

Background: CD166 is overexpressed and regarded as a valuable prognostic marker in tumors.
Results: An autoregulatory feedback between PI3K/AKT and CD166 was revealed, and YAP was identified as a CD166 downstream effecter.
Conclusion: CD166 is regulated by PI3K/AKT to exert its anti-apoptotic role via YAP.
Significance: The relationship between CD166 and YAP provides new therapeutic insights into liver cancer.
Keywords: Akt PKB, CREB, Hepatocellular Carcinoma, Membrane Proteins, Signal Transduction
Abstract
Cluster of differentiation 166 (CD166 or Alcam) is a cell surface molecule that can be greatly induced in liver cancer cells after serum deprivation, suggesting its role in influencing cell survival. However, whether and how CD166 acts as an anti-apoptotic regulator needs to be further investigated. Here, we report that gene silencing of CD166 promoted apoptosis via down-regulation of Bcl-2 in liver cancer cells. PI3K/AKT signaling was found to up-regulate CD166 expression independently of transcription. We also revealed that CD166 promoted both AKT expression and activity, thus providing a novel positive regulatory feedback between PI3K/AKT signaling and CD166. Moreover, Yes-associated protein (YAP) was identified as a CD166 downstream effecter, which can partly rescue CD166 knockdown-induced apoptosis and reduced in vivo cancer cell growth. Mechanically, CD166 modulated YAP expression and activity through at least two different ways, transcriptional regulation of YAP through cAMP-response element-binding protein and post-transcriptional control of YAP stability through inhibition to AMOT130. We also showed that CD9 enhanced CD166-mediated regulation of YAP via a mechanism involving facilitating CD166-CD166 homophilic interaction. Tissue microarray analysis revealed that CD166 and YAP were up-regulated and closely correlated in liver cancer samples, demonstrating the importance of their relationship. Taken together, this work summarizes a novel link between CD166 and YAP, explores the interplay among related important signaling pathways, and may lead to more effective therapeutic strategies for liver cancer.
Introduction
The treatment options for liver cancer are extremely limited mainly because the mechanisms of pathogenesis of this kind of disease are not completely known. Cluster of differentiation 166 (CD166)4 is a cell surface member of the immunoglobulin superfamily (1), which is overexpressed and regarded as a valuable prognostic marker of disease progression and poor survival in several types of epithelial tumors (2–4). In recent years, CD166 has been identified experimentally as a putative cancer stem cell marker in various cancers with a high capacity for sphere and xenograft formation (5–7). CD166-CD166 interactions are crucial to the survival and primary site maintenance of cancer cells (8). Furthermore, CD166 gene silencing decreases the concentration of Bcl-2 and increases levels of apoptosis (poly(ADP-ribose) polymerase and active caspase-7) (8); therefore, CD166 may also play an important role in protecting cancer cells against apoptosis. Although CD166 is closely related to various cancers, including those of the digestive system, whether and how CD166 exerts its role in liver cancer remains poorly understood. In our previous study, we observed that activation of anti-apoptotic canonical NF-κB signaling greatly induces CD166 expression in liver cancer cells after serum deprivation, a condition that inhibits cell growth and leads to apoptosis (9), suggesting that CD166 may also influence cell survival in liver cancer cells. However, the mechanism underlying how CD166 acts as an anti-apoptotic regulator needs to be further investigated.
Recently, dysfunction of Yes-associated protein (YAP) has been linked to hepatocarcinogenesis (10). Amplification of the YAP gene and induction of YAP in liver cancer have previously been reported to contribute to hepatocyte malignant transformation and tumor progression (11). Clinical studies also revealed that YAP is an independent predictor associated with poor disease survival in liver cancers (12). Overexpression of YAP resulted in resistance against doxorubicin-induced apoptosis in liver cancer cell lines, whereas suppression of the endogenous YAP expression by RNA interference demonstrated the reverse effect (11). Also, YAP-controlled expression of connective tissue growth factor (CTGF) reduces sensitivity of liver cancer cells toward tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis (13). Our previous studies also support the conclusion that YAP plays critical roles in protecting liver cancer cells from apoptosis (14, 15); however, the upstream regulation of YAP anti-apoptotic function is still largely unknown.
In this study, we found that PI3K/AKT up-regulated CD166 expression independently of transcription. Moreover, we revealed that CD166 promoted both AKT expression and activity, thus providing a positive regulatory feedback between PI3K/AKT signaling and CD166 in liver cancer cells. Our data also showed that CD166 exerted its anti-apoptotic role mainly through enhancing YAP function, demonstrating that CD166 is an upstream regulator of YAP. In addition, we found that CD166 and YAP were closely correlated in liver cancer samples, suggesting the importance of their relationship. Taken together, this work summarizes a novel link between two major oncoproteins and a potential mechanism for liver tumorigenesis.
EXPERIMENTAL PROCEDURES
Cell Culture and Vectors
HepG2, Bel-7402, SMMC-7721, QSG-7701, and HL-7702 cells were cultured in DMEM. Cells were treated by doxorubicin (0.5 μg/ml; Sigma-Aldrich), wortmannin (50 μm; Cayman, Ann Arbor, MI), LY294002 (20 μm; Cell Signaling Technology (CST), Boston, MA), actinomycin D (10 μg/ml; Beyotime, Haimen, China), or MG132 (25 μm; Cayman) 5–24 h before harvest. shRNAs against CD9 (TRCN0000057472) and CD166 (shRNA-1, TRCN0000150706) were purchased from Open Biosystems (Huntsville, AL). shRNAs against AMOT130 and AKT were cloned into pLKO.1 lentiviral vectors. The cDNA fragments encoding human AKT and CD9 were purchased from Origene (Beijing, China) and subcloned into pGIPZ-based lentiviral vector (14). CD6 expression vector was purchased from Origene. CD166-HA/FLAG was cloned into pCDNA3.1(+) vector, and the primers used are listed in Tables 1 and 2. Protein-expressing vectors, including CD166 (without tag), YAP (with or without FLAG tag), AMOT130-HA, pTEN-HA, and Ub-HA, as well as shRNA targeting YAP were obtained from previous studies (9, 14–16).
TABLE 1.
Primers for protein expression vectors
| Name | 5′–3′ |
|---|---|
| CD166-HA(pcdna3.1)-F | GTACGGATCCCACCAAGAAGGAGGAGGAAT |
| CD166-HA(pcdna3.1)-R | GTACCTCGAGTTAAGCGTAGTCTGGGACGTCGTATGGGTAGGCTTCAGTTTTGTGATTGT |
| CD166-FLAG(pCDNA3.1)-F | GCTAGGATCC ATGGAATCCAAGGGGGCCAGTT |
| CD166-FLAG(pCDNA3.1)-R | GACTCTCGAGTTACTTGTCATCGTCATCCTTGTAATCGGCTTCAGTTTTGTGATTGTT |
| CD9-MYC(pGIPZ2a)-F | GTACGCTAGCCCGGTCAAAGGAGGCACCAAG |
| CD9-MYC(pGIPZ2a)-R | GCTAGCGGCCGCTTACAGATCTTCTTCAGAAATAAGTTTTTGTTCGACCATCTCGCGGTTCCGC |
| AKT-MYC(pGIPZ2a)-F | GGTC GCTAGCAGCGACGTGGCTATTGTGAA |
| AKT-MYC(pGIPZ2a)-R | GTATCCTGCAGGTTACAGATCTTCTTCAGAAATAAGTTTTTGTTCGGCCGTGCCGCTGGCCGAT |
TABLE 2.
Primers for lentiviral shRNA
| Name | 5′–3′ |
|---|---|
| AKT-shRNA1-F | CCGGAACTCCTCAAGAATGATGGCACTCGAGTGCCATCATTCTTGAGGAGTTTTTTTG |
| AKT-shRNA1-R | AATTCAAAAAAACTCCTCAAGAATGATGGCACTCGAGTGCCATCATTCTTGAGGAGTT |
| AMOT-sh1-F | CCGGAAGGTGACTACTTTGAAATAACTCGAGTTATTTCAAAGTAGTCACCTTTTTTTG |
| AMOT-sh1-R | AATTCAAAAAAAGGTGACTACTTTGAAATAACTCGAGTTATTTCAAAGTAGTCACCTT |
| AMOT-sh2-F | CCGGAAGCCATCCTCTGCTTCTTATCTCGAGATAAGAAGCAGAGGATGGCTTTTTTTG |
| AMOT-sh2-R | AATTCAAAAAAAGCCATCCTCTGCTTCTTATCTCGAGATAAGAAGCAGAGGATGGCTT |
| CD166-sh2-F | CCGGTCAAGCAACCATCTAAACCTGCTCGAGCAGGTTTAGATGGTTGCTTGATTTTTG |
| CD166-sh2-R | AATTCAAAAATCAAGCAACCATCTAAACCTGCTCGAGCAGGTTTAGATGGTTGCTTGA |
Immunohistochemistry (IHC), Immunofluorescence, and Western Blotting (WB)
For IHC, human liver cancer tissue microarray (TMA) slides were purchased from U.S. Biomax (Rockville, MD). Following deparaffinization and rehydration of the tissue sections, antigen retrieval was performed at 100 °C for 2 h with Tris-EDTA buffer, pH 6.0 (Beyotime). Endogenous peroxidase was blocked with 3% peroxide for 20 min, followed by additional rinses in PBS for 3 × 5 min. Sections were then blocked in a buffer containing 5% BSA and 0.1% Triton X-100 and incubated overnight in primary antibodies against CD166 (Epitomics (Burlingame, CA), catalogue no. 3133) or YAP (Epitomics, catalogue no. 2060). Signal detection was accomplished by the Vectastain ABC kit (Vector Labs, Burlingame, CA). Sections were scored using a semiquantitative scale for each individual tumor tissue on the array slide as follows: +, weak staining (i.e. 20–40% of cells showing weak to intermediate intensity staining); ++, strong staining (i.e. ≥ 10% of cells showing very intense staining or >50% of cells showing weak to moderately intense staining in an appropriate subcellular distribution); +++, very strong staining (i.e. ≥30% of cells showing very intense staining or >80% of cells showing moderately intense staining). Scoring results were simplified into +, ++, and +++ categories. Statistical analysis was done using χ2 analysis, and a p value of <0.05 was considered statistically significant. For xenograft tissues, the IHC procedure used was the same as described above. The primary antibodies used were as follows: anti-CD133 (Hua'an (Hangzhou, China), catalogue no. 0804-5), anti-CD166 (Epitomics, catalogue no. 3133), and anti-Ki67 (Abcam (Cambridge, UK), catalogue no. Ab15580).
For immunofluorescence, cells were fixed by 4% paraformaldehyde for 15 min, washed with PBS and blocking buffer (3% FBS + 1% heat-inactivated sheep serum + 0.1% Triton X-100), and then incubated overnight at 4 °C in primary antibodies against YAP (CST, catalogue no. 4912), cleaved caspase-3 (CST, catalogue no. 9664), or HA (CST, catalogue no. 2367). Alexa-Fluor-488 or -555 fluorescent conjugated secondary antibodies (Invitrogen) were used for detection.
For WB, proteins were resolved on SDS-polyacrylamide gels followed by standard WB. primary antibodies used were as follows: CD166 (Epitomics, catalogue no. 3133), FLAG (Sigma (catalogue no. F3165) or CST (catalogue no. 2368)), HA (CST, catalogue no. 3724 or 2367), Myc (CST, catalogue no. 2278 or 2276), YAP (Epitomics, catalogue no. 2060), ubiquitin (CST, catalogue no. 3933), CREB (CST, catalogue no. 9197), p-CREB (CST, catalogue no. 9198), p38 (CST, catalogue no. 9218), phospho-p38 (CST, catalogue no. 4511), CTGF (Santa Cruz Biotechnology, Inc., catalogue no. sc-373936), LATS1 (CST, catalogue no. 3477), phospho-LATS1 (CST, catalogue no. 9157), AMOT130 (Abcam (Hong Kong, China), catalogue no. ab85143), AKT (CST, Catalogue number 4691), p-AKT (CST, catalogue no. 4060), PDK1 (CST, catalogue no. 3062), phospho-PDK1 (CST, catalogue no. 3438), p-AKT substrate (CST, Catalogue no. 9614), phospho-PKA substrate (CST, catalogue no. 9624), GAPDH (CST, catalogue no. 5174), VEGFR1 (Epitomics, catalogue no. 1303), VEGFR2 (Epitomics, catalogue no. 2296), CD133 (Hua'an, catalogue no. 0804-5), Nanog (Epitomics, catalogue no. 3369), Sox2 (Epitomics, catalogue no. 2683), c-Myc (CST, catalogue no. 5605), Oct4 (Epitomics, catalogue no. 2876), or BIRC5 (CST, catalogue no. 2808).
Cell Proliferation, Caspase-3/7 Activity, and Quantitative RT-PCR (qPCR)
Cell proliferation was measured by an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide-based proliferation assay, as described before (14, 15). The caspase-3/7 activity was determined using the Caspase-Glo 3/7 assay system (Promega, Madison, WI). Quantitative RT-PCR was performed as described previously (14, 15). Primers used for qPCR are listed in Table 3.
TABLE 3.
Primers for qPCR
| Name | 5′–3′ |
|---|---|
| GAPDH-F | ATCATCCCTGCCTCTACTGG |
| GAPDH-R | GTCAGGTCCACCACTGACAC |
| YAP-F | CCTCGTTTTGCCATGAACCAG |
| YAP-R | GTTCTTGCTGTTTCAGCCGCAG |
Immunoprecipitation
For immunoprecipitation, cells were washed with PBS and subsequently lysed in Western/IP lysis buffer (Beyotime). Protein lysates were centrifuged at 14,000 × g for 10 min to pellet debris. After preclearing for 1 h with 50 μl of protein A/G-Sepharose (Invitrogen), the supernatants were incubated at 4 °C overnight with 3 μg of antibodies, as indicated, cross-linked to protein A/G-Sepharose beads. Beads were washed five times with lysis buffer, resuspended in SDS loading buffer, and analyzed by WB analysis with antibodies as indicated.
Luciferase Reporter Analysis
The luciferase reporter constructs containing the YAP or HULC promoter region were obtained from our previous study (15) and then co-transfected with a Renilla luciferase expression plasmid into cells. Luciferase activities were analyzed using a Dual-Luciferase reporter kit (Promega).
Detection of HBsAg and HCV-IgG
Serum hepatitis B surface antigen (HBsAg) was tested by a reagent from Roche Applied Science using a Cobas® e601 immunoassay analyzer. Serum HCV-IgG was tested by reagent from Ortho Clinical Diagnostics using a VITROS® 3600 immunoassay analyzer. Results were determined automatically by the software as a cut-off index value by comparing the electrochemiluminescence signal obtained from the reaction product of the sample with the signal of the cut-off value previously obtained by calibration, and cut-off index > 1 was regarded as positive (+).
Xenograft Mouse Model
5 × 106 Bel-7402 cells expressing shRNA or protein, as indicated, were subcutaneously injected into the athymic nude mice (Bikai, Shanghai, China). Tumor size was measured every 6 days using a caliper, and the tumor volume was calculated as 0.5 × L × W2, with L indicating length and W indicating width. The mice were euthanized at 30 days after injection.
In Vivo Metastasis Assays
To produce experimental lung metastasis, 1 × 106 Bel-7402 cells were injected into the tail veins of 8-week-old female athymic nude mice. After 4 weeks, the mice were necropsied, and the lungs were removed.
Statistical Analysis
Tests to examine differences between groups included analysis by Student's t test and χ2 test. p < 0.05 was regarded as statistical significance.
RESULTS
CD166 Was Overexpressed and Anti-apoptotic in Liver Cancer Cells
IHC results indicated that CD166 was highly overexpressed in liver cancer tissues compared with their adjacent normal tissues from all 10 patients tested who underwent curative surgery for liver cancer (Fig. 1A). Also, CD166 had a relatively higher level in established liver cancer cell lines (Bel-7402, SMMC-7721, and HepG2) compared with that in normal hepatic cell lines (HL-7702 and QSG-7701) (Fig. 1B), suggesting the importance of CD166 during liver cancer development.
FIGURE 1.

CD166 was overexpressed in liver cancer and protected cells from apoptosis. A, representative IHC images of CD166 staining in liver cancer and adjacent normal liver tissues. B, CD166 expression pattern in different cell lines as measured by Western blotting. S, shorter exposure; L, longer exposure. C, phase-contrast images of Bel-7402 cells expressing shRNA against GFP (Control) or CD166 (sh1). Cells were plated at a density of 5,000 cells/well and imaged 6 days later. Scale bar, 60 μm (left). Cells were then quantified, and the data are shown as mean ± S.D. (error bars) from three independent tests (right). p < 0.01 indicates statistic significance. D, Western blots of Bcl-2 in control (infected with GFP-shRNA) and Bel-7402 cells with CD166 knocked down (infected with sh1 and sh2, respectively). E, CD166 knockdown-induced apoptosis as measured by cleaved caspase-3 staining. Cells were plated at a density of 2,000 cells/well and harvested for immunofluorescence 24 h later. Areas indicated by a rectangular box were enlarged to show cleaved caspase-3 localization. Scale bar, 30 μm (top). Cleaved caspase-3-positive cells were then quantified, and the data are shown as mean ± S.D. from three independent tests (bottom). p < 0.01 indicates statistical significance. F, knockdown efficiency of CD166-specific shRNA (sh1) as measured in Bel-7402 and SMMC-7721 cells by Western blotting. G, knockdown of CD166 enhanced doxorubicin-induced apoptosis as measured by caspase-3/7 activity in control (infected by GFP-shRNA) and Bel-7402 cells with CD166 knocked down (infected by CD166-sh1) treated with DMSO or doxorubicin (0.5 μg/ml). p < 0.01 indicates statistic significance. H, in vivo metastasis analysis indicated no difference in the lung before and after overexpression of CD166. 1 × 106 control (transfected with empty plasmid) or Bel-7402 cells with CD166 overexpressed (transfected with CD166-expressing plasmid) were injected into tail veins of 8-week-old nude mice, which were sacrificed 4 weeks later. The mice were then necropsied, and the lungs were removed. I, Western blots of VEGFR1/2 in control (infected with GFP-shRNA) and Bel-7402 cells with CD166 knocked down (infected with CD166-sh1).
Because CD166 can be significantly induced under serum deprivation, a condition that leads to apoptosis (9), we hypothesized that CD166 is an apoptosis regulator. We found that Bel-7402 cells with CD166 knocked down became unhealthy, with both decreased cell numbers (Fig. 1C) and decreased Bcl-2 expression (Fig. 1D), and showed apoptotic morphology and nuclear segmentation with labeling of cleaved caspase-3 (Fig. 1E), an apoptosis phenomenon similar to that described previously (17). The high knockdown efficiency of CD166-sh1 was then verified by Western blotting assay in both Bel-7402 and SMMC-7721 cells (Fig. 1F). We also detected that CD166 knockdown markedly increased caspase-3/7 activity both at basal level and under doxorubicin treatment in Bel-7402 cells (Fig. 1G), suggesting that CD166 is critical for cell survival in liver cancer cells.
We reported previously that overexpression of CD166 inhibits cell mobility, as measured by wound healing and a Transwell assay (9). Unfortunately, experimental in vivo metastasis analysis showed no obvious surface-visible metastatic tumors formed by either control or Bel-7402 cells with CD166 overexpressed in lungs (Fig. 1H). In addition, Bel-7402 cells with CD166 overexpressed exhibited an enhanced ability to adhere with each other compared with the control (data not shown), indicating that CD166 is a cell surface adhesion molecule that has a negative role in cell mobility and metastasis. Meanwhile, we also ruled out the possibility that CD166 affects angiogenesis, because angiogenesis markers VEGFR1 and VEGFR2 (18) remained unchanged before and after knockdown of CD166 (Fig. 1I).
An Autoregulatory Feedback between PI3K/AKT and CD166
Because anti-apoptotic NF-κB activates CD166 gene transcription (9) and PI3K/AKT is an upstream regulator of NF-κB signaling (19, 20), we postulated that AKT may regulate CD166. By using two PI3K inhibitors, LY294002 and wortmannin, we found that both p-AKT and CD166 proteins were significantly reduced (Fig. 2A). In mammals, AKT1, AKT2, and AKT3 have a similar domain and function (21). We found that AKT1 was predominately expressed in HEK-293T, HL-7702, SMMC-7721, and Bel-7402 cells, because the AKT1 expression pattern was similar to that of pan-AKT (AKT1/2/3) (Fig. 2B). Thus, we mainly focused on AKT1 (hereafter referred to as AKT) in the following study. Through gain and loss of function analysis, we found that CD166 protein can be reduced by knockdown of AKT (Fig. 2C, a′), whereas it is induced by ectopically expressed AKT (Fig. 2C, b′). By overexpressing the endogenous PI3K/AKT inhibitor pTEN, CD166 was also markedly inhibited (Fig. 2C, c′). Unexpectedly, both mRNA level and promoter activity of CD166 were not affected after inhibition of PI3K/AKT by LY294002 (Fig. 2D), suggesting that PI3K/AKT only regulates CD166 protein and not its mRNA. Also, we excluded a role of NF-κB signaling in the regulation of CD166 by AKT.
FIGURE 2.

Regulatory feedback between PI3K/AKT and CD166. A, inhibition of PI3K/AKT reduced CD166. Shown are Western blots of proteins as indicated in cells treated with DMSO, LY294002 (20 μm), or wortmannin (50 μm) for 24 h. B, AKT expression patterns in different cell lines. Shown are Western blots of AKT1 as well as AKT1/2/3 (pan-AKT) in different cell lines as indicated. C (a′–c′), AKT-regulated CD166. Western blots of CD166 in control and cells with either AKT knockdown (a′), AKT overexpressed (b′), or pTEN-HA overexpressed (c′). D, PI3K/AKT did not affect YAP gene transcription. YAP mRNA was detected by qPCR in Bel-7402 and HepG2 cells treated with either DMSO or LY294002 (20 μm) for 24 h (top). PI3K/AKT did not affect CD166 promoter activity. Luciferase activities from −1,992 to +170 relative to the transcription start site of the CD166 gene were measured in Bel-7402 and HepG2 cells treated with either DMSO or LY294002 (20 μm) for 24 h. Firefly luciferase activity was normalized to that of Reniila (bottom). E (a′–c′), CD166-induced AKT expression. Western blots of p-AKT/AKT in control (transfected with empty vector) and HL-7702 cells with CD166 overexpressed (a′). Shown are Western blots of proteins as indicated in control cells (infected with GFP shRNA) and cells with CD166 knocked down (infected with CD166-sh1 (b′) and CD166-sh2 (c′), respectively). F, CD166-induced AKT phosphorylation activity. Shown are Western blots of p-AKT substrate using an antibody specific for p-AKT substrate in control (infected with GFP shRNA) and Bel-7402 cells with CD166 knocked down (infected with CD166-sh1; left). S, shorter exposure; L, longer exposure. Western blots of p-AKT substrate in control (transfected with empty vector) and HL-7702 cells with CD166 overexpressed (right). G, AKT bound to CD166. CD166-HA was co-transfected with AKT-Myc into Bel-7402 cells, as indicated. CD166 and AKT associations were examined by reciprocal co-IP, as indicated. H, endogenous CD166 was immunoprecipitated with anti-AKT antibodies (ab), and co-immunoprecipitation of CD166 was shown by anti-CD166 Western blot. A control IgG was used as the negative control for immunoprecipitation. S, shorter exposure; L, longer exposure.
To explore whether there was an autoregulatory feedback between CD166 and PI3K/AKT, CD166 was overexpressed in low CD166-expressing normal hepatic cell line, HL-7702 (Fig. 1B), and we found that both p-AKT and total AKT were induced (Fig. 2E, a′). Because the ratio between p-AKT and total AKT was similar before and after overexpression of CD166 (Fig. 2E, a′), the induction of p-AKT was largely due to the up-regulation of total AKT. Moreover, when CD166 was knocked down by sh1 in liver cancer cell lines, Bel-7402 and SMMC-7721, p-AKT, total AKT, and phosphorylation of its downstream target, p70S6K, were significantly reduced; however, its upstream regulator phospho-PDK1 and total PDK1 remained unchanged (Fig. 2E, b′), suggesting that CD166 regulates p-AKT/AKT independently of PDK1. Similar results were also obtained by CD166-sh2, which excluded the off-target effect of small RNA (Fig. 2E, c′).
By using a newly developed mAb that recognizes endogenous proteins containing phospho-Ser/Thr preceded by Arg at positions −5 and −3 (RXRXX(S*/T*) (asterisks indicating phosphorylation) regarded as p-AKT substrate), we found that phosphorylation of AKT substrates was greatly inhibited by CD166 knockdown in Bel-7402 cells, whereas it was enhanced by CD166 overexpression in HL-7702 cells (Fig. 2F), indicating that CD166 is a positive regulator of PI3K/AKT signaling.
To confirm the interaction between CD166 and AKT, we performed reciprocal co-IP experiments and found that exogenous CD166-HA could be readily pulled down by AKT-Myc in Bel-7402 cells, and vice versa (Fig. 2G). Co-IP for endogenous CD166 and AKT proteins in Bel-7402 cells also demonstrated that these two proteins readily co-immunoprecipitated (Fig. 2H).
CD166 Regulated YAP
Because YAP is also an apoptosis regulator in liver cancer cells (14, 15), and CD166 is a membrane protein, we hypothesized that CD166 may be an upstream regulator of YAP. We found that YAP protein and its well known target, CTGF, and BIRC5 were greatly reduced after knockdown of CD166 by sh1 and sh2, respectively (Fig. 3, A and B), whereas the key regulator of the Hippo pathway, phospho-LATS1, and total LATS1 remained unchanged (Fig. 3A). Furthermore, both BIRC5 and YAP could be significantly up-regulated by overexpression of CD166 (Fig. 3, C and D). The above data manifested that the regulation of YAP and its targets by CD166 is independent of the canonical Hippo pathway. However, knockdown of YAP had no effect on CD166 expression (Fig. 3E), suggesting that there is no direct feedback between YAP and CD166. We also found that a relatively higher level of endogenous YAP correlated with a higher level of CD166 in Bel-7402 cells compared with SMMC-7721 cells (Fig. 3F). A previous study (22) described that YAP is stable at low cell density but unstable at high cell density in NIH3T3 cells. We also observed this phenomenon in Huh7 and HepG2 cells, and the down-regulation patterns of YAP and CD166 were similar (Fig. 3G). It was previously reported that transcription of the CD166 gene is controlled by NF-κB p50/p65 (9), and CD166 is a target gene of AKT (Fig. 2). To explain why the expression of CD166 protein was down-regulated when the cell density was increased, we tested if the expression of p50/p65 as well as phosphorylation levels of AKT substrates could be changed. As shown in Fig. 3H, only the phosphorylation levels of AKT substrates were down-regulated, whereas no significant changes were detected for p50/p65 in high density cell culture compared with the low density one, suggesting that the down-regulation of CD166 protein in high density cell culture is via inactivation of AKT. In addition, inhibition of PI3K/AKT by chemical inhibitors reduced not only CD166 but also YAP expression (Fig. 3I), suggesting that down-regulation of YAP may result from down-regulation of CD166.
FIGURE 3.

YAP depended on CD166. A and B, knockdown of CD166 (infected with CD166-sh1 (A) and CD166-sh2 (B), respectively) reduced YAP as well as its target gene expression as measured by Western blotting. C, overexpression of CD166 induced BIRC5 expression. Shown are Western blots of proteins as indicated in control (transfected with empty vector) and Bel-7402 cells with CD166 overexpressed. D, CD166 dose-dependently induced YAP expression as measured by Western blotting in HL-7702 cells. E, YAP did not affect CD166 expression. Shown are Western blots of CD166 and YAP in Bel-7402 and SMMC-7721 cells with or without YAP knocked down (infected with shRNA against YAP). F, endogenous expression patterns of YAP and CD166 in Bel-7402 and SMMC-7721 cells. G, high density culture of cells induced down-regulation of YAP and CD166 as measured by Western blotting in Huh7 and HepG2 cells. H, increased cell density reduced p-AKT substrate level. Western blots of proteins as indicated in lower or higher density cultured Huh7 cells. I, inhibition of PI3K/AKT inhibited both YAP and CD166 expression. Shown are Western blots of YAP and CD166 in Bel-7402 and SMMC-7721 cells treated with DMSO, LY294002 (20 μm), or wortmannin (50 μm) for 24 h. S, shorter exposure; L, longer exposure. J, knockdown of CD166 reduced YAP activity. Luciferase activities from the pUAS-LUC/TEAD-Gal4 system were measured in control cells (infected with GFP shRNA) and cells with CD166 knocked down (infected with CD166-sh1). K, CD166 facilitated YAP/TEAD interaction. In CD166-HA-transfected Bel-7402 cells, YAP was immunoprecipitated with an anti-FLAG antibody, and Western blotting analysis of TEAD was done by an anti-Myc antibody. L, YAP intracellular localization in Bel-7402 cells with or without CD166-HA overexpressed. Scale bar, 15 μm. M, no direct association between YAP and CD166. CD166-HA was co-transfected with YAP-FLAG into Bel-7402 cells as indicated. YAP and CD166 associations were examined by co-IP as indicated. Error bars, S.D.
The TEAD transcription factors mediate YAP-dependent downstream transcriptional activation (23–24). We examined whether CD166 affects YAP-TEAD interaction. We found that CD166 knockdown inhibited the activity of a YAP-dependent TEAD-luciferase reporter (14) in both Bel-7402 and SMMC-7721 cells (Fig. 3J). Furthermore, the ability of YAP to bind TEAD was dose-dependently enhanced after overexpression of CD166 (Fig. 3K). However, YAP was still mainly retained in the nucleus in CD166-overexpressing Bel-7402 cells compared with normal ones (Fig. 3L), suggesting that CD166 does not affect YAP subcellular localization. Also, no direct interaction between CD166 and YAP was detected (Fig. 3M).
The Expression Pattern of CD166 and YAP as Well as Related Clinical Features in Liver Cancer Samples
To further uncover the relationship between CD166 and YAP, we performed IHC using TMA on greater than 400 human liver cancer samples and found that both CD166 and YAP proteins were highly expressed in a subset of liver cancers and closely correlated (Fig. 4A). We then analyzed the correlation between the expression of CD166 and the clinical features of the patients. On the basis of the clinical information accompanied with the TMA (provided by U.S. Biomax), we found a positive correlation between tumor stage and CD166 expression (p < 0.01) (i.e. liver cancer patients with a higher tumor stage of disease can have a higher CD166 expression level) (Fig. 4B, top), whereas no significant correlation was detected between histologic grade and the expression of CD166 (p > 0.05) (Fig. 4B, bottom).
FIGURE 4.
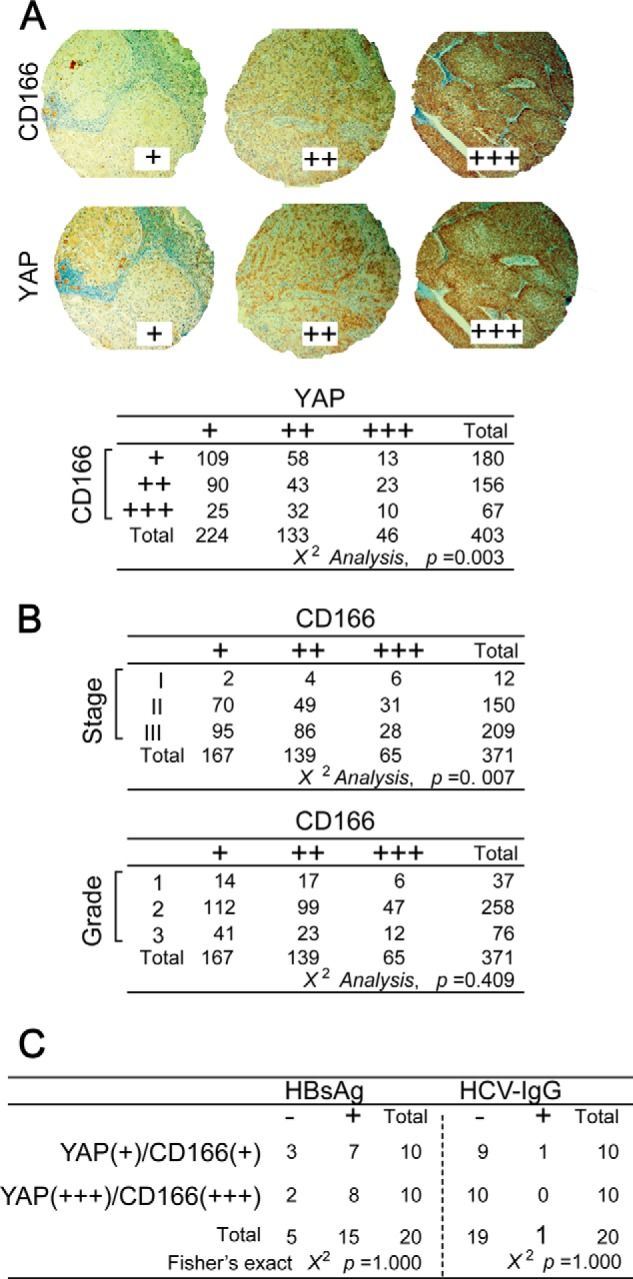
The expression pattern of CD166 and YAP as well as related clinical features in liver cancer samples. A, representative IHC images of CD166 and YAP staining from the TMA analysis (top). Statistical analysis of the TMA data using the χ2 test is shown at the bottom. B, tumor stage but not histologic grade was correlated with CD166 expression in liver cancer samples as analyzed by the χ2 test. C, no correlation between CD166-dependent YAP activation and hepatitis B virus or HCV infection as analyzed by Fisher's exact χ2 test. Serum HBsAg and HCV IgG were tested from the same patients whose liver cancer tissues were used for IHC determination for CD166 and YAP.
It is well recognized that the liver cancer develops with virus infection. Through IHC staining, we separated patients into two groups (each had 10 patients), one with a “+++” score for both CD166 and YAP, according to the principles in Fig. 4A, and another with a “+” score for both CD166 and YAP. Then the serum HBsAg (a current hepatitis B virus infection marker) and HCV IgG (an HCV infection marker) were each tested in these patients. Unfortunately, no significant statistical significance (p = 1.000) was detected between the expression of CD166 and YAP and the serum HBsAg or HCV-IgG (Fig. 4C), suggesting no close correlation between CD166-dependent YAP activation and hepatitis B virus or HCV infection in liver cancer patients.
CD166 Regulated YAP Gene Transcription via CREB
To explore how CD166 regulates YAP, we first tested the YAP mRNA levels before and after knockdown of CD166. As shown in Fig. 5A, YAP mRNA was reduced when CD166 was knocked down, suggesting that CD166 regulates YAP gene transcription. Because we previously reported (15) that CREB enhances YAP gene transcription, we tested whether CD166 affects CREB. As shown in Fig. 5B, reduction of CREB resulted from knockdown of CD166 (infected by CD166-sh1); however, phosphorylated and total p38/MAPK14, which contributes to CREB degradation, were not affected. In addition, down-regulation of p-CREB was largely due to down-regulation of total CREB, because the ratio of p-CREB/total CREB was almost unchanged before and after CD166 was knocked down (Fig. 5C). To rule out the off-target effects by CD166-sh1, we used CD166-sh2 and found that both p-CREB and CREB were reduced (Fig. 5D). Gain of function analysis provided evidence that CREB could be induced in a CD166 dose-dependent manner (Fig. 5E). However, CD166 did not affect the CREB upstream regulator, PKA activity, because the phosphorylation of PKA substrate was not changed before and after knockdown of CD166 (Fig. 5F).
FIGURE 5.

CD166 regulated YAP gene transcription via CREB. A, YAP mRNA levels were detected by qPCR without (control; infected with GFP shRNA) or with CD166 knocked down (infected with CD166-sh1) in Bel-7402 or SMMC-7721 cells. B, CD166 regulated CREB. Shown are Western blots of proteins as indicated in control cells (infected with GFP shRNA) and Bel-7402 or SMMC-7721 cells with CD166 knockdown (infected with CD166-sh1). C, quantification of p-CREB/total CREB ratio from B. D, knockdown of CD166 by sh2 reduced both p-CREB and total CREB in Bel-7402 cells. E, CD166 dose-dependently increased CREB expression in HL-7702 cells. S, shorter exposure; L, longer exposure. F, CD166 did not affect PKA activity. Phospho-PKA substrates were tested in control (infected with GFP shRNA) and Bel-7402 or SMMC-7721 cells with CD166 knocked down (infected with CD166-sh1). G, CD166 regulated HULC and YAP promoter activities. Promoter activities were tested from luciferase constructs as indicated in control (infected with GFP shRNA) and SMMC-7721 (top) or Bel-7402 (bottom) cells with CD166 knocked down (infected with CD166-sh1). Error bars, S.D.
Using the HULC promoter luciferase reporter system (15), we confirmed our hypothesis that CD166 regulates CREB activity. We found that promoter activities from wild type (WT; with cAMP-response element (CRE)) were greatly inhibited, whereas no obvious changes were detected from the mutant (MUT; without CRE) one in cells with CD166 knocked down compared with the control (Fig. 5G). Furthermore, luciferase activity from the YAP promoter reporter, which contains CRE, was also reduced after knockdown of CD166 (Fig. 5G). Collectively, these experiments demonstrated that CD166 may play an important role in maintaining CREB protein expression and activity.
CD166 Protected YAP Protein from Degradation
Next, we tested whether CD166 affects YAP stability. As shown in Fig. 6A, when Bel-7402 cells were treated with universal transcription inhibitor, actinomycin D, YAP protein was dramatically reduced, which may result from transcription inhibition (Fig. 6B, top). However, actinomycin D-induced YAP protein degradation was much slower when CD166 was ectopically expressed (Fig. 6, A and B, bottom), suggesting that CD166 up-regulates YAP protein expression not only transcriptionally, but also post-transcriptionally.
FIGURE 6.
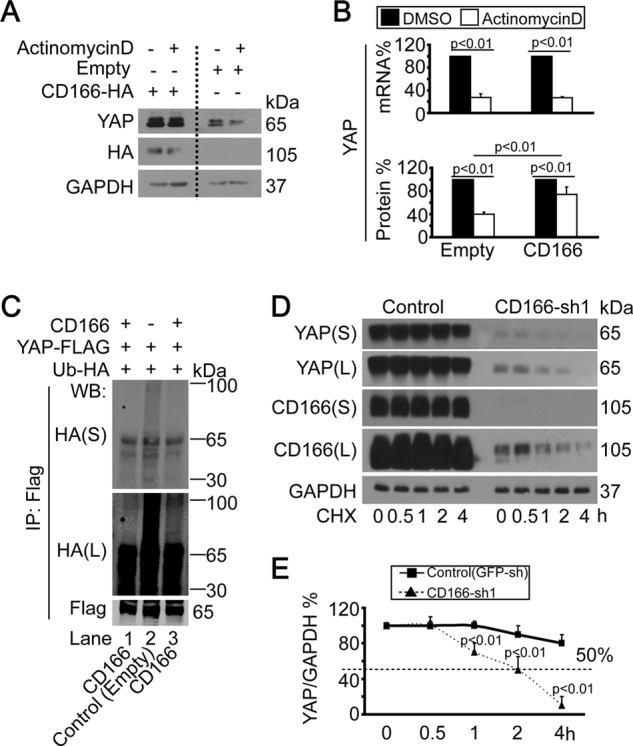
CD166 enhanced YAP protein stability. A, CD166 decreased YAP degradation by actinomycin D. YAP was detected in empty plasmid vector or CD166-expressing plasmid-transfected Bel-7402 cells with or without (DMSO) actinomycin D (10 μg/ml) treatment. B, relative YAP expression was detected by qPCR (top) and Western blotting (bottom) under the same conditions as in A. Protein levels were regarded as the ratio of YAP/GAPDH. C, CD166 reduced YAP ubiquitination. Bel-7402 cells transfected with expressing constructs as indicated were treated with MG132 (25 μm) for 5 h before harvest. Exogenous YAP-FLAG was immunoprecipitated, and Western blotting analysis was done using anti-FLAG or anti-HA antibody. S, shorter exposure; L, longer exposure. D and E, CD166 protected YAP from degradation. Protein synthesis was blocked by treatment of cycloheximide (CHX; 50 μg/ml) for the indicated time in control (infected with GFP shRNA) and Bel-7402 cells with CD166 knocked down (infected with CD166-sh1) (D). Relative YAP protein levels were quantified by YAP/GAPDH ratio (E). Error bars, S.D.
In addition, in Bel-7402 cells with CD166 overexpressed, we detected less accumulation of ubiquitinated YAP compared with the control (Fig. 6C, lanes 1 and 3 versus lane 2). When Bel-7402 cells were treated with the protein synthesis inhibitor cycloheximide (CHX), the half-life time of YAP was more than 4 h. However, YAP degraded much faster, with a half-life time of about 2 h, when CD166 was knocked down (Fig. 6, D and E), suggesting that CD166 is also a regulator of YAP protein stability.
CD166 Inhibited AMOT130
CD166 regulation of YAP was independent of LATS (Fig. 3A). The fact that AMOT130 inhibition of YAP activity is also LATS-independent (16) led us to link CD166 and AMOT130 together. We found that both YAP expression and activity were greatly reduced after overexpression of AMOT130, whereas the expression of CD166 and p-AKT/total AKT remained unchanged (Fig. 7A) (data not shown). We also found that AMOT130 facilitated YAP translocation from the nucleus to the cytoplasm (Fig. 7B), suggesting that AMOT130 inhibits YAP activity through cytoplasm retention of YAP. Moreover, induction of YAP was detected after knockdown of AMOT130 (Fig. 7C), further supporting our conclusion that AMOT130 is a critical regulator of YAP expression.
FIGURE 7.
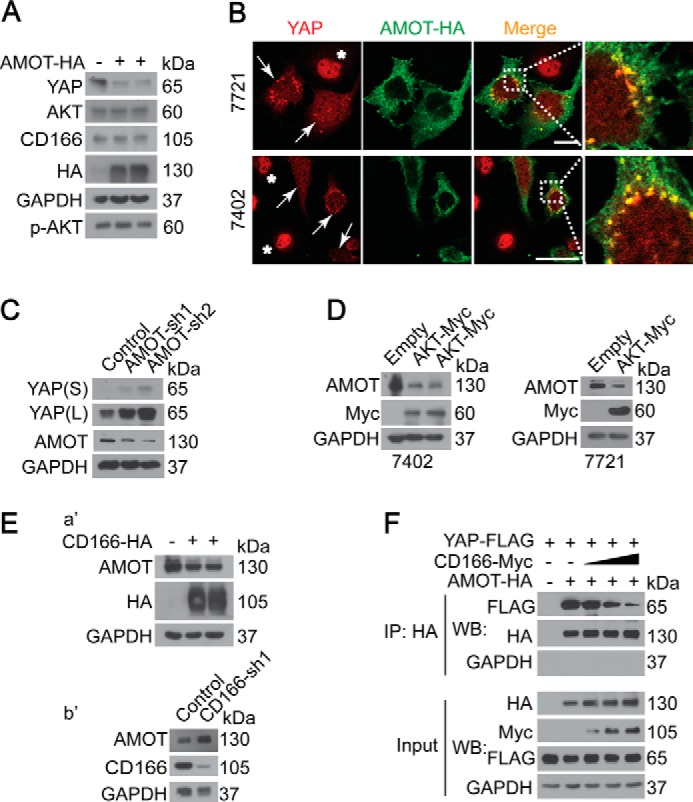
CD166 protected YAP via down-regulation of AMOT130. A and B, AMOT130 regulated YAP expression by affecting its localization. YAP expression and localization were detected by Western blotting (Bel-7402 cells) (A) and microscopic analysis (Bel-7402 and SMMC-7721 cells) (B). Arrows, cells transfected with AMOT130-HA; *, cells without successful transfection. C, YAP expression in control (infected with GFP shRNA) and Bel-7402 cells with AMOT130 knocked down (infected with sh1 and sh2, respectively) by Western blotting. D, AKT reduced AMOT130. Western blots of AMOT in control (transfected with empty plasmid) and cells as indicated with AKT overexpressed (transfected with AKT-Myc-expressing plasmid). E (a′ and b′) Western blots of AMOT130 in Bel-7402 cells with or without CD166 overexpressed (a′) or knocked down (b′). F, CD166 blocked AMOT130 interaction with YAP. In CD166-Myc-transfected Bel-7402 cells, AMOT130 was immunoprecipitated with an anti-HA antibody, and Western blotting analysis of YAP was done by an anti-FLAG antibody.
Because CD166 was regulated by AKT (Fig. 2), we then tested if AKT affects AMOT130 expression. We found that AMOT130 was greatly reduced after overexpression of AKT (Fig. 7D) in both Bel-7402 and SMMC-7721 cells, suggesting that CD166 is a mediator between AKT and AMOT130. To further confirm the relationship between CD166 and AMOT130, gain and loss of function analysis was employed. As shown in Fig. 7E, AMOT130 was down-regulated by CD166 overexpression (Fig. 7E, a′), whereas it was up-regulated by CD166 knockdown (Fig. 7E, b′).
Because the inhibition of YAP by AMOT130 is dependent on their interaction (16), we then tested if CD166 affects AMOT130-YAP interaction. We found that the interaction between AMOT130-YAP was dissociated after CD166 overexpression in a dose-dependent manner (Fig. 7F), suggesting that CD166 blocks not only AMOT130 expression but also its activity.
CD9 Enhanced the Effect of CD166 on YAP
CD166 mediates its function through either homophilic (CD166-CD166) or heterophilic (CD166-CD6) interactions (25). Recently, another cell surface molecule, CD9, was reported to be involved in the enhancement of CD166 function (25). Thus, we tested whether CD6 or CD9 synergizes CD166 in regulating YAP. It was found that overexpression of CD9 but not CD6 was able to induce YAP protein expression through reduction of AMOT130 (Fig. 8, A and B). However, CD9 did not affect either protein or mRNA expression of CD166 (Fig. 8, B and C). Although YAP protein was down-regulated in both Bel-7402 and SMMC-7721 cells with CD9 knocked down (Fig. 8C), YAP mRNA was not affected (Fig. 8D), suggesting that the regulation of YAP by CD9 is at the protein level.
FIGURE 8.

CD9 interacted with CD166 to up-regulate YAP. A, Western blots of AMOT130 and YAP in Bel-7402 cells with or without CD6-FLAG or CD9-FLAG overexpressed. B, CD9 increased YAP without affecting CD166 expression. Shown are Western blots of proteins as indicated in control cells (transfected with empty plasmid) and SMMC-7721 cells with CD9 overexpressed (transfected with CD9-Myc-expressing plasmid). C, knockdown of CD9 decreased YAP expression as measured by Western blotting. D, CD9 did not affect YAP transcription as measured by qPCR with or without CD9 knocked down (control; infected with GFP-shRNA). E, CD9 bound to CD166. CD166-HA was co-transfected with CD9-Myc into Bel-7402 cells. CD166 and CD9 associations were examined by reciprocal co-IP as indicated. F, CD9 facilitated self-binding of CD166. In CD9-Myc-transfected Bel-7402 cells, CD166-HA was immunoprecipitated with an anti-HA antibody, and Western blotting analysis of CD166-FLAG was done by an anti-FLAG antibody. Error bars, S.D.
Furthermore, the interaction between CD9 and CD166 in liver cancer cells was confirmed by performing reciprocal co-IP experiments, and it was found that exogenous CD166-HA could be readily pulled down by CD9-Myc, and vice versa (Fig. 8E). Interestingly, we found that CD9-enhanced CD166-mediated regulation of YAP was via a mechanism involving augmented clustering of CD166 molecules because overexpression of CD9 facilitated CD166-HA-CD166-FLAG homophilic interaction (Fig. 8F).
YAP Rescued CD166 Knockdown-induced Pro-apoptotic Outcome
We then tested the effect of YAP and CD166 on apoptosis. It was found that CD166 knockdown-induced pro-apoptotic effects, including decreased Bcl-2 expression and in vitro and in vivo tumor cell growth (Fig. 9, A, C, and D), as well as increased caspase-3/7 activity (Fig. 9B) could be partially rescued by simultaneous ectopic expression of YAP (Fig. 9, A and D), indicating that CD166 exerts its anti-apoptotic role at least in part via YAP.
FIGURE 9.

YAP rescued effects of CD166 depletion. A, YAP rescued CD166 knockdown-induced down-regulation of Bcl-2. Shown are Western blots of Bcl-2 in Bel-7402 cells with CD166 knocked down (infected with CD166-sh1) with or without simultaneous overexpression of YAP (transfected with YAP-FLAG-expressing plasmids). B, knockdown of CD166 (infected with CD166-sh1) induced apoptosis, which can be rescued by overexpression of YAP (infected with YAP-expressing lentivirus), as measured by caspase-3/7 activity. C, relative cell proliferation measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide in cells expressing shRNA against CD166 (CD166-sh1) with or without ectopic expression of YAP (infected with YAP-expressing lentivirus). D, ectopic expression of YAP rescued inhibitory tumor growth by silencing of CD166 in vivo. Tumor volumes were measured for 24 days after subcutaneous injection of different cells as indicated. n = 5 per group. p < 0.05 or 0.01 indicates statistic significance. E, representative IHC images of control and xenograft tissues with CD166 knocked down from D stained using anti-CD133 and anti-Ki67 antibodies. Scale bar, 200 μm. F, Western blots of stem cell markers as indicated in control cells (infected with GFP-shRNA) and Bel-7402 cells with CD166 knocked down (infected with CD166-sh1). Error bars, S.D.
CD166 and CD133 are cell surface markers that have recently been associated with cancer stem cells (6, 26). To find out whether CD166 is dedicated to stem cell formation and cell proliferation in vivo, we stained the sections from the xenografts using anti-CD133 and anti-Ki67 antibodies. It was detected that CD133 was significantly down-regulated, whereas Ki67 expression remained unchanged in CD166-knocked down xenograft compared with the control (Fig. 9E), suggesting that CD166 may influence liver cancer stem cell formation but has no effect on cell proliferation. Data from Western blotting also demonstrated that stem cell markers CD133 and c-Myc could be down-regulated in Bel-7402 cells with CD166 knocked down compared with the control (Fig. 9F). Surprisingly, other canonical stem cell markers Nanog, Sox2, and Oct-4 (the anti-Nanog, Sox2, and Oct-4 antibodies had been verified in HEK-293T cells) were undetectable in Bel-7402 cells (Fig. 9F). Collectively, CD166 and CD133 may also be used as stem cell markers in liver cancer cells, and CD166 may play an important role in liver cancer stem cell formation. The data also suggested that the positive effect of CD166 on in vitro and in vivo cell growth may not be through the regulation of cell proliferation.
DISCUSSION
In the present study, we showed that CD166 was a critical anti-apoptotic regulator in liver cancer cells. We also showed that CD166 was not only an effecter but also a stimulator of PI3K/AKT signaling; thereby, we revealed another positive autoregulatory feedback loop. Moreover, YAP was identified as a CD166 downstream effecter because this protein can partly rescue CD166 knockdown-induced apoptosis and reduced cell growth both in vitro and in vivo (Fig. 9, A–D).
AKT provides several layers of protection from apoptosis, making it an attractive target for therapeutic intervention in the treatment of malignant disease. AKT has been reported to promote YAP localization to the cytoplasm, resulting in loss from the nucleus, where it functions as a coactivator of p73, a transcription factor that mediates induction of Bax expression (27). Bax, which counters death repressor activity of Bcl-2, forms heterodimers with Bcl-2 in vivo, and the ratio of Bcl-2 to Bax determines survival or death following apoptotic stimulus (28). Knockdown of AKT target, CD166-induced apoptosis may result from the reduction the ratio of Bcl-2 to Bax because Bcl-2 could be down-regulated, whereas Bax remained unchanged and almost undetectable after silencing of CD166 (Fig. 1D) (data not shown). Furthermore, YAP intracellular localization could not be affected by CD166 (Fig. 3L). These data experimentally excluded the involvement of p73 and Bax in the AKT-CD166-YAP anti-apoptotic axis and provided new insights into the interaction between YAP and AKT and their anti-apoptotic roles in liver cancer cells.
We previously reported that NF-κB signaling regulates CD166 gene transcription via promoter binding (9). Emerging evidence has uncovered that PI3K/AKT is an upstream regulator of NF-κB (19–20). However, AKT-controlled CD166 protein expression was independent of transcription (Fig. 2, A, C, and D). Moreover, down-regulation of CD166 protein induced by increased cell density (Fig. 3G) was an outcome of inhibition of AKT but not of NF-κB (Fig. 3H). Thereby, we propose that the control of CD166 protein by AKT may not be via NF-κB but rather through protein-protein interaction with AKT (Fig. 2, G and H). Interestingly, stabilization of protein can be enhanced by AKT-dependent phosphorylation (29, 30). To the best of our knowledge, there is no direct evidence demonstrating that CD166 can be phosphorylated by AKT. The mechanism underlying the regulation of the stability of CD166 by AKT should be explored and discovered in the future.
On the basis of the fact that the anti-apoptotic role of CD166 in liver cancer cells was at least in part through induction of both YAP expression and activity (Fig. 3, A–K), we propose a model in which preferentially high expression of CD166 in liver cancer provides additional regulatory input into YAP-mediated anti-apoptotic gene transcription. Interestingly, a report suggests that one of YAP targets, CTGF, contributes to liver cancer cell dedifferentiation, in vivo cell growth, and, most importantly, resistance toward doxorubicin and TRAIL-mediated apoptosis (13). It is currently unknown whether other anti-apoptotic genes are involved in the YAP regulatory program; however, it is likely that the complex regulatory network of CD166-YAP is critical for liver cancer development.
Our study pointed to CD166 as a critical integrating point to connect PI3K/AKT, Hippo/YAP, and PKA/CREB signaling pathways in liver cancer cells (Fig. 10). The functional interactions between PI3K/AKT and Hippo/YAP signaling in cancers are probably complex and dependent on context (31, 32). The effects of PKA/CREB on Hippo/YAP also seem controversial in development and liver cancers. PKA stimulates LATS kinases and YAP phosphorylation in Drosophila and developing chick embryos (33, 34); however, previous studies from our laboratory and others suggest that the PKA downstream effecter, CREB, is able to activate YAP gene transcription through promoter binding in liver cancer cells (15, 35). Interestingly, YAP can also protect CREB from degradation (15). In our view, YAP is a multifunctional liver cancer oncogenic regulator, which acts as not only a transcription co-activator but also a protein adaptor because of our previous studies showing that YAP can exert its role in tumorigenesis through interaction with other key oncoproteins, such as TRIB2, CREB, MEK1, SRSF1, and c-Myc (14–15, 36–38).
FIGURE 10.
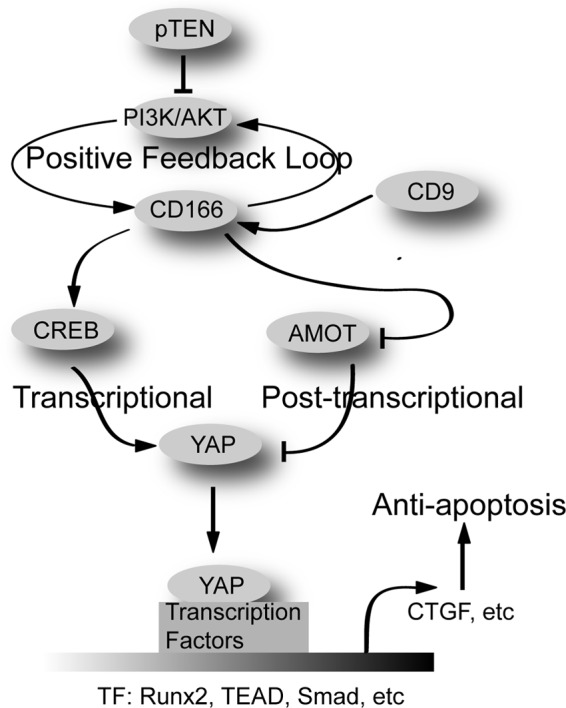
Possible mechanism underlying how CD166 exerts its role in apoptosis through YAP.
In summary, our data indicate that PI3K/AKT/CD166 modulated YAP expression and activity through at least two different mechanisms, transcriptional regulation of the YAP gene through CREB and post-transcriptional control of YAP protein stability through inhibition to AMOT130 (Fig. 10). Further exploration of the interplay among these important signaling pathways may lead to more effective therapeutic strategies for liver cancer.
This work was supported by China National 973 projects (Grants 20111812 and 20110402), Natural Science Foundation of China Grant 81301689, the Shanghai Tenth People's Hospital Climbing Training Program (to J. W.), and a China Central High School basic research-specific grant (to J. X.).
- CD166
- cluster of differentiation 166
- YAP
- Yes-associated protein
- TMA
- tissue microarray analysis
- CTGF
- connective tissue growth factor
- TRAIL
- tumor necrosis factor-related apoptosis-inducing ligand
- qPCR
- quantitative RT-PCR
- HBsAg
- hepatitis B surface antigen
- CREB
- cAMP-response element-binding protein
- CRE
- cAMP-response element
- IHC
- immunohistochemistry
- IP
- immunoprecipitation
- WB
- Western blot
- p-AKT and p-CREB
- phospho-AKT and phospho-CREB, respectively
- HCV
- hepatitis C virus
- TEAD
- TEA domain.
REFERENCES
- 1. van Kempen L. C., Nelissen J. M., Degen W. G., Torensma R., Weidle U. H., Bloemers H. P., Figdor C. G., Swart G. W. (2001) Molecular basis for the homophilic activated leukocyte cell adhesion molecule (ALCAM)-ALCAM interaction. J. Biol. Chem. 276, 25783–25790 [DOI] [PubMed] [Google Scholar]
- 2. Weichert W., Knösel T., Bellach J., Dietel M., Kristiansen G. (2004) ALCAM/CD166 is overexpressed in colorectal carcinoma and correlates with shortened patient survival. J. Clin. Pathol. 57, 1160–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burkhardt M., Mayordomo E., Winzer K. J., Fritzsche F., Gansukh T., Pahl S., Weichert W., Denkert C., Guski H., Dietel M., Kristiansen G. (2006) Cytoplasmic overexpression of ALCAM is prognostic of disease progression in breast cancer. J. Clin. Pathol. 59, 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kahlert C., Weber H., Mogler C., Bergmann F., Schirmacher P., Kenngott H. G., Matterne U., Mollberg N., Rahbari N. N., Hinz U., Koch M., Aigner M., Weitz J. (2009) Increased expression of ALCAM/CD166 in pancreatic cancer is an independent prognostic marker for poor survival and early tumour relapse. Br. J. Cancer 101, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dalerba P., Dylla S. J., Park I. K., Liu R., Wang X., Cho R. W., Hoey T., Gurney A., Huang E. H., Simeone D. M., Shelton A. A., Parmiani G., Castelli C., Clarke M. F. (2007) Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. U.S.A. 104, 10158–10163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levin T. G., Powell A. E., Davies P. S., Silk A. D., Dismuke A. D., Anderson E. C., Swain J. R., Wong M. H. (2010) Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology 139, 2072–2082.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiao J., Hindoyan A., Wang S., Tran L. M., Goldstein A. S., Lawson D., Chen D., Li Y., Guo C., Zhang B., Fazli L., Gleave M., Witte O. N., Garraway I. P., Wu H. (2012) Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PLoS One 7, e42564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jezierska A., Matysiak W., Motyl T. (2006) ALCAM/CD166 protects breast cancer cells against apoptosis and autophagy. Med. Sci. Monit. 12, BR263–BR273 [PubMed] [Google Scholar]
- 9. Wang J., Gu Z., Ni P., Qiao Y., Chen C., Liu X., Lin J., Chen N., Fan Q. (2011) NF-κB P50/P65 hetero-dimer mediates differential regulation of CD166/ALCAM expression via interaction with micoRNA-9 after serum deprivation, providing evidence for a novel negative auto-regulatory loop. Nucleic Acids Res. 39, 6440–6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zeng Q., Hong W. (2008) The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell 13, 188–192 [DOI] [PubMed] [Google Scholar]
- 11. Huo X., Zhang Q., Liu A. M., Tang C., Gong Y., Bian J., Luk J. M., Xu Z., Chen J. (2013) Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar carcinoma. Oncol. Rep. 29, 840–846 [DOI] [PubMed] [Google Scholar]
- 12. Xu M. Z., Yao T. J., Lee N. P., Ng I. O., Chan Y. T., Zender L., Lowe S. W., Poon R. T., Luk J. M. (2009) Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 115, 4576–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Urtasun R., Latasa M. U., Demartis M. I., Balzani S., Goñi S., Garcia-Irigoyen O., Elizalde M., Azcona M., Pascale R.M., Feo F., Bioulac-Sage P., Balabaud C., Muntané J., Prieto J., Berasain C., Avila M. A. (2011) Connective tissue growth factor autocriny in human hepatocellular carcinoma. Oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 54, 2149–2158 [DOI] [PubMed] [Google Scholar]
- 14. Wang J., Park J. S., Wei Y., Rajurkar M., Cotton J. L., Fan Q., Lewis B. C., Ji H., Mao J. (2013) TRIB2 acts downstream of Wnt/TCF in liver cancer cells to regulate YAP and C/EBPα function. Mol. Cell 51, 211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang J., Ma L., Weng W., Qiao Y., Zhang Y., He J., Wang H., Xiao W., Li L., Chu Q., Pan Q., Yu Y., Sun F. (2013) Mutual interaction between YAP and CREB promotes tumorigenesis in liver cancer. Hepatology 58, 1011–1020 [DOI] [PubMed] [Google Scholar]
- 16. Zhao B., Li L., Lu Q., Wang L. H., Liu C. Y., Lei Q., Guan K. L. (2011) Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 25, 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gown A. M., Willingham M. C. (2002) Improved detection of apoptotic cells in archival paraffin sections. Immunohistochemistry using antibodies to cleaved caspase 3. J. Histochem. Cytochem. 50, 449–454 [DOI] [PubMed] [Google Scholar]
- 18. Llovet J. M., Bruix J. (2008) Molecular targeted therapies in hepatocellular carcinoma. Hepatology 48, 1312–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Graham T. R., Odero-Marah V. A., Chung L. W., Agrawal K. C., Davis R., Abdel-Mageed A. B. (2009) PI3K/Akt-dependent transcriptional regulation and activation of BMP-2-Smad signaling by NF-κB in metastatic prostate cancer cells. Prostate 69, 168–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng J. C., Chou C. H., Kuo M. L., Hsieh C. Y. (2006) Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-κB signal transduction pathway. Oncogene 25, 7009–7018 [DOI] [PubMed] [Google Scholar]
- 21. Yang Z. Z., Tschopp O., Di-Poï N., Bruder E., Baudry A., Dümmler B., Wahli W., Hemmings B. A. (2005) Dosage-dependent effects of Akt1/protein kinase Bα (PKBα) and Akt3/PKBγ on thymus, skin, and cardiovascular and nervous system development in mice. Mol. Cell. Biol. 25, 10407–10418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao B., Li L., Tumaneng K., Wang C. Y., Guan K. L. (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(β-TRCP). Genes Dev. 24, 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pan D. (2010) The Hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao B., Tumaneng K., Guan K. L. (2011) The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 13, 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilsanz A., Sánchez-Martín L., Gutiérrez-López M. D., Ovalle S., Machado-Pineda Y., Reyes R., Swart G. W., Figdor C. G., Lafuente E. M., Cabañas C. (2013) ALCAM/CD166 adhesive function is regulated by the tetraspanin CD9. Cell. Mol. Life Sci. 70, 475–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lugli A., Iezzi G., Hostettler I., Muraro M. G., Mele V., Tornillo L., Carafa V., Spagnoli G., Terracciano L., Zlobec I. (2010) Prognostic impact of the expression of putative cancer stem cell markers CD133, CD166, CD44s, EpCAM, and ALDH1 in colorectal cancer. Br. J. Cancer 103, 382–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Basu S., Totty N. F., Irwin M. S., Sudol M., Downward J. (2003) Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 11, 11–23 [DOI] [PubMed] [Google Scholar]
- 28. Oltvai Z. N., Milliman C. L., Korsmeyer S. J. (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74, 609–619 [DOI] [PubMed] [Google Scholar]
- 29. Li Y., Dowbenko D., Lasky L. A. (2002) AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 277, 11352–11361 [DOI] [PubMed] [Google Scholar]
- 30. Feng J., Tamaskovic R., Yang Z., Brazil D. P., Merlo A., Hess D., Hemmings B. A. (2004) Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 279, 35510–35517 [DOI] [PubMed] [Google Scholar]
- 31. Fan R., Kim N. G., Gumbiner B. M. (2013) Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl. Acad. Sci. U.S.A. 110, 2569–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fernandez-L A., Squatrito M., Northcott P., Awan A., Holland E. C., Taylor M. D., Nahlé Z., Kenney A. M. (2012) Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2-mediated Akt activation. Oncogene 31, 1923–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu F. X., Zhang Y., Park H. W., Jewell J. L., Chen Q., Deng Y., Pan D., Taylor S. S., Lai Z. C., Guan K. L. (2013) Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 27, 1223–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim M., Kim M., Lee S., Kuninaka S., Saya H., Lee H., Lee S., Lim D. S. (2013) cAMP/PKA signalling reinforces the LATS-YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J. 32, 1543–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang T., Zhang J., You X., Liu Q., Du Y., Gao Y., Shan C., Kong G., Wang Y., Yang X., Ye L., Zhang X. (2012) Hepatitis B virus X protein modulates oncogene Yes-associated protein by CREB to promote growth of hepatoma cells. Hepatology 56, 2051–2059 [DOI] [PubMed] [Google Scholar]
- 36. Li L., Wang J., Zhang Y., Zhang Y., Ma L., Weng W., Qiao Y., Xiao W., Wang H., Yu W., Pan Q., He Y., Sun F. (2013) MEK1 promotes YAP and their interaction is critical for tumorigenesis in liver cancer. FEBS Lett. 587, 3921–3927 [DOI] [PubMed] [Google Scholar]
- 37. Xiao W., Wang J., Ou C., Zhang Y., Ma L., Weng W., Pan Q., Sun F. (2013) Mutual interaction between YAP and c-Myc is critical for carcinogenesis in liver cancer. Biochem. Biophys. Res. Commun. 439, 167–172 [DOI] [PubMed] [Google Scholar]
- 38. Wang J., Wang H., Zhang. Y., Zhen N., Zhang L., Qiao. Y., Weng W., Liu X., Ma L., Xiao W., Yu W., Chu Q., Pan Q., Sun F. (2014) Mutual inhibition between YAP and SRSF1 maintains long non-coding RNA, Malat1-induced tumourigenesis in liver cancer. Cell. Signal. 10.1016/j.cellsig.2014.01.022 [DOI] [PubMed] [Google Scholar]