

Background: The mechanism and significance of phosphoinositide metabolism during heart stress stimulations are not clear.
Results: Norepinephrine and angiotensin II increase cardiac phosphatidylinositol 4,5-bisphosphate levels via an enhanced interaction between phosphatidylinositol 4-kinase IIIβ and PKC, which correlate with a maintained systolic function.
Conclusion: Cardiac phosphoinositide turnover is enhanced.
Significance: A novel mechanism of phosphoinositide metabolism is described for modulation of cardiac function.
Keywords: Cardiac Hypertrophy, G Protein-coupled Receptors (GPCR), Phosphatidylinositol Kinase, Phospholipid Metabolism, Signal Transduction
Abstract
The seemly paradoxical Gq agonist-stimulated phosphoinositide production has long been known, but the underlying mechanism and its physiological significance are not known. In this study, we studied cardiac phosphoinositide levels in both cells and whole animals under the stimulation of norepinephrine (NE), angiotensin II (Ang II), and other physiologically relevant interventions. The results demonstrated that activation of membrane receptors related to NE or Ang II caused an initial increase and a later fall in phosphatidylinositol 4,5-bisphosphate (PIP2) levels in the primary cultured cardiomyocytes from adult rats. The possible mechanism underlying this increase in PIP2 was found to be through an enhanced activity of phosphatidylinositol 4-kinase IIIβ, which was mediated by an up-regulated interaction between phosphatidylinositol 4-kinase IIIβ and PKC; the increased activity of phosphatidylinositol 4-phosphate 5-kinase γ was also involved for NE-induced increase of PIP2. When the systolic functions of the NE/Ang II-treated cells were measured, a maintained or failed contractility was found to be correlated with a rise or fall in corresponding PIP2 levels. In two animal models of cardiac hypertrophy, PIP2 levels were significantly reduced in hypertrophic hearts induced by isoprenaline but not in those induced by swimming exercise. This study describes a novel mechanism for phosphoinositide metabolism and modulation of cardiac function.
Introduction
Phosphoinositides are important membrane phospholipids that serve important cellular functions. Among them, phosphatidylinositol 4,5-bisphosphate (PIP2)3 is a minor but central phosphoinositide that anchors and modulates numerous important signaling and cytoskeletal proteins (1). PIP2 regulates diverse cellular activities, including endocytosis, exocytosis, cell migration, focal adhesion formation, angiogenesis, transcriptional regulation, and mRNA processing (2, 3). Furthermore, the activities of numerous membrane proteins including ion channels and transporters are also regulated by PIP2 (4, 5).
Phosphoinositides and PIP2 levels in the cell membrane are dynamically regulated (6, 7). One of the well established cellular events involved in PIP2 dynamics is mediated by the activation of the Gq protein-coupled receptor (GqPCR): the stimulation of GqPCR activates phospholipase C (PLC), which in turn, hydrolyzes PIP2 into inositol 1,4,5-trisphosphate and diacylglycerol (8). Meanwhile, most PIP2 is synthesized at the membrane by the sequential phosphorylation of phosphatidylinositol and phosphatidylinositol phosphate (PIP) (9). However, the seemly paradoxical Gq agonist-stimulated phosphoinositide production has been known since the early 1950s (10–12). In pancreas and brain cortex slices, the synthesis of phosphoinositides is increased many fold by acetylcholine (10). Furthermore, total PIP levels in guinea pig heart increase with the activation of Gq/PLC-dependent pathways by carbachol and phenylephrine (13). In superior cervical ganglion neurons, enhanced phosphatidylinositol 4-kinase activity is thought to be responsible for compensated membrane PIP2 levels when challenged with bradykinin (14). Furthermore, rather than a global dynamic regulation, PIP2 hydrolysis in the cell membrane might be a localized event. Distinct pools of membrane PIP2 have been suggested early in the development of the concept of phosphoinositide signaling (6), but the exact mechanisms of formation and maintenance of such pools, indeed even their existence, remain controversial. However, such mechanisms are supported by the fact that membrane PIP2 is characterized by low lateral mobility (15), which suggests that a signaling pathway that utilizes PIP2 could use PIP2 turnover machinery locally. It is still an intriguing issue as to how phosphoinositide metabolism is modulated in the event of Gq/PLC pathway activation.
The GqPCR-PLC-PIP2 signaling pathway is arguably the most important cell signaling pathway involved in cardiac hypertrophy and failure (8). It is well established that activation of Gq/PLC signaling via several extracellular signals directly stimulates the hypertrophic response, and blocking the Gq/PLC pathway in vivo can partially attenuate the hypertrophy induced by pressure overload (16). Some important hypertrophic agonists, including norepinephrine (NE) and angiotensin II (Ang II), are well established GqPCR agonists (17); their involvement in the development of different types of cardiac hypertrophy has been well documented (8, 16, 18, 19).
In this study, we studied the modulation and mechanism of cardiac phosphoinositides by NE and Ang II at both the cellular and whole animal levels. We describe an agonist-induced increase in both PIP and PIP2 levels and present a novel mechanism for these increases to be the result of the enhanced activity of PI4KIIIβ, mediated by an up-regulated interaction between PI4KIIIβ and PKC; furthermore, for NE, an increased activity of phosphatidylinositol 4-phosphate 5-kinase γ was also involved. This enhanced PIP2 turnover is correlated with maintained cardiac systolic function in stimulated cardiac hypertrophy.
EXPERIMENTAL PROCEDURES
Cardiomyocyte Isolation and Stimulation with Agonists
Cardiomyocytes were isolated from male Sprague-Dawley rats (200–250 g). The rats were anesthetized (sodium pentobarbital, 120 mg/kg), and then the hearts were rapidly excised and mounted on a Langendorff apparatus. The hearts were perfused for 1 min with Ca2+-free Tyrode's solution (pH 7.4), containing 140 mm NaCl, 5.4 mm KCl, 1.0 mm MgCl2, 10 mm HEPES, and 10 mm glucose; gassed with 95% O2, 5% CO2; and kept at 37 °C. The hearts were then transferred to the perfusion medium containing 0.05% collagenase II, 0.1% BSA, and 50 μm CaCl2. At the end of a 24-min recirculation period, the ventricle was cut into small pieces to obtain cardiomyocytes that were suspended for 20 min each in buffers containing gradually increasing extracellular Ca2+ concentrations (250, 500, and 750 μm) to a final concentration of 1 mm containing 1% BSA. The final pellet was resuspended in medium 199 (Invitrogen 12340) and plated onto a 6-well plate at 4.5 × 105 cells/well in a 5% CO2 humidified incubator at 37 °C.
After a 24-h incubation, the cells were subjected to treatments with 5 μm NE or 3 μm Ang II for variable times. Further experiments were carried out in which cardiomyocytes were pretreated for 30 min with prazosin (3 μm), losartan (3 μm), propranolol (3 μm), U73122 (0.8 μm), U73343 (0.8 μm), bisindolylmaleimide-1 (Bis-1, 2 μm), H89 (30 μm), wortmannin (10 μm), or cycloheximide (CHX, 10 μm) followed by incubation with NE or Ang II.
Lipid Extraction and Membrane PIP2 Assay
Cellular lipids were extracted from cardiomyocytes (4.5 × 105 cells/sample) after different treatments. The cell pellets were first washed with 0.75 ml of 0.5 m TCA and 0.5 ml of 5% TCA with 1 mm EDTA and were then resuspended in 0.325 ml of chloroform, methanol, 12 n HCl (40:80:1, v/v/v) for 15 min at room temperature and vortexed vigorously four times during this period. Then 0.125 ml of chloroform and 0.225 ml of 0.1 n HCl were added to the cells, which were then vortexed for 2 min and centrifuged (1,500 rpm for 5 min). The bottom layer was transferred to another tube for drying in a vacuum dryer. This method of lipid extraction is modified from the protocol described previously (20).
The dried lipid samples were reconstituted with chloroform/methanol/water (5:5:1 v/v/v), vortexed, and sonicated in an icy water bath for 5 min. Phospholipids in the samples were then analyzed using TLC (21, 22). One-microliter aliquots of the reconstituted lipid samples were subjected to analysis on TLC plates (GF254). The mobile phase for TLC was chloroform, methanol, 4 n NH4OH (45:35:10, v/v/v). Phospholipids were visualized with iodine vapor. PIP and PIP2 were confirmed by mass spectrometry, and the images of the spots were analyzed by densitometry analysis software.
Lipid-Protein Overlay Assay
For this assay, a PIP2 mass strip kit (Echlon Biosciences) was used. Three microliters of phospholipid from each cardiomyocyte sample (per sample/15 μl in 80:80:1 chloroform, methanol, 1 n HCl) was spotted onto a nitrocellulose membrane and dried. Membranes were blocked in PBS containing 3% BSA at room temperature for 1 h. The procedure followed the manufacturer's protocol. Images of the spots were analyzed by densitometry analysis software.
Co-immunoprecipitation and Western Blot
Cardiomyocytes (0.9–1 × 106 cells/sample) were harvested in lysis buffer. One hundred microliters of cell lysate were incubated with 0.5 μg of antibody and gently rocked for 12 h at 4 °C. Then 20 μl of protein G beads (GE Biotech) were added with gentle rocking for 2 h at 4 °C. After washing five times with washing buffer (PBS buffer, protease inhibitors, 0.5% Nonidet P-40, and 0.1% Triton X-100), the co-immunoprecipitated samples were subjected to SDS-PAGE and immunoblot analysis using appropriate antibodies. Cell lysates incubated with protein G beads alone were routinely used as a negative control. Studies were repeated at least three times, and the antibodies used for immunoprecipitation and immunoblot were also exchanged.
The proteins was mixed with loading buffer (10% glycerol, 50 mm Tris-HCl, 2% SDS, 5% mercaptoethanol, and 0.02% bromphenol blue), heat-denatured at 95 °C for 5 min, and subjected to SDS-PAGE. The proteins resolved by 10% SDS-PAGE were transferred to nitrocellulose membranes (Millipore) in transfer buffer (20% methanol, 15.6 mm Tris-base, and 120 mm glycine) for 3 h at 100 V and were probed with anti-PIP5Kγ or anti-PI4KIIIβ antibodies (1:500 dilution; Millipore) or PKC isoforms antibodies (1:500 dilution; Epitomics), overnight at 4 °C. Nonspecific binding was blocked with 1.5% (w/v) evaporated skimmed milk (Difco) in TBS (154 mm NaCl, 10 mm Tris-base). Anti-rabbit or anti-mouse secondary antibodies conjugated to IRDye700DX (1:5000 dilution; Rockland) were used to probe primary antibodies. Protein bands were detected and quantified on an Odyssey two-color infrared imaging system (LI-COR Biosciences).
Contraction Measurements
The cardiomyocytes were transferred to a chamber mounted on the stage of an inverted microscope and superfused with Tyrode's solution (1.8 mm Ca2+) at room temperature. The cells were field stimulated with a 4-ms bipolar, supramaximal pulse stimulator (IonOptix, Milton, MA), and the frequency of the stimulation was chosen to be 1 Hz (15 V). Prior to recording, the cells were stimulated for 3 min at a constant pacing rate to allow them to adapt to the new frequency and reach steady state (23). Sarcomere shortening was recorded using an IonOptix system, and recordings were analyzed using the IonOptix analysis package (IonOptix, Milton, MA).
Rat Cardiac Hypertrophic Models
Swimming exercise was used to establish the physiological hypertrophic model. The rats (180 ± 20 g) underwent adaptive training for the first 3 days; then the training period was increased to 2 h/day, 6 days/week. The training procedure normally lasted for 8 weeks. To establish the pathological hypertrophic model, the isoproterenol was injected (subcutaneously 5 mg/kg/day) for 1 week (24–26).
Statistical Analysis
All results are reported as the means ± S.E. Comparisons of two groups only were accomplished using the unpaired Student's t test. Experiments with more than two groups were compared by one-way analysis of variance. All data were analyzed using Origin 7.5, and the figures were plotted using Adobe Illustrator CS4.
RESULTS
NE and Ang II Increase PIP2 Levels in Cardiomyocytes
NE and Ang II are well known GqPCR agonists that are expected to induce membrane PIP2 hydrolysis. We first tested the effects of these agonists on total PIP2 abundance in cultured cardiomyocytes from adult rats. It has been shown that incubation of adult cardiomyocytes with NE (5 μm) results in a rapid activation of the Gq/PLC pathway with a maximal effect occurring at 2 h (27), whereas Ang II needed a longer time (24 h) but a lower working concentration (28, 29). After incubation with NE (5 μm, 2 h) or Ang II (3 μm, 24 h), the total abundance of PIP2 in cardiomyocytes (from 4.5 × 105 cells), measured using TLC, was found to be significantly increased by 19 ± 6 and 18 ± 5%, respectively (Fig. 1A). We also used a lipid-protein overlay assay (30) (Fig. 1B), which confirmed the TLC results. We tried shorter incubation times with Ang II (2 h) or NE (20 min) treatment but failed to observe any prominent effects on PIP2 levels (see Fig. 5A). Specific antagonists of the α-adrenergic receptor (α1) prazosin or Ang II receptor (AT1) losartan (3 μm) abolished the effects of NE and Ang II, respectively (Fig. 1C), indicating the involvement of GqPCR pathway.
FIGURE 1.
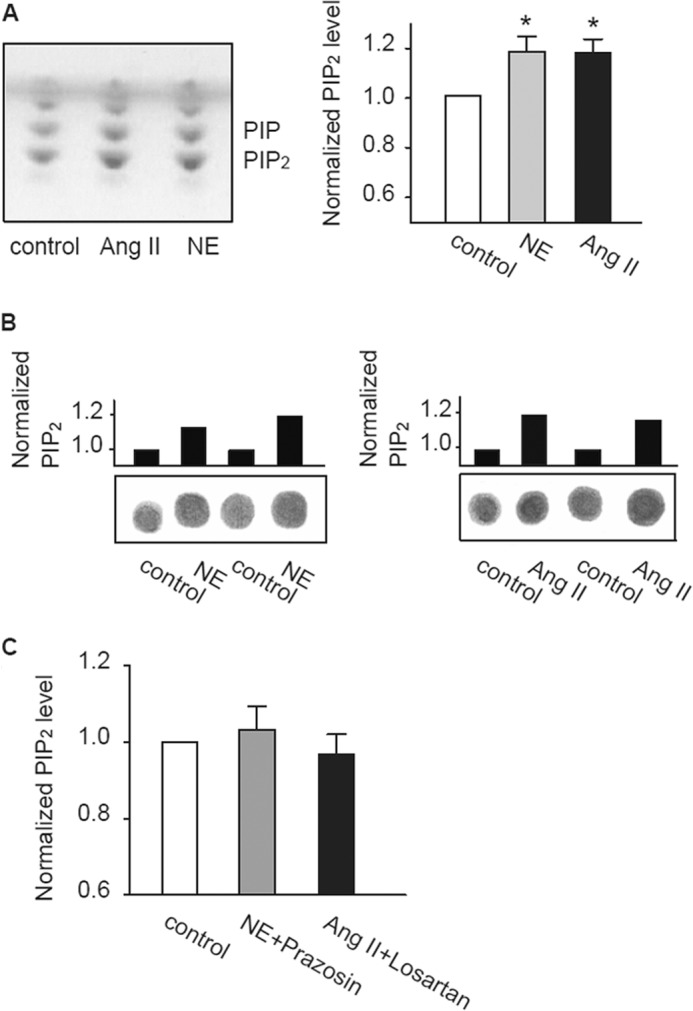
The effects of NE and Ang II on PIP2 levels in cultured adult rat cardiomyocytes. A, total PIP2 levels in adult rat cardiomyocytes treated with Ang II (3 μm, 24 h) or NE (5 μm, 2 h) measured by TLC. Iodine staining of phospholipids is shown in the left panel. Normalized PIP2 levels obtained from TLC analysis were shown in the right panel. *, p < 0.01 versus control (n = 17 animals). B, representative results of the lipid-protein overlay assay of PIP2 levels in adult rat cardiomyocytes. C, the effect of prazosin (3 μm) and losartan (3 μm) on the NE- and Ang II-induced increase in total PIP2, respectively (n = 7 animals).
FIGURE 5.
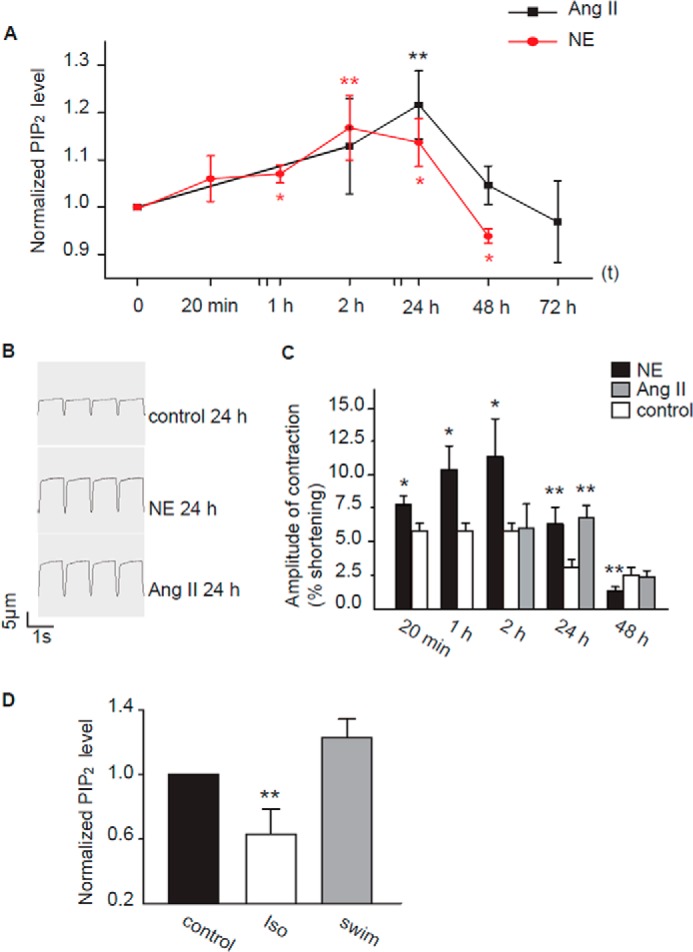
Correlation of PIP2 level with cardiomyocyte function and different hypertrophic hearts. A, time course for the effects of NE and Ang II on PIP2 level of cardiomyocytes. NE (5 μm, red circles) and Ang II (3 μm, black squares) were administered to cultured rat cardiomyocytes for the periods of time indicated (n = 6–15 animals). B and C, the time course of the effects of NE and Ang II on cardiomyocyte systolic function. B, a recording sample of sarcomere shortening; myocytes were first incubated with either NE (5 μm) or Ang II (3 μm) for the periods of times indicated. C, the time course of the effects of NE and Ang II on cardiomyocyte systolic function. The data show the percentages of cell length shortening. The control values were from the cells treated with solvent for the same periods of time as indicated (n = 11–17 animals). D, total PIP2 levels in rat hearts from Iso- and swimming-induced cardiac hypertrophy (n = 4 animals). *, p < 0.05; **, p < 0.01 versus control.
PLC, PKC, and PI4K Are Involved in the Increase in PIP2 Level
It has long been known that Gq agonists can stimulate phosphoinositide production, but the mechanism is not clear (10–13). We made an effort to understand this mechanism.
In parallel with changes in PIP2 levels, PIP levels were also increased after treatment with NE or Ang II (Fig. 2, B and D), indicating an increased activity of PI4K (31). PLC and PKC are signaling molecules of GqPCR, and PI4K is the key enzyme for PIP2 synthesis. The roles of PLC, PKC, and PI4K were examined for their involvement in the observed effects of NE and Ang II on phosphoinositide production. The PLC inhibitor U73122 (0.8 μm; inactive analog U73343, 0.8 μm, was used as a control), the PKC inhibitor Bis-1 (2 μm), and the PI4K inhibitor wortmannin (10 μm) were used to assess the roles of these molecules. Cardiomyocytes were treated with these inhibitors for 30 min before being treatment with NE or Ang II. Panels A and B of Fig. 2 show the results of NE-treated cells, and panels C and D show the results of Ang II-treated cells. Pretreatment of cardiomyocytes with U73122, Bis-1, and wortmannin prevented the NE- or Ang II-induced increase in PIP and PIP2. These results indicate that both the Gq/PLC signaling pathway and PI4K are involved in the NE- and Ang II-induced increase of phosphoinositides in cardiomyocytes.
FIGURE 2.
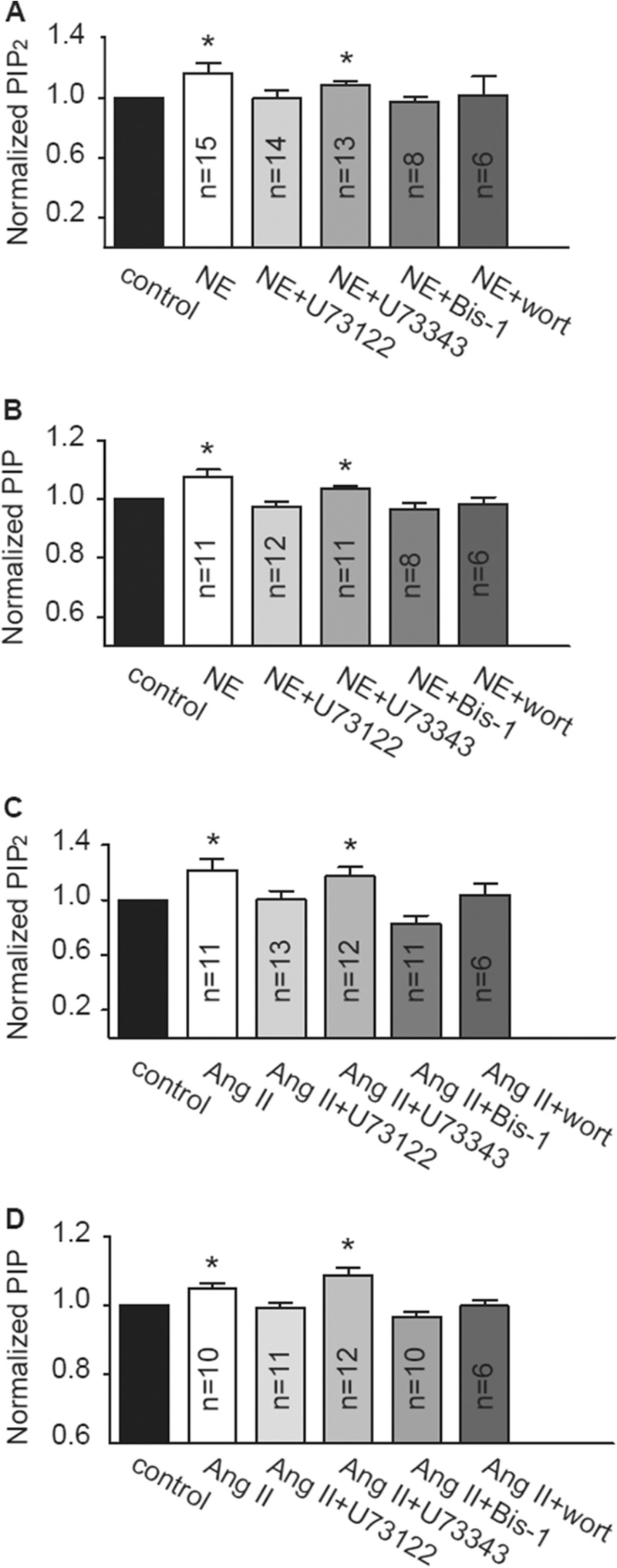
The effects of blockers of PLC, PKC, and PI4K on the NE- and Ang II-induced increase in PIP2 and PIP levels. A–D, the effects of U73122 (0.8 μm), U73343 (0.8 μm), Bis-1 (2 μm), and wortmannin (wort, 10 μm) on the NE-induced (A and B) or Ang II-induced (C and D) increase in total PIP2 and PIP levels in cardiomyocytes. All blockers were added 30 min before either NE or Ang II was given. PIP and PIP2 were extracted after incubation with NE (2 h) or Ang II (24 h). These results were confirmed using lipid-protein overlay assay. *, p < 0.05 versus control.
The Interaction between PI4KIIIβ and PKC Is Responsible for Enhanced PIP2 Synthesis
Four mammalian PI4Ks have been cloned and characterized; two isoforms of type III PI4Ks are sensitive to inhibition by high micromolar concentrations of wortmannin, and two isoforms of type II PI4Ks are wortmannin-insensitive but can be inhibited by low micromolar concentrations of adenosine (9, 32). We first tested the effect of PIK93, a specific blocker of PI4KIIIβ, which can inhibit the activity of PI4KIIIβ by ∼90% at a concentration of 250 nm (33). PIK93 abolished the NE- and Ang II-induced increases in PIP2 and PIP (Fig. 3A) and reduced the levels of PIP slightly (Fig. 3A). Similar to wortmannin (13) but contrary to adenosine, PIK93 had no effect on the basal level of PIP2 (Fig. 3A). We then tested the effect of adenosine. Adenosine (2 μm) greatly reduced basal PIP2 and PIP levels. Under this influence, total PIP2 and PIP levels (basal plus NE induced) in the presence of NE and adenosine were not significantly different from control PIP2 and PIP levels (basal only) in the absence of NE and adenosine. These results suggest that adenosine does not inhibit NE-induced increases in phosphoinositides and may even enhance the effect of NE, considering that the absolute increase in phosphoinositides induced by NE was greater in the presence of adenosine than in the absence of NE (Fig. 3B). A similar conclusion could also be made for the effect of adenosine on Ang II-induced effects on the phosphoinositides (Fig. 3B). These results suggest that PI4KIII is mostly involved in NE- and Ang II-induced increases in phosphoinositide production.
FIGURE 3.

NE, Ang II, and PMA enhanced the interaction between PKC and PI4KIIIβ. A and B, the effects of PIK93 (A, 250 nm) and adenosine (B, 2 μm) on basal and NE-induced (2 h) or Ang II-induced (24 h) increase in PIP and PIP2 in primary cultured cardiomyocytes (n = 5–7 animals): adenosine (2 μm, 2 h), but not PIK93 (250 nm, 2 h), reduced basal PIP2 levels in primary cultured cardiomyocytes. C, co-immunoprecipitation of PKC isoforms and PI4KIIIα. D and E, representative results of co-immunoprecipitation experiments for interactions between PKCα and PI4KIIIβ (D) or between PKCβII and PI4KIIIβ (E) are shown in the left panels. Protein lysates from cardiomyocytes, which were treated with NE (5 μm), Ang II (3 μm), or Iso (0.5 μm) for 2 h, or PMA (0.5 μm) for 20 min, were immunoprecipitated and blotted with anti-PI4KIIIβ, anti-PKCα, or anti-PKCβII antibodies as indicated. The right panels show summary data for the normalized relative intensity of the precipitates (n = 7 animals). *, p < 0.05 versus control. IP, immunoprecipitation.
We then further investigated how PI4KIII is involved in phosphoinositide production. Our previous study in Xenopus oocytes demonstrated that activation of PKC increases the activity of PI4KIII through an enhanced interaction between PKC and PI4KIII (21, 34). Because PKC is also involved in the NE- and Ang II-induced increase in phosphoinositides in this study (Fig. 2), we reasoned that a similar mechanism could exist in cardiomyocytes. We thus studied the interaction of PI4K with two PKC isoforms: PKCα and PKCβII. A previous study suggested that both PKCα and PKCβII could interact with PI4KIIIβ in Xenopus oocytes (21, 34). For the cardiomocytes studied here, co-immunoprecipitation results showed that both PKC isoforms α (Fig. 3D) and βII (Fig. 3E) were able to interact with PI4KIIIβ, but not with PI4KIIIα (Fig. 3C). Furthermore, the interaction between PKC and PI4KIIIβ was enhanced after treatment of cardiomyocytes with NE (5 μm) or Ang II (3 μm) for 2 h or PMA (a potent PKC activator, 0.5 μm) for 20 min, yet the expression of PI4KIIIβ did not change markedly. Thus, although both isoforms (α and β) of PI4KIII are known to supply the PI4P substrate during GqPCR-activated PLC signaling (35), only PI4KIIIβ was able to interact with activated PKC. Furthermore, treatment of cardiomyocytes with isoprenaline (Iso, 0.5 μm, 2 h), which selectively activates β-adrenergic receptors, did not affect the interaction between PKC and PI4K. These results suggest that activation of the Gq/PLC pathway (and subsequent activation of PKC) could enhance the interaction between PKC and PI4KIIIβ, which contributes to increased cellular PIP2 levels.
Role of PIP5K in the NE-induced PIP2 Increase
NE, unlike Ang II, apart from activating the Gq/PLC/PKC pathway, also activates the Gs/cAMP/PKA pathway mediated by β-adrenergic receptors (36, 37). This difference may explain the fact that the NE-induced increase of PIP2 occurs faster than the Ang II-induced does. We thus tested whether the Gs/cAMP/PKA pathway is also involved in the NE-induced PIP2 increase.
For this, we compared the effects of NE and Ang II on the expression of PIP5K protein, a key kinase in PIP2 synthesis (38). Among the three isoforms (α, β, and γ) of PIP5K, the expression of the γ, but not the α and β isoforms (data not shown), was found in cardiomyocytes to be up-regulated after stimulation with NE (5 μm, 2 h), Iso (0.5 μm, 2 h), Ang II (3 μm, 2 h), and PMA (0.5 μm, 20 min). However, the effect of NE and Iso on PIP5Kγ was significantly greater than that of Ang II or PMA (Fig. 4A). Consistent with the involvement of the β receptor/cAMP/PKA pathway, the enhanced expression of PIP5Kγ by NE was abolished by propranolol (a β receptor blocker) and H89 (a PKA blocker), but not by prazosin (an α1 receptor blocker) and Bis-1 (a PKC blocker) (Fig. 4B). Consistent with these results, propranolol abolished the NE-induced increase in PIP2 (Fig. 4C), and Iso induced an increase in PIP2 levels, although it was less efficient than NE (11 ± 3% versus 19 ± 6%; Fig. 4C). These results suggest that activation of β-adrenergic receptors and the activation of the downstream signaling pathway by NE led to an increased expression of PIP5Kγ, which contributes to the increased synthesis of PIP2.
FIGURE 4.
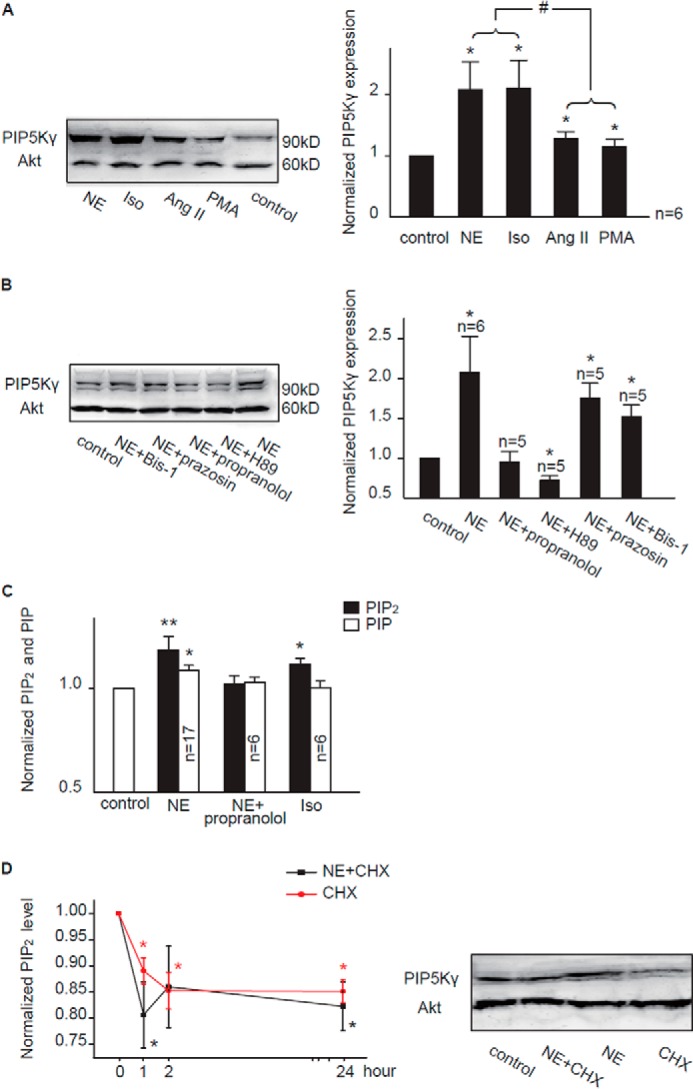
Activation of the β-adrenergic receptor increases expression of PIP5Kγ and PIP2 levels. A and B, representative Western blots of membrane proteins from cardiomyocytes probed with antibodies against PIP5Kγ and Akt are shown in the left panels. Normalized data from densitometry analyses are shown in the right panels. *, p < 0.05 versus control. #, p < 0.05. The Western blot band intensity of each group was first normalized to each corresponding Akt band intensity. The concentrations of the drugs used were as follows: NE (5 μm), Iso (0.5 μm), Ang II (3 μm), PMA (0.5 μm), propranolol (3 μm), H89 (30 μm), prazosin (3 μm), and Bis-1 (2 μm). C, the effect of Iso on PIP2 levels and the effect of propranolol on the NE-induced increase of PIP2 levels. D, left panel, effect of CHX (10 μm) on the NE-induced increase in PIP2 levels; right panel, effect of CHX (10 μm) on PIP5Kγ expression. CHX was able to down-regulate cardiac PIP5Kγ expression with or without NE. The summary data were normalized to the corresponding control values, which were obtained from cells treated with solvent alone for the same period of time as for CHX treatment (n = 5 animals). *, p < 0.05; **, p < 0.01 versus control.
The above results suggest a relatively rapid turnover of PIP5Kγ. It has been reported that lipid kinases like PIP5K turned over rapidly enough to alter cellular PIP2 levels within 1 h (39). Nonetheless, we tested the effects of CHX, a protein synthesis inhibitor. In the presence of CHX, the NE-induced increase in total PIP2 and PIP5Kγ expression in cardiomyocytes was inhibited (Fig. 4D).
PIP2 Level Is Correlated with the Cardiac Function
Finally, we studied whether the NE- and Ang II-induced effects on PIP2 turnover are associated with their effects on the function of cardiomyocytes. Both NE and Ang II have long been known to be associated with cardiac hypertrophy and heart failure, which often manifest an altered cardiac function. We found that a prolonged incubation of cardiomyocytes with NE or Ang II resulted in a pattern of PIP2 changes, which correlated with altered cardiomyocyte systolic function. When cardiomyocytes were incubated with NE (5 μm) or Ang II (3 μm) for up to 72 h (the maximal incubation time for NE was 48 h, because many cells died after a 72-h treatment), both NE and Ang II induced a gradual increase in PIP2 levels within the first 24 h, but afterward, the effects of NE and Ang II began to recede, and PIP2 levels fell back to control or even lower levels (Fig. 5A). The systolic function of the cultured cardiomyocytes was assessed by measuring sarcomere shortening induced by an electrical stimulation (for details see “Experimental Procedures”), which represents the amplitude of cardiomyocyte contraction (Fig. 5B). In our hands, cultured cardiomyocytes can maintain their systolic function up to 48 h, although the contraction gradually weakened with time. When applied to cultured cardiomyocytes up to 48 h, NE (5 μm) or Ang II (3 μm) affected the contraction of cardiomyocytes with a pattern and time course similar to the effect of NE and Ang II on PIP2 levels: the contraction gradually increased within a 24-h incubation with either NE or Ang II but fell back to control levels after a 48-h incubation (Fig. 5C). In the cells treated with NE for 48 h, contraction actually weakened to levels lower than the control.
To further investigate the relationship between PIP2 level and cardiac function, we studied changes in PIP2 level in intact hearts from two rat models of cardiac hypertrophy: swimming-induced compensatory physiological hypertrophy and Iso-induced pathological hypertrophy. The systolic and diastolic functions of these two models were first assessed (data not shown), and the cardiac functions of the swimming rats were better maintained in comparison with those of the Iso-injected rats. As shown in Fig. 5D, the PIP2 levels in the Iso-induced hearts were significantly reduced compared with the time-matched controls; PIP2 levels in swimming-induced hearts were increased compared with control hearts by 23 ± 12%, although the increase did not reach statistical significance. The above results suggest that maintained PIP2 levels in the membrane of cardiomyocytes are important for compensatory cardiac function.
DISCUSSION
Activation of GqPCR is expected to hydrolyze PIP2 into inositol 1,4,5-trisphosphate and diacylglycerol; thus, membrane PIP2 levels would be expected to fall. However, in this study, we demonstrated that activation of α1 receptor (and β1 receptor) or AT1 receptor with NE and Ang II, respectively, did not reduce but increased cellular PIP2 levels in primary cultured adult rat cardiomyocytes. These results, combined with earlier findings in pancreas, brain (10–12), and hearts (13), point to a general phosphoinositide metabolism event in the cell: activation of GqPCR not only initiates the hydrolysis of PIP2 but also simultaneously starts the biochemical mechanisms that favor the increased resynthesis of PIP2.
Importantly, we provided a novel mechanism for this GqPCR-mediated phosphoinositide production: PKC mediates the enhancement of PI4K activity. First, using specific blockers, we found that two downstream signaling molecules, PLC and PKC, were indispensable to the NE/Ang II-induced increase in PIP2 levels. Thus, it seems that the agonist-induced activation of the Gq/PLC pathway and the hydrolysis of PIP2 itself initiate the compensatory mechanism of enhanced PIP2 synthesis, a handy mechanism for maintaining global PIP2 stabilization. We then demonstrated that the key to the coupling of PIP2 hydrolysis and enhanced synthesis was the interaction between PKC, the downstream product of PIP2 hydrolysis, and PI4K; activation of PKC by the Gq-PLC-PIP2 pathway increases its interaction with PI4K, which increases the activity of the latter.
We went further to show that only PI4KIIIβ, but not all PI4Ks, selectively participates in this PKC-mediated event. Mammalian cells express two classes of PI4Ks, termed types II and III, and each class contains α- and β-isoforms (40). The type III kinases are inhibited by micromolar concentrations of wortmannin (9). The fact that wortmannin and PIK93, a specific blocker of PI4KIIIβ, abolished the NE/Ang II-induced PIP2 increase but did not affect basal PIP2 levels suggests that NE/Ang II selectively activates the type III kinases, more specifically, PI4KIIIβ. On the other hand, adenosine, a proposed type II kinase inhibitor, reduced basal PIP2 levels but did not affect the NE/Ang II-induced PIP2 increase. These results suggest that PIP2 in the membrane could come from different pools that are synthesized by different PI4Ks. The type III PI4Ks come into play only when phosphoinositides begin to be depleted in the membrane with continued PLC activation (41). As membrane trafficking to the cell membrane is increased during GqPCR activation, membrane insertion during receptor activation likely brings PI4Ks to the surface membrane, along with other materials required for the synthesis of PIP2. In our previous study, we reported that an enhanced interaction between PKC and PI4KIIIβ is responsible for the PKC-mediated activation of PIP and PIP2 synthesis in Xenopus oocytes (34). In this study, we confirmed that a similar mechanism exists in cardiomyocytes, with an important role linking the Gq/PLC pathway and membrane phosphoinositide metabolism.
Although NE is more often considered to be an agonist of α-adrenergic receptors, it does activate β-adrenergic receptors (19, 36), which also contribute to phosphoinositide production. However, the β receptor-mediated PIP2 increase was primarily via the increased expression of PIP5Kγ. This result implies that the expression of PIP5Kγ is regulated within minutes of NE action. Although we did not find markedly increased expression of PI4K (only its increased interaction with PKC) in this study, we did find increased expression of PI4K in Xenopus oocytes when treated with PMA within tens of minutes (34). These results are not surprising because enzymes of crucial significance in metabolic and growth processes frequently have short half-lives because their function is to appear promptly when there is expression of a vital process and to decline rapidly when their role is completed. Involvement of the rapid turnover of PIP5Kγ in NE-induced synthesis in this current study was manifested by the effect of CHX, a protein synthesis inhibitor.
β receptor is a Gs-coupled receptor, the activation of which usually leads to the activation of cAMP/PKA pathway. Not surprisingly, the increased expression of PIP5Kγ induced by NE was blocked by PKA blocker H89. Although Ang II and PMA also increased the expression of PIP5Kγ, their effects were much weaker than those of NE or Iso. The weak effect brought by Ang II and PMA on PIP5Kγ expression is likely the result of another mechanism: there have been reports suggesting that cross-talk between the cAMP/PKA pathway and PKC activation might regulate PIP5K activity (38).
What then is the physiological significance of the NE/Ang II-induced modulation of cardiomyocyte PIP2? Because the activities of PLC and PKC are essential to NE/Ang II-induced cardiomyocyte hypertrophy (8, 42), PIP2 may play an important role in the hypertrophic remodeling process through the functional modulation of signaling proteins and ion channels (43). Several transporters and ion channels families in cardiac myocytes, including KATP channels, and Na+-Ca2+ exchangers (44), GIRK/KAch (45), and KCNQ/IKs (46), need PIP2 for their physiological functions. Importantly, these ion channels are modulated by GqPCR agonists, including phenylephrine and Ang II, through modulation of membrane PIP2 (47). It is not clear whether the PIP2-mediated modulation of these ion channels and transporters directly contributes to the modulation of cardiac function in this study. Nonetheless, in this study, the measured systolic function correlated well with the PIP2 levels in cultured cardiomyocytes treated with NE or Ang II. Furthermore, the apparent disparity in cardiac PIP2 level between the two hypertrophic animal models indicates that PIP2 has different metabolic patterns in different functional states of hypertrophic processes. It has already been reported that wortmannin can induce heart failure (48–50). Thus, blocking the resynthesis of PIP2 with wortmannin may accelerate the process of cardiomyocyte hypertrophy. Overall, the results indicate that the increase in cellular PIP2 levels is a possible compensatory mechanism related to maintaining normal cardiomyocyte systolic function. Our present results suggest that membrane PIP2 could be a crucial factor in determining compensatory capability, at least systolic function, in hearts under stress.
This work supported by National Natural Science Foundation of China Grant 31270882 (to H. Z.) and National Basic Research Program of China Grant 2013CB531302 (to H. Z.).
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- GqPCR
- Gq protein-coupled receptor
- PLC
- phospholipase C
- PIP
- phosphatidylinositol 4-phosphate
- PI4K
- phosphatidylinositol 4-kinase
- NE
- norepinephrine
- Ang II
- angiotensin II
- Bis-1
- bisindolylmaleimide-1
- PMA
- phorbol 12-myristate 13-acetate
- Iso
- isoprenaline
- PKA
- protein kinase A
- PIP5K
- phosphatidylinositol 4-phosphate 5-kinase
- CHX
- cycloheximide.
REFERENCES
- 1. Gamper N., Shapiro M. S. (2007) Target-specific PIP2 signalling. How might it work? J. Physiol. 582, 967–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rajala R. V., Anderson R. E. (2010) Focus on molecules. Phosphatidylinositol-4,5-bisphosphate (PIP2). Exp. Eye Res. 91, 324–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mellman D. L., Anderson R. A. (2009) A novel gene expression pathway regulated by nuclear phosphoinositides. Adv. Enzyme Regul. 49, 11–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hansen S. B., Tao X., MacKinnon R. (2011) Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. 477, 495–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suh B. C., Hille B. (2008) PIP2 is a necessary cofactor for ion channel function. How and why? Annu. Rev. Biophys. 37, 175–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hilgemann D. W. (2007) Local PIP2 signals. When, where, and how? Pflugers Arch. 455, 55–67 [DOI] [PubMed] [Google Scholar]
- 7. McLaughlin S., Wang J., Gambhir A., Murray D. (2002) PIP2 and proteins. Interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175 [DOI] [PubMed] [Google Scholar]
- 8. Jalili T., Takeishi Y., Walsh R. A. (1999) Signal transduction during cardiac hypertrophy. The role of Gαq, PLCβI, and PKC. Cardiovasc. Res. 44, 5–9 [DOI] [PubMed] [Google Scholar]
- 9. Clayton E. L., Minogue S., Waugh M. G. (2013) Phosphatidylinositol 4-kinases and PI4P metabolism in the nervous system. Roles in psychiatric and neurological diseases. Mol. Neurobiol. 47, 361–372 [DOI] [PubMed] [Google Scholar]
- 10. Hokin L. E., Hokin M. R. (1955) Effects of acetylcholine on the turnover of phosphoryl units in individual phospholipids of pancreas slices and brain cortex slices. Biochim. Biophys. Acta 18, 102–110 [DOI] [PubMed] [Google Scholar]
- 11. Hokin L. E., Hokin M. R. (1956) Metabolism of phospholipids in vitro. Can. J. Biochem. Physiol. 34, 349–360 [PubMed] [Google Scholar]
- 12. Hokin L. E., Huebner D. (1967) Radioautographic localization of the increased synthesis of phosphatidylinositol in response to pancreozymin or acetylcholine in guinea pig pancreas slices. J. Cell Biol. 33, 521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nasuhoglu C., Feng S., Mao Y., Shammat I., Yamamato M., Earnest S., Lemmon M., Hilgemann D. W. (2002) Modulation of cardiac PIP2 by cardioactive hormones and other physiologically relevant interventions. Am. J. Physiol. Cell Physiol. 283, C223–C234 [DOI] [PubMed] [Google Scholar]
- 14. Winks J. S., Hughes S., Filippov A. K., Tatulian L., Abogadie F. C., Brown D. A., Marsh S. J. (2005) Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J. Neurosci. 25, 3400–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cho H., Kim Y. A., Ho W. K. (2006) Phosphate number and acyl chain length determine the subcellular location and lateral mobility of phosphoinositides. Mol. Cells 22, 97–103 [PubMed] [Google Scholar]
- 16. Wang Y. (2001) Signal transduction in cardiac hypertrophy-dissecting compensatory versus pathological pathways utilizing a transgenic approach. Curr. Opin. Pharmacol. 1, 134–140 [DOI] [PubMed] [Google Scholar]
- 17. Clerk A., Sugden P. H. (1999) Activation of protein kinase cascades in the heart by hypertrophic G protein-coupled receptor agonists. Am. J. Cardiol. 83, 64H–69H [DOI] [PubMed] [Google Scholar]
- 18. Adams J. W., Sakata Y., Davis M. G., Sah V. P., Wang Y., Liggett S. B., Chien K. R., Brown J. H., Dorn G. W., 2nd (1998) Enhanced Gαq signaling. A common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc. Natl. Acad. Sci. U.S.A. 95, 10140–10145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Colucci W. S. (1998) The effects of norepinephrine on myocardial biology. Implications for the therapy of heart failure. Clin. Cardiol. 21, I20–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gray A., Olsson H., Batty I. H., Priganica L., Peter Downes C. (2003) Nonradioactive methods for the assay of phosphoinositide 3-kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal. Biochem. 313, 234–245 [DOI] [PubMed] [Google Scholar]
- 21. Zhang X., Chen X., Jia C., Geng X., Du X., Zhang H. (2010) Depolarization increases phosphatidylinositol (PI) 4,5-bisphosphate level and KCNQ currents through PI 4-kinase mechanisms. J. Biol. Chem. 285, 9402–9409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu C., Watras J., Loew L. M. (2003) Kinetic analysis of receptor-activated phosphoinositide turnover. J. Cell Biol. 161, 779–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Banyasz T., Lozinskiy I., Payne C. E., Edelmann S., Norton B., Chen B., Chen-Izu Y., Izu L. T., Balke C. W. (2008) Transformation of adult rat cardiac myocytes in primary culture. Exp. Physiol. 93, 370–382 [DOI] [PubMed] [Google Scholar]
- 24. Nakayama H., Bodi I., Maillet M., DeSantiago J., Domeier T. L., Mikoshiba K., Lorenz J. N., Blatter L. A., Bers D. M., Molkentin J. D. (2010) The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ. Res. 107, 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Q., Wilkins B. J., Lee Y. J., Ichijo H., Molkentin J. D. (2006) Direct interaction and reciprocal regulation between ASK1 and calcineurin-NFAT control cardiomyocyte death and growth. Mol. Cell. Biol. 26, 3785–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wilkins B. J., Dai Y. S., Bueno O. F., Parsons S. A., Xu J., Plank D. M., Jones F., Kimball T. R., Molkentin J. D. (2004) Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 94, 110–118 [DOI] [PubMed] [Google Scholar]
- 27. Singal T., Dhalla N. S., Tappia P. S. (2006) Norepinephrine-induced changes in gene expression of phospholipase C in cardiomyocytes. J. Mol. Cell. Cardiol. 41, 126–137 [DOI] [PubMed] [Google Scholar]
- 28. Vanamala S. K., Gopinath S., Gondi C. S., Rao J. S. (2009) Effect of human umbilical cord blood cells on Ang-II-induced hypertrophy in mice. Biochem. Biophys. Res. Commun. 386, 386–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Min L., Sim M. K., Xu X. G. (2000) Effects of des-aspartate-angiotensin I on angiotensin II-induced incorporation of phenylalanine and thymidine in cultured rat cardiomyocytes and aortic smooth muscle cells. Regul Pept. 95, 93–97 [DOI] [PubMed] [Google Scholar]
- 30. Watt S. A., Kular G., Fleming I. N., Downes C. P., Lucocq J. M. (2002) Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase Cδ1. Biochem. J. 363, 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yue J., Liu J., Shen X. (2001) Inhibition of phosphatidylinositol 4-kinase results in a significant reduced respiratory burst in formyl-methionyl-leucyl-phenylalanine-stimulated human neutrophils. J. Biol. Chem. 276, 49093–49099 [DOI] [PubMed] [Google Scholar]
- 32. Minogue S., Waugh M. G. (2012) The phosphatidylinositol 4-kinases. Don't call it a comeback. Subcell. Biochem. 58, 1–24 [DOI] [PubMed] [Google Scholar]
- 33. Tóth B., Balla A., Ma H., Knight Z. A., Shokat K. M., Balla T. (2006) Phosphatidylinositol 4-kinase IIIβ regulates the transport of ceramide between the endoplasmic reticulum and Golgi. J. Biol. Chem. 281, 36369–36377 [DOI] [PubMed] [Google Scholar]
- 34. Chen X., Zhang X., Jia C., Xu J., Gao H., Zhang G., Du X., Zhang H. (2011) Membrane depolarization increases membrane PtdIns(4,5)P2 levels through mechanisms involving PKCβII and PI4 kinase. J. Biol. Chem. 286, 39760–39767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Balla A., Tuymetova G., Tsiomenko A., Várnai P., Balla T. (2005) A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-IIIα. Studies with the PH domains of the oxysterol binding protein and FAPP1. Mol. Biol. Cell. 16, 1282–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Singh K., Xiao L., Remondino A., Sawyer D. B., Colucci W. S. (2001) Adrenergic regulation of cardiac myocyte apoptosis. J. Cell. Physiol. 189, 257–265 [DOI] [PubMed] [Google Scholar]
- 37. Nicolas C. S., Park K. H., El Harchi A., Camonis J., Kass R. S., Escande D., Mérot J., Loussouarn G., Le Bouffant F., Baró I. (2008) IKs response to protein kinase A-dependent KCNQ1 phosphorylation requires direct interaction with microtubules. Cardiovasc. Res. 79, 427–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van den Bout I., Divecha N. (2009) PIP5K-driven PtdIns(4,5)P2 synthesis. Regulation and cellular functions. J. Cell Sci. 122, 3837–3850 [DOI] [PubMed] [Google Scholar]
- 39. Singhal R. L., Prajda N., Yeh Y. A., Weber G. (1994) 1-Phosphatidylinositol 4-phosphate 5-kinase (EC 2.7.1.68). A proliferation- and malignancy-linked signal transduction enzyme. Cancer Res. 54, 5574–5578 [PubMed] [Google Scholar]
- 40. Balla A., Balla T. (2006) Phosphatidylinositol 4-kinases. Old enzymes with emerging functions. Trends Cell Biol. 16, 351–361 [DOI] [PubMed] [Google Scholar]
- 41. Yaradanakul A., Feng S., Shen C., Lariccia V., Lin M. J., Yang J., Kang T. M., Dong P., Yin H. L., Albanesi J. P., Hilgemann D. W. (2007) Dual control of cardiac Na+ Ca2+ exchange by PIP2. Electrophysiological analysis of direct and indirect mechanisms. J. Physiol. 582, 991–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dent M. R., Aroutiounova N., Dhalla N. S., Tappia P. S. (2006) Losartan attenuates phospholipase C isozyme gene expression in hypertrophied hearts due to volume overload. J. Cell. Mol. Med. 10, 470–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Howes A. L., Arthur J. F., Zhang T., Miyamoto S., Adams J. W., Dorn G. W., 2nd, Woodcock E. A., Brown J. H. (2003) Akt-mediated cardiomyocyte survival pathways are compromised by Gαq-induced phosphoinositide 4,5-bisphosphate depletion. J. Biol. Chem. 278, 40343–40351 [DOI] [PubMed] [Google Scholar]
- 44. Hilgemann D. W., Ball R. (1996) Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 273, 956–959 [DOI] [PubMed] [Google Scholar]
- 45. Kobrinsky E., Mirshahi T., Zhang H., Jin T., Logothetis D. E. (2000) Receptor-mediated hydrolysis of plasma membrane messenger PIP2 leads to K+-current desensitization. Nat. Cell Biol. 2, 507–514 [DOI] [PubMed] [Google Scholar]
- 46. Ding W. G., Toyoda F., Matsuura H. (2004) Regulation of cardiac IKs potassium current by membrane phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 279, 50726–50734 [DOI] [PubMed] [Google Scholar]
- 47. Cho H., Lee D., Lee S. H., Ho W. K. (2005) Receptor-induced depletion of phosphatidylinositol 4,5-bisphosphate inhibits inwardly rectifying K+ channels in a receptor-specific manner. Proc. Natl. Acad. Sci. U.S.A. 102, 4643–4648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nienaber J. J., Tachibana H., Naga Prasad S. V., Esposito G., Wu D., Mao L., Rockman H. A. (2003) Inhibition of receptor-localized PI3K preserves cardiac β-adrenergic receptor function and ameliorates pressure overload heart failure. J. Clin. Invest. 112, 1067–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Naga Prasad S. V., Perrino C., Rockman H. A. (2003) Role of phosphoinositide 3-kinase in cardiac function and heart failure. Trends Cardiovasc. Med. 13, 206–212 [DOI] [PubMed] [Google Scholar]
- 50. Fujio Y., Nguyen T., Wencker D., Kitsis R. N., Walsh K. (2000) Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 101, 660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]