

Background: The regulatory mechanism of the DLC1 tumor suppressor protein is unclear.
Results: Structure-function analysis revealed determinants for the selectivity, activity, and inhibition of DLC1 RhoGAP function.
Conclusion: p120RasGAP competitively and selectively inhibits DLC1 by targeting its catalytic arginine finger.
Significance: This mechanistic study emphasizes the importance of the functional inter-relationships of GTPase-activating proteins mediating cross-talk between the Ras and Rho pathways.
Keywords: GTPase, Kinetics, Molecular Modeling, SH3 Domains, Structural Biology, Arginine Finger, DLC1, Deleted in Liver Cancer, RhoGAP, p120RasGAP
Abstract
The three deleted in liver cancer genes (DLC1–3) encode Rho-specific GTPase-activating proteins (RhoGAPs). Their expression is frequently silenced in a variety of cancers. The RhoGAP activity, which is required for full DLC-dependent tumor suppressor activity, can be inhibited by the Src homology 3 (SH3) domain of a Ras-specific GAP (p120RasGAP). Here, we comprehensively investigated the molecular mechanism underlying cross-talk between two distinct regulators of small GTP-binding proteins using structural and biochemical methods. We demonstrate that only the SH3 domain of p120 selectively inhibits the RhoGAP activity of all three DLC isoforms as compared with a large set of other representative SH3 or RhoGAP proteins. Structural and mutational analyses provide new insights into a putative interaction mode of the p120 SH3 domain with the DLC1 RhoGAP domain that is atypical and does not follow the classical PXXP-directed interaction. Hence, p120 associates with the DLC1 RhoGAP domain by targeting the catalytic arginine finger and thus by competitively and very potently inhibiting RhoGAP activity. The novel findings of this study shed light on the molecular mechanisms underlying the DLC inhibitory effects of p120 and suggest a functional cross-talk between Ras and Rho proteins at the level of regulatory proteins.
Introduction
The Ras and Rho families of small GTP-binding proteins are key transducers of a variety of cellular processes ranging from reorganization of the cytoskeleton to transcriptional regulation and control of cell growth and survival (1). Loss of the control mechanisms and aberrant activation of Ras and Rho proteins are one of the most common molecular alterations found in cancer cells promoting tumor growth and metastasis (2–5). Ras signaling stimulates diverse pathways and signals toward Rho proteins, which are known to be required for cell transformation by oncogenic Ras (6–8). Emerging evidence suggests that the GTPase-activating proteins (GAPs),5 in particular p120RasGAP (also known as RAS p21 protein activator 1 or RASA1; here called p120) and the Rho-specific p190ARhoGAP (also known as ARHGAP35; here called p190), p200RhoGAP (also known as ARHGAP32, p250GAP, GC-GAP, Rics, or Grit) and deleted in liver cancer 1 (DLC1; also known as ARHGAP7, p122RhoGAP, or STARD12), act as a linker between Ras and Rho signaling pathways (9–11). GAPs are multifaceted and multifunctional molecules (12, 13) and are the principal inactivators of Ras and Rho signaling. They utilize a catalytic “arginine finger” to stimulate the inefficient intrinsic GTP hydrolysis reaction of these small GTP-binding proteins by several orders of magnitude (14).
Frequent loss of DLC1 gene expression was first described in liver cancer (15) and later in breast, colon, gastric, prostate, cervical, esophageal, and other cancers (16–18). DLC1 RhoGAP function is required for the maintenance of cell morphology and the coordination of cell migration (11, 19–21). DLC1 and its isoforms DLC2 (also known as ARHGAP37 or STARD13) and DLC3 (also known as ARHGAP38 or STARD8) consist of an N-terminal sterile α motif (SAM) domain, a central phosphorylation region followed by the catalytic RhoGAP domain, and a C-terminal steroidogenic acute regulatory related lipid transfer (START) domain (see Fig. 1A) (22, 23). The SAM and GAP domains are linked by a serine-containing region, which contains a recognition motif for the phosphoserine/phosphothreonine-binding 14-3-3 adaptor proteins (22). DLC1 has been reported to interact with tensin, talin, focal adhesion kinase, and α-catenin (22, 24–29) and with lipids (30). However, the precise mechanism of DLC1 regulation remains unclear.
FIGURE 1.

Schematic representation of domain organization and designed fragments of GAP (A) and SH3 domain-containing proteins (B) used in this study. The numbers indicate the N and C termini of the amino acids of the respective fragments. BAR, Bin/Amphiphysin/Rvs; C1, cysteine-rich region; CC, coiled coil; DH, Dbl homology domain; FF, double phenylalanine; P, proline-rich; PH, pleckstrin homology; PSET, proline, serine, glutamic acid, and threonine; RGS, regulator of G-protein signaling; Sec14, secretion and cell surface growth 14.
An emerging theme is that RhoGAPs, such as the OPHN1 and GRAF1 (31, 32) and p50RhoGAP (33–36), require activation through the relief of autoinhibitory elements. These elements are collectively membrane-binding modules, including BAR (Bin/Amphiphysin/Rvs), PH (pleckstrin homology), C1, and Sec14 domains (31–33, 36). The SAM domain of DLC1 has been suggested to act as an autoinhibitory domain of DLC1 RhoGAP activity in vitro and in vivo. SAM domain-deleted DLC1 displayed enhanced catalytic activity for RhoA (20). However, it is still unclear how such an autoregulatory mechanism of DLC1 may operate.
p120 contains multiple domains with different functions (see Fig. 1B) (37). Whereas the C terminus of p120 with the catalytic GAP activity is responsible for Ras inactivation (38–40), its N-terminal Src homology 2 and 3 (SH2 and SH3) domains have been suggested to possess an effector function (41–44). p120 functionally modulates Rho signaling by direct binding to two Rho-specific GAPs, p190 and DLC1 (9, 11, 45). The association of p120 with the tyrosine phosphorylated p190 via its SH2 domain promotes Rho inactivation (45–47). Thus, p120 positively regulates the RhoGAP function of p190. Another mechanism, which connects the Ras and Rho pathways and regulates the actin cytoskeleton, is dependent on the p120 SH3 domain and controls Rho activation (41). This mechanism was later revealed to involve DLC1 but not p190 (11). Here, the p120 SH3 domain (called p120SH3) binds to the RhoGAP domain of DLC1 (called DLC1GAP) and inhibits the DLC1-dependent Rho inactivation (11). Hereby, p120 acts as a negative regulator not only for Ras but also for the GAP activity of DLC1. However, the molecular mechanisms underlying these cross-talk phenomena have not yet been elucidated.
In this study, we have explored the regulatory mechanism of DLC1 at the molecular level, in particular its trans-inhibition by p120SH3. We have characterized the selectivity of the interaction between the DLC1GAP and p120SH3 using a large number of purified SH3 and RhoGAP proteins and identified structural and functional determinants for the DLC1-p120 interaction. This study provides deep insights into the underlying regulatory cross-talk between the Rho and Ras family of small GTP-binding proteins.
EXPERIMENTAL PROCEDURES
Constructs
Human AbrGAP (aa 559–822), DLC1fl (aa 1–1091), DLC1GAP (aa 609–878), DLC1SAM (aa 1–96), DLC1START (aa 880–1079), DLC2GAP (aa 644–916), DLC3GAP (aa 620–890), GRAF1GAP (aa 383–583), MgcRacGAP (aa 343–620), NadrinGAP (aa 245–499), OPHN1GAP (aa 375–583), p50GAP (aa 198–439), p190GAP (aa 1250–1513), N-terminal truncated p120Δn128 (aa 129–1047); SH2-SH3-SH2-encoding p120SH2-3-2 (aa 129–447), p120SH3 (aa 275–350), SrcSH3 (aa 77–140), and human RhoA (aa 1–181), Cdc42 (aa 1–178), and Rac1 (aa 1–184) were amplified by standard PCR and cloned in pGEX-4T1 and pGEX-4T1-NTEV, respectively. Constructs of SH3 domain of Crk1SH3 (aa 131–191), Grb2SH3-1 (aa 1–55), Grb2SH3-2 (aa 159–217), Nck1SH3-1 (aa 5–60), Nck1SH3-2 (aa 109–163), and Nck1SH3-3 (aa 173–262) were created as described previously (48).
Site-directed Mutagenesis
Point mutations N311R; L313A; W319G; and N311R,L313A,W319G in p120SH3 and R677A in DLC1GAP were generated using the QuikChangeTM site-directed mutagenesis kit (Stratagene) and confirmed by DNA sequencing.
Proteins
Escherichia coli BL21(DE3) pLysS, BL21(DE3) CodonPlus-RIL, and Rosetta(DE3) strains containing the respective plasmids (see constructs) were grown to an A600 of 0.7 (37 °C at 140 rpm) and induced with 0.1 mm isopropyl β-d-thiogalactopyranoside overnight at 25 °C as described before (49, 50). All proteins were isolated in a first step as glutathione S-transferase (GST) fusion proteins by affinity chromatography on a GSH-agarose column and in a second step by size exclusion chromatography (Superdex S75 or S200) after proteolytic cleavage of GST. GTP-binding proteins without nucleotide (nucleotide-free form) or with tetramethylrhodamine-conjugated GTP (tamraGTP) were prepared as described before (49, 50). Concentrations of proteins were determined by Bradford assay or absorbance at 280 nm using the extinction coefficient deduced from the protein sequence. Purified proteins were snap frozen in liquid nitrogen and stored at −80 °C.
Analytical Size Exclusion Chromatography (aSEC)
aSEC for the detection of complex formation was performed for DLC1GAP and p120SH3 on a Superdex 75 column (10/300) using buffer containing 30 mm HEPES, pH 7.6, 5 mm MgCl2, 150 mm NaCl, and 3 mm DTT. 10 μm DLC1GAP was incubated with 15 μm p120SH3 for 5 min at 4 °C in the same buffer in a total volume of 150 μl. Before loading to an aSEC column, samples were spun at 13,000 rpm at 4 °C to remove any particulate impurities. The flow rate was maintained at 0.5 ml/min, and 500-μl fractions were collected. Peak fractions were visualized by 15% SDS-PAGE and subsequent Coomassie Blue staining.
Kinetics Measurements
All fluorescence measurements were performed at 25 °C in a buffer containing 30 mm Tris-HCl, pH 7.5, 10 mm K2HPO4/KH2PO4, pH 7.4, 10 mm MgCl2, and 3 mm DTT. The tamraGTP hydrolysis of Rho proteins (0.2 μm) was measured in the absence and presence of different amounts of respective GAP proteins as described previously (49, 52). Fast kinetics (<1000 s) were performed with a Hi-Tech Scientific SF-61 stopped-flow instrument with a mercury xenon light source and TgK Scientific Kinetic Studio software (version 2.19). An excitation wavelength of 545 nm was used for tamra. Emission was detected through a cutoff filter of 570 nm. Slow kinetics (>1000 s) were measured on a PerkinElmer Life Sciences spectrofluorometer (LS50B) using an excitation wavelength of 545 nm and an emission wavelength of 583 nm. Data were evaluated by single exponential fitting with the GraFit program to obtain the observed rate constant (kobs) for the respective reaction as described before (49, 52).
Isothermal Titration Calorimetry (ITC) Measurements
The interaction of DLC1GAP and p120SH3 and analysis of DLC1GAP variant and different p120SH3 variants were studied by ITC (MicroCalTM VP-ITC microcalorimeter) as described (48). All measurements were carried out in 30 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm MgCl2, and 1 mm tris(2-carboxyethyl)phosphine hydrochloride. The data were analyzed using Origin 7.0 software provided by the manufacturer.
Structural Analysis
To obtain insight into the residues responsible for the binding of the SH3 domain of p120 and RhoGAP domain of DLC1, docking of their corresponding structures (Protein Data Bank code 2J05 (53) and Protein Data Bank code 3KUQ, respectively), was performed with the program PatchDock (54). From the 20 best scored models, we selected the lowest energy model, which also has the Arg finger Arg-677 at the interface, and used it for further refinement with the program CHARMM (55). As the arginine finger is assumed to be crucial for the formation of the complex, we thoroughly explored its conformation in the course of refinement. Torsion angles of its side chain were additionally set up according to the Dynameomics rotamer library (56), and the energy of each complex was minimized by 2000 steps using the adapted basis Newton-Raphson method.
RESULTS
Low GAP Activities of the DLC Isoforms
Real time kinetic measurements of the RhoGAP activities of the DLC isoforms toward Cdc42, Rac1, and RhoA were performed using purified RhoGAP domains of the DLC proteins (Fig. 1) and fluorescent tamraGTP. This GTP analog is sensitive toward conformational changes induced by GTP hydrolysis (52). As shown in Fig. 2A, the very slow intrinsic tamraGTP hydrolysis of Cdc42 (inset) was markedly increased in the presence of the RhoGAP domain of DLC1 (DLC1GAP). Similar experiments were performed under the same conditions with Rac1 and RhoA (Fig. 2B). Observed rate constants (kobs) of respective DLC1GAP activities are presented in comparison with intrinsic hydrolysis rates as bars in Fig. 2B. DLC1GAP exhibited the highest activity for RhoA (1,650-fold) and Cdc42 (332-fold) and the lowest activity for Rac1 (75-fold). We next focused on the differences among the DLC isoforms and measured the activities of DLC2 and DLC3 for Cdc42 (Fig. 2C). Obtained data show that DLC2 and DLC3 exhibit 78- and 11-fold lower GAP activities, respectively, as compared with that of DLC1. Our results indicate that the DLC family members are inefficient GAPs, at least in vitro, with catalytic activities that are several orders of magnitude lower than the activities of the RhoGAPs p50 and p190 (Fig. 2C) or other highly efficient RhoGAPs, such as GRAF1 or OPHN1 (32).
FIGURE 2.
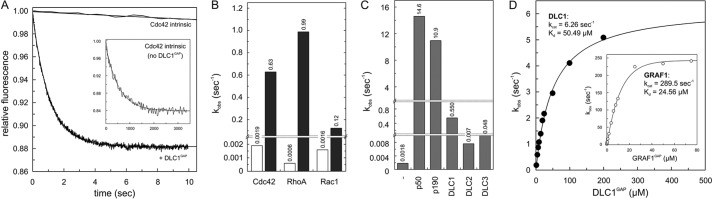
Inefficient GAP activities of the DLC isoforms. A, Cdc42-tamraGTP (0.2 μm) was rapidly mixed with 5 μm DLC1GAP to monitor the GAP-stimulated tamraGTP hydrolysis reaction of Cdc42 in real time. Note the very slow intrinsic GTPase reaction of Cdc42 (inset) that was measured in the absence of GAP. Rate constants (kobs) were obtained by single exponential fitting of the data. B, the kobs values of GTP hydrolysis of Rho proteins (0.2 μm) measured in the presence of DLC1GAP (5 μm) are represented as a column chart. Calculated -fold activation values were obtained by dividing the kobs values of GAP-stimulated reactions by the kobs values of the intrinsic reactions of respective GTPases. For convenience, the kobs values are given above the bar charts. C, measured GAP activities of DLC1, DLC2, and DLC3 (5 μm, respectively) toward Cdc42 (0.2 μm) were very low as compared with p150 and p190. D, the GTP hydrolysis of Cdc42 (0.2 μm) was measured in the presence of increasing concentrations of the respective GAP domains of DLC1 and GRAF1 (inset). The dependence of the kobs values of the GAP-stimulated GTP hydrolysis plotted on the concentrations of DLC1GAP and GRAF1 was fitted by a hyperbolic curve to obtain the kinetic parameters (kcat and Kd).
A comparison of the obtained data on the DLC isoforms with those of other RhoGAP family members raised the question of whether the extremely low GAP activities of DLC proteins stem from effects on either binding affinity (Kd) or catalytic activity (kcat). Therefore, we measured the kinetics of tamraGTP hydrolysis of Cdc42 at increasing concentrations of DLC1GAP and GRAF1GAP. The rate constants (kobs) of the fitted single exponential decays increased in a hyperbolic manner as a function of GAP concentrations as described previously (52, 57). We used Cdc42 in most experiments because of a large change in fluorescence upon tamraGTP hydrolysis as compared with Rac1 and RhoA. Fitting a hyperbolic curve to the points according to Equation 1 led to the corresponding kinetic parameters Kd and kcat (Fig. 2D).
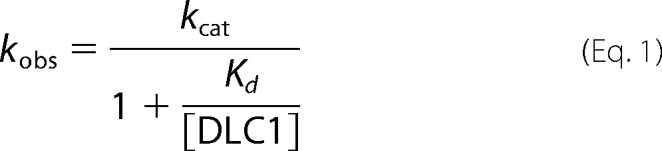 |
Unlike the relatively similar Kd values, there was a large difference in the kcat values for the GTP hydrolysis reaction: 6.26 s−1 for DLC1GAPcompared with 289 s−1 for the highly efficient GRAF1GAP. These data clearly indicate that the very low GAP activity of the DLC proteins relies more on the catalytic activity than on the binding affinity to Cdc42.
Insights into cis-Regulatory Modules of DLC1 Function
To examine the influence of other domains of DLC1 (Fig. 1A) on its GAP activity, we further measured tamraGTP hydrolysis of Cdc42 stimulated by full-length DLC1 (DLC1fl). As shown in Fig. 3A, DLC1fl exhibited a strongly reduced GAP activity as compared with the isolated DLC1GAP. The kobs values obtained from single turnover kinetic data were 0.02 and 0.47 s−1, respectively, and reveal that the DLC1fl activity was 23.5-fold lower than that of DLC1GAP (Fig. 3B). This result strongly supports the previous notion that other regions of DLC1, such as the SAM domain (20), may undergo an intramolecular interaction with the GAP domain and thus contribute to its autoinhibition in a cis-inhibitory manner.
FIGURE 3.

cis-Acting regulation of DLC1GAP activity. A, kinetics of the tamraGTP hydrolysis reaction of Cdc42 (0.2 μm) stimulated by DLC1fl (5 μm) was much slower (inset) than that stimulated by DLCGAP (5 μm). B, the kobs values, illustrated as a bar chart, showed that the GAP activity of DLC1fl is reduced by 23.5-fold as compared with that of the DLC1GAP but not completely inhibited as compared with the intrinsic GTPase reaction. For convenience, the kobs values are given above the bar charts. C, the activity of DLC1GAP (10 μm) on tamraGTP hydrolysis of Cdc42 (0.2 μm) was not significantly changed in the presence of a 100-fold excess of SAM, START, or both domains (1 mm, respectively).
To analyze whether the autoinhibitory effect is caused by N- and C-terminal SAM and/or START domains of DLC1 (Fig. 1A), we purified these domains and measured their effects on the DLC1GAP activity in vitro. Using high concentrations of SAM, START, or both (up to a 100-fold molar excess above the GAP domain), we did not observe any significant inhibition of the DLC1GAP activity using tamraGTP hydrolysis of Cdc42 (Fig. 3C). The fact that the isolated SAM and START domains did not reveal any GAP-inhibitory activity strongly suggests that the autoinhibitory mechanism of DLC1 may require additional regions of the full-length protein. One possibility is the serine-rich 14-3-3 binding region between the SAM and the GAP domains (Fig. 1A).
p120 SH3 as a Potent trans-Inhibitory Factor of the DLC1GAP Activity
The SH3 domain of p120 has been reported as a novel binding partner of DLC1 with GAP-inhibitory and growth suppression activity (11). To monitor this effect in real time, DLC1GAP activity was measured in the absence and presence of purified p120SH3 under the same conditions as in the experiments described above (Fig. 2). As shown in Fig. 4A, DLC1GAP-stimulated tamraGTP hydrolysis of Cdc42 was drastically reduced using a 10-fold excess of p120SH3 over the DLC1GAP concentration. The respective kobs value of 0.63 for DLC1GAP activity was reduced by 83-fold in the presence of p120SH3 to 0.0076 s−1 (Fig. 4B), which is close to the intrinsic tamraGTP hydrolysis of Cdc42 (0.02 s−1). These measurements were also performed for RhoA and Rac1 using the same conditions as for Cdc42 (Fig. 4B). Similarly, 247- and 15.5-fold reductions of the DLC1GAP activity for RhoA and Rac1, respectively, were determined in the presence of a 10-fold molar excess of p120SH3. An explanation for this large variation may be the significant differences in DLC1GAP binding affinity for the three members of the Rho family.
FIGURE 4.

p120SH3 as a potent inhibitor of the DLC GAP function. A, kinetics of the tamraGTP hydrolysis reaction of Cdc42 (0.2 μm) stimulated by DLC1GAP (5 μm) was reduced in the presence of a 10-fold excess of p120SH3 (50 μm). The complete reaction is shown in the inset. B, DLC1GAP activities toward Cdc42, RhoA, and Rac1, measured under the same conditions as in A, are strongly inhibited by p120SH3. For convenience, the kobs values are given above the bar charts. C, DLC3GAP (5 μm) was not inhibited by p120SH3 (50 and 500 μm) as efficiently as DLC1GAP and DLC2GAP (5 μm, respectively). D, p120SH2-3-2 and p120Δn128 (40 μm) inhibited the activity of DLCGAP (10 μm) but not as efficiently as p120SH3 (40 μm).
In the next step, we analyzed the inhibitory effect of p120SH3 on the GAP activity of DLC2 and DLC3 toward Cdc42. Fig. 4C shows that the catalytic GAP activity of purified DLC2GAP and DLC3GAP was also inhibited in the presence of p120SH3 but not as drastically as in the case of DLC1GAP. The next question we addressed was whether the SH3 domain is freely accessible to exert its inhibitory effect or whether other domains of p120 also play a role in the inhibition of DLC GAP activity (Fig. 1). Therefore, we purified the SH2-SH3-SH2-encompassing p120SH2-3-2 and N-terminal truncated p120Δn128 proteins and analyzed their DLC1GAP inhibitory effects in direct comparison with isolated p120SH3. Larger p120 fragments inhibited the DLC1GAP activity but to a 19- and 10-fold lower extent than p120SH3 (Fig. 4D).
Taken together, our in vitro data demonstrate that (i) p120SH3 acts as a potent trans-inhibitory factor of the GAP activity of the DLC isoforms and (ii) the SH3 domain of p120 is not completely unmasked (freely accessible) in the presence of other p120 domains, especially the adjacent SH2 domains. Whether the N-terminal 128 amino acids play a role in this regard remains unclear. Full-length p120 could not be purified due to its instability.
Highly Selective Interaction between p120SH3 and DLC1GAP
The next issue we addressed was the selectivity of the p120SH3 toward DLC1GAP. Therefore, we purified seven additional RhoGAP and SH3 domains of other proteins (Fig. 1). We measured the effect of p120SH3 on the GAP activity of Abr, GRAF1, MgcRacGAP, Nadrin, OPHN1, p50, and p190 on the one hand and the effects of the SH3 domains of Crk1, c-Src, Grb2 (N- and C-terminal SH3 domains), and Nck1 (all three SH3 domains) on the DLC1GAP activity on the other hand. As summarized in Fig. 5, neither did p120SH3 inhibit the activity of other GAPs of the Rho family (Fig. 5A) nor was the DLC1GAP activity affected by the presence of other SH3 domains (Fig. 5B). These data clearly demonstrate that the p120SH3-mediated trans-inhibition of DLC isoforms is highly selective.
FIGURE 5.

Highly selective interaction between p120SH3 and DLC1GAP. A, p120SH3-inhibiting effect on seven additional RhoGAPs (2 μm, respectively) was measured using the tamraGTP hydrolysis reaction of Cdc42 (0.2 μm) and p120SH3 (20 and 200 μm, respectively). p120SH3 inhibited only DLC1GAP but not the other RhoGAPs. For convenience, the kobs values are given above the bar charts. B, the effect of seven additional SH3 proteins (100 μm, respectively) on inhibiting DLC1GAP (10 μm) was measured. Only p120SH3 inhibited DLC1GAP but not the other SH3 domains.
Potent DLC1 Inhibition Due to High Affinity p120SH3-DLC1GAP Complex Formation
In the next step, we characterized in more detail the interaction between p120SH3 and DLC1GAP as well as the inhibition of the DLC1GAP activity induced by p120SH3 using different qualitative and quantitative biophysical and biochemical methods. aSEC is an accurate and simple method to visualize high affinity protein-protein interactions. p120SH3 (9 kDa) and DLC1GAP (31 kDa) alone and a mixture of both proteins were loaded on a Superdex 75 (10/300) column, and eluted peak fractions were analyzed by SDS-PAGE. Data summarized in Fig. 6A clearly illustrate that a mixture of p120SH3 and DLC1GAP shift the elution profile of the respective protein domains to an elution volume of 10.5 ml, indicating the formation of a complex between both proteins. We next determined the inhibitory potency of p120SH3 by measuring DLC1GAP activity at increasing concentrations of p120SH3. An inhibitory constant (Ki) of 0.61 μm was calculated by fitting the Morrison equation for a tight binding inhibitor (58) to individual kobs values plotted against different p120SH3 concentrations (Fig. 6B). Furthermore, we measured the dissociation constant of the p120SH3-DLC1GAP interaction using ITC. The results shown in Fig. 6C allowed the determination of a stoichiometry of 1:1 and a dissociation constant (Kd) of 0.6 μm for the binding of p120SH3 to DLC1GAP (Fig. 6C); this value nicely resembles the Ki value obtained from inhibition kinetics (Fig. 6B). This binding affinity is remarkably high and unexpected considering the low micromolar range affinities of SH3 domains for their PXXP-containing proteins (59). Taken together, these data strongly suggest that the mode of the p120SH3-DLC1GAP interaction most likely differs from the conventional SH3 interaction with PXXP loop motifs as recently published (48).
FIGURE 6.

High affinity interaction between p120SH3 and DLC1GAP. A, co-elution of a mixture of DLC1GAP (10 μm) and p120SH3 (15 μm) (open circles) from a Superdex 75 (10/300) as shown by SDS-PAGE (15%) and Coomassie Brilliant Blue (CBB) staining (inset) indicates their complex formation. B, the activity of DLC1GAP (20 μm) toward Cdc42 (0.2 μm) was measured at increasing concentrations of p120SH3, and the obtained kobs values were plotted against increasing concentrations of the inhibitor p120SH3. The Ki value was obtained by non-linear regression based on the Morrison equation for tight binding inhibitors (58). C, ITC analysis was performed by titrating DLC1GAP (20 μm) with p120SH3 (400 μm). Kd is the dissociation constant, and n is the stoichiometry.
Structural Insight into a Putative Binding Mode between p120SH3 and DLC1GAP
The high nanomolar affinity of p120SH3 for DLC1GAP and the absence of a PXXP motif in DLC1GAP strongly support the notion that the p120SH3-DLC1GAP interaction is mediated via a novel binding mechanism. To gain insight into the structural basis of this interaction, we first performed protein-protein docking of available crystal structures of p120SH3 (Protein Data Bank code 2J05) (53) and DLC1GAP (Protein Data Bank code 3KUQ) using the PatchDock program (54). The model of the complex ranked as the first among 20 resulting models fulfilled the criteria for a close proximity of p120SH3 to the catalytic arginine finger (Arg-677) of the DLC1GAP domain and was thus selected for refinement by molecular modeling methods. Inspecting the refined model, we identified three potential DLC1GAP binding residues of p120SH3 (Asn-311, Leu-313, and Trp-319) that were closest to the catalytic Arg-677 of DLC1GAP (Fig. 7A). We proposed that mutation of these residues may impair binding of the SH3 domain, which otherwise masks the arginine finger of DLC1GAP. Catalytic arginine is known to stabilize the transition intermediate state of the hydrolysis reaction in the active center of Rho proteins (Fig. 7B) (14, 60). This assumption also suggests that p120 competitively inhibits DLC1 GAP function.
FIGURE 7.
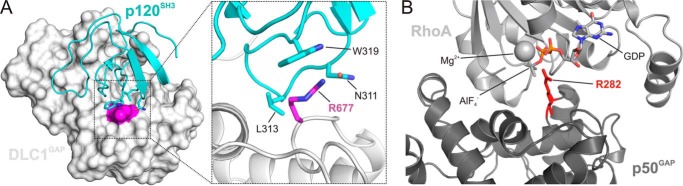
Structural insight into a putative binding mode between p120SH3 and DLC1GAP. A, molecular docking analyses were performed between the available crystal structures of p120SH3 (Protein Data Bank code 2J05) (53) and DLC1GAP (Protein Data Bank code 3KUQ) using the program PatchDock (54). In the best ranked and refined model, p120SH3 was located in close proximity of the catalytic arginine finger (Arg-677; magenta) of DLC1GAP. In this model, p120SH3 supplied three amino acids (Asn-311, Leu-313, and Trp-319) to directly contact the catalytic core of DLC1GAP, especially Arg-677, and mask its accessibility to the Rho proteins. B, p50GAP provides an arginine finger (Arg-282; red) in the active site of RhoA to stabilize the transition state of the GTP hydrolysis reaction (Protein Data Bank code 1TX4) (60). GDP-AlF4− mimics the transition state of the GTP hydrolysis reaction.
To validate our assumption, we performed mutational analysis of the above mentioned key residues at the p120SH3-DLC1GAP interface: N311R, L313A, and W319G in p120SH3 (single, double, and triple single point mutations) and R677A in DLC1GAP. Expectedly, DLC1GAP with the catalytic arginine finger substituted to alanine was deficient in stimulating tamraGTP hydrolysis of Cdc42 (data not shown) and most remarkably in associating with p120SH3 (Fig. 8, A and B). The latter was examined using two independent methods, ITC and aSEC. Reciprocally, p120SH3(N311R,L313A,W319G) was almost disabled in inhibiting DLC1GAP activity (Fig. 8E), most probably due to its inability to bind to DLC1GAP (Fig. 8, C and D). The analysis of the single point mutations revealed that W319G substitution had a minor effect on the association with (data not shown) and on the inhibition of DLCGAP (Fig. 8E). p120SH3(N311R,L313A) on the other hand significantly abolished both the inhibitory effect of p120SH3 (Fig. 8E) and the complex formation with DLC1GAP (data not shown) as compared with wild-type p120SH3. Taken together, our mutational and biochemical analyses support the in silico structural model (Fig. 7A) and provide new insight into how p120SH3 may bind and inhibit the catalytic activity of DLC1GAP.
FIGURE 8.

Loss of p120-DLC1 interaction by mutational analysis. No interaction was observed between DLC1GAP(R677A) and p120SH3(WT) (A and B) and DLC1GAP(WT) and p120SH3(N311R,L313A,W319G) (C and D). Loss of interaction and of inhibition was measured by ITC (A and C) and aSEC (B and D) as compared with the p120SH3(WT)-DLC1GAP(WT) interaction shown in Fig. 6. E, the activity of DLC1GAP (25 μm) in stimulating tamraGTP hydrolysis of Cdc42 (0.2 μm) was measured in the presence of p120SH3 variants (125 μm), respectively. For convenience, the kobs values are given above the bar charts.
DISCUSSION
In this study, we have elucidated the molecular mechanism of how the RasGAP p120 selectively acts as a negative regulator of the RhoGAP activity of DLC1. We have shown that p120SH3, by utilizing a novel binding mode, selectively undergoes a high affinity interaction with the RhoGAP domain of DLC1 and effectively inhibits its GAP activity by targeting its catalytic arginine finger. Interestingly, p120SH3 acts on the DLC isoforms but not on seven other representative members of the RhoGAP family. Our data together support the notion of a functional cross-talk between Ras and Rho proteins at the level of regulatory proteins (11, 45).
In contrast to the molecular mechanism of Rho protein inactivation by GAPs, which is well established (14, 61), it is still unclear how GAPs themselves are regulated. Different mechanisms are implicated in the regulation of GAPs, such as regulation by protein phosphorylation, proteolytic degradation, intramolecular autoinhibition, and changes in subcellular localization or protein complex formation (62, 63). “Intramolecular inhibition” (also called “autoinhibition,” “cis-inhibition,” “autoinhibitory loop,” “autoregulation,” and “bistable switch”) of biological molecules is a fundamental control mechanism in nature and is an emerging theme in the regulation of different kinds of proteins, including the regulators of small GTP-binding proteins themselves. Besides the guanine nucleotide exchange factors (64–69), GAPs also have been reported to require activation through the relief of autoinhibitory elements (20, 31–33, 35, 36). Kim et al. (20) have shown that DLC1fl has a reduced GAP activity and have proposed that the N-terminal SAM domain may be a cis-inhibitory element contributing to DLC1 autoinhibition. Our real time kinetic experiments, however, have shown that neither isolated SAM or START alone nor both domains in combination are directly responsible for the observed DLC1fl autoinhibition in a cell-free system (Fig. 3). Taken together, it rather seems plausible that other regions, probably together with SAM and START domains, are involved in the autoinhibition of DLC1. In addition, it is important to note that release of the autoinhibitory loop of DLC1 is most likely subjected to posttranslational modifications (21, 70) and interactions with other proteins (16, 28, 34) along with changes in subcellular localization (30), collectively contribute to the regulation of DLC1 GAP activity in intact cells. In this context, PKD-mediated phosphorylation (70) and 14-3-3 binding and cytosolic sequestration (22) are good examples for the regulation DLC1 function.
Functional characterization and structural elucidation of the trans-inhibitory mechanism of DLC1 mediated by the Ras-specific GAP p120 protein (11) was the central theme of this study. Our data clearly revealed that the GAP activity of not only DLC1 but also that of DLC2 and DLC3 was almost completely abolished in the presence of the SH3 domain of p120 (Fig. 4). We showed that larger fragments of p120, such as p120SH2-3-2 and the almost full-length p120Δn128, inhibit the DLC GAP function but strikingly not to the same extent as seen for the isolated SH3 domain (Fig. 4D). These data indicate that only a freely accessible and exposed SH3 domain of p120, most probably following an upstream signal and in a defined subcellular environment (11, 37), is able to potently inhibit DLC proteins. One of the p120 binding partners is p190, which has been proposed to induce a conformational change in p120 by binding to its SH2 domains and exposing the adjacent SH3 domain for additional protein interactions with additional proteins (47), one of which is most likely DLC1.
Several studies have shown that DLC1 is able to inactivate Cdc42 and the Rho isoforms (RhoA, RhoB, and RhoC) but not Rac1 in vitro (20, 71–73). DLC1GAP activity toward other members of the Rho family has not yet been published. Our preliminary data showed that the DLC proteins are active in vitro on almost all members of the Rho family that are able to hydrolyze GTP.6
Chan et al. (74) have shown an increased level of RhoA-GTP in DLC2-null mice but not in samples from control mice. Consistently, the overexpression of DLC isoforms has been shown to lead to inactivation of RhoA and to the reduction of actin stress fiber formation (75, 76), suggesting that DLC proteins are Rho-selective GAPs and the role of the DLC trans-inhibitory protein p120 is to retain Rho proteins in their active GTP-bound states. Contrary to DLC proteins, p120 binding is part of the p190 activation process that controls inactivation of Rho-type proteins (45, 47, 77). A prerequisite for this interaction is phosphorylation of p190 at tyrosine 1105, which is a target of the p120 SH2 domains (77). In this regard, p120 oppositely controls the activities of two different RhoGAPs and obviously two different Rho/Rho effector systems; one is left activated, and the other is switched off.
SH3 domain-containing cellular signaling proteins mediate interactions via specific proline-containing peptides. The SH3 domain of p120 has been discussed recently to interact with other proteins in a PXXP motif-independent manner (48). In silico analysis revealed that the GAP domain of DLC1 does not possess a proline-rich region and therefore, unlike classical PXXP motif-recognizing SH3 domains, the interaction mode of the p120 SH3 domain is atypical and utilizes different amino acids to bind and mask the catalytic arginine finger of the GAP domain of DLC1. The Ser/Thr kinases Aurora A and Aurora B are other examples in addition to DLC1 for negative modulation of biological processes by p120 (78). The SH3 domain of p120 binds to the catalytic domain of Aurora kinases that inhibits their kinase activity. These interactions also do not involve a proline-rich consensus sequence. Two accessible hydrophobic regions of p120 SH3 have been suggested to function as binding sites for protein interaction (79). Our study supports this notion as we have shown that mutation of three amino acids close to one of these proposed binding sites indeed diminished the DLC1GAP binding and inhibiting ability of p120 SH3.
We demonstrated that the interaction between p120SH3 and DLC1GAP displays at least three remarkable characteristics, namely high affinity, high selectively, and a non-canonical binding mode. The high affinity interaction of 0.6 μm is striking because the binding constants of SH3 domains for proline-rich motifs in their target proteins are mostly in the micromolar range (48, 59). The very few examples of high affinity binding of SH3 domains are those between Mona/Gads and SLP-76 (80), C3G and c-Crk (51), and Grb2 and Wrch1 (48).
CONCLUSION
Mechanistic and structural insights into selectivity, activity, and regulation of DLC1 presented in this study shed light on the role of the multifunctional, regulatory signaling molecule p120RasGAP. It is evident that p120 acts in addition to its RasGAP domain, which is required to switch off Ras signal transduction, as an “effector” conversely controlling, via its SH2 domains and a non-canonical SH3 domain, the RhoGAP activities of the DLC and p190 proteins and hence Rho signal transduction. Interestingly, p120 interacts, in addition to DLC1 and p190, with a third RhoGAP, called p200RhoGAP. In contrast to p190 and DLC1, which are downstream of p120, p200RhoGAP has been proposed to bind to the p120 SH3 domain via its very C-terminal proline-rich region and to sequestrate its RasGAP function from inactivating Ras (10). These examples nicely illustrate the interdependence of the Ras and Rho signaling pathways and underline the multifunctional and multifaceted nature of regulatory proteins beyond their critical GAP functions.
Acknowledgments
We thank Linda van Aelst, Katrin Rittinger, Olivier Dorseui, Gerard Gacon, Alan Hall, Tony Pawson, and Jeffrey Settleman for sharing reagents with us that proved indispensable for the work.
This work was supported in part by the German Research Foundation (Deutsche Forschungsgemeinschaft (DFG)) through the Collaborative Research Center 974 (SFB 974) “Communication and Systems Relevance during Liver Injury and Regeneration,” the International Research Training Group 1902 (IRGT1902), and Project Grant AH 92/5-1; the National Genome Research Network Plus program of the German Ministry of Science and Education (Bundesministerium für Bildung und Forschung Grant 01GS08100); and the International North Rhine-Westphalia Research School BioStruct granted by the Ministry of Innovation, Science and Research of the State North Rhine-Westphalia, the Heinrich Heine University of Düsseldorf, and the Entrepreneur Foundation at the Heinrich Heine University of Düsseldorf.
M. Jaiswal, E. Amin, and R. Dvorsky, unpublished data.
- GAP
- GTPase-activating protein
- DLC
- deleted in liver cancer
- SH
- Src homology
- SAM
- sterile α motif
- START
- steroidogenic acute regulatory related lipid transfer
- aa
- amino acids
- tamra
- tetramethylrhodamine
- aSEC
- analytical size exclusion chromatography
- ITC
- isothermal titration calorimetry.
REFERENCES
- 1. Aznar S., Lacal J. C. (2001) Searching new targets for anticancer drug design: the families of Ras and Rho GTPases and their effectors. Prog. Nucleic Acid Res. Mol. Biol. 67, 193–234 [DOI] [PubMed] [Google Scholar]
- 2. Fritz G., Just I., Kaina B. (1999) Rho GTPases are over-expressed in human tumors. Int. J. Cancer 81, 682–687 [DOI] [PubMed] [Google Scholar]
- 3. Pruitt K., Der C. J. (2001) Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett. 171, 1–10 [DOI] [PubMed] [Google Scholar]
- 4. Zondag G. C., Evers E. E., ten Klooster J. P., Janssen L., van der Kammen R. A., Collard J. G. (2000) Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial-mesenchymal transition. J. Cell Biol. 149, 775–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sahai E., Marshall C. J. (2002) RHO-GTPases and cancer. Nat. Rev. Cancer 2, 133–142 [DOI] [PubMed] [Google Scholar]
- 6. Khosravi-Far R., Campbell S., Rossman K. L., Der C. J. (1998) Increasing complexity of Ras signal transduction: involvement of Rho family proteins. Adv. Cancer Res. 72, 57–107 [DOI] [PubMed] [Google Scholar]
- 7. Coleman M. L., Marshall C. J., Olson M. F. (2004) RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat. Rev. Mol. Cell Biol. 5, 355–366 [DOI] [PubMed] [Google Scholar]
- 8. Karnoub A. E., Weinberg R. A. (2008) Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 9, 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Asnaghi L., Vass W. C., Quadri R., Day P. M., Qian X., Braverman R., Papageorge A. G., Lowy D. R. (2010) E-cadherin negatively regulates neoplastic growth in non-small cell lung cancer: role of Rho GTPases. Oncogene 29, 2760–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shang X., Moon S. Y., Zheng Y. (2007) p200 RhoGAP promotes cell proliferation by mediating cross-talk between Ras and Rho signaling pathways. J. Biol. Chem. 282, 8801–8811 [DOI] [PubMed] [Google Scholar]
- 11. Yang X. Y., Guan M., Vigil D., Der C. J., Lowy D. R., Popescu N. C. (2009) p120Ras-GAP binds the DLC1 Rho-GAP tumor suppressor protein and inhibits its RhoA GTPase and growth-suppressing activities. Oncogene 28, 1401–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scheffzek K., Ahmadian M. R. (2005) GTPase activating proteins: structural and functional insights 18 years after discovery. Cell. Mol. Life Sci. 62, 3014–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ligeti E., Welti S., Scheffzek K. (2012) Inhibition and termination of physiological responses by GTPase activating proteins. Physiol. Rev. 92, 237–272 [DOI] [PubMed] [Google Scholar]
- 14. Scheffzek K., Ahmadian M. R., Wittinghofer A. (1998) GTPase-activating proteins: helping hands to complement an active site. Trends Biochem. Sci. 23, 257–262 [DOI] [PubMed] [Google Scholar]
- 15. Yuan B. Z., Miller M. J., Keck C. L., Zimonjic D. B., Thorgeirsson S. S., Popescu N. C. (1998) Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 58, 2196–2199 [PubMed] [Google Scholar]
- 16. Durkin M. E., Yuan B. Z., Zhou X., Zimonjic D. B., Lowy D. R., Thorgeirsson S. S., Popescu N. C. (2007) DLC-1: a Rho GTPase-activating protein and tumour suppressor. J. Cell. Mol. Med. 11, 1185–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liao Y. C., Lo S. H. (2008) Deleted in liver cancer-1 (DLC-1): a tumor suppressor not just for liver. Int. J. Biochem. Cell Biol. 40, 843–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zimonjic D. B., Popescu N. C. (2012) Role of DLC1 tumor suppressor gene and MYC oncogene in pathogenesis of human hepatocellular carcinoma: potential prospects for combined targeted therapeutics (review). Int. J. Oncol. 41, 393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holeiter G., Heering J., Erlmann P., Schmid S., Jähne R., Olayioye M. A. (2008) Deleted in liver cancer 1 controls cell migration through a Dia1-dependent signaling pathway. Cancer Res. 68, 8743–8751 [DOI] [PubMed] [Google Scholar]
- 20. Kim T. Y., Healy K. D., Der C. J., Sciaky N., Bang Y. J., Juliano R. L. (2008) Effects of structure of Rho GTPase-activating protein DLC-1 on cell morphology and migration. J. Biol. Chem. 283, 32762–32770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim T. Y., Vigil D., Der C. J., Juliano R. L. (2009) Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer Metastasis Rev. 28, 77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scholz R. P., Regner J., Theil A., Erlmann P., Holeiter G., Jähne R., Schmid S., Hausser A., Olayioye M. A. (2009) DLC1 interacts with 14-3-3 proteins to inhibit RhoGAP activity and block nucleocytoplasmic shuttling. J. Cell Sci. 122, 92–102 [DOI] [PubMed] [Google Scholar]
- 23. Lukasik D., Wilczek E., Wasiutynski A., Gornicka B. (2011) Deleted in liver cancer protein family in human malignancies (review). Oncol. Lett. 2, 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yam J. W., Ko F. C., Chan C. Y., Jin D. Y., Ng I. O. (2006) Interaction of deleted in liver cancer 1 with tensin2 in caveolae and implications in tumor suppression. Cancer Res. 66, 8367–8372 [DOI] [PubMed] [Google Scholar]
- 25. Liao Y. C., Si L., deVere White R. W., Lo S. H. (2007) The phosphotyrosine-independent interaction of DLC-1 and the SH2 domain of cten regulates focal adhesion localization and growth suppression activity of DLC-1. J. Cell Biol. 176, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qian X., Li G., Asmussen H. K., Asnaghi L., Vass W. C., Braverman R., Yamada K. M., Popescu N. C., Papageorge A. G., Lowy D. R. (2007) Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc. Natl. Acad. Sci. U.S.A. 104, 9012–9017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan L. K., Ko F. C., Ng I. O., Yam J. W. (2009) Deleted in liver cancer 1 (DLC1) utilizes a novel binding site for Tensin2 PTB domain interaction and is required for tumor-suppressive function. PLoS One 4, e5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tripathi V., Popescu N. C., Zimonjic D. B. (2013) DLC1 induces expression of E-cadherin in prostate cancer cells through Rho pathway and suppresses invasion. Oncogene 4, 1–10 [DOI] [PubMed] [Google Scholar]
- 29. Li G., Du X., Vass W. C., Papageorge A. G., Lowy D. R., Qian X. (2011) Full activity of the deleted in liver cancer 1 (DLC1) tumor suppressor depends on an LD-like motif that binds talin and focal adhesion kinase (FAK). Proc. Natl. Acad. Sci. U.S.A. 108, 17129–17134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Erlmann P., Schmid S., Horenkamp F. A., Geyer M., Pomorski T. G., Olayioye M. A. (2009) DLC1 activation requires lipid interaction through a polybasic region preceding the RhoGAP domain. Mol. Biol. Cell 20, 4400–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fauchereau F., Herbrand U., Chafey P., Eberth A., Koulakoff A., Vinet M. C., Ahmadian M. R., Chelly J., Billuart P. (2003) The RhoGAP activity of OPHN1, a new F-actin-binding protein, is negatively controlled by its amino-terminal domain. Mol. Cell. Neurosci. 23, 574–586 [DOI] [PubMed] [Google Scholar]
- 32. Eberth A., Ahmadian M. R. (2009) In vitro GEF and GAP assays. Curr. Protoc. Cell Biol. Chapter 14, Unit 14.19 [DOI] [PubMed] [Google Scholar]
- 33. Colón-González F., Leskow F. C., Kazanietz M. G. (2008) Identification of an autoinhibitory mechanism that restricts C1 domain-mediated activation of the Rac-GAP α2-chimaerin. J. Biol. Chem. 283, 35247–35257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jian X., Brown P., Schuck P., Gruschus J. M., Balbo A., Hinshaw J. E., Randazzo P. A. (2009) Autoinhibition of Arf GTPase-activating protein activity by the BAR domain in ASAP1. J. Biol. Chem. 284, 1652–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou Y. T., Chew L. L., Lin S. C., Low B. C. (2010) The BNIP-2 and Cdc42GAP homology (BCH) domain of p50RhoGAP/Cdc42GAP sequesters RhoA from inactivation by the adjacent GTPase-activating protein domain. Mol. Biol. Cell 21, 3232–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moskwa P., Paclet M. H., Dagher M. C., Ligeti E. (2005) Autoinhibition of p50 Rho GTPase-activating protein (GAP) is released by prenylated small GTPases. J. Biol. Chem. 280, 6716–6720 [DOI] [PubMed] [Google Scholar]
- 37. Pamonsinlapatham P., Hadj-Slimane R., Lepelletier Y., Allain B., Toccafondi M., Garbay C., Raynaud F. (2009) p120-Ras GTPase activating protein (RasGAP): a multi-interacting protein in downstream signaling. Biochimie 91, 320–328 [DOI] [PubMed] [Google Scholar]
- 38. Ahmadian M. R., Hoffmann U., Goody R. S., Wittinghofer A. (1997) Individual rate constants for the interaction of Ras proteins with GTPase-activating proteins determined by fluorescence spectroscopy. Biochemistry 36, 4535–4541 [DOI] [PubMed] [Google Scholar]
- 39. Ahmadian M. R., Stege P., Scheffzek K., Wittinghofer A. (1997) Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat. Struct. Biol. 4, 686–689 [DOI] [PubMed] [Google Scholar]
- 40. Scheffzek K., Ahmadian M. R., Kabsch W., Wiesmüller L., Lautwein A., Schmitz F., Wittinghofer A. (1997) The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277, 333–338 [DOI] [PubMed] [Google Scholar]
- 41. Leblanc V., Tocque B., Delumeau I. (1998) Ras-GAP controls Rho-mediated cytoskeletal reorganization through its SH3 domain. Mol. Cell. Biol. 18, 5567–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chan P. C., Chen H. C. (2012) p120RasGAP-mediated activation of c-Src is critical for oncogenic Ras to induce tumor invasion. Cancer Res. 72, 2405–2415 [DOI] [PubMed] [Google Scholar]
- 43. Clark G. J., Der C. J. (1995) Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res. Treat. 35, 133–144 [DOI] [PubMed] [Google Scholar]
- 44. Clark G. J., Westwick J. K., Der C. J. (1997) p120 GAP modulates Ras activation of Jun kinases and transformation. J. Biol. Chem. 272, 1677–1681 [DOI] [PubMed] [Google Scholar]
- 45. Herbrand U., Ahmadian M. R. (2006) p190-RhoGAP as an integral component of the Tiam1/Rac1-induced downregulation of Rho. Biol. Chem. 387, 311–317 [DOI] [PubMed] [Google Scholar]
- 46. Wang Z., Tung P. S., Moran M. F. (1996) Association of p120 ras GAP with endocytic components and colocalization with epidermal growth factor (EGF) receptor in response to EGF stimulation. Cell Growth Differ. 7, 123–133 [PubMed] [Google Scholar]
- 47. Hu K. Q., Settleman J. (1997) Tandem SH2 binding sites mediate the RasGAP-RhoGAP interaction: a conformational mechanism for SH3 domain regulation. EMBO J. 16, 473–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Risse S. L., Vaz B., Burton M. F., Aspenström P., Piekorz R. P., Brunsveld L., Ahmadian M. R. (2013) SH3-mediated targeting of Wrch1/RhoU by multiple adaptor proteins. Biol. Chem. 394, 421–432 [DOI] [PubMed] [Google Scholar]
- 49. Jaiswal M., Dubey B. N., Koessmeier K. T., Gremer L., Ahmadian M. R. (2012) Biochemical assays to characterize Rho GTPases. Methods Mol. Biol. 827, 37–58 [DOI] [PubMed] [Google Scholar]
- 50. Eberth A., Lundmark R., Gremer L., Dvorsky R., Koessmeier K. T., McMahon H. T., Ahmadian M. R. (2009) A BAR domain-mediated autoinhibitory mechanism for RhoGAPs of the GRAF family. Biochem. J. 417, 371–377 [DOI] [PubMed] [Google Scholar]
- 51. Wu X., Knudsen B., Feller S. M., Zheng J., Sali A., Cowburn D., Hanafusa H., Kuriyan J. (1995) Structural basis for the specific interaction of lysine-containing proline-rich peptides with the N-terminal SH3 domain of c-Crk. Structure 3, 215–226 [DOI] [PubMed] [Google Scholar]
- 52. Eberth A., Dvorsky R., Becker C. F., Beste A., Goody R. S., Ahmadian M. R. (2005) Monitoring the real-time kinetics of the hydrolysis reaction of guanine nucleotide-binding proteins. Biol. Chem. 386, 1105–1114 [DOI] [PubMed] [Google Scholar]
- 53. Ross B., Kristensen O., Favre D., Walicki J., Kastrup J. S., Widmann C., Gajhede M. (2007) High resolution crystal structures of the p120 RasGAP SH3 domain. Biochem. Biophys. Res. Commun. 353, 463–468 [DOI] [PubMed] [Google Scholar]
- 54. Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H. J. (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, W363–W367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Scouras A. D., Daggett V. (2011) The Dynameomics rotamer library: amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water. Protein Sci. 20, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eccleston J. F., Moore K. J., Morgan L., Skinner R. H., Lowe P. N. (1993) Kinetics of interaction between normal and proline 12 Ras and the GTPase-activating proteins, p120-GAP and neurofibromin. The significance of the intrinsic GTPase rate in determining the transforming ability of ras. J. Biol. Chem. 268, 27012–27019 [PubMed] [Google Scholar]
- 58. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 59. Kärkkäinen S., Hiipakka M., Wang J. H., Kleino I., Vähä-Jaakkola M., Renkema G. H., Liss M., Wagner R., Saksela K. (2006) Identification of preferred protein interactions by phage-display of the human Src homology-3 proteome. EMBO Rep. 7, 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rittinger K., Walker P. A., Eccleston J. F., Smerdon S. J., Gamblin S. J. (1997) Structure at 1.65 Å of RhoA and its GTPase-activating protein in complex with a transition-state analogue. Nature 389, 758–762 [DOI] [PubMed] [Google Scholar]
- 61. Dvorsky R., Ahmadian M. R. (2004) Always look on the bright site of Rho: structural implications for a conserved intermolecular interface. EMBO Rep. 5, 1130–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moon S. Y., Zheng Y. (2003) Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 13, 13–22 [DOI] [PubMed] [Google Scholar]
- 63. Tcherkezian J., Lamarche-Vane N. (2007) Current knowledge of the large RhoGAP family of proteins. Biol. Cell 99, 67–86 [DOI] [PubMed] [Google Scholar]
- 64. Rossman K. L., Der C. J., Sondek J. (2005) GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 65. Aittaleb M., Boguth C. A., Tesmer J. J. (2010) Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol. Pharmacol. 77, 111–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. DiNitto J. P., Lee M. T., Malaby A. W., Lambright D. G. (2010) Specificity and membrane partitioning of Grsp1 signaling complexes with Grp1 family Arf exchange factors. Biochemistry 49, 6083–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gureasko J., Kuchment O., Makino D. L., Sondermann H., Bar-Sagi D., Kuriyan J. (2010) Role of the histone domain in the autoinhibition and activation of the Ras activator Son of Sevenless. Proc. Natl. Acad. Sci. U.S.A. 107, 3430–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jaiswal M., Gremer L., Dvorsky R., Haeusler L. C., Cirstea I. C., Uhlenbrock K., Ahmadian M. R. (2011) Mechanistic insights into specificity, activity, and regulatory elements of the regulator of G-protein signaling (RGS)-containing Rho-specific guanine nucleotide exchange factors (GEFs) p115, PDZ-RhoGEF (PRG), and leukemia-associated RhoGEF (LARG). J. Biol. Chem. 286, 18202–18212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mitin N., Betts L., Yohe M. E., Der C. J., Sondek J., Rossman K. L. (2007) Release of autoinhibition of ASEF by APC leads to CDC42 activation and tumor suppression. Nat. Struct. Mol. Biol. 14, 814–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Scholz R. P., Gustafsson J. O., Hoffmann P., Jaiswal M., Ahmadian M. R., Eisler S. A., Erlmann P., Schmid S., Hausser A., Olayioye M. A. (2011) The tumor suppressor protein DLC1 is regulated by PKD-mediated GAP domain phosphorylation. Exp. Cell Res. 317, 496–503 [DOI] [PubMed] [Google Scholar]
- 71. Ching Y. P., Wong C. M., Chan S. F., Leung T. H., Ng D. C., Jin D. Y., Ng I. O. (2003) Deleted in liver cancer (DLC) 2 encodes a RhoGAP protein with growth suppressor function and is underexpressed in hepatocellular carcinoma. J. Biol. Chem. 278, 10824–10830 [DOI] [PubMed] [Google Scholar]
- 72. Wong C. M., Lee J. M., Ching Y. P., Jin D. Y., Ng I. O. (2003) Genetic and epigenetic alterations of DLC-1 gene in hepatocellular carcinoma. Cancer Res. 63, 7646–7651 [PubMed] [Google Scholar]
- 73. Healy K. D., Hodgson L., Kim T. Y., Shutes A., Maddileti S., Juliano R. L., Hahn K. M., Harden T. K., Bang Y. J., Der C. J. (2008) DLC-1 suppresses non-small cell lung cancer growth and invasion by RhoGAP-dependent and independent mechanisms. Mol. Carcinog. 47, 326–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chan F. K., Chung S. S., Ng I. O., Chung S. K. (2012) The RhoA GTPase-activating protein DLC2 modulates RhoA activity and hyperalgesia to noxious thermal and inflammatory stimuli. Neurosignals 20, 112–126 [DOI] [PubMed] [Google Scholar]
- 75. Leung T. H., Ching Y. P., Yam J. W., Wong C. M., Yau T. O., Jin D. Y., Ng I. O. (2005) Deleted in liver cancer 2 (DLC2) suppresses cell transformation by means of inhibition of RhoA activity. Proc. Natl. Acad. Sci. U.S.A. 102, 15207–15212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kawai K., Kiyota M., Seike J., Deki Y., Yagisawa H. (2007) START-GAP3/DLC3 is a GAP for RhoA and Cdc42 and is localized in focal adhesions regulating cell morphology. Biochem. Biophys. Res. Commun. 364, 783–789 [DOI] [PubMed] [Google Scholar]
- 77. Roof R. W., Haskell M. D., Dukes B. D., Sherman N., Kinter M., Parsons S. J. (1998) Phosphotyrosine (p-Tyr)-dependent and -independent mechanisms of p190 RhoGAP-p120 RasGAP interaction: Tyr 1105 of p190, a substrate for c-Src, is the sole p-Tyr mediator of complex formation. Mol. Cell. Biol. 18, 7052–7063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gigoux V., L'Hoste S., Raynaud F., Camonis J., Garbay C. (2002) Identification of Aurora kinases as RasGAP Src homology 3 domain-binding proteins. J. Biol. Chem. 277, 23742–23746 [DOI] [PubMed] [Google Scholar]
- 79. Yang Y. S., Garbay C., Duchesne M., Cornille F., Jullian N., Fromage N., Tocque B., Roques B. P. (1994) Solution structure of GAP SH3 domain by 1H NMR and spatial arrangement of essential Ras signaling-involved sequence. EMBO J. 13, 1270–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Harkiolaki M., Lewitzky M., Gilbert R. J., Jones E. Y., Bourette R. P., Mouchiroud G., Sondermann H., Moarefi I., Feller S. M. (2003) Structural basis for SH3 domain-mediated high-affinity binding between Mona/Gads and SLP-76. EMBO J. 22, 2571–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]