

Background: Hyaluronan metabolism is altered in idiopathic pulmonary arterial hypertension lung tissue.
Results: This hyaluronan is of a pathological form covalently modified by heavy chains from inter-α-inhibitor.
Conclusion: The distribution of this matrix in regions of abnormal vascular remodeling implies a mechanistic role.
Significance: Pathological hyaluronan matrices may direct inflammatory events and vascular changes in this disease.
Keywords: Extracellular Matrix, Glycobiology, Hyaluronate, Lung, Proteoglycan, TSG-6, Heavy Chain, Hyaluronan, Inter-α-inhibitor, Tumor Necrosis Factor-stimulated Gene 6
Abstract
We previously reported an altered hyaluronan (HA) metabolism in idiopathic pulmonary arterial hypertension (IPAH) lung tissue and cultured smooth muscle cells. Hyaluronan was present in the smooth muscle cell layer surrounding the pulmonary vasculature and in plexigenic lesions. Additionally, cultured pulmonary artery smooth muscle cells produced spontaneous HA “cable” structures, without additional stimuli, that were leukocyte-adhesive. We now present evidence that the HA that accumulates in IPAH plexigenic lesions is a pathological form of HA in which heavy chains (HCs) from the serum-derived proteoglycan inter-α-inhibitor are covalently attached to the HA backbone to form a pathological HC-HA complex. CD45-positive leukocytes were identified within these HC-HA matrices. Elevated mRNA levels of the enzyme that transfers HCs to HA, known as tumor necrosis factor-stimulated gene 6, were detected in IPAH lung tissue.
Introduction
Idiopathic pulmonary arterial hypertension (IPAH)3 is a progressive disease that leads to deterioration in cardiopulmonary function and premature death from right ventricular failure. The pathogenesis of IPAH is not fully understood, although it is typified by (i) cell proliferation, (ii) vascular remodeling, and (iii) inflammatory cell recruitment (1). During remodeling, medial and intimal thickening results in arterial luminal occlusion, often followed by an angiogenic event whereby the occluded vessel buds and branches into an abnormal vascular complex known as a plexigenic lesion. We have previously reported increased hyaluronan (HA) deposition in the extracellular matrix surrounding plexigenic lesions in IPAH lungs (2). We tested the hypothesis that the HA that accumulates in these regions is an abnormal form of HA characterized by its covalent substitution with heavy chains (HCs) from inter-α-inhibitor (IαI). This abnormal form of HA (i.e. HC-HA) is known to increase the avidity of leukocytes to HA matrices.
EXPERIMENTAL PROCEDURES
IPAH Lung Tissue and Serum
Histological paraffin section staining was performed on postmortem lungs. Tissues processed for HC-HA Western blot analysis were obtained from lungs explanted at the time of lung transplantation. Serum was collected from live patients.
Hyaluronidase Extraction of HCs from HC-HA Complexes
Pieces of human lung tissue were cut with a scalpel and transferred to pre-weighed 1.5-ml tubes that were pre-chilled on dry ice. Weights of the lung tissue were recorded, and pre-chilled PBS was added to the tubes such that 100 μl of cold PBS was added for every 30 mg of tissue. The tissue was minced with scissors, on ice, in PBS for approximately 1 min. A 50-μl aliquot of the minced tissue suspension was transferred to two pre-chilled 1.5-ml new tubes. Streptomyces hyaluronidase (10 μl of a 0.5 turbidity unit/ml stock; product 100740-1; Seikagaku, East Falmouth, MA) was added to one of these tubes, and PBS (10 μl) was added to the other. The tubes were incubated on ice for 30 min and then centrifuged at 13,200 rpm at 4 °C for 5 min. The supernatants were transferred to pre-chilled 1.5-ml new tubes and incubated for another 30 min at 37 °C. Then 25 μl of digests was added per lane on 4–15% Mini-PROTEAN TGX gels (Bio-Rad) and blotted using the Bio-Rad nitrocellulose and Trans-Blot Turbo System. The blots were blocked for 1 h with Li-Cor blocking buffer (927-40000; Li-Cor, Lincoln, NE). The blots were then probed with a rabbit polyclonal antibody against IαI (A0301; Dako North America, Carpinteria, CA, 1:8000 dilution). The secondary was IRDYE anti-rabbit 800CW (926-32211; Li-Cor) used at 1:15,000. The blots were washed and imaged on an Odyssey Infrared Imaging System (Li-Cor).
Immunofluorescence
Paraffin-embedded sections from IPAH and normal human lung tissue were deparaffinized and blocked for 30 min in PBS with 1% BSA. HA, IαI, and the common leukocyte antigen (CD45) were simultaneously labeled with the following primary antibodies/binding proteins in the blocking solution: (i) HA was labeled with a biotinylated hyaluronan-binding protein (5 μg/ml, 385911; EMD/Millipore, Gibbstown, NJ); (ii) IαI was labeled with a rabbit polyclonal antibody against IαI (1:100); and (iii) CD45 was labeled with a mouse monoclonal antibody (1:100, M070; Dako, Denmark). In one experiment, a paraffin human lung section was probed with an antibody against bikunin (Abcam, Cambridge, MA; ab153806) following antigen retrieval in a citrate buffer, pH 6.0. After a 45-min incubation at room temperature, the slides were washed in PBS three times, 10 min each wash. The following secondary antibodies/binding proteins were simultaneously applied in the blocking solution: (i) streptavidin conjugated to Alexa Fluor 633 (1:500, S21375; Invitrogen); (ii) Cy3 donkey anti-rabbit (1:400, 711-165-152; Jackson ImmunoResearch); (iii) donkey anti-mouse Alexa Fluor 488 (1:250, A21202; Invitrogen). After a 45-min incubation at room temperature, the slides were washed in PBS three times, 10 min each wash. Vectashield fluorescent mounting medium, with DAPI (H-1200; Vector Laboratories, Burlingame, CA) was applied previous to placing the coverslip. Imaging was done by confocal (see Figs. 2 and 3) and standard (see Fig. 4) fluorescent microscopy. Grayscale images were pseudocolored using ImageJ software (National Institutes of Health, Bethesda, MD).
FIGURE 2.

Localization of the HC-HA complex in plexigenic lesions of pulmonary arterial hypertension patients. Paraffin lung sections from normal and IPAH patients were probed with an antibody against IαI (red) and a HA-binding protein (green). DAPI-stained nuclei are shown in blue. A–C, in normal lungs, HA is deposited at relatively low levels in the connective tissue surrounding blood vessels (*) and airways (#). D–F, IPAH lungs have higher levels of HA and IαI in regions of vascular remodeling and plexigenic lesions (white box). G–I, the white boxed areas of D–F are shown at higher magnification (×40). J–L, a serial section in G–I was probed with only secondary antibodies as a negative control. Scale bars in A and J, 200 μm. Magnification is indicated in A, D, G, and J. n = 3 from three different IPAH patients.
FIGURE 3.

Localization of the HC-HA complex in plexigenic lesions of pulmonary arterial hypertension patients. Paraffin lung section from an IPAH patient was probed with an antibody against IαI (red) and a HA-binding protein (green). DAPI-stained nuclei are shown in blue. The plexigenic lesion is shown at magnification of ×20 (A–C), ×40 (D–F), and ×63 (G–I). Scale bar in A, 200 μm.
FIGURE 4.

The HCs of IαI co-localize with HA in plexigenic lesions, not with intact IαI itself. Paraffin lung section from an IPAH patient was probed with a polyclonal antibody against IαI (red) and a HA-binding protein (green) (A–C), or an antibody against bikunin (red) and a HA-binding protein (green) (D–F). DAPI-stained nuclei are shown in blue. Negative controls probed with only secondary antibodies are shown in G–I. Scale bar in A, 300 μm. n = 1.
Detection of HC-HA in Serum
Aliquots (1.25 μl) of serum from normal and IPAH patients were treated ± Streptomyces hyaluronidase (1 μl of a 0.5 turbidity unit/ml stock) in a 25-μl reaction volume of PBS for 18 h. The entire 25-μl digest was loaded onto a single lane of an SDS-polyacrylamide gel, blotted, and probed with a rabbit polyclonal antibody against IαI (1:8000 dilution). In some instances, an aliquot of artificially made HC-HA was added to the reaction mixture as a positive control and the volume adjusted to 25 μl. Artificial HC-HA was made by incubating the following in a 1.5-ml tube at 37 °C for 4 h: 234 μl of water, 12 μl of high molecular mass HA (80190; 1.7 kDa; Lifecore Biomedical, Chaska, MN), 12 μl of normal human serum, 12 μl of TSG-6, and 30 μl of 10× PBS containing 10 mm MgCl2. TSG-6 activity was stopped by adding 6 μl of 0.5 m EDTA (pH 8.0). An aliquot (25 μl) of this preparation was loaded onto an SDS-polyacrylamide gel followed by 1:2 dilutions to demonstrate our limit of detection.
cDNA Synthesis and Real Time Quantitative PCR (RTQ-PCR)
RTQ-PCR was used to quantitatively measure TSG-6 mRNA expression in control and IPAH whole lung tissues. RNA was isolated from whole lung tissues obtained from explanted lungs. For cDNA synthesis, 1 μg of each RNA sample was digested with DNase I (Roche Diagnostics), and the respective mRNA probes were synthesized using an oligo(dT) primer and MLV (Invitrogen). SYBR Green technology (Applied Biosystems) was used for all RTQ-PCR experiments. Reactions were done in an iCycler Thermal cycler (Bio-Rad) in a total volume of 20 μl (Table 1). Expressions of TSG-6 and HCs mRNA relative to the housekeeping gene GAPDH were calculated using the ΔΔCt method (3). The PCR conditions were 1 cycle at 60 °C for 2 min, 1 cycle at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 65 °C for 1 min for all primers.
TABLE 1.
Primer used in RTQ-RT-PCR
Shown are the names, sequences, source sequence accession numbers, and relative positions of the primers used for RTQ-RT-PCR. The annealing temperature for all RTQ-RT-PCR primers was 65 °C. The letter h in front of primer names designates primers specific for human genes. fwd, forward primer; rev, reverse primer.
RESULTS
The Pathological Heavy Chain Modification of Hyaluronan Is Present in the Lung Tissue of Patients with Idiopathic Pulmonary Arterial Hypertension
We minced normal and IPAH lung tissue in PBS on ice. The minced tissue was centrifuged, and the supernatant was separated from the pellet. The pellet contained the insoluble cellular material and extracellular matrix. The supernatant contained soluble factors, such as IαI, present in serum. An aliquot of Streptomyces hyaluronidase was added to half of the extract, and an aliquot of PBS was added to the other half (as a negative control). By itself, HC-HA is too large to enter the gel. Hyaluronidase digests HA, releasing free HCs from HC-HA that appear as a single band at approximately 83 kDa. These extracts were analyzed by Western blotting (Fig. 1), probing the blot with a polyclonal antibody against IαI that recognizes the HCs of IαI. HCs were predominantly present in the IPAH cellular/matrix pellet treated with hyaluronidase, implying that they were attached to HA in the extracellular matrix. Lesser amounts were found in the IPAH supernatants treated with hyaluronidase. In contrast, no HCs were found in the supernatant of the control extract when treated with hyaluronidase, and only a faint HC band was observed in the cellular/matrix pellet when treated with hyaluronidase. This experiment was repeated with two other control and IPAH lung tissues from different human subjects with similar effects, demonstrating a 4.37-fold increase in HC-HA in IPAH lung tissue compared with control lung (p = 0.030). These data were quantified and are presented in graph format in Fig. 1B.
FIGURE 1.

Hyaluronidase extraction of heavy chains from the lungs of IPAH patients. Portions of normal and IPAH lungs were minced on ice in PBS, and the insoluble pellet was separated from the supernatant (Sup) by centrifugation. These two fractions (i.e. pellet and supernatant) were treated ± Streptomyces hyaluronidase, and aliquots of the extract were analyzed by Western blotting, probing the blot with an antibody against IαI (green). The diagrams to the right of the gel portray IαI, pre-IαI, and free HCs from IαI (top to bottom). The green diagram portrays the heavy chains of IαI. The blue diagram portrays bikunin. The black line connecting bikunin with the heavy chains is a chondroitin sulfate chain. Similar results were seen with two other normal and two other IPAH patients. These were quantified and are presented graphically in B. Molecular mass standards are shown in red on the far left and between lanes 4 and 5. n = 3 from three different IPAH patients. Error bars represent S.D.
The Heavy Chain-Hyaluronan Complex Is Present in Plexigenic Lesions
Plexigenic lesions are a unique pathology associated with IPAH. They are a product of the extensive vascular remodeling that occurs, culminating in arterial luminal exclusion. As a result of this occlusion, the occluded vessel buds and branches into an abnormal vascular complex known as a plexigenic lesion. An example of a plexigenic lesion is marked by a white box in Fig. 2, D–F. This figure portrays normal (A–C) and IPAH (D–L) lung tissue probed with a biotinylated hyaluronan-binding protein and an antibody against IαI that recognizes the HCs of IαI. Whereas HA was found throughout IPAH lung tissue, staining with the IαI antibody was much more regional (Fig. 2, D–F). Specifically, we observed strong IαI staining within plexigenic lesions (enlarged G–I). This staining co-localized with HA in the submucosa region of these lesions. No co-localization of HA with IαI was observed around the airways or vasculature of normal lung tissue (Fig. 2, A–C). These results have been confirmed in lung tissues from two other IPAH and normal subjects. Another plexigenic lesion from an IPAH patient is shown in Fig. 3.
The co-localization of an antibody against IαI with HA in plexigenic lesions suggests that HA is covalently modified with HCs from IαI, but it could also occur if IαI, itself, is simply within the same region as HA. This is a plausible interpretation because higher levels of IαI and pre-IαI are present in IPAH tissue (Fig. 1), most likely the result of a greater infusion of serum into IPAH tissue compared with controls. To test these two hypotheses, we probed a parallel IPAH lung section with an antibody against bikunin alone, determining its co-localization with HA in a plexigenic lesion (Fig. 4). Although there was some overlap in bikunin staining with HA, co-localization of HA with a polyclonal antibody that has immunoreactivity to both bikunin and the heavy chains of IαI was more pronounced. We conclude that most of the regions where the IαI antibody co-localizes with HA is representative of HC-HA and not simply HA co-localizing with intact IαI. It should be acknowledged that the extent to which HC-HA versus the higher levels of intact IαI contributes to the primary pathology of IPAH cannot be absolutely determined through this study. However, the leukocyte adhesive qualities of HC-HA (4, 5) make it more likely that HC-HA, rather than IαI itself, contributes to the primary pathology of IPAH.
Presence of Leukocytes Embedded within Heavy Chain-Hyaluronan Matrices in Plexigenic Lesions
One of the known functions of the HC modification of HA is that it promotes leukocyte adhesion to HA matrices (4, 5). Thus, we tested the hypothesis that leukocytes might accumulate in HC-HA-enriched regions. To do this, we probed IPAH lung tissue with HA-binding protein (green), an antibody against IαI (red), and an antibody against the common leukocyte antigen CD45 (magenta) that functions as a generic leukocyte marker. As can be seen in Fig. 5, CD45-positive leukocytes were found embedded within the HC-HA matrices of a plexigenic lesion. These results were repeated in two other IPAH subjects' lung tissues.
FIGURE 5.

Co-localization of leukocytes in plexigenic lesions containing HC-HA. A paraffin lung section from an IPAH patient was probed with an antibody against IαI (red), HA-binding protein (green), and CD45 (magenta) as a generic marker for leukocytes. DAPI-stained nuclei are shown in blue. Scale bar in D, 100 μm. n = 1.
Undetectable Levels of the Heavy Chain-Hyaluronan Complex in IPAH Serum
The presence of HC-HA in serum from patients with rheumatoid arthritis has been demonstrated previously (6). In this instance, the levels of HC-HA were found to correlate with clinical signs of joint inflammation. We wanted to determine whether HC-HA levels were also high in IPAH serum. We measured HC-HA content of plasma from patients with IPAH (Fig. 6C), but none was observed. For a positive control, we artificially made HC-HA by transferring HCs from serum IαI to high molecular mass HA using recombinant TSG-6 (Fig. 6A). We tested the limit of detection for this artificially made HC-HA in a dilution series (Fig. 6A) and then spiked a portion of normal and IPAH serum with a dilute amount of HC-HA from the dilution series to demonstrate the limit of detection (Fig. 6B). We conclude that, if HC-HA is present in IPAH serum, it is below the limit of detection.
FIGURE 6.

HC-HA complex is absent in IPAH serum. Western blots were probed with an antibody against IαI (green). Molecular mass standards are shown in red. A portrays a dilution series of artificially made HC-HA treated with Streptomyces hyaluronidase (HAase) overnight at 37 °C (lanes 4–8). Lane 3 portrays HC-HA not digested with HAase. Lane 2 portrays only normal human serum, representing 1 μl of serum. In lanes 3 and 4, 1 μl of serum was used as the source of IαI for HC transfer to HA. In panel B, 1-μl aliquots of normal (lanes 2–4) and IPAH sera (lanes 5–7) were treated with (+) and without (−) HAase overnight at 37 °C. In lanes 4 and 7, the reaction mixture was spiked with the same amount of HC-HA as in lane 7 of panel A, to function as a positive control and demonstrate the limit of detection. Panel C portrays the serum from two more normal and IPAH patients, treated with and without HAase. n = 3 from three different IPAH and control patients.
TSG-6 Gene Expression Is Increased in IPAH Lung Tissue
Because the enzymatic product of TSG-6 (i.e. HC-HA) was found in IPAH lung tissue (Figs. 1–4), we used real time PCR to measure the gene expression of the enzyme itself (i.e. TSG-6). Fig. 7 demonstrates a statistically significant 6.26-fold increase in TSG-6 gene expression in IPAH lung tissue compared with normal tissue.
FIGURE 7.

TSG-6 gene expression is increased in IPAH. Real-time quantitative PCR SYBR Green analysis of normal (control) and IPAH lung tissue was performed. Relative expression values ± experimental error (error bars) were calculated using GAPDH as a control for the amount of cDNA. n = 3 from three different IPAH and control patients.
DISCUSSION
In this paper we presented biochemical and immunofluorescence evidence that the HA that accumulates in plexigenic regions of IPAH lungs is a pathological type of HA containing covalently bound HCs from the serum-derived proteoglycan IαI. Fluorescent microscopy, showing the regional distribution of HC-HA, was observed in specific regions throughout the IPAH lungs and was not limited to the plexigenic lesion. Although there was a correlation between the distribution of HC-HA and the distribution of leukocytes in the lung tissue, this correlation was not absolute. There were some regions with HC-HA deposits that had little or no leukocytes present and other regions that had significant patches of leukocytes present in the absence of HC-HA. We have shown that monocytic leukocytes “cap” CD44 on their cell surface and degrade HC-HA when bound to it (7). Thus, it is possible that HC-HA, in leukocyte-dense regions of IPAH lung tissue, may have been digested, giving a negative result. The major HA receptor, CD44, has also been localized to the endothelial cells of microvessels within plexigenic lesions (8).
Considering the regional distribution of HC-HA in the vascular wall of plexigenic lesions, it is likely that vascular smooth muscle cells are the origin for the HA deposits within plexigenic lesions. Factors that stimulate cells to make leukocyte-adhesive HA “cable” structures include endoplasmic reticulum stress (9), viral infections (10, 11), hyperglycemia (12), and adrenergic receptor stimulation (13). Pro-inflammatory cytokines up-regulated in IPAH include TNF-α, IL-1β, IL-6, etc. Studies are underway to investigate the effect of these cytokines on pulmonary smooth muscle cell induction of HA synthesis (data not shown).
The relatively high gene expression of TSG-6 in IPAH lung tissue implies that one or more cell populations are actively producing this enzyme. The acronym TSG-6 stands for “tumor necrosis factor stimulated gene-6” because it was originally identified as a novel gene induced in human fibroblasts treated with TNF-α (14). Thus, the origin of TSG-6 in IPAH lung tissue could be the vascular smooth muscle cells. Elevated levels of TNF-α have been reported during episodes of pulmonary arterial hypertension (15, 16). Additionally, agonists of TNF-α have been shown to attenuate monocrotaline-induced pulmonary arterial hypertension (17). Thus, it is possible that the elevated TSG-6 gene expression in IPAH tissue is caused by the elevated levels of TNF-α, as has been reported previously (16, 17). Because monocytes are a primary source of TNF-α in inflamed tissues, one model is that monocytes release this cytokine, which acts upon vascular smooth muscle cells to induce TSG-6 secretion and the formation of HC-HA in IPAH lung tissue. Furthermore, TSG-6, itself, may stimulate HA synthesis in IPAH tissue, as we have described in airway smooth muscle cells and in asthmatic airways (5).
We presented data that HC-HA levels in IPAH serum were below the limit of detection. Our inability to detect HC-HA in IPAH serum, while others detected it in serum from rheumatoid arthritis patients, could be because our Western blotting methodology is not as sensitive as ELISA. We have previously demonstrated that HA, itself, is present in IPAH plasma at significantly higher levels than in normal plasma (18). Perhaps more likely, the reason why HC-HA is detectable in rheumatoid arthritis serum, but not in IPAH serum, is because the inflammation in rheumatoid arthritis is systemic, whereas in IPAH, it is primarily localized to the lung, and in particular, to the pulmonary vasculature.
The role of inflammation in IPAH is increasingly recognized (8, 19, 20). We, and others, have shown that the presence of HCs on HA significantly promotes leukocyte adhesion to HA matrices (4, 5). Our findings provide a possible mechanistic explanation for the inflammation described in IPAH lungs, especially around plexigenic lesions (21). It is reasonable to speculate that the co-localization of leukocytes in HC-HA-enriched regions may be the result of the increased leukocyte avidity to HC-HA matrices compared with HA alone. There is evidence that leukocyte adhesion to HC-HA matrices activates them, resulting in CD44 capping and a phagocytic phenotype (7). The overall effect of HC-HA on leukocyte activation and IPAH pathology remains to be determined.
The role of HC-HA in the etiology of IPAH remains to be demonstrated. We have presented evidence that HC-HA is present at higher levels in IPAH tissue than in normal tissue. The regional distribution of HC-HA within vascular pathologies, such as plexigenic lesions, and the co-localization of leukocytes in these regions, suggest that HC-HA may direct inflammatory events, cell proliferation, and vascular remodeling in IPAH (Fig. 8).
FIGURE 8.
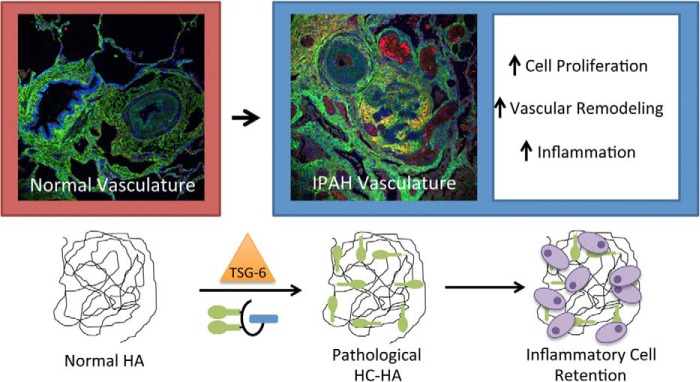
Model for the role of the pathological heavy chain modification of hyaluronan in IPAH. Under normal conditions, HA is present in the submucosa of pulmonary vasculature as a very large, linear glycosaminoglycan of the extracellular matrix, lacking any protein modifications. During IPAH, the enzyme TSG-6 covalently transfers HCs from IαI to HA in the submucosa of pulmonary vasculature, forming the pathological HC-HA matrix. This structure promotes inflammatory cell adhesion and retention in the submucosa region of the vasculature. It may also have a role in vascular remodeling and cell proliferation, leading to the occlusion of pulmonary arteries and the formation of plexigenic lesions.
Acknowledgment
We thank the Cleveland Clinic Program of Excellence in Glycoscience (PEG) Resource Core for assistance with the hyaluronan fluorescent microscopy and heavy chain extraction from human lung tissue.
This work was supported, in whole or in part, by National Institutes of Health Grants P01HL107147, HL081064, HL103453, and HL060917 from the NHLBI.
- IPAH
- idiopathic pulmonary arterial hypertension
- HA
- hyaluronan
- HC
- heavy chain
- IαI
- inter-α-inhibitor
- TSG-6
- tumor necrosis factor-stimulated gene 6.
REFERENCES
- 1. Wilkins M. R. (2012) Pulmonary hypertension: the science behind the disease spectrum. Eur. Respir. Rev. 21, 19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aytekin M., Comhair S. A., de la Motte C., Bandyopadhyay S. K., Farver C. F., Hascall V. C., Erzurum S. C., Dweik R. A. (2008) High levels of hyaluronan in idiopathic pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L789–L799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔC(T) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 4. Zhuo L., Kanamori A., Kannagi R., Itano N., Wu J., Hamaguchi M., Ishiguro N., Kimata K. (2006) SHAP potentiates the CD44-mediated leukocyte adhesion to the hyaluronan substratum. J. Biol. Chem. 281, 20303–20314 [DOI] [PubMed] [Google Scholar]
- 5. Lauer M. E., Cheng G., Swaidani S., Aronica M. A., Weigel P. H., Hascall V. C. (2013) Tumor necrosis factor-stimulated gene-6 (TSG-6) amplifies hyaluronan synthesis by airway smooth muscle cells. J. Biol. Chem. 288, 423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kida D., Yoneda M., Miyaura S., Ishimaru T., Yoshida Y., Ito T., Ishiguro N., Iwata H., Kimata K. (1999) The SHAP-HA complex in sera from patients with rheumatoid arthritis and osteoarthritis. J. Rheumatol. 26, 1230–1238 [PubMed] [Google Scholar]
- 7. Wang A., de la Motte C., Lauer M., Hascall V. (2011) Hyaluronan matrices in pathobiological processes. FEBS J. 278, 1412–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ohta-Ogo K., Hao H., Ishibashi-Ueda H., Hirota S., Nakamura K., Ohe T., Ito H. (2012) CD44 expression in plexiform lesions of idiopathic pulmonary arterial hypertension. Pathol. Int. 62, 219–225 [DOI] [PubMed] [Google Scholar]
- 9. Majors A. K., Austin R. C., de la Motte C. A., Pyeritz R. E., Hascall V. C., Kessler S. P., Sen G., Strong S. A. (2003) Endoplasmic reticulum stress induces hyaluronan deposition and leukocyte adhesion. J. Biol. Chem. 278, 47223–47231 [DOI] [PubMed] [Google Scholar]
- 10. de La Motte C. A., Hascall V. C., Calabro A., Yen-Lieberman B., Strong S. A. (1999) Mononuclear leukocytes preferentially bind via CD44 to hyaluronan on human intestinal mucosal smooth muscle cells after virus infection or treatment with poly(I·C). J. Biol. Chem. 274, 30747–30755 [DOI] [PubMed] [Google Scholar]
- 11. de la Motte C. A., Hascall V. C., Drazba J., Bandyopadhyay S. K., Strong S. A. (2003) Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyinosinic acid:polycytidylic acid: inter-α-trypsin inhibitor is crucial to structure and function. Am. J. Pathol. 163, 121–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang A., Hascall V. C. (2004) Hyaluronan structures synthesized by rat mesangial cells in response to hyperglycemia induce monocyte adhesion. J. Biol. Chem. 279, 10279–10285 [DOI] [PubMed] [Google Scholar]
- 13. Shi T., Duan Z. H., Papay R., Pluskota E., Gaivin R. J., de la Motte C. A., Plow E. F., Perez D. M. (2006) Novel α1-adrenergic receptor signaling pathways: secreted factors and interactions with the extracellular matrix. Mol. Pharmacol. 70, 129–142 [DOI] [PubMed] [Google Scholar]
- 14. Lee T. H., Lee G. W., Ziff E. B., Vilcek J. (1990) Isolation and characterization of eight tumor necrosis factor-induced gene sequences from human fibroblasts. Mol. Cell. Biol. 10, 1982–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leeper-Woodford S. K., Carey P. D., Byrne K., Jenkins J. K., Fisher B. J., Blocher C., Sugerman H. J., Fowler A. A., 3rd. (1991) Tumor necrosis factor: α and β subtypes appear in circulation during onset of sepsis-induced lung injury. Am. Rev. Resp. Dis. 143, 1076–1082 [DOI] [PubMed] [Google Scholar]
- 16. Lei Y., Zhen J., Ming X. L., Jian H. K. (2002) Induction of higher expression of IL-β and TNF-α, lower expression of IL-10 and cyclic guanosine monophosphate by pulmonary arterial hypertension following cardiopulmonary bypass. Asian J. Surg. 25, 203–208 [DOI] [PubMed] [Google Scholar]
- 17. Wang Q., Zuo X. R., Wang Y. Y., Xie W. P., Wang H., Zhang M. (2013) Monocrotaline-induced pulmonary arterial hypertension is attenuated by TNF-α antagonists via the suppression of TNF-α expression and NF-κB pathway in rats. Vascul. Pharmacol. 58, 71–77 [DOI] [PubMed] [Google Scholar]
- 18. Kalay N., Elcik D., Canatan H., Kaya M. G., Yarlioglues M., Oguzhan A., Dweik R. A., Aytekin M. (2013) Elevated plasma hyaluronan levels in pulmonary hypertension. Tohoku J. Exp. Med. 230, 7–11 [DOI] [PubMed] [Google Scholar]
- 19. El Chami H., Hassoun P. M. (2012) Immune and inflammatory mechanisms in pulmonary arterial hypertension. Prog. Cardiovasc. Dis. 55, 218–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Price L. C., Wort S. J., Perros F., Dorfmüller P., Huertas A., Montani D., Cohen-Kaminsky S., Humbert M. (2012) Inflammation in pulmonary arterial hypertension. Chest 141, 210–221 [DOI] [PubMed] [Google Scholar]
- 21. Stacher E., Graham B. B., Hunt J. M., Gandjeva A., Groshong S. D., McLaughlin V. V., Jessup M., Grizzle W. E., Aldred M. A., Cool C. D., Tuder R. M. (2012) Modern age pathology of pulmonary arterial hypertension. Am. J. Resp. Crit. Care Med. 186, 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]