma membrane. Ena-VASP complexes, anti-capping proteins, are found at the tip of filopodia. BAIAP2 induces filopodia formation through its I-BAR domain (see Table 1 and Fig. 6), inducing cluster formation of phosphatidylinositol 4,5-bisphosphate at the membrane and activating phosphatidylinositol 4,5-bisphosphate-binding proteins such as WASL (46). BAIAP2 activity is inhibited by threonine phosphorylation, leading to 14-3-3 protein binding (47).
TABLE 1.
Conserved domains most frequently observed interacting with huntingtin in the primary network
| Domain | Accession | No. | Associated proteins (gene_symbol) |
|---|---|---|---|
| WW superfamily | cl00157 | 1325 | APBB2,TCERG1, WBP4, FNBP4, SETD2, WAC, PRPF40A |
| FF superfamily | cl02610 | 170 | TCERG1, PRPF40A |
| zf-H2C2_2 superfamily | cl16282 | 144 | SP3, KLF11, ZNF655, ZNF675 |
| SH2 superfamily | cl15255 | 132 | PIK3R1, PIK3R2, PIK3R3 |
| SH3 | cd00174 | 123 | PRMT2, PTK6, SRGAP3, BAIAP2, SORBS1, SRGAP2, OSTF1, SRGAP1 |
| Dynamitin | pfam04912 | 50 | DCTN2 |
| NrdE_NrdA | TIGR02506 | 46 | USP9X |
| DnaJ | cd06257 | 43 | DNAJC11, DNAJC4, DNAJA3, DNAJC21 |
| Ubiquitin-associated superfamily | cl00153 | 42 | UBAC1, NUB1, OPTN |
| Pyruvate_kinase superfamily | cl09155 | 24 | PKM |
| NR_DBD_like superfamily | cl02596 | 21 | PPARG |
| Snf7 superfamily | cl02305 | 21 | CHMP4B |
| ARS2 superfamily | cl04863 | 17 | SRRT |
| ANK | cd00204 | 16 | OSTF1, NFKB1 |
| RhoGAP superfamily | cl02570 | 16 | ARHGAP24, ARHGAP25, SRGAP1 |
| DHR2_DOCK superfamily | cl06123 | 14 | DOCK9, DOCK11 |
| 14-3-3 superfamily | cl02098 | 12 | YWHAB |
| ARM | cd00020 | 12 | KPNA3 |
| Mre11_DNA_bind superfamily | cl04421 | 12 | MRE11A |
| zf-C2H2_jaz superfamily | cl09952 | 11 | DNAJC21 |
| BAR superfamily | cl12013 | 10 | BAIAP2, PACSIN1 |
| TBD superfamily | cl15118 | 10 | TANK |
| DDT superfamily | cl02674 | 7 | BAZ1A |
| VHS_ENTH_ANTH superfamily | cl02544 | 7 | HIP1 |
| P4Ha_N superfamily | cl07084 | 6 | P4HA1 |
| PH-like superfamily | cl00273 | 6 | DNM2, DNM1 |
| SAM_superfamily | cl15755 | 6 | SASH1 |
| TACC | pfam05010 | 6 | TACC1 |
| FEZ superfamily | cl06682 | 5 | FEZ1 |
| zf-CCCH superfamily | cl11592 | 5 | MKRN2 |
| MT superfamily | cl15084 | 4 | DYNC1H1 |
| vWFA superfamily | cl00057 | 4 | PSMD4 |
| API5 superfamily | cl09392 | 3 | API5 |
| HLH | cd00083 | 3 | SREBF2 |
| KRAB_A-box superfamily | cl02581 | 3 | ZNF133 |
| Med15 | pfam09606 | 3 | MED15 |
| Adaptin_N | pfam01602 | 2 | AP2A2 |
| BTB superfamily | cl02518 | 2 | ZBTB16 |
| Coatamer_β_C superfamily | cl06658 | 2 | COPB1 |
| CULLIN superfamily | cl10551 | 2 | CUL5 |
| ERM | pfam00769 | 2 | EZR |
| NGN_Euk | cd09888 | 2 | SUPT5H |
| PRP40 | COG5104 | 2 | PRPF40A, TCERG1 |
| Pyrophosphatase superfamily | cl00217 | 2 | PPA2 |
| RING superfamily | cl15348 | 2 | MKRN2 |
| TPR | cd00189 | 2 | GTF3C3 |
FIGURE 6.

Domain-domain interactions of primary partners in the Rho signaling module. Schematic representation of primary partners of HTT from Rho GTPase signaling pathway. Rectangles and associated labels indicate conserved domains identified from Y2H sequence analysis. Horizontal black lines indicate the coverage of the domain by positive clone sequences; numbers to right of black lines and thickness indicate the number of independent Y2H observations. Arrows indicate interactions with other conserved domains (vertical labels) contained in interacting proteins of family members (horizontal labels). Red lines indicate coverage of positive sequence for HTT interaction regions. Thickness and numbers to right indicate number of independent observations of Y2H interaction of protein with HTT.
EZR is a member of ERM (ezrin, radixin, and moesin) proteins. ERM proteins mediate the attachment of the membrane to actin filaments and are activated by phosphorylation. They are involved in membrane organization and signal transduction (48). Phosphorylated forms of ERM proteins are found at the site of filopodial shafts in axonal growth cones and are dephosphorylated in response to signals that cause growth cone collapse and loss of filopodia (49).
Domain Interactions in HDNet
HDNet is derived from 9423 unbiased observations of individual binary protein interactions (i.e. Y2H-positive yeast colonies). Because the bait and prey libraries used in this study were prepared from cDNA fragments, Y2H interactions reported here are between protein fragments. The average insert size in our Y2H libraries is 481 bp (±140 bp). This corresponds to an average protein fragment size of 160 amino acids. We used the conserved domain database (50) to identify conserved domains contained in protein coding regions for both primary and secondary HTT interacting partners. Of these, specific conserved protein domains could be identified in 6541 (35%) activation domain fusions and 3903 (21%) binding domain fusions. In 2268 (12%) cases conserved domains were identified for both activation domain and DNA binding domain in a binary pair (supplemental Table S5). To characterize the domain-domain interaction information in HDNet, we determined frequency of domains present in the protein sequences mediating binary interaction with HTT. Table 1 shows the conserved domains most frequently present in primary partners interacting directly with HTT. Only those domains observed interacting with HTT two or more times are included. The WW domain was found as the most frequent conserved domain interacting with HTT. The WW domain is a short module of 40 amino acids that mediate protein-protein interaction with PRR and thus are likely to interact with the PRR of HTT (51). Binding of TCERG1, APBB members, WBP4, and SETD2 with HTT is known to involve WW-PRR domain interactions (11, 52–54). It has also been shown that interactions involving the PRR of HTT can be affected polyQ expansion (55), indicating potential roles in HD pathogenesis (56). The FF domain was also found at high frequency among primary interactors. Composed of three α-helices, FF domains are present in RNA regulatory proteins and Rho GTPase regulatory proteins and are often found adjacent to WW domains (57, 58). High frequency of interactors with DNA binding domains such as helix-loop-helix and zinc finger domains are consistent with the role of mutant HTT in direct effects causing transcriptional dysregulation (36, 59). Src homology 2 and 3 (SH2 and SH3) domains were also found at high frequency in primary partner sequences responsible for their interaction with HTT. The SH2 domain is mainly found in adaptor proteins and recognizes tyrosine-phosphorylated sites involved in intracellular signaling cascades (60). SH3 domains are also present in adaptor proteins that link partners with their respective PRRs (61). Like the WW domain, SH3 has been implicated in HD pathogenesis through its interaction with the PRR of HTT. Two domain superfamilies are linked to protein degradation, DnaJ and ubiquitin-associated domains. Proteins containing these domains have been implicated in mutant HTT toxicity and clearance of HTT aggregates (62–64).
The presence of domains like RhoGAP and DHR2_DOCK (dock homology region, a guanine exchange factor domain) indicates a potential role of HTT in activity of small GTPases such as RAC1 or CDC42. BAR domain superfamily is responsible for membrane remodeling, invagination or protrusion, in collaboration with nucleation-promoting factors. BAR domain superfamily can be divided into three families, based on structure and functions as follows: N-BAR and F-BAR domains form homodimers that bind concave membrane surface, creating a membrane tubular invagination, whereas the I-BAR domain is responsible for membrane protrusion (e.g. lamellipodia/filopodia) (65). These findings are in agreement with results showing enrichments for Rho family GTPases and actin signaling pathways. This analysis adds a new insight into PPI data, more precise resolution for HTT protein interactions at pathway and network levels. As an example, we represented primary partners of Rho GTPase modules (Fig. 6) with their interactions with candidate conserved domains based on our domain analyses. Results showed that BAIAP2 seems to interact mostly through its SH3 domain with HTT. The BAR domain of BAIAP2 is a member of I-BAR, or IMD (IRSp53-MIM homology domain). BAIAP2 interacts through IMD with small GTPases (CDC42, RAC1, and RHOF) and with downstream effectors like EPS8, Mena, and WAVE family members through its SH3 domain for actin remodeling (45, 66–70). Analysis of interactions of the BAR domain of BAIAP2 highlighted PDZ domains present in MAGI (membrane-associated guanylate kinase, WW and PDZ domain containing) and DLG (discs, large homolog), SH3 domain (present in EPS8), and RhoGEF domains (RASGRF1 and FGD5). This is in agreement with the role of BAIAP2 in modulating actin cytoskeleton and membrane protrusion through small GTPases signaling. Domain analysis shows that HTT interacts with the ERM domain of EZR. We also note that all three members of PIK3R subunits interact with HTT through their first SH2 domain. These data support a model of HTT acting as a large scaffolding protein interacting with multiple components of signaling and structural regulators of cytoskeletal and membrane remodeling factors at sites of cell movement.
Modifiers of HD Toxicity in the Rho GTPase Pathway
Our group and others have shown that physical partners of HTT are good candidates as potential modifiers of polyglutamine toxicity (15, 16, 71). As a first step of functional analysis, we asked whether manipulating levels of components of the Rho GTPases subnetwork were modifiers of mutant HTT cytotoxicity. To do this, we used an assay based on immortalized mouse striatal cell Hdh7Q/7Q and Hdh111Q/111Q lines (29) to evaluate the effect of knockdown of candidates on mutant HTT-specific caspase activation upon serum withdrawal (28).
Because it is a significantly enriched canonical pathway with a high levels of transcriptional dysregulation (Figs. 3 and 4), we decided to focus on components of the RhoGTPase signaling module for functional studies. This module contains seven primary HIPs (Fig. 4). Of these, NFKB1 and cAMP-response element-binding protein-binding protein are known to be modifiers of HTT toxicity (72, 73). These were not studied further. Of the remaining primary partners in this subnetwork, siRNA-mediated knockdown of three of the five tested proteins resulted in significant modification of toxicity in this cell model. Knockdown of BAIAP2 showed the most significant reduction of mutant HTT toxicity (Fig. 7), without any effect on caspase activity in the Hdh7Q/7Q cell line (Fig. 8). Intriguingly, expression of BAIAP2 is down-regulated in caudate of grade 0–2 HD patients. This could suggest a toxic role of the interaction between BAIAP2 and mutant HTT, with a compensatory effect of down-regulation in diseased neurons. Knockdown of EZR showed also a rescue effect on mutant HTT toxicity, without any effect on Hdh7Q/7Q cells. In this case, gene expression profiling of HD caudate showed an increase of EZR mRNA levels, suggesting that expression of EZR could be detrimental for HD neurons. Knockdown of regulatory subunits of PI3K seemed to enhance mutant HTT toxicity, with a significant effect of PIK3R1 knockdown effect in Hdh111Q/111Q cells, without any effect on Hdh7Q/7Q cells.
FIGURE 7.
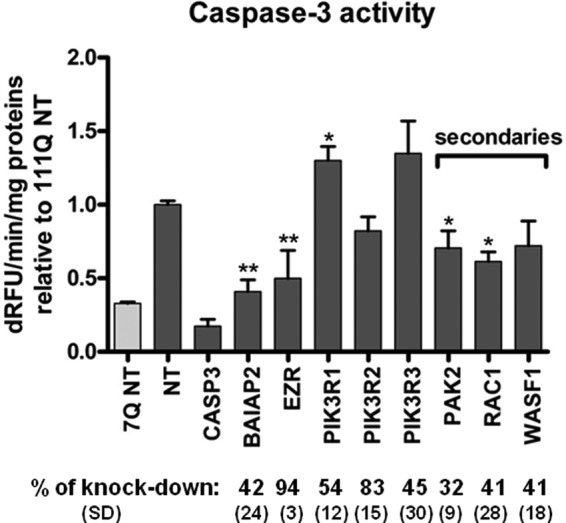
Rho family GTPase signaling components modify mutant HTT-induced toxicity. Caspase 3/7 activity following siRNA-mediated knockdown of Rho family GTPase signaling components in STHdh111Q/111Q cells (n = 3, *, p value<0.05; **, p value <0.01, t test). Data are represented as relative value to STHdh111Q/111Q treated with nontargeting siRNA (NT). Percent protein knockdown and standard deviations as determined by Western blot (n = 3) are indicated. dRFU, change in relative fluorescence units.
FIGURE 8.
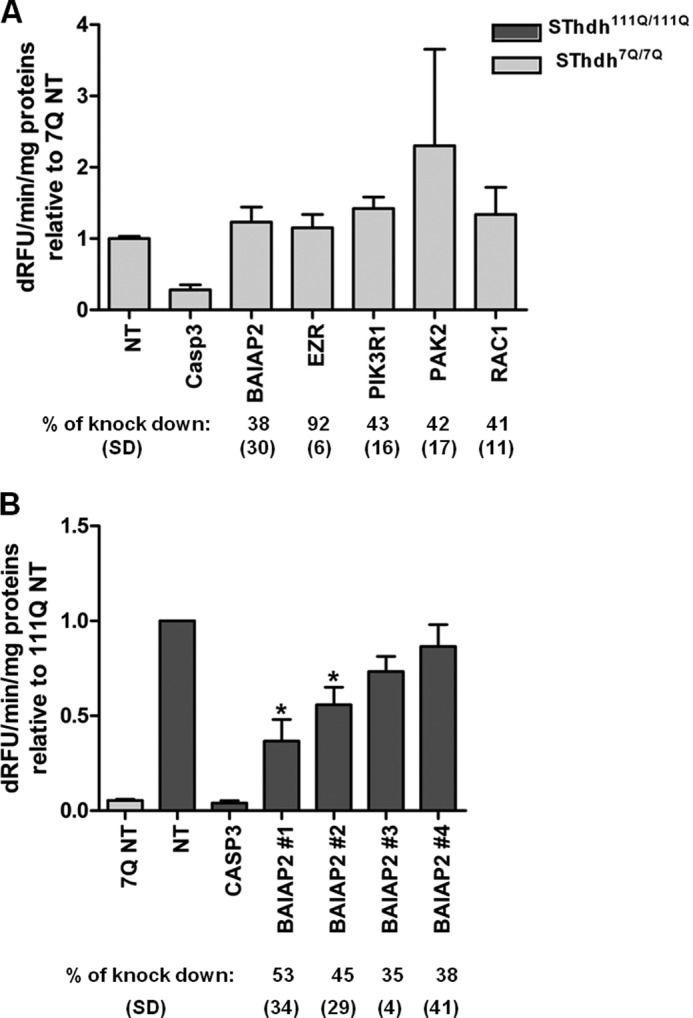
Modifier effects of Rho family GTPase signaling components is specific for mutant HTT-induced toxicity. A, caspase 3/7 assay following siRNA-mediated knockdown of Rho family GTPase signaling components in STHdh7Q/7Q cells (n = 3). Data are represented as relative to STHdh7Q/7Q treated with nontargeting siRNA (NT). Percent protein knockdown and standard deviations as determined by Western blot (n = 3) are indicated. B, caspase assay following BAIAP2 knockdown in STHdh111Q/111Q cells using individual siRNAs from deconvoluted pools (n = 3, *, p value<0.05, t test). Percent protein knockdown and standard deviations as determined by Western blot (n = 3) are indicated. dRFU, change in relative fluorescence units.
EZR, BAIAP2, and PI3K proteins are keys components of the signaling pathway through Rho family GTPases, but they are also involved in actin cytoskeletal organization and are present in the IPA actin cytoskeleton signaling pathway (Fig. 4). We examined secondary partners that were common between these two modules and asked whether they could modulate mutant HTT toxicity in striatal cell lines. PAK2 (p21-activated kinase 2) is a serine/threonine kinase that links small GTPases CDC42 and RAC1 to cytoskeleton reorganization (74). siRNA-mediated knockdown of RAC1 and PAK2 showed significant reduction of mutant HTT toxicity, without any effect on caspase activity in the Hdh7Q/7Q cell line. Overall, the observed suppressor effect of knockdown of RAC1 and PAK2 (and WASF1 which trended toward suppression) confirmed the involvement of BAIAP2 and EZR in mediating mutant HTT toxicity.
We then examined the functional significance of the interaction between HTT and Rho GTPase pathway members. Out of the numerous cell processes requiring components of this signaling pathway, cell migration, cell adhesion, and neurite outgrowth were of particular interest in regard to HD. One of the first molecular steps for these cellular processes is structural reorganization of the plasma membrane. Various plasma membrane structures, including invaginations (caveolae and clathrin-coated pits) and membrane protrusions, such as lamellipodia and filopodia, are involved in cell morphogenesis, endocytosis, cell migration, and neuritogenesis. Filopodia are cylindrical extensions of the plasma membrane that contain bundles of parallel actin filaments at their core (75). Filopodia are thought to act as sensors, probing their microenvironment and regulating cell protrusion and cell migration in response to extracellular cues. Key components of focal adhesions, such as integrin, cadherin, PTK2 (focal adhesion kinase), and Talin, are found in the tips of filopodia (76, 77). These cytoskeletal-membrane structures are necessary for neurite formation and outgrowth in cortical neurons (78, 79), as well as synaptogenesis (80). It has been shown that inhibition of Rac1 GTPase function decreases the number of axonal filopodia as well as at growth cone formation in sensory neurons, whereas inhibition of Cdc42 function inhibits filopodia formation only at growth cones (81). In these filopodial structures, BAIAP2 plays a central role as a linker between actin filaments, through its IMD domain and plasma membrane (82), and has been shown to regulate filopodia formation (47).
We confirmed the binary physical interaction between BAIAP2 and HTT observed with Y2H using co-immunoprecipitation methods. Using HEK293T cells overexpressing an HTT fragment (1–558) fused to GFP and BAIAP2 fused to V5 tag, we observed co-immunoprecipitation of normal (23Q) and mutant (135Q) HTT with BAIAP2 (Fig. 9A). We were also able to show a reciprocal co-immunoprecipitation of BAIAP2 with HTT fragment (Fig. 9B). Moreover, using NIH-3T3 cells, we showed that both nonexpanded and mutant HTT co-localized with BAIAP2 and filamentous actin at the tip of filopodial structures (Fig. 9C).
FIGURE 9.

HTT and BAIAP2 interact and co-localize in cells. A, co-immunoprecipitation of GFP-tagged HTT fragments with V5-tagged BAIAP2 expressed in HEK293T cells. BAIAP2 was immunoprecipitated with anti-V5, and HTT was probed with a GFP antibody. A lysate transfected with empty vector was performed in parallel to confirm a specific protein interaction. 10% of input for each protein lysate (left) and immunoprecipitation (IP) results (right) are shown for each protein. B, reciprocal co-immunoprecipitation of V5-tagged BAIAP2 with GFP-tagged HTT. HTT was immunoprecipitated with anti-GFP. A native IgG pulldown was performed as a control (NS). C, co-localization (left panel) in actin-positive (red, center left) filopodia of BAIAP2 (purple, center right) and HTT (green, right) in NIH-3T3 cells. Nuclei were labeled with DAPI (blue).
We next investigated the function of HTT in filopodia formation in the NIH-3T3 cell model. We co-transfected mouse fibroblasts with full-length HTT, normal (17Q), or mutant (138Q), or an empty vector control, along with V5-tagged BAIAP2 or V5-tagged LacZ (control). We observed that ectopic expression of BAIAP2 led to a significant increase in the number of actin-positive filopodia in NIH-3T3 cells when co-transfected with empty control. Overexpression of normal full-length HTT did not interfere with BAIAP2-induced increase of filopodia. However, expression of mutant HTT completely abolished this effect (Fig. 10A). This result suggests that the presence of mutant HTT inhibits the induction of filopodia formation by BAIAP2. Additionally, we showed that siRNA-mediated knockdown of BAIAP2 decreased the number of filopodia-like protrusions in mouse embryonic fibroblasts, without any effect of normal HTT overexpression (Fig. 10B). The presence of mutant HTT showed a trend to abolish this decrease. Filopodia are dynamic structures; elongation and retraction of the finger-like sensors are not yet completely understood. Rho family members have been shown to be involved in both formation and stabilization of these structures, and it would be of great interest to study dynamics of filopodia in vitro and in vivo, particularly at the neuronal, axonal, and synaptic level. These experiments demonstrate that huntingtin interacts both physically and genetically with the filopodial protein BAIAP2. Furthermore, mutant HTT can interfere with the ability of BAIAP2 to modulate filopodial morphogenesis.
FIGURE 10.

Mutant HTT interferes with BAIAP2-induced filopodia formation. A, NIH-3T3 cells were co-transfected with V5-tagged BAIAP2 or V5-tagged LacZ (control) along with full-length HTT (17Q) or HTT (138Q) or with an empty vector (control). Cells were fixed and stained with rhodamine-phalloidin, anti-V5, and anti-HTT. The number of actin-positive filopodia were counted in V5- and in HTT-positive cells, 15 cells per experiment with n = 3 (*, p value<0.05, t test). The relative numbers of filopodia in each condition are indicated. B, NIH-3T3 cells were co-transfected with BAIAP2 siRNA or nontargeting siRNA (control) along with full-length HTT (17Q), or HTT (138Q), or with an empty vector as (control). Cells were fixed and stained with rhodamine-phalloidin and anti-HTT. Number of actin-positive filopodia were counted in HTT-positive cells, 15 cells per experiment with n = 3 (*, p value<0.05; **, p value<0.01, t test). The relative numbers of filopodia in each condition are indicated. C and D, expression levels of BAIAP2 and HTT were analyzed by immunoblotting with indicated antibodies, using β-actin as a loading control, for overexpression (C) and knockdown (D) of BAIAP2. E and F, representative images of rhodamine-phalloidin labeled cells co-transfected with BAIAP2 and HTT 17Q (E) or 138Q (F), and their respective digitized images.
DISCUSSION
Here, we describe HDNet, a protein interaction network that provides a genome-scale resource for elucidating normal functions of HTT, novel pathogenic mechanisms of mutant HTT, as well as deeper mechanistic insights into known pathways affected in HD. Partners of HTT identified in our screen, at both primary and secondary levels of interaction, showed enhanced interconnectivity as compared with other proteins in the context of HPRD, an independently derived and curated reference protein interaction network. We demonstrated that this connectivity is not due to node degree effects or annotation biases in HPRD. We conclude that independent validation of enhanced connectivity of HDNet arises from shared functional properties relevant to the functions of HTT as well as the pathogenic process in Huntington disease.
HDNet is significantly enriched in proteins that function in pathways known to be involved in HD, such as protein homeostasis, cytoskeleton, and vesicle trafficking (Fig. 3). These pathways were recently discovered and described as preferentially enriched in HTT partners from two interaction screens by affinity purification followed by mass spectrometry from brain of HD mouse models (14, 17). Here, we demonstrate that expanding the HD network with secondary interacting partners and combining enriched functional modules with gene expression dysregulation data from human brain tissue highlights signaling by Rho family GTPases and actin remodeling as being processes in HTT function and HD pathogenesis.
HDNet is a protein interaction network defined at the protein domain resolution. Identification of binding domain underlying protein interaction networks enhances our mechanistic understanding of how normal and pathological mechanisms may operate at the molecular level (83). This study used a Y2H method based on construction of binding domain and activation domain libraries from cDNA fragments. Therefore, interactions reported here are between protein fragments rather than between full-length proteins (16, 21). In many cases these fragments were shown to contain specific conserved protein domains (as defined by the NCBI conserved domain database). Knowledge of specific domain-domain interactions provides a higher resolution understanding of how specific proteins are interacting with HTT itself as well as HTT binding partners. With regard to HD pathogenesis, identification of binding domains of HTT partners is of high interest. Some studies suggest that the normal role of polyQ regions in proteins would be to stabilize PPI, and expansion of polyQ could have a deleterious effect through a gain of abnormal interactions or impairments of protein interaction dynamics (84). Recently, the role of the proline-rich region of HTT has been highlighted in different studies. Intrabodies targeting PRR were shown to reduce toxicity of mutant HTT and increase turnover of mutant HTT (85). It was also shown that deletion of the PRR domain in full-length HTT caused late onset learning and memory deficits in transgenic mice (86). This suggests that protein interaction with the PRR domain of HTT can affect both its normal and toxic activities as well as turnover. Partners of HTT containing SH3 or WW domains in the binding sequences could thus represent an interesting group of modifiers. We also note that a number of proteins with SH3 domains are involved in cell motility pathways. These are SRGAP1, SRGAP2, SRGAP3, and BAIAP2.
Based on analysis of canonical pathways represented in HDNet and gene expression changes observed in HD brain, we focused on RhoGTPase signaling to validate a specific functional module as being involved in HTT function and toxicity. The potent loss-of-function suppression of HTT toxicity observed in STHdh111Q/111Q cells upon knockdown of BAIAP2 further focused our attention on filopodia components as modifiers of mutant HTT toxicity in cell models. These structures are essential for cell adhesion, cell migration, and neurite outgrowth (87). Here, we show that expression of mutant HTT impairs BAIAP2-induced filopodia formation. This phenomenon is observed for induction of filopodia formation following BAIAP2 overexpression, as well as a decrease of formation of these structures following knockdown of BAIAP2. This suggests that mutant HTT can impair dynamic regulation of filopodia by BAIAP2. The regulation of BAIAP2-induced filopodial formation remains to be elucidated, as well as the specific involvement of this process in neuron-specific pathways. BAIAP2 is also a binding partner of PDZ domain-containing proteins such as DGL-4 (PSD-95) in the postsynaptic density indicating that the HTT-BAIAP2 interaction may influence both pre- and post-synaptic processes (88, 89). BAIAP2 has also has been shown to interact with ATN1 (atrophin-1), the protein responsible for DRPLA, a polyQ expansion disease with pathological and clinical similarities to HD (90). Identifying the protein composition of filopodia neurons affected in HD, and deciphering the involvement of HTT in these dynamic structures could help elucidate novel mechanisms of neuronal dysfunction. In human brain, previous studies showed marked morphological changes in dendritic structures and branching of medium spiny neurons in post-mortem HD brain (91, 92). Our data indicate that dynamic organization of neuronal processes as well as sites of cell-cell contacts may be a primary pathogenic mechanism in HD. Furthermore, we provide evidence that components of the Rho GTPase signaling cascade such as BAIAP2 can be directly affected by mutant HTT and are therefore candidates for HTT-mediated defects in cell morphology. The protein machinery regulating cytoskeleton-membrane interactions and filopodia formation represents key targets for further studies with regard to neurodevelopmental pathways and synaptic homeostasis in Huntington disease.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants NS055247 and GM084432 (to R. E. H.). This work was also supported by a grant from CHDI Foundation Inc. (to R. E. H.).
This article was selected as a Paper of the Week.

This article contains supplemental Tables S1–S5.
- HD
- Huntington disease
- HIP
- huntingtin-interacting protein
- HPRD
- Human Protein Resource Database
- IPA
- ingenuity pathway analysis
- Y2H
- yeast two-hybrid
- HDNet
- huntingtin protein interaction network
- PRR
- proline-rich region
- SH
- Src homology
- BAR
- Bin-Amphiphysin-Rvs
- PPI
- protein-protein interaction
- ILK
- integrin-linked kinase
- RAR
- retinoic acid receptor.
REFERENCES
- 1. The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 [DOI] [PubMed] [Google Scholar]
- 2. Reiner A., Albin R. L., Anderson K. D., D'Amato C. J., Penney J. B., Young A. B. (1988) Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 85, 5733–5737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walker F. O. (2007) Huntington's disease. Lancet 369, 218–228 [DOI] [PubMed] [Google Scholar]
- 4. Borrell-Pagès M., Zala D., Humbert S., Saudou F. (2006) Huntington's disease: from huntingtin function and dysfunction to therapeutic strategies. Cell. Mol. Life Sci. 63, 2642–2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zuccato C., Valenza M., Cattaneo E. (2010) Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol. Rev. 90, 905–981 [DOI] [PubMed] [Google Scholar]
- 6. Wellington C. L., Ellerby L. M., Gutekunst C. A., Rogers D., Warby S., Graham R. K., Loubser O., van Raamsdonk J., Singaraja R., Yang Y. Z., Gafni J., Bredesen D., Hersch S. M., Leavitt B. R., Roy S., Nicholson D. W., Hayden M. R. (2002) Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J. Neurosci. 22, 7862–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Graham R. K., Deng Y., Slow E. J., Haigh B., Bissada N., Lu G., Pearson J., Shehadeh J., Bertram L., Murphy Z., Warby S. C., Doty C. N., Roy S., Wellington C. L., Leavitt B. R., Raymond L. A., Nicholson D. W., Hayden M. R. (2006) Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125, 1179–1191 [DOI] [PubMed] [Google Scholar]
- 8. Ratovitski T., Gucek M., Jiang H., Chighladze E., Waldron E., D'Ambola J., Hou Z., Liang Y., Poirier M. A., Hirschhorn R. R., Graham R., Hayden M. R., Cole R. N., Ross C. A. (2009) Mutant huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J. Biol. Chem. 284, 10855–10867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun B., Fan W., Balciunas A., Cooper J. K., Bitan G., Steavenson S., Denis P. E., Young Y., Adler B., Daugherty L., Manoukian R., Elliott G., Shen W., Talvenheimo J., Teplow D. B., Haniu M., Haldankar R., Wypych J., Ross C. A., Citron M., Richards W. G. (2002) Polyglutamine repeat length-dependent proteolysis of huntingtin. Neurobiol. Dis. 11, 111–122 [DOI] [PubMed] [Google Scholar]
- 10. Boutell J. M., Thomas P., Neal J. W., Weston V. J., Duce J., Harper P. S., Jones A. L. (1999) Aberrant interactions of transcriptional repressor proteins with the Huntington's disease gene product, huntingtin. Hum. Mol. Genet. 8, 1647–1655 [DOI] [PubMed] [Google Scholar]
- 11. Holbert S., Denghien I., Kiechle T., Rosenblatt A., Wellington C., Hayden M. R., Margolis R. L., Ross C. A., Dausset J., Ferrante R. J., Néri C. (2001) The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 98, 1811–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Steffan J. S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y. Z., Gohler H., Wanker E. E., Bates G. P., Housman D. E., Thompson L. M. (2000) The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. U.S.A. 97, 6763–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caviston J. P., Ross J. L., Antony S. M., Tokito M., Holzbaur E. L. (2007) Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. U.S.A. 104, 10045–10050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Culver B. P., Savas J. N., Park S. K., Choi J. H., Zheng S., Zeitlin S. O., Yates J. R., 3rd, Tanese N. (2012) Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J. Biol. Chem. 287, 21599–21614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goehler H., Lalowski M., Stelzl U., Waelter S., Stroedicke M., Worm U., Droege A., Lindenberg K. S., Knoblich M., Haenig C., Herbst M., Suopanki J., Scherzinger E., Abraham C., Bauer B., Hasenbank R., Fritzsche A., Ludewig A. H., Büssow K., Buessow K., Coleman S. H., Gutekunst C. A., Landwehrmeyer B. G., Lehrach H., Wanker E. E. (2004) A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol. Cell 15, 853–865 [DOI] [PubMed] [Google Scholar]
- 16. Kaltenbach L. S., Romero E., Becklin R. R., Chettier R., Bell R., Phansalkar A., Strand A., Torcassi C., Savage J., Hurlburt A., Cha G. H., Ukani L., Chepanoske C. L., Zhen Y., Sahasrabudhe S., Olson J., Kurschner C., Ellerby L. M., Peltier J. M., Botas J., Hughes R. E. (2007) Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 3, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shirasaki D. I., Greiner E. R., Al-Ramahi I., Gray M., Boontheung P., Geschwind D. H., Botas J., Coppola G., Horvath S., Loo J. A., Yang X. W. (2012) Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 75, 41–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bell R., Hubbard A., Chettier R., Chen D., Miller J. P., Kapahi P., Tarnopolsky M., Sahasrabuhde S., Melov S., Hughes R. E. (2009) A human protein interaction network shows conservation of aging processes between human and invertebrate species. PLoS Genet. 5, e1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bandyopadhyay S., Chiang C. Y., Srivastava J., Gersten M., White S., Bell R., Kurschner C., Martin C., Smoot M., Sahasrabudhe S., Barber D. L., Chanda S. K., Ideker T. (2010) A human MAP kinase interactome. Nat. Methods 7, 801–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ge H., Walhout A. J., Vidal M. (2003) Integrating 'omic' information: a bridge between genomics and systems biology. Trends Genet. 19, 551–560 [DOI] [PubMed] [Google Scholar]
- 21. LaCount D. J., Vignali M., Chettier R., Phansalkar A., Bell R., Hesselberth J. R., Schoenfeld L. W., Ota I., Sahasrabudhe S., Kurschner C., Fields S., Hughes R. E. (2005) A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 438, 103–107 [DOI] [PubMed] [Google Scholar]
- 22. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keshava Prasad T. S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., Balakrishnan L., Marimuthu A., Banerjee S., Somanathan D. S., Sebastian A., Rani S., Ray S., Harrys Kishore C. J., Kanth S., Ahmed M., Kashyap M. K., Mohmood R., Ramachandra Y. L., Krishna V., Rahiman B. A., Mohan S., Ranganathan P., Ramabadran S., Chaerkady R., Pandey A. (2009) Human Protein Reference Database–2009 update. Nucleic Acids Res. 37, D767–D772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hodges A., Strand A. D., Aragaki A. K., Kuhn A., Sengstag T., Hughes G., Elliston L. A., Hartog C., Goldstein D. R., Thu D., Hollingsworth Z. R., Collin F., Synek B., Holmans P. A., Young A. B., Wexler N. S., Delorenzi M., Kooperberg C., Augood S. J., Faull R. L., Olson J. M., Jones L., Luthi-Carter R. (2006) Regional and cellular gene expression changes in human Huntington's disease brain. Hum. Mol. Genet. 15, 965–977 [DOI] [PubMed] [Google Scholar]
- 25. Kuhn A., Goldstein D. R., Hodges A., Strand A. D., Sengstag T., Kooperberg C., Becanovic K., Pouladi M. A., Sathasivam K., Cha J. H., Hannan A. J., Hayden M. R., Leavitt B. R., Dunnett S. B., Ferrante R. J., Albin R., Shelbourne P., Delorenzi M., Augood S. J., Faull R. L., Olson J. M., Bates G. P., Jones L., Luthi-Carter R. (2007) Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 16, 1845–1861 [DOI] [PubMed] [Google Scholar]
- 26. Miller J. P., Yates B. E., Al-Ramahi I., Berman A. E., Sanhueza M., Kim E., de Haro M., DeGiacomo F., Torcassi C., Holcomb J., Gafni J., Mooney S. D., Botas J., Ellerby L. M., Hughes R. E. (2012) A genome-scale RNA-interference screen identifies RRAS signaling as a pathologic feature of Huntington's disease. PLoS Genet. 8, e1003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Campeau E., Ruhl V. E., Rodier F., Smith C. L., Rahmberg B. L., Fuss J. O., Campisi J., Yaswen P., Cooper P. K., Kaufman P. D. (2009) A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller J. P., Holcomb J., Al-Ramahi I., de Haro M., Gafni J., Zhang N., Kim E., Sanhueza M., Torcassi C., Kwak S., Botas J., Hughes R. E., Ellerby L. M. (2010) Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington's disease. Neuron 67, 199–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trettel F., Rigamonti D., Hilditch-Maguire P., Wheeler V. C., Sharp A. H., Persichetti F., Cattaneo E., MacDonald M. E. (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 9, 2799–2809 [DOI] [PubMed] [Google Scholar]
- 30. Barabási A. L., Gulbahce N., Loscalzo J. (2011) Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yuan T. L., Cantley L. C. (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rommel C., Camps M., Ji H. (2007) PI3Kδ and PI3Kγ: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat. Rev. Immunol. 7, 191–201 [DOI] [PubMed] [Google Scholar]
- 33. Lee J. Y., Kim Y. R., Park J., Kim S. (2012) Inositol polyphosphate multikinase signaling in the regulation of metabolism. Ann. N.Y. Acad. Sci. 1271, 68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chong Z. Z., Shang Y. C., Wang S., Maiese K. (2012) A critical kinase cascade in neurological disorders: PI 3-K, Akt, and mTOR. Future Neurol. 7, 733–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ravikumar B., Vacher C., Berger Z., Davies J. E., Luo S., Oroz L. G., Scaravilli F., Easton D. F., Duden R., O'Kane C. J., Rubinsztein D. C. (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 [DOI] [PubMed] [Google Scholar]
- 36. Seredenina T., Luthi-Carter R. (2012) What have we learned from gene expression profiles in Huntington's disease? Neurobiol. Dis. 45, 83–98 [DOI] [PubMed] [Google Scholar]
- 37. Borgonovo J. E., Troncoso M., Lucas J. J., Sosa M. A. (2013) Mutant huntingtin affects endocytosis in striatal cells by altering the binding of AP-2 to membranes. Exp. Neurol. 241, 75–83 [DOI] [PubMed] [Google Scholar]
- 38. Trushina E., Singh R. D., Dyer R. B., Cao S., Shah V. H., Parton R. G., Pagano R. E., McMurray C. T. (2006) Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum. Mol. Genet. 15, 3578–3591 [DOI] [PubMed] [Google Scholar]
- 39. Lange A., Wickström S. A., Jakobson M., Zent R., Sainio K., Fässler R. (2009) Integrin-linked kinase is an adaptor with essential functions during mouse development. Nature 461, 1002–1006 [DOI] [PubMed] [Google Scholar]
- 40. Qin J., Wu C. (2012) ILK: a pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr. Opin. Cell Biol. 24, 607–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bauer P. O., Wong H. K., Oyama F., Goswami A., Okuno M., Kino Y., Miyazaki H., Nukina N. (2009) Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J. Biol. Chem. 284, 13153–13164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shao J., Welch W. J., Diamond M. I. (2008) ROCK and PRK-2 mediate the inhibitory effect of Y-27632 on polyglutamine aggregation. FEBS Lett. 582, 1637–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. De Las Rivas J., Fontanillo C. (2010) Protein-protein interactions essentials: key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 6, e1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Buchsbaum R. J. (2007) Rho activation at a glance. J. Cell Sci. 120, 1149–1152 [DOI] [PubMed] [Google Scholar]
- 45. Miki H., Yamaguchi H., Suetsugu S., Takenawa T. (2000) IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature 408, 732–735 [DOI] [PubMed] [Google Scholar]
- 46. Mattila P. K., Pykäläinen A., Saarikangas J., Paavilainen V. O., Vihinen H., Jokitalo E., Lappalainen P. (2007) Missing-in-metastasis and IRSp53 deform PI(4,5)P2-rich membranes by an inverse BAR domain-like mechanism. J. Cell Biol. 176, 953–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robens J. M., Yeow-Fong L., Ng E., Hall C., Manser E. (2010) Regulation of IRSp53-dependent filopodial dynamics by antagonism between 14-3-3 binding and SH3-mediated localization. Mol. Cell. Biol. 30, 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Neisch A. L., Fehon R. G. (2011) Ezrin, Radixin and Moesin: key regulators of membrane-cortex interactions and signaling. Curr. Opin. Cell Biol. 23, 377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marsick B. M., Roche F. K., Letourneau P. C. (2012) Repulsive axon guidance cues ephrin-A2 and slit3 stop protrusion of the growth cone leading margin concurrently with inhibition of ADF/cofilin and ERM proteins. Cytoskeleton 69, 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marchler-Bauer A., Lu S., Anderson J. B., Chitsaz F., Derbyshire M. K., DeWeese-Scott C., Fong J. H., Geer L. Y., Geer R. C., Gonzales N. R., Gwadz M., Hurwitz D. I., Jackson J. D., Ke Z., Lanczycki C. J., Lu F., Marchler G. H., Mullokandov M., Omelchenko M. V., Robertson C. L., Song J. S., Thanki N., Yamashita R. A., Zhang D., Zhang N., Zheng C., Bryant S. H. (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bedford M. T., Reed R., Leder P. (1998) WW domain-mediated interactions reveal a spliceosome-associated protein that binds a third class of proline-rich motif: the proline glycine and methionine-rich motif. Proc. Natl. Acad. Sci. U.S.A. 95, 10602–10607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chow W. N., Luk H. W., Chan H. Y., Lau K. F. (2012) Degradation of mutant huntingtin via the ubiquitin/proteasome system is modulated by FE65. Biochem. J. 443, 681–689 [DOI] [PubMed] [Google Scholar]
- 53. Faber P. W., Barnes G. T., Srinidhi J., Chen J., Gusella J. F., MacDonald M. E. (1998) Huntingtin interacts with a family of WW domain proteins. Hum. Mol. Genet. 7, 1463–1474 [DOI] [PubMed] [Google Scholar]
- 54. Passani L. A., Bedford M. T., Faber P. W., McGinnis K. M., Sharp A. H., Gusella J. F., Vonsattel J. P., MacDonald M. E. (2000) Huntingtin's WW domain partners in Huntington's disease post-mortem brain fulfill genetic criteria for direct involvement in Huntington's disease pathogenesis. Hum. Mol. Genet. 9, 2175–2182 [DOI] [PubMed] [Google Scholar]
- 55. Jiang Y. J., Che M. X., Yuan J. Q., Xie Y. Y., Yan X. Z., Hu H. Y. (2011) Interaction with polyglutamine-expanded huntingtin alters cellular distribution and RNA processing of huntingtin yeast two-hybrid protein A (HYPA). J. Biol. Chem. 286, 25236–25245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zheng S., Ghitani N., Blackburn J. S., Liu J. P., Zeitlin S. O. (2012) A series of N-terminal epitope tagged Hdh knock-in alleles expressing normal and mutant huntingtin: their application to understanding the effect of increasing the length of normal Huntingtin's polyglutamine stretch on CAG140 mouse model pathogenesis. Mol. Brain 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Allen M., Friedler A., Schon O., Bycroft M. (2002) The structure of an FF domain from human HYPA/FBP11. J. Mol. Biol. 323, 411–416 [DOI] [PubMed] [Google Scholar]
- 58. Bedford M. T., Leder P. (1999) The FF domain: a novel motif that often accompanies WW domains. Trends Biochem. Sci. 24, 264–265 [DOI] [PubMed] [Google Scholar]
- 59. Chiang M. C., Chen C. M., Lee M. R., Chen H. W., Chen H. M., Wu Y. S., Hung C. H., Kang J. J., Chang C. P., Chang C., Wu Y. R., Tsai Y. S., Chern Y. (2010) Modulation of energy deficiency in Huntington's disease via activation of the peroxisome proliferator-activated receptor γ. Hum. Mol. Genet. 19, 4043–4058 [DOI] [PubMed] [Google Scholar]
- 60. Huang H., Li L., Wu C., Schibli D., Colwill K., Ma S., Li C., Roy P., Ho K., Songyang Z., Pawson T., Gao Y., Li S. S. (2008) Defining the specificity space of the human SRC homology 2 domain. Mol. Cell. Proteomics 7, 768–784 [DOI] [PubMed] [Google Scholar]
- 61. Morton C. J., Campbell I. D. (1994) SH3 domains. Molecular ‘Velcro’. Curr. Biol. 4, 615–617 [DOI] [PubMed] [Google Scholar]
- 62. Chuang J. Z., Zhou H., Zhu M., Li S. H., Li X. J., Sung C. H. (2002) Characterization of a brain-enriched chaperone, MRJ, that inhibits Huntingtin aggregation and toxicity independently. J. Biol. Chem. 277, 19831–19838 [DOI] [PubMed] [Google Scholar]
- 63. Lu B., Al-Ramahi I., Valencia A., Wang Q., Berenshteyn F., Yang H., Gallego-Flores T., Ichcho S., Lacoste A., Hild M., Difiglia M., Botas J., Palacino J. (2013) Identification of NUB1 as a suppressor of mutant Huntington toxicity via enhanced protein clearance. Nat. Neurosci. 16, 562–570 [DOI] [PubMed] [Google Scholar]
- 64. Muchowski P. J., Schaffar G., Sittler A., Wanker E. E., Hayer-Hartl M. K., Hartl F. U. (2000) Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 97, 7841–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Suetsugu S., Toyooka K., Senju Y. (2010) Subcellular membrane curvature mediated by the BAR domain superfamily proteins. Semin. Cell Dev. Biol. 21, 340–349 [DOI] [PubMed] [Google Scholar]
- 66. Disanza A., Mantoani S., Hertzog M., Gerboth S., Frittoli E., Steffen A., Berhoerster K., Kreienkamp H. J., Milanesi F., Di Fiore P. P., Ciliberto A., Stradal T. E., Scita G. (2006) Regulation of cell shape by Cdc42 is mediated by the synergic actin-bundling activity of the Eps8-IRSp53 complex. Nat. Cell Biol. 8, 1337–1347 [DOI] [PubMed] [Google Scholar]
- 67. Krugmann S., Jordens I., Gevaert K., Driessens M., Vandekerckhove J., Hall A. (2001) Cdc42 induces filopodia by promoting the formation of an IRSp53:Mena complex. Curr. Biol. 11, 1645–1655 [DOI] [PubMed] [Google Scholar]
- 68. Lim K. B., Bu W., Goh W. I., Koh E., Ong S. H., Pawson T., Sudhaharan T., Ahmed S. (2008) The Cdc42 effector IRSp53 generates filopodia by coupling membrane protrusion with actin dynamics. J. Biol. Chem. 283, 20454–20472 [DOI] [PubMed] [Google Scholar]
- 69. Nakagawa H., Miki H., Nozumi M., Takenawa T., Miyamoto S., Wehland J., Small J. V. (2003) IRSp53 is colocalised with WAVE2 at the tips of protruding lamellipodia and filopodia independently of Mena. J. Cell Sci. 116, 2577–2583 [DOI] [PubMed] [Google Scholar]
- 70. Suetsugu S., Murayama K., Sakamoto A., Hanawa-Suetsugu K., Seto A., Oikawa T., Mishima C., Shirouzu M., Takenawa T., Yokoyama S. (2006) The RAC binding domain/IRSp53-MIM homology domain of IRSp53 induces RAC-dependent membrane deformation. J. Biol. Chem. 281, 35347–35358 [DOI] [PubMed] [Google Scholar]
- 71. Li S. H., Li X. J. (2004) Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 20, 146–154 [DOI] [PubMed] [Google Scholar]
- 72. Jiang H., Poirier M. A., Liang Y., Pei Z., Weiskittel C. E., Smith W. W., DeFranco D. B., Ross C. A. (2006) Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 23, 543–551 [DOI] [PubMed] [Google Scholar]
- 73. Yu Z., Zhou D., Cheng G., Mattson M. P. (2000) Neuroprotective role for the p50 subunit of NF-κB in an experimental model of Huntington's disease. J. Mol. Neurosci. 15, 31–44 [DOI] [PubMed] [Google Scholar]
- 74. Manser E., Leung T., Salihuddin H., Zhao Z. S., Lim L. (1994) A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 367, 40–46 [DOI] [PubMed] [Google Scholar]
- 75. Mattila P. K., Lappalainen P. (2008) Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 9, 446–454 [DOI] [PubMed] [Google Scholar]
- 76. Galbraith C. G., Yamada K. M., Galbraith J. A. (2007) Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science 315, 992–995 [DOI] [PubMed] [Google Scholar]
- 77. Vasioukhin V., Bauer C., Yin M., Fuchs E. (2000) Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell 100, 209–219 [DOI] [PubMed] [Google Scholar]
- 78. Dent E. W., Kwiatkowski A. V., Mebane L. M., Philippar U., Barzik M., Rubinson D. A., Gupton S., Van Veen J. E., Furman C., Zhang J., Alberts A. S., Mori S., Gertler F. B. (2007) Filopodia are required for cortical neurite initiation. Nat. Cell Biol. 9, 1347–1359 [DOI] [PubMed] [Google Scholar]
- 79. Kwiatkowski A. V., Rubinson D. A., Dent E. W., Edward van Veen J., Leslie J. D., Zhang J., Mebane L. M., Philippar U., Pinheiro E. M., Burds A. A., Bronson R. T., Mori S., Fässler R., Gertler F. B. (2007) Ena/VASP is Required for neuritogenesis in the developing cortex. Neuron 56, 441–455 [DOI] [PubMed] [Google Scholar]
- 80. Menna E., Fossati G., Scita G., Matteoli M. (2011) From filopodia to synapses: the role of actin-capping and anti-capping proteins. Eur. J. Neurosci. 34, 1655–1662 [DOI] [PubMed] [Google Scholar]
- 81. Spillane M., Ketschek A., Donnelly C. J., Pacheco A., Twiss J. L., Gallo G. (2012) Nerve growth factor-induced formation of axonal filopodia and collateral branches involves the intra-axonal synthesis of regulators of the actin-nucleating Arp2/3 complex. J. Neurosci. 32, 17671–17689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Millard T. H., Bompard G., Heung M. Y., Dafforn T. R., Scott D. J., Machesky L. M., Fütterer K. (2005) Structural basis of filopodia formation induced by the IRSp53/MIM homology domain of human IRSp53. EMBO J. 24, 240–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ngounou Wetie A. G., Sokolowska I., Woods A. G., Roy U., Deinhardt K., Darie C. C. (2013) Protein-protein interactions: switch from classical methods to proteomics and bioinformatics-based approaches. Cell. Mol. Life Sci. 71, 205–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schaefer M. H., Wanker E. E., Andrade-Navarro M. A. (2012) Evolution and function of CAG/polyglutamine repeats in protein-protein interaction networks. Nucleic Acids Res. 40, 4273–4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Southwell A. L., Khoshnan A., Dunn D. E., Bugg C. W., Lo D. C., Patterson P. H. (2008) Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity. J. Neurosci. 28, 9013–9020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Neveklovska M., Clabough E. B., Steffan J. S., Zeitlin S. O. (2012) Deletion of the huntingtin proline-rich region does not significantly affect normal huntingtin function in mice. J. Huntingtons Dis. 1, 71–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ahmed S., Goh W. I., Bu W. (2010) I-BAR domains, IRSp53 and filopodium formation. Semin. Cell Dev. Biol. 21, 350–356 [DOI] [PubMed] [Google Scholar]
- 88. Hori K., Yasuda H., Konno D., Maruoka H., Tsumoto T., Sobue K. (2005) NMDA receptor-dependent synaptic translocation of insulin receptor substrate p53 via protein kinase C signaling. J. Neurosci. 25, 2670–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Soltau M., Berhörster K., Kindler S., Buck F., Richter D., Kreienkamp H. J. (2004) Insulin receptor substrate of 53 kDa links postsynaptic shank to PSD-95. J. Neurochem. 90, 659–665 [DOI] [PubMed] [Google Scholar]
- 90. Thomas E. A., Foye P. E., Alvarez C. E., Usui H., Sutcliffe J. G. (2001) Insulin receptor substrate protein p53 localization in rats suggests mechanism for specific polyglutamine neurodegeneration. Neurosci. Lett. 309, 145–148 [DOI] [PubMed] [Google Scholar]
- 91. Ferrante R. J., Kowall N. W., Richardson E. P., Jr. (1991) Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J. Neurosci. 11, 3877–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Graveland G. A., Williams R. S., DiFiglia M. (1985) Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science 227, 770–773 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.