

Background: The oxicams are anti-inflammatory drugs targeting the cyclooxygenase enzymes.
Results: Crystal complexes of mCOX-2·isoxicam, mCOX-2·meloxicam, and oCOX-1·meloxicam are solved.
Conclusion: Oxicams bind to the cyclooxygenase active sites in a novel mode.
Significance: The first structural description of cyclooxygenase-oxicam complexes reveal a new binding pocket of inhibitors to cyclooxygenases.
Keywords: Cyclooxygenase (COX) Pathway, Drug Action, Enzyme Structure, Protein Drug Interactions, X-ray Crystallography
Abstract
Oxicams are widely used nonsteroidal anti-inflammatory drugs (NSAIDs), but little is known about the molecular basis of the interaction with their target enzymes, the cyclooxygenases (COX). Isoxicam is a nonselective inhibitor of COX-1 and COX-2 whereas meloxicam displays some selectivity for COX-2. Here we report crystal complexes of COX-2 with isoxicam and meloxicam at 2.0 and 2.45 angstroms, respectively, and a crystal complex of COX-1 with meloxicam at 2.4 angstroms. These structures reveal that the oxicams bind to the active site of COX-2 using a binding pose not seen with other NSAIDs through two highly coordinated water molecules. The 4-hydroxyl group on the thiazine ring partners with Ser-530 via hydrogen bonding, and the heteroatom of the carboxamide ring of the oxicam scaffold interacts with Tyr-385 and Ser-530 through a highly coordinated water molecule. The nitrogen atom of the thiazine and the oxygen atom of the carboxamide bind to Arg-120 and Tyr-355 via another highly ordered water molecule. The rotation of Leu-531 in the structure opens a novel binding pocket, which is not utilized for the binding of other NSAIDs. In addition, a detailed study of meloxicam·COX-2 interactions revealed that mutation of Val-434 to Ile significantly reduces inhibition by meloxicam due to subtle changes around Phe-518, giving rise to the preferential inhibition of COX-2 over COX-1.
Introduction
Two prostaglandin endoperoxide synthases, cyclooxygenase-1 and cyclooxygenase-2, are involved in prostaglandin biosynthesis and are the main targets for nonsteroidal anti-inflammatory drugs (NSAIDs)2 (1). COX-1 is constitutively expressed, whereas COX-2 is induced in inflammation and other pathological conditions (2). The structural details of NSAIDs bound to the cyclooxygenase channel of COX-1 and COX-2 have been elucidated by x-ray crystallographic studies (3–12). The classic NSAIDs, including indomethacin and profens, typically bind within the cyclooxygenase channel via ionic interactions between the carboxylate group of the NSAIDs and the side chain of Arg-120 and via H-bonding interactions with the side chain of Tyr-355 (Fig. 1A). These residues are located at the base of the active site and comprise part of the gate that separates the active site from the ligand access channel (6, 10, 13). In contrast, diclofenac H-bonds through its carboxylate with the catalytic Tyr-385 residue as well as Ser-530 at the apex of the active site (Fig. 1B) (11). Both binding modes mentioned above are observed in the interaction of the substrate arachidonic acid (AA) in the active site of COX-2; one molecule is bound in a productive conformation in which the carboxylic acid group of AA forms hydrogen bonding interactions with Arg-120 and Tyr-355 (14, 15); the other molecule of AA is bound in a nonproductive conformation in which the carboxylate group interacts with Tyr-385 and Ser-530 (16). A third binding mode exhibited by diarylheterocycles, such as celecoxib and rofecoxib, takes advantage of a side pocket present in COX-2 but not COX-1 off the main cyclooxygenase channel (6, 7, 12).
FIGURE 1.

Binding modes of NSAIDs in the COX-2 active site. A, mCOX-2·naproxen complex (PDB ID 3NT1) reveals the H-bonding interactions (dashes) between naproxen (yellow) and the constriction site of mCOX-2. B, COX-2·diclofenac complex (PDB ID 1PXX) showing the H-bonding interactions (dashes) between diclofenac (magenta) and Ser-530 and Tyr-385. C, chemical structures of oxicams and other representative NSAIDs.
Oxicams are a class of NSAIDs structurally related to 2-methyl-1,2-benzothiazine-enolamide-1,1-dioxides and were introduced to the market in the 1980s (17). Groups or fragments of the lead compounds were substituted with moieties of similar stereo-electronic features to improve pharmacological efficacy, leading to the development of several marketed drugs (18). Meloxicam was approved in 2000 as a moderately selective COX-2 inhibitor and is among the most prescribed drugs through its use for various arthritic conditions and postoperative inflammation (19). Additionally, droxicam and ampiroxicam were developed as prodrugs of piroxicam for better administration and pharmacological properties (20, 21).
Structurally, the oxicams are distinct from other classes of NSAIDs, containing a fused thiazine dioxide ring and an extended carboxamide substitution (Fig. 1C). The oxicam scaffold exhibits considerable flexibility and exists as several different protonation tautomers through the keto/enol equilibrium (22). Two independent computational studies suggested that oxicam binding to COX-2 might exhibit an inverted mode where the sulfonyl dioxide group in the thiazine interacts with Tyr-385 and Ser-530 as seen with the carboxylic group of diclofenac (23, 24). However, this model fails to explain the fact that mutation of Arg-120 to Ala and Tyr-355 to Phe abolish inhibition by piroxicam (11) and does not explain why oxicams are mainly reported as nonselective COX inhibitors with some preference for COX-2.
Thus, we utilized x-ray crystallography to study the binding mode of oxicams, and here we report the first crystal structure determination of murine COX-oxicam complexes. The oxicams bind to the cyclooxygenase active site via a novel binding mode that utilizes multiple interactions with residues throughout the active site and includes two tightly bound water molecules. In addition, our site-directed mutagenesis studies reveal why meloxicam exhibits some selectivity for COX-2 over COX-1.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Murine cyclooxygenase-2 and its mutants were expressed in a baculovirus-insect cell expression system as described (11). Site-directed mutagenesis was performed on a pvL-1393 plasmid bearing the cDNA of murine COX-2 as previously described (11). Mutagenesis was performed using a QuikChange mutagenesis kit from Stratagene. The resulting plasmids bearing the native cDNA of COX-2 or mutants were co-transfected with linearized BaculoGold DNA to generate the recombinant baculovirus. Protein expression was performed in Sf-21 insect cells by infection for 48 h. The cell pellets were harvested, lysed, and purified by sequential ion exchange and size exclusion chromatography to >95% purity. Ovine COX-1 was purified as previously described from sheep seminal vesicles (25).
COX Inhibition Assay
Cyclooxygenase inhibition was monitored by O2 consumption using a Clark-type O2 sensitive electrode (Hansatech Pentney, Norfolk, England) as described previously (26). Calibration was achieved using a N2 saturated solution to establish a zero O2 level within the reaction chamber prior to experimental measurements. All final 1-ml assay solutions contained 100 nm purified protein, 2 equivalents of hematin, 100 mm Tris-Cl, pH 8.0, 5 mm phenol, and 2% dimethyl sulfoxide. The inhibitor was delivered by an airtight syringe in dimethyl sulfoxide and incubated with protein for 12 min at room temperature and an additional 3 min at 37 °C in the chamber before the addition of AA. Oxygen consumption was monitored for 2 min, and the reaction rates were reported as maximal rates occurring after a lag phase (27).
Crystallization of mCOX-2 and oCOX-1 Complexes
Crystallization of murine cyclooxygenase-2 was performed as previously described (4). Briefly, purified mCOX-2 protein reconstituted with a 2-fold molar excess of Fe3+-protoprophyrin IX was dialyzed overnight at 4 °C against 20 mm sodium phosphate buffer, pH 6.7, 100 mm NaCl, 0.6% (w/v) β-OG, and 0.1% NaN3. The concentration of β-OG was adjusted to 1.2%, and 10-fold molar excess of inhibitors from 25 mm dimethyl sulfoxide stocks were added to protein samples for 20–30 min prior to the crystallization setup. Crystallization was conducted using the hanging drop method by mixing 3 μl of the protein-inhibitor complex with 3 μl of crystallization solution containing 50 mm EPPS, pH 8.0, 120 mm MgCl2, 22–26% PEG MME-550 against reservoir solutions comprising 50 mm EPPS, pH 8.0, 120 mm MgCl2, 22–26% PEG MME-550. Crystals appeared in approximately 5 days and grew to full size in approximately 3 weeks. The crystals were then transferred into a solution of 50 mm EPPS, pH 8.0, 120 mm MgCl2, 28% PEG MME-550 for about 30 s and flash frozen in liquid nitrogen for shipment and data collection.
Crystallization of cyclooxygenase-1 was conducted via the sitting drop method as previously described (5). Ovine COX-1 was reconstituted with a 1.5-fold excess of hemin and then incubated with 0.1 mm meloxicam prior to crystallization. Sitting drop vapor diffusion was conducted by mixing 3 μl of complex with 3 μl of buffer composed of 0.64 m sodium citrate, 0.3–0.9 m LiCl, 0.3% (w/v) β-OG, and 1 mm NaN3 against a reservoir containing 0.68–0.88 m sodium citrate, 0.3–0.6 m LiCl, and 1 mm NaN3 at 20 °C. After 2–3 weeks, COX-1 crystals appeared. The crystals were briefly soaked in a solution containing 1.0 m sodium citrate, 1.0 m LiCl, 0.15% (w/v) β-OG, and 1 mm sodium malonate as a cryoprotectant and stored in liquid nitrogen for later data collection.
X-ray Data Collection, Structure Determination, and Refinement
Ovine COX-1·meloxicam complex and COX-2 complexes were collected using the synchrotron radiation x-ray source with 100 K liquid nitrogen streaming at beamline DNC-CAT and 24-ID-C in the Advance Photon Source at Argonne National Laboratory, respectively. Diffraction data were collected and processed with XDS (28). COX-1 complex was determined as P65 space group whereas two COX-2 complexes were determined as P21212 space group. Initial phases were determined by molecular replacement using monomer coordinates (1Q4G for COX-1, 3NT1 for COX-2) with Phaser (29). A set of random selected data (Rfree set) was set aside for test and quality control. A twin operator (h, -h-k, -l) was applied for the COX-1 complex. The models were improved with several rounds of model building against the remaining data with F ≥ 0 as working data in COOT (30) and Phenix (31). Global noncrystallographic symmetry was applied during the refinement and released after the first few cycles of refinement. Ligand constraints were computed using the PRODRG server (32). Water molecules were added during the last cycles of refinement. TLS refinement was applied to COX-2 complexes in the last cycle of refinement (33). The potential of phase bias was excluded by simulated annealing using Phenix (34). The values of the Ramachandran plot for the final refinement of the structure were obtained by use of the Phenix suite (97% favored and 0.09% outliers for COX-2·isoxicam complex, 97% favored and no outliers for COX-2·meloxicam complex, 95% favored and 0.18% outliers for COX-1·meloxicam complex) (31). X-ray data collection and structural refinement statistics are reported in Table 1. Among the monomers in the asymmetric unit, no significant differences were observed, and all illustrations were prepared using the coordinates of one monomer with PyMOL (Schrödinger, LLC).
TABLE 1.
Statistics of x-ray data collection and structure refinement
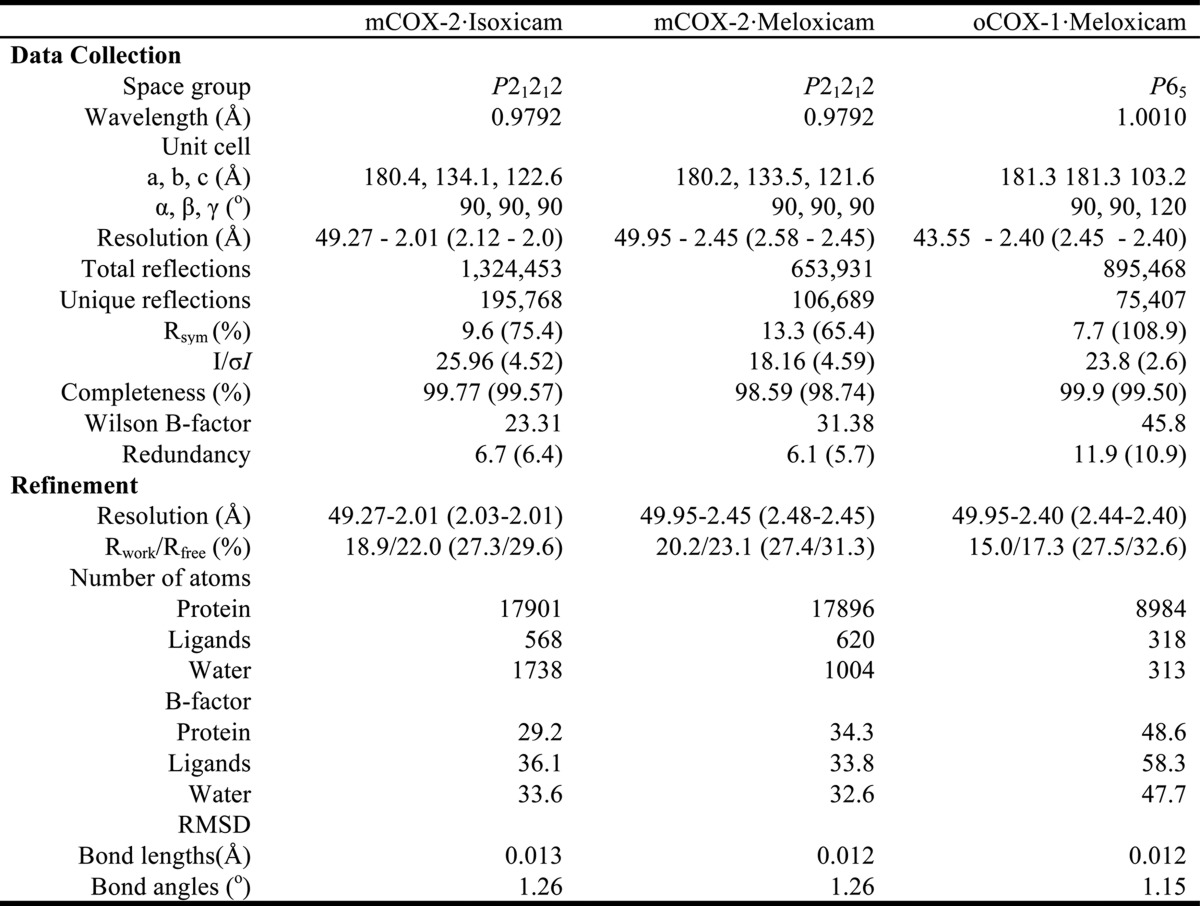
Number of crystals for each data set = 1; the values in parentheses are for the highest resolution shell; Rsym = , R = , where Fo and Fc are the observed and calculated structure factors, and Rfree sets are 5.0%, 5.0% and 4.0%, respectively.
RESULTS
Isoxicam Binds to the COX-2 Active Site Utilizing a Novel Binding Pose
The murine COX-2·isoxicam complex was crystallized in the space group P21212 and diffracted to 2.0 Å using previously described conditions (4). The electron density map of the COX-2·isoxicam complex was of excellent quality to build in considerable amounts of COX-2 (residues 33–583) and other protein substructural elements including protoporphyrin IX, the glycosylation sites at Asn-68, Asn-144, Asn-410, and four β-OG detergent molecules after molecular replacement and structural refinement. Four detergent molecules are located in the outer shell of the protein. There are four molecules adjacent to Lys-180 on the side of each monomer, which are shared with four other monomers from symmetric mates, counting as two detergent molecules for each asymmetric unit. The other two detergent molecules are outside of Tyr-91 in helix B in monomers B and C. No detergent molecules were observed in the lobby region or close to helix D. The overall structure, composed of the epidermal growth factor domain, membrane binding domain, and the larger catalytic domain, is similar to the high resolution crystal structure of the mCOX-2·naproxen complex (PDB ID 3NT1), and few changes were found among the four monomers in the asymmetric unit with a root mean square deviation from 0.16 to 0.21 Å for the backbone atoms. As such, the conformation of isoxicam in monomer A was utilized to describe the details associated with COX interactions and comparisons with other COX complexes. The residues are labeled according to the ovine COX-1 numbering system for clarity (6).
Well defined electron densities accounting for isoxicam were observed in the COX active site of all four monomers in one asymmetric unit (Fig. 2A). Isoxicam binds in the COX channel in a strikingly different manner compared with other reported mCOX·NSAID complexes (Fig. 3). Isoxicam establishes a planar conformation between helix 6 and helix 17 in the active site, adapting a configuration with an intramolecular hydrogen bond between the nitrogen atom from the carboxamide and the 4-hydroxyl oxygen of the benzothiazine. The drug interfaces with the hydrophobic COX channel mainly through van der Waals interactions. The only immediate polar interaction between isoxicam and COX is a hydrogen bond between the 4-hydroxyl group from benzothiazine ring and Ser-530 with an effective distance of approximately 2.7 Å.
FIGURE 2.

Stereo-diagram of the electron density maps around isoxicam and meloxicam. A, a simulated annealing composite omit map in the active site of mCOX-2·isoxicam complex. B, a simulated annealing composite omit map in the active site of mCOX-2·meloxicam complex. C, a simulated annealing composite omit map in the active site of oCOX-1·meloxicam complex. The simulated annealing omit maps (34) are contoured at 1.5 σ in blue mesh; the key residues are illustrated with a stick representation of the final model (carbon in green, oxygen in red, isoxicam in orange); the molecule of isoxicam is colored in orange, and meloxicam is colored in wheat and yellow.
FIGURE 3.
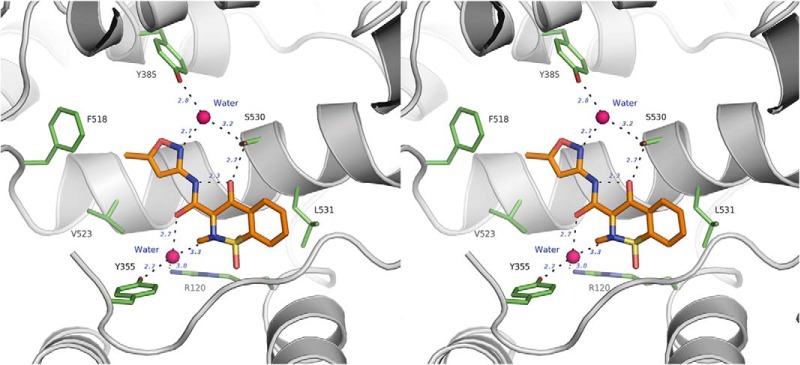
Isoxicam binds to COX-2 in a novel pose via a two-water-mediated network (stereo-diagram). The H-bonds are illustrated as dashes, the key residues are in green sticks, waters are in deep pink, and isoxicam is in orange.
In contrast, the sulfonyl dioxide, the hypothesized binding candidate for the interaction with Tyr-385 and Ser-530 in simulations (23, 24), is located ∼3 Å above the constriction site, exhibiting few direct interactions with COX-2. One of the dioxide oxygens may form a weak hydrogen bond with the backbone oxygen of Ala-527 with a distance of 3.5 Å, whereas the other oxygen of the dioxide may sterically interfere with the side chain of Val-116 with a closest distance of 3.1 Å. In response to this interference, the thiazine ring displays a twisted envelope conformation in which the sulfur and the 2-hetero nitrogen are off the plane in opposite directions. The 2-methyl substituent points into the pocket composed of Tyr-366, Val-349, and Leu-359, whereas the other four atoms of the thiazine ring remain in the plane. The carboxamide moiety is inserted into the hydrophobic pocket comprising Leu-384, Tyr-385, Trp-387, Phe-518, Met-522, and Val-523. The rest of the molecule interacts with COX mainly through hydrophobic interactions.
Interestingly, two highly ordered water molecules in the active site provide additional polar bridges between isoxicam and COX residues besides the 4-OH–Ser-530 interaction (Figs. 2 and 3). The first water, which is located immediately above the constriction site of COX, is approximately 3.0 Å away from both the oxygen of Tyr-355 and the guanidino nitrogen of Arg-120, indicating that two hydrogen bonds are formed. Meanwhile, the same water molecule is within H-bond distances of the oxygen of the carboxamide and the nitrogen atom in the thiazine ring of the inhibitor. At the apex of the active site, another water lies between Tyr-385 and Ser-530 and the nitrogen atom of the isoxazole ring from isoxicam.
The Binding of Isoxicam to COX-2 Involves Side Chain Rotation of Leu-531 and Structural Movement of Helix D
The isoxicam complex reveals a small difference in the backbones of helices C and D in the membrane binding domain and the side chain of Leu-531 in helix 17 compared with the 1.7 Å resolution mCOX-2·naproxen complex (PDB ID 3NT1). Due to steric hindrance between Val-116 and one oxygen of the dioxide of isoxicam, helix D of COX-2 moves over 1.0 Å to accommodate the benzyl ring of the benzothiazine without further changes in the orientation of the corresponding residues in the helix (Fig. 4). This sort of movement was observed in the complexes of COX-2·AA and COX-2·1-AG (3MDL) but to a lesser extent. This accommodation is also seen in helix C, which experiences a 0.5 Å shift of the backbone from the original position. This flexibility of membrane binding domain helices is consistent with the observations made in the complex of human COX-2 with a zomepirac derivative, RS57067 (12).
FIGURE 4.
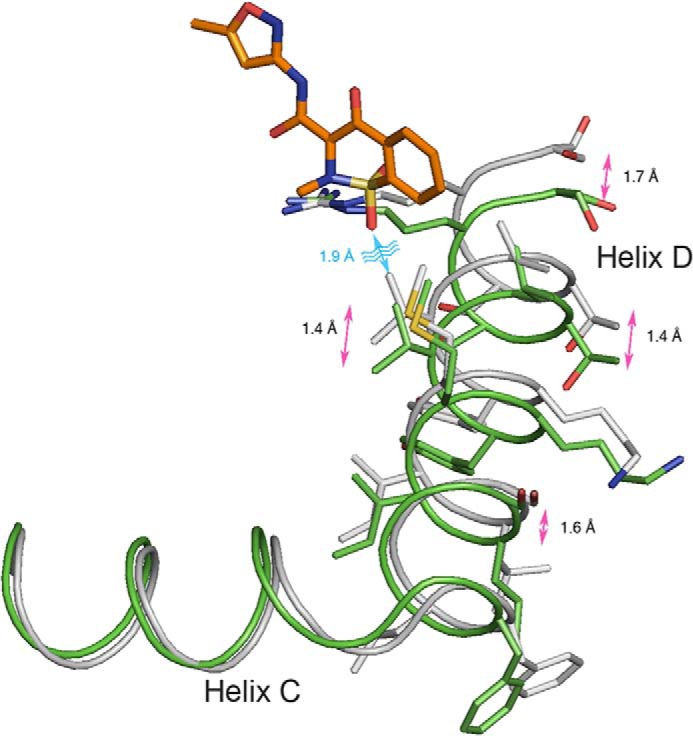
Structural changes in helices C and D of COX-2 upon the binding of isoxicam. The backbone atoms of monomer A from mCOX-2·naproxen (PDB ID 3NT1) and isoxicam complexes are aligned. The helices are in a tube representation, with the naproxen complex in gray and the isoxicam complex in green.
In contrast, helix 17, which also interacts with isoxicam, does not move. However, the side chain of Leu-531 in this helix displays a different rotamer (Fig. 5). The Cδ atom of Leu-531 shifts approximately 3.5 Å away from the active site toward the dimer interface, resulting in a possible CH-π interaction between the Cβ atom of Leu-531 and the aromatic ring of the inhibitor with a distance of approximately 4.2 Å. This movement opens a new hydrophobic pocket composed of Met-113, Val-116, Leu-117, Ile-345, Val-349, Leu-531, Leu-534, and Met-535, which accommodates the rigid fused benzyl ring of isoxicam (Fig. 5).
FIGURE 5.

Leu-531 opens a new binding pocket for isoxicam in the mCOX-2 active site. A, stereo-diagram of the surface representation around isoxicam (orange sticks and balls). B, same view of the binding pocket around the naproxen (yellow sticks and balls) complex (PDB ID 3NT1). C, binding pocket around 1-AG (yellow sticks and balls) complex (PDB ID 3MDL). The interacting residues are in green, and Leu-531 is highlighted in blue; the surrounding surfaces are shown in semi-transparent gray.
Meloxicam Displays Two Conformations upon Binding to COX-2
Meloxicam, an analog of isoxicam, is a 5-fold preferential inhibitor of COX-2 over COX-1 (18). To investigate why meloxicam is a preferential COX-2 inhibitor while isoxicam is a nonselective one, we determined the mCOX-2·meloxicam complex by x-ray crystallography. The 2.45 Å resolution structure is of sufficient quality to build in most of the components including the EGF domain, membrane binding domain, the catalytic domain, and glycosylation sites at Asn-68, Asn-144, and Asn-410. However, the detailed orientation of the carboxamide moiety of meloxicam could not be determined. Both the sulfur and nitrogen atoms of the thiazole ring are good candidates to form a similar hydrogen bonding network between the coordinated water and the catalytic apex. The electron density around the methyl group on the thiazole ring is insufficient to differentiate the conformation in the complex (Fig. 2B). This observation suggests that two conformations are present in the mCOX-2·meloxicam complex. In the first conformation the methyl group points down toward the entry of the active site. The nitrogen of the thiazole ring is approximately 2.8 Å away from the water, whereas the sulfur atom interacts with Val-523 and Phe-518 in this conformation. In the second conformation the methyl group points up and binds in a hydrophobic pocket comprising Leu-384, Trp-387, Phe-518, and Met-522. In this conformation, the sulfur of the thiazole group interacts with the water molecule at an effective distance of 3.3 Å. We were unable to rule out either conformation based on the quality of the electron density map or the chemical plausibility for satisfying hydrogen bonding and hydrophobic interactions. Thus, both conformations are included in the final structure (Fig. 6). Other than this conformational flexibility, meloxicam binds to the COX active site in the same mode as isoxicam including hydrogen bonding to Ser-530, hydrogen bonding to two coordinated waters complexed to Tyr-385/Ser-530 and Arg-120/Tyr-355, and the new hydrophobic pocket generated by the movement of Leu-531.
FIGURE 6.
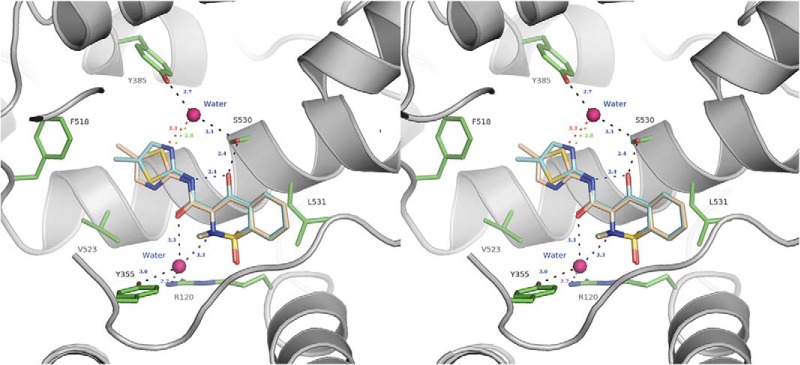
Stereo-diagram of meloxicam bound to the mCOX-2 active site. The protein residues are shown as green sticks and balls, the coordinated waters are in spheres, and the methyl-pointing-down conformation of meloxicam is colored in cyan and a green dash for the interaction with the water; the methyl-pointing-up conformation is in wheat with the corresponding H-bond in red. Other hydrogen bonds are illustrated as black dashes.
Subtle Structural Features around Phe-518 Differentiate the Preference of Meloxicam for COX-2 over COX-1, Providing New Structural Insights on Selectivity
The structure of ovine COX-1 in complex with meloxicam was solved at 2.4 Å resolution. The binding pose of meloxicam in the COX-1 active was similar to that of meloxicam bound to COX-2 (Table 1 and Figs. 2 and 7). In our in vitro assay, meloxicam exhibits an IC50 of 150 nm toward wild-type recombinant mouse COX-2 whereas it exhibits an IC50 of 990 nm for native ovine COX-1 (Fig. 7), which is consistent with literature reports (18). Structurally, COX-1 and COX-2 share most secondary features with minor changes in the active site that include I434V, H513R, and I523V COX-1→COX-2 substitutions (6, 35). The triple COX-2 mutant of V434I/R513H/V523I displayed a reduced inhibition by meloxicam with an IC50 of 1.22 μm, which is similar to wild-type ovine COX-1 (Fig. 7). As Arg-513 is located in the side pocket of COX-2 and barely interacts with meloxicam in our crystal structure, we focused inhibition studies on the mCOX-2 mutants of V434I and V523I. The V523I mutant exhibited potent inhibition by meloxicam with an IC50 of 174 nm. In contrast, meloxicam inhibited the V434I mutant with an IC50 of 1.4 μm. This observation reveals that mutation of Val-434 to Ile at position 434 in murine COX-2 is sufficient to decrease the potency of meloxicam COX-2 inhibition to that of ovine COX-1 (Fig. 7). The additional carbon in the side chain of Ile-434 in the secondary shell in COX-1 causes Phe-518 to move toward the main channel further than in COX-2 in which a valine is located at the same position (6). Thus, the selectivity of meloxicam for COX-2 is as a result of the subtle structural features in the neighborhood of Phe-518, where different rotamers of Phe-518 are presented in COX-1 and COX-2 structures due to the difference of secondary shell residues Ile-434 for COX-1 and Val-434 for COX-2.
FIGURE 7.
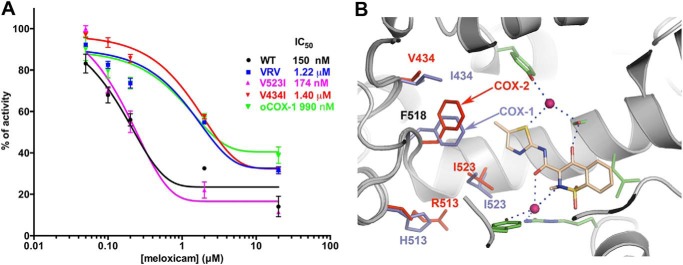
Inhibition of mCOX-2 and mutants by meloxicam. A, inhibition profiles of meloxicam toward oCOX-1, mCOX-2, and its mutants. B, structural difference in the active site between oCOX-1 (slate sticks) and mCOX-2 (red sticks) highlighting the changes in residues 434, 518, 513, and 523. Points in A are from triplicate measurements and are depicted as the mean ± S.E. (error bars). The backbone atoms of oCOX-1·meloxicam structure are aligned with those of the mCOX-2·meloxicam complex. Only the up conformation of meloxicam from mCOX-2 complex is represented for clarity.
DISCUSSION
Oxicams, a structurally unique class of NSAIDs, are widely used drugs, yet little information is available about the molecular basis for their inhibition of cyclooxygenases. In this study, we report for the first time the crystal complexes of isoxicam and meloxicam bound to mCOX-2. Oxicams bind in the cyclooxygenase channel with a novel pose compared with previously reported NSAID binding modes, in which the carboxylic acid of the inhibitor interacts either in the canonical mode with Arg-120 and Tyr-355 (6, 10) or in the inverted binding mode with Tyr-385 and Ser-530 (11). Instead, oxicams interact with all four of these residues, Arg-120, Tyr-355, Tyr-385, and Ser-530. However, aside from a direct hydrogen bond between the 4-hydroxyl group of the benzothiazine moiety and Ser-530, the oxicams do not directly interact with any of these residues by ion pairing or hydrogen bonding. Instead, they interact indirectly by bridging with two tightly bound waters: a tetrahedrally coordinated water bound to Arg-120 and Tyr-355 and a trigonally coordinated water bound to Tyr-385 and Ser-530. This explains why none of the S530A, R120A, or Y355F COX-2 mutants is inhibited by piroxicam (11). In addition, the water bridge between the carboxamide and Ser-530 and Tyr-385 provides the first structural explanation for the observation that all oxicam drugs contain a heteroatom at the 1′ position in the carboxamide moiety.
The binding of oxicams is accompanied by changes in protein conformation, illustrating an induced fit mechanism, which is rare in the interaction between COX and NSAIDs. This includes the rotation of the side chain of Leu-531 and a modest movement of helix D. The rotamer of Leu-531 in the oxicam complexes is similar to that observed in the nonproductive conformations of mCOX-2·AA (16), mCOX-2·1-AG (36), and G533V-AA complexes (37), but such a rotation has not been detected in the binding of any NSAIDs. In the case of isoxicam, it opens an entirely new subpocket in the COX active site (Fig. 5), which may represent a target region for drug discovery. It is worth noting that the difference in orientation of Leu-531 in the complexes of oxicams or 1-AG is mainly a result of structural fluctuations among the complexes and differences in quality of the electron densities around the region in the two complexes (36). The second isoxicam-induced protein conformation is the over 1.0 Å movement of helix D as a response to steric hindrance between the oxygen of the sulfonyl dioxide and the side chain of Val-116. This further validates the flexibility of the membrane-binding domain of COX-2 (12).
The determination of this novel binding mode provides exact interactions between oxicams and COX-2 and amends the previous computational models for oxicams (10, 24). The difficulty of predicting the oxicam-protein interactions arises for several reasons: the oxicams may exist in several tautomers (22); the two highly coordinated water molecules in the active site are nearly impossible to predict, as the representation and role that water plays in ligand-protein interactions reduces the effectiveness of docking predictions (38); and the recognition of the movement of Leu-531 in COX-2 complexes was only recently reported (36, 37). These complex perturbations in the COX-2 active site present an array of computational problems that complicate the prediction of oxicam binding modes.
Despite the fact that most structure-activity relationship studies of oxicams were completed before the discovery of COX-2 and before the recognition of the importance of COX-2 in inflammation, several lines of evidence indicate that the binding mode described here fits well with published studies and the pharmacological characterizations of oxicams (39–41). The methyl substituent at the 2 position of the benzothiazine is optimal for anti-inflammatory activity (39). This methyl group interacts with residues Leu-359, Tyr-355, and Val-349. Replacement of this methyl group with any other substituent (e.g. H, benzyl, allyl, ethyl, propyl) either removes the interaction between the protein residues or introduces steric bulk that prevents binding in this region. Thus, such substituents were less active than the 2-methyl compounds (39, 41). Among analogs of 3-carboxamides with anilide substituents, meta-substituted anilides exhibit more potent activity than para-substituted anilides. The meta-substitution pattern mimics the methyl-up conformation of meloxicam and fits quite well within the pocket formed by Leu-384, Tyr-385, Met-522, Phe-518, and Trp-387 in our crystal structures. Finally, in a study of thieno[2,3,-e]thiazine analogs, the 2-pyridyl carboxamide (tenoxicam) presented better activity compared with isomeric 3- or 4-pyridyl amides, consistent with the essential role of a H-bond acceptor for ligand binding to the water coordinated to Tyr-385 and Ser-530 (40).
Meloxicam displays modest COX-2 selectivity with an IC50 of 150 nm for COX-2 and an IC50 of 990 nm for COX-1 in our in vitro assay. Our site-directed mutagenesis studies suggest that the preference of meloxicam for COX-2 is not due to the substitution of Ile-523 in COX-1 for Val-523 in COX-2, which accounts for the selectivity of inhibition by rofecoxib and celecoxib, but rather subtle changes around Phe-518 caused by the substitution of isoleucine to valine at residue 434 in COX-2 (see Fig. 7). The additional methylene group of Ile-434 in oCOX-1 pushes Phe-518 into the main channel of the cyclooxygenase active site and shrinks the available space close to the thiazole ring of meloxicam. This difference in the orientation of Phe-518 between COX-1 and COX-2 structures was first described in the complex of COX-2 with SC-558, a diaryl heterocyclic COX-2 selective inhibitor (6). It was suggested that the insertion of the phenylsulfonamide group into the side pocket of COX-2 is facilitated by the movement of Phe-518 to accommodate the movement of Val-523. This hypothesis was not tested with mutagenesis experiments. The potential clashes between the 4′-methyl group of meloxicam and Phe-518 suggest a direct interaction of Phe-518 with inhibitor in the case of meloxicam as opposed to protein movement in the case SC-558. We confirmed this hypothesis by the described site-directed mutagenesis studies. Thus, subtle environmental differences around Phe-518 adjust the COX-2 selectivity of different classes of inhibitors.
Taken together, the crystal structure of recombinant murine COX-2 in complex with isoxicam and meloxicam presented here provides the first structural description for the binding mode of the oxicam class of NSAIDs and reveals a novel binding mode in the cyclooxygenase channel via a two-water-mediated network characterized by interactions with previously identified binding residues throughout the main channel of the active site. Moreover, this binding mode requires the movement of Leu-531 to accommodate the inhibitor. This is the first time Leu-531 has been identified as an interacting residue in NSAID binding although it is involved in binding certain substrates. In addition, mutagenesis studies indicate that the molecular basis for the preferential inhibition of COX-2 by meloxicam is due to the subtle differences around Phe-518 in the active site. Finally, this structural description of COX-2·oxicam complexes explains previous structure-activity studies of oxicams, guides new substitutions within the oxicam scaffold, and identifies a new pocket around Leu-531 for development of novel NSAIDs.
This work was supported, in whole or in part, by National Institutes of Health Research and Training Grants CA089450 (to L. J. M.), GM15431 (to L. J. M.), and DA031572 (to D. J. H.). This work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by National Institutes of Health Grant P41 GM103403 from the NIGMS. Use of the Advanced Photon Source, an Office of Science User Facility operated for the United States Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the United States DOE under Contract DE-AC02-06CH11357.
The atomic coordinates and structure factors (codes 4M10, 4M11, and 4O1Z) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NSAID
- nonsteroidal anti-inflammatory drug
- AA
- arachidonic acid
- β-OG
- n-octyl-β-d-glucopyranoside
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid
- mCOX-2
- murine cyclooxygenase-2
- oCOX-1
- ovine cyclooxygenase-1
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation
- 1-AG
- 1 arachidonoylglycerol.
REFERENCES
- 1. Marnett L. J. (2002) Recent developments in cyclooxygenase inhibition. Prostaglandins Other Lipid Mediat. 68, 153–164 [DOI] [PubMed] [Google Scholar]
- 2. Smith C. J., Zhang Y., Koboldt C. M., Muhammad J., Zweifel B. S., Shaffer A., Talley J. J., Masferrer J. L., Seibert K., Isakson P. C. (1998) Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. U.S.A. 95, 13313–13318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Duggan K. C., Walters M. J., Musee J., Harp J. M., Kiefer J. R., Oates J. A., Marnett L. J. (2010) Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J. Biol. Chem. 285, 34950–34959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Windsor M. A., Hermanson D. J., Kingsley P. J., Xu S., Crews B. C., Ho W., Keenan C. M., Banerjee S., Sharkey K. A., Marnett L. J. (2012) Substrate-selective inhibition of cyclooxygenase-2: development and evaluation of achiral profen probes. ACS Med. Chem. Lett. 3, 759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harman C. A., Turman M. V., Kozak K. R., Marnett L. J., Smith W. L., Garavito R. M. (2007) Structural basis of enantioselective inhibition of cyclooxygenase-1 by S-α-substituted indomethacin ethanolamides. J. Biol. Chem. 282, 28096–28105 [DOI] [PubMed] [Google Scholar]
- 6. Kurumbail R. G., Stevens A. M., Gierse J. K., McDonald J. J., Stegeman R. A., Pak J. Y., Gildehaus D., Miyashiro J. M., Penning T. D., Seibert K., Isakson P. C., Stallings W. C. (1996) Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 384, 644–648 [DOI] [PubMed] [Google Scholar]
- 7. Wang J. L., Limburg D., Graneto M. J., Springer J., Hamper J. R., Liao S., Pawlitz J. L., Kurumbail R. G., Maziasz T., Talley J. J., Kiefer J. R., Carter J. (2010) The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: the second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 20, 7159–7163 [DOI] [PubMed] [Google Scholar]
- 8. Selinsky B. S., Gupta K., Sharkey C. T., Loll P. J. (2001) Structural analysis of NSAID binding by prostaglandin H2 synthase: time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry 40, 5172–5180 [DOI] [PubMed] [Google Scholar]
- 9. Picot D., Loll P. J., Garavito R. M. (1994) The x-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 367, 243–249 [DOI] [PubMed] [Google Scholar]
- 10. Loll P. J., Picot D., Garavito R. M. (1995) The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat. Struct. Biol. 2, 637–643 [DOI] [PubMed] [Google Scholar]
- 11. Rowlinson S. W., Kiefer J. R., Prusakiewicz J. J., Pawlitz J. L., Kozak K. R., Kalgutkar A. S., Stallings W. C., Kurumbail R. G., Marnett L. J. (2003) A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J. Biol. Chem. 278, 45763–45769 [DOI] [PubMed] [Google Scholar]
- 12. Luong C., Miller A., Barnett J., Chow J., Ramesha C., Browner M. F. (1996) Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 3, 927–933 [DOI] [PubMed] [Google Scholar]
- 13. Prusakiewicz J. J., Duggan K. C., Rouzer C. A., Marnett L. J. (2009) Differential sensitivity and mechanism of inhibition of COX-2 oxygenation of arachidonic acid and 2-arachidonoylglycerol by ibuprofen and mefenamic acid. Biochemistry 48, 7353–7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malkowski M. G., Ginell S. L., Smith W. L., Garavito R. M. (2000) The productive conformation of arachidonic acid bound to prostaglandin synthase. Science 289, 1933–1937 [DOI] [PubMed] [Google Scholar]
- 15. Vecchio A. J., Simmons D. M., Malkowski M. G. (2010) Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 285, 22152–22163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kiefer J. R., Pawlitz J. L., Moreland K. T., Stegeman R. A., Hood W. F., Gierse J. K., Stevens A. M., Goodwin D. C., Rowlinson S. W., Marnett L. J., Stallings W. C., Kurumbail R. G. (2000) Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 405, 97–101 [DOI] [PubMed] [Google Scholar]
- 17. Davies N. M., Skjodt N. M. (1999) Clinical pharmacokinetics of meloxicam: a cyclo-oxygenase-2 preferential nonsteroidal anti-inflammatory drug. Clin. Pharmacokinet. 36, 115–126 [DOI] [PubMed] [Google Scholar]
- 18. Noble S., Balfour J. A. (1996) Meloxicam. Drugs 51, 424–430; discussion 431–432 [DOI] [PubMed] [Google Scholar]
- 19. Fleischmann R., Iqbal I., Slobodin G. (2002) Meloxicam. Expert Opin. Pharmacother. 3, 1501–1512 [DOI] [PubMed] [Google Scholar]
- 20. Farré A. J., Colombo M., Fort M., Gutierrez B., Rodríguez L., Roser R. (1986) Pharmacological properties of droxicam, a new non-steroidal anti-inflammatory agent. Methods. Find. Exp. Clin. Pharmacol. 8, 407–422 [PubMed] [Google Scholar]
- 21. Carty T. J., Marfat A., Moore P. F., Falkner F. C., Twomey T. M., Weissman A. (1993) Ampiroxicam, an anti-inflammatory agent which is a prodrug of piroxicam. Agents Actions 39, 157–165 [DOI] [PubMed] [Google Scholar]
- 22. Ho J., Coote M. L., Franco-Pérez M., Gómez-Balderas R. (2010) First-principles prediction of the pKas of anti-inflammatory oxicams. J. Phys. Chem. A 114, 11992–12003 [DOI] [PubMed] [Google Scholar]
- 23. Park H., Lee S. (2005) Free energy perturbation approach to the critical assessment of selective cyclooxygenase-2 inhibitors. J. Comput. Aided Mol. Des. 19, 17–31 [DOI] [PubMed] [Google Scholar]
- 24. Kothekar V., Sahi S., Srinivasan M., Mohan A., Mishra J. (2001) Recognition of cyclooxygenase-2 (COX-2) active site by NSAIDs: a computer modelling study. Indian J. Biochem. Biophys. 38, 56–63 [PubMed] [Google Scholar]
- 25. Odenwaller R., Chen Y. N., Marnett L. J. (1990) Preparation and proteolytic cleavage of apoprostaglandin endoperoxide synthase. Methods Enzymol. 187, 479–485 [DOI] [PubMed] [Google Scholar]
- 26. Kalgutkar A. S., Kozak K. R., Crews B. C., Hochgesang G. P., Jr., Marnett L. J. (1998) Covalent modification of cyclooxygenase-2 (COX-2) by 2-acetoxyphenyl alkyl sulfides, a new class of selective COX-2 inactivators. J. Med. Chem. 41, 4800–4818 [DOI] [PubMed] [Google Scholar]
- 27. Dong L., Vecchio A. J., Sharma N. P., Jurban B. J., Malkowski M. G., Smith W. L. (2011) Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J. Biol. Chem. 286, 19035–19046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of COOT. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Phenix: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schüttelkopf A. W., van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 33. Winn M. D., Isupov M. N., Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 34. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., Adams P. D. (2008) Iterative model building, structure refinement and density modification with the Phenix AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gupta K., Selinsky B. S., Kaub C. J., Katz A. K., Loll P. J. (2004) The 2.0 Å resolution crystal structure of prostaglandin H-2 synthase-1: structural insights into an unusual peroxidase. J. Mol. Biol. 335, 503–518 [DOI] [PubMed] [Google Scholar]
- 36. Vecchio A. J., Malkowski M. G. (2011) The structural basis of endocannabinoid oxygenation by cyclooxygenase-2. J. Biol. Chem. 286, 20736–20745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vecchio A. J., Orlando B. J., Nandagiri R., Malkowski M. G. (2012) Investigating substrate promiscuity in cyclooxygenase-2: the role of Arg-120 and residues lining the hydrophobic groove. J. Biol. Chem. 287, 24619–24630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Forli S., Olson A. J. (2012) A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J. Med. Chem. 55, 623–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lombardino J. G., Wiseman E. H., McLamore W. M. (1971) Synthesis and antiinflammatory activity of some 3-carboxamides of 2-alkyl-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide. J. Med. Chem. 14, 1171–1175 [DOI] [PubMed] [Google Scholar]
- 40. Binder D., Hromatka O., Geissler F., Schmied K., Noe C. R., Burri K., Pfister R., Strub K., Zeller P. (1987) Analogues and derivatives of tenoxicam. 1. Synthesis and antiinflammatory activity of analogues with different residues on the ring nitrogen and the amide nitrogen. J. Med. Chem. 30, 678–682 [DOI] [PubMed] [Google Scholar]
- 41. Lazer E. S., Miao C. K., Cywin C. L., Sorcek R., Wong H. C., Meng Z., Potocki I., Hoermann M., Snow R. J., Tschantz M. A., Kelly T. A., McNeil D. W., Coutts S. J., Churchill L., Graham A. G., David E., Grob P. M., Engel W., Meier H., Trummlitz G. (1997) Effect of structural modification of enol-carboxamide-type nonsteroidal antiinflammatory drugs on COX-2/COX-1 selectivity. J. Med. Chem. 40, 980–989 [DOI] [PubMed] [Google Scholar]