

Background: Arp2/3 and the upstream modulator, WAVE2, regulate actin branching and polymerization.
Results: HIV triggers WAVE2 phosphorylation for Arp2/3 activity, which is essential for nuclear migration.
Conclusion: Arp2/3 and WAVE2 are cellular cofactors hijacked by HIV for intracellular migration.
Significance: HIV-mediated WAVE2-Arp2/3 activity may serve as novel therapeutic targets.
Keywords: HIV-1, Immunology, Microbiology, Signal Transduction, Virology, Arp2/3, CCR5, CD4, Wav2
Abstract
The human immunodeficiency virus type 1 (HIV-1) initiates receptor signaling and early actin dynamics during viral entry. This process is required for viral infection of primary targets such as resting CD4 T cells. WAVE2 is a component of a multiprotein complex linking receptor signaling to dynamic remodeling of the actin cytoskeleton. WAVE2 directly activates Arp2/3, leading to actin nucleation and filament branching. Although several bacterial and viral pathogens target Arp2/3 for intracellular mobility, it remains unknown whether HIV-1 actively modulates the Arp2/3 complex through virus-mediated receptor signal transduction. Here we report that HIV-1 triggers WAVE2 phosphorylation at serine 351 through gp120 binding to the chemokine coreceptor CXCR4 or CCR5 during entry. This phosphorylation event involves both Gαi-dependent and -independent pathways, and is conserved both in X4 and R5 viral infection of resting CD4 T cells and primary macrophages. We further demonstrate that inhibition of WAVE2-mediated Arp2/3 activity through stable shRNA knockdown of Arp3 dramatically diminished HIV-1 infection of CD4 T cells, preventing viral nuclear migration. Inhibition of Arp2/3 through a specific inhibitor, CK548, also drastically inhibited HIV-1 nuclear migration and infection of CD4 T cells. Our results suggest that Arp2/3 and the upstream regulator, WAVE2, are essential co-factors hijacked by HIV for intracellular migration, and may serve as novel targets to prevent HIV transmission.
Introduction
The heptameric Arp2/3 complex is a major actin branching and nucleation factor that regulates the actin cytoskeleton. The Arp2/3 complex is activated by nucleation-promoting factors (NPFs)2 such as N-WASP, WASP, and WAVE proteins through a unique WCA domain (1). During Arp2/3 activation, the NPF's WCA domain engages the Arp2/3 complex and brings Arp2 and Arp3 subunits together to create a nascent actin filament nucleus suitable for filament growth (1). This process of actin nucleation and branching is important for cell motility and receptor trafficking (1, 2). In addition, this Arp2/3-mediated actin activity is also frequently hijacked by bacterial and viral pathogens to propel intracellular migration (3–6). In this process, bacteria and viruses often encode and utilize the WCA domain to mimic NPFs for Arp2/3 activation and actin dynamics (3–5). The Human Immunodeficiency Virus Type 1 (HIV-1) also depends on actin dynamics to infect blood CD4 T cells (7–11). During viral entry, the virus initiates chemokine coreceptor signaling and early actin dynamics to facilitate intracellular migration (7). Nevertheless, although targeting actin or Arp2/3 can inhibit HIV (6, 12, 13), no HIV virion proteins contain a WCA domain, and it is unclear how the virus actively engages the Arp2/3 complex. We asked whether HIV is capable of modulating Arp2/3 and upstream NPFs through chemokine coreceptor binding and signal transduction.
Among NPFs, WAVE2 is a component of a multiprotein complex linking receptor signaling to dynamic remodeling of the actin cytoskeleton (14). WAVE2 directly activates Arp2/3, leading to actin nucleation and filament branching (1). Thus, we tested possible activation of WAVE2 by HIV during the infection of blood resting CD4 T cells. Here we report that HIV-1 triggers WAVE2 phosphorylation at serine 351 through gp120 binding to the chemokine coreceptor CXCR4 or CCR5 during entry. This phosphorylation event involves both Gαi-dependent and -independent pathways, and is conserved both in X4 and R5 viral infection of resting CD4 T cells and primary macrophages. We further demonstrate that inhibition of WAVE2-mediated Arp2/3 activity through stable shRNA knockdown of Arp3 dramatically diminished HIV-1 infection of CD4 T cells, preventing viral nuclear migration. Inhibition of Arp2/3 through a specific inhibitor, CK548 (15), also drastically inhibited HIV-1 nuclear migration and infection of CD4 T cells. Our results suggest that Arp2/3 and the upstream regulator, WAVE2, are essential co-factors hijacked by HIV for intracellular migration, and may serve as novel targets to prevent HIV transmission.
EXPERIMENTAL PROCEDURES
Isolation of Resting CD4 T Cells and Monocytes from Peripheral Blood
All protocols involving human subjects were reviewed and approved by the George Mason University Institutional Review Board. Resting CD4 T cells were purified from peripheral blood of HIV-1 negative donors by two rounds of negative selection as previously described (16). Briefly, for the first-round depletion, we used monoclonal antibodies against human CD14, CD56 and HLA-DR, DP, and DQ (BD Biosciences). For the second-round depletion, we used monoclonal antibodies against human CD8, CD11b, and CD19 (BD Biosciences). Antibody-bound cells were depleted by using Dynabeads Pan Mouse IgG (Invitrogen). For further negative selection of the memory and naïve CD4 T cell subsets, monoclonal antibody against either CD45RA (0.02 μl per million cells) or CD45RO (0.1 μl per million cells) (BD Biosciences) was added during the second round of depletion. Purified cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), penicillin (50 units/ml) (Invitrogen), and streptomycin (50 μg/ml) (Invitrogen). Cells were rested overnight before infection or treatment. Macrophages were differentiated from human monocytes from the peripheral blood of HIV-1 negative donors. Briefly, two million peripheral blood mononuclear cells were plated into each well of six-well plates in serum-free RPMI 1640 medium for 1 h. Adherent cells were cultured in RPMI 1640 plus 10% heat-inactivated fetal bovine serum (FBS) with 10 ng/ml macrophage colony stimulating factor (M-CSF) (R&D System, Minneapolis, MN) for 2 weeks with medium change for every 2 days.
Virus Preparation and Infection
Virus stocks of HIV-1NL4–3, HIV-1NL4–3(AD8), and HIV-1(Yu2) were prepared by transfection of HeLa cells with cloned proviral DNA as described (16). Single-cycle virus HIV-1(VSV-G) and HIV-1(Env) were prepared as previously described (17). Levels of p24 in the viral supernatant were measured in triplicate on the same ELISA plates using an in-house ELISA Kit. Viral titer (TCID50) was determined on the Rev-dependent GFP indicator cell Rev-CEM (18, 19).
For viral infection, unless otherwise specified, 103.5 to 104.5 TCID50 units of HIV-1 were used to infect 106 cells. For infection, resting CD4 T cells were incubated with the virus for 2 h, washed once, and then resuspended into fresh medium (106 cells per ml) and incubated for 5 days without stimulation. Cells were activated with anti-CD3/CD28 magnetic beads at 4 beads per cell. Culture supernatant (100 μl) was taken every 2 days or daily after stimulation. Cells were removed by centrifugation, and supernatant saved for p24 ELISA. Fresh medium was added when needed. CEM-SS cells, either carrying shRNA knockdown or not, were similarly infected for 2 h, washed twice, and then resuspended into fresh medium (2 × 105 cells per ml). Culture supernatant was taken for p24 ELISA. Rev-CEM cells were also similarly infected, and viral infection was measured by flow cytometry (FACSCalibur, BD Biosciences) of GFP-positive cells. To exclude drug cytotoxicity, propidium iodide (PI) (2 μg/ml, Fluka) was added into the cell suspension prior to flow cytometry, and only viable cells (PI negative) were used for measuring GFP expression.
Surface Staining of CD4 and CXCR4
Cells were stained with FITC-labeled monoclonal antibody against human CD4 (clone PRA-T4) or CXCR4 (clone 12G5) (BD Biosciences). Cells were stained on ice in PBS + 0.1% BSA for 30 min, washed with cold PBS-0.5% BSA, and then analyzed on a FACSCalibur (BD Biosciences).
Chemotaxis Assay
A half million Jurkat T cells were resuspended into 100 μl of RPMI 1640 medium and then added to the upper chamber of a 24-well transwell plate (Corning). The lower chamber was filled with 600 μl of medium premixed with SDF-1 (40 ng/ml). The plate was incubated at 37 °C for 2 h, and then the upper chamber was removed, and cells in the lower chamber were counted. Where indicated, CK548 and CK636 (Sigma) were added to the culture supernatant for 2 h prior to the assay along with a DMSO control.
Viral Entry Assays
The BlaM-Vpr-based viral entry assay was performed as previously described (7, 20). We also used a Nef-luciferase-based entry assay as described (21). Briefly, cells (1 × 106) were infected with 200 ng of Nef-luciferase containing viruses at 37 °C for 2 h, and then washed three times with medium. Cells were resuspended in 0.1 ml of luciferase assay buffer (Promega), and luciferase activity was measured in live cells using a GloMax-Multi Detection System (Promega).
For measuring the entry of Nef-luciferase tagged HIV-1(VSV-G), cells (2 × 105) were infected with 500 μl of virus for 2 h at 37 °C. Cells were also pretreated with 20 mm NH4Cl for 1 h at 37 °C, and infected in the presence of 20 mm NH4Cl for 2 h. Afterward, cells were washed twice with 3 ml of ice-cold, serum-free RPMI, and once with 1 ml of PBS. Cells were resuspended in 0.1 ml of luciferase assay buffer (Promega), and luciferase activity was measured. Data were normalized to within-experiment NTC averages from three separate experiments, with five reads from each experiment.
Viral Budding Assays
The viral budding assay was performed as follows: CEM-SS cells were infected with HIV-1(Env), a single-cycle HIV-1 virus pseudotyped with the NL4–3 gp160 (17). Cells were infected for 12–18 h, washed for three times, and then treated with CK548 at different dosages. Viral budding was monitored by harvesting and measuring viral p24 in the supernatant in the presence of CK548.
shRNA Arp3 Knockdown
Plasmids (pLKO.1-puro) carrying Arp3 shRNA (clone TRCN0000029382), and a non-targeting shRNA (NTC) were purchased from Sigma-Aldrich. For transient Arp3 knockdown, 1 million CEM-SS cells were electroporated with a total of 2 μg of plasmid DNA (shRNA-Arp3, shRNA-NTC, or a combination of both in various ratios) using nucleofector and Nucleofector Kit R (Lonza). Electroporation was carried out following protocols recommended by the manufacturer. Electroporated cells were cultured and infected with HIV at 72 h post-electroporation. For lentiviral vector-mediated stable knockdown of Arp3, viral particles were assembled by transfection of HEK 293T cells along with pCMVΔ8.2 and pHCMV-G using Lipofectamine 2000 (Invitrogen). Viral particles were concentrated 20–40-fold using centrifugal concentrators (Cole-Parmer). CEM-SS cells were infected for 6 h at 37 °C and 5% CO2 and cultured for an additional 48 h prior to antibiotic selection using 10 μg/ml puromycin (Invitrogen). Clones were acquired by limiting dilution, culturing the cells in 50% v/v CEM-SS-conditioned RPMI.
Western Blotting
One million cells were lysed in 100 μl of NuPAGE LDS Sample Buffer (Invitrogen). Cell lysates were sonicated and separated in 4–12% Bis-Tris polyacrylamide gels (Invitorgen) and transferred to nitrocellulose membranes (Invitrogen). The membranes were washed in TBST for 3 min and then blocked for 30 min at room temperature using either 5% skim milk in TBS plus 0.2% Tween 20 or LiCor Blocking Buffer (LiCor). Membranes were incubated with an anti-Phospho-S351 WAVE2 antibody (Millipore) (1: 1000 dilution in LiCor Blocking Buffer) or an anti-GAPDH antibody (Abcam) (1:1000 dilution in 2.5% skim milk) overnight at 4 °C. For staining with secondary antibodies, IRDye 800CW Goat anti-Rabbit IgG (LiCor) or Rabbit Anti-Goat IgG DyLight 680-labeled (KPL) secondary antibodies were used and diluted 1:5000 in LiCor Blocking Buffer. The blots were incubated for 1 h at 4 °C, washed three times for 15 min, and scanned with Odyssey Infrared Imager (Li-cor Biosciences).
WAVE2 was similarly stained with a 1:1000 dilution of rabbit anti-WAVE2 (Cell Signaling Technology) in Tris-buffered saline (TBS) with 0.2% Tween-20 (TBS-T) with 2.5% skim milk (w/v) overnight at 4 °C. After three 15 min washes in TBS-T, samples were stained for 1 h with DyLight 800-conjugated goat anti-rabbit (KPL) at a dilution of 1:5000 and at room temperature. Subsequently, blots were washed 3 times for 15 min each and imaged on a LiCor Odyssey Infrared Imager. Arp3 was stained with a 1:1000 dilution of Arp3 monoclonal antibody (Cell Signaling) in TBS-T with 2.5% skim milk overnight at 4 °C. For secondary staining, a 1:5000 dilution of HRP-conjugated Goat anti-rabbit antibodies were used. Chemiluminescent signal was generated using SuperSignal Femto HRP substrate (Thermo Scientific). GAPDH was stained with goat anti-GAPDH (Abcam, ab9483) as a 1:1000 dilution in TBS-T with 2.5% skim milk overnight at 4 °C. Secondary staining was performed using 1:5000 dilution of Rabbit Anti-Goat IgG DyLight 680 (KPL, 072-06-13-06) in TBS-T with 2.5% skim milk for 1 h at room temperature. IR-dye conjugated stains were imaged on an ODYSSEY imaging system (Li-Cor Biosciences).
In Vitro Actin Bead Assay
Carboxylated polystyrene 4.5 μm diameter microsphere (Polysciences, Warrington, PA) were coated with 8.5 μm GST tagged VCA by incubation for 1 h at room temperature. Particles were pelleted by low speed centrifugation and resuspended in storage buffer (10 mm HEPES pH 7.8, 0.1 mm KCl, 1 mm MgCl2, 1 mm ATP, 0.1 mm CaCl2, 0.01% NaN3) containing 1 mg/ml bovine serum albumin (BSA, Sigma-Aldrich) to block subsequent nonspecific binding. Particles were stored at 4 °C for up to 1 week. For reconstitution of bead motility, glass slides and coverslips were cleaned and blocked overnight in 1% BSA at 4 °C and dried in air before use. We placed 16 μl of reaction mixture on a BSA coated slide, covered with a BSA coated coverslip, and sealed the chamber with VALAP. Labeled and unlabeled Ca-ATP actin were diluted to the desired labeled fraction, mixed 9:1 with 10× magnesium exchange buffer (10× ME: 10 mm EGTA, 1 mm MgCl2) and incubated on ice for 2 min to form 4× final concentrations of Mg-ATP actin. We placed 8 μl of Mg-ATP actin at the bottom of a 1.5 ml Eppendorf tube and added 7 μl of motility protein mixtures with or with no Arp2/3 complex inhibitor and 1 μl of coated nanofibers or beads on the side of the tube. We washed both drops together with 16 μl 2× TIRF buffer (2x: 100 mm KCl, 2 mm MgCl2, 2 mm EGTA, 20 mm imidazole, pH 7.0, 200 mm DTT, 0.4 mm ATP, 30 mm glucose, 0.25% 1500 cP methylcellulose, 40 μg/ml catalase, 200 μg/ml glucose oxidase) and placed the reaction mixture in slide-coverslip as described above. For Image acquisition and processing, actin fluorescence was observed with a 60 × 1.49 NA TIRF objective on an Olympus IX2 inverted microscope. Images were captured with a Retiga EXi cooled CCD camera (QImaging) using SlideBook image acquisition software (Intelligent Imaging Innovations, Inc). All subsequent image-processing steps were performed in ImageJ, available at http://rsbweb.nih.gov/ij. Epi-fluorescence microscopy images were unprocessed. Images were cropped for publication.
Quantitative Real-time PCR
Viral DNA quantification was carried out using the Bio-Rad iQ5 real-time PCR detection system, utilizing the forward primer 5′LTR-U5, the reverse primer 3′ gag, and the probe FAM-U5/gag (7). Pre-qualified, full-length proviral plasmid pNL4–3 was used as the DNA standard. Viral DNA and 2-LTR circles in shRNA lentiviral vector- transduced cells (shNTC, shArp-12, and shArp-13) were measured as described previously (8). For measuring 2-LTR-circles (22), the DNA was amplified by real-time PCR with primers and probe MH536, MH535, and MH603 (23, 24). For real-time PCR quantification of the nef transcripts, total cellular RNA was extracted with SV Total RNA isolation system (Promega), and then reverse transcribed into cDNA using random decamers and M-MLV reverse transcriptase as previously described (16). Nef cDNA was further quantified by real time PCR using primers 5′ Nef (5′-GGCGGCGACTGGAAGAA-3′), 3′ Rev (5′-AGGTGGGTTGCTTTGATAGAGAAG-3′), and the probe Nef/Rev (5′-FAM-CGGAGACAGCGACGAAGAGCTCATC-TAMRA-3′).
Conjugation of Antibodies to Magnetic Beads
Monoclonal antibodies against human CD3 (clone UCHT1) and CD28 (clone CD28.2) were from BD Pharmingen (BD Biosciences). For conjugation, 10 μg of antibodies were conjugated with 4 × 108 Dynabeads Pan Mouse IgG (Invitrogen) for 30 min at room temperature. Free antibodies were washed away with PBS + 0.5% BSA, and the magnetic beads were resuspended in 1 ml of PBS + 0.5% BSA.
Confocal Microscopy
Stained cells were imaged using a Zeiss Laser Scanning Microscope, LSM 510 META, with a 40 NA 1.3 or 60 NA 1.4 oil DIC Plan-Neofluar objective. Samples were excited with the 488 nm laser line for FITC. Images were processed and analyzed with LSM 510 META software.
Statistical Analysis
All statistical analyses were performed by unpaired, two-sample t test assuming a normal distribution.
RESULTS
HIV-1 Infection Triggers WAVE2 Phosphorylation
To detect possible WAVE2 activation by HIV-1, we used WAVE2 serine 351 as a convenient marker, as WAVE2 activity is regulated through multiple serine phosphorylations (25, 26). We observed a rapid phosphorylation of WAVE2 in resting CD4 T cells following infection with HIV-1 (Fig. 1A). In prior studies, activation of the actin modulator cofilin was linked to Gαi-dependent chemokine coreceptor signaling by HIV gp120 binding (7, 8). We pretreated resting CD4 T cells with pertussis toxin (PTX), an inhibitor that uncouples Gαi from the chemokine coreceptor. PTX treatment largely eliminated late signal transduction to WAVE2, but not early signal transduction (Fig. 1B), suggesting the involvement of an early Gαi-independent and late Gαi-dependent pathway. Signaling from both Gαi and Gαq has been known to be involved in HIV infection (7, 8, 27, 28). Given that HIV triggers higher levels of actin activity to promote latent infection of resting memory CD4 T cells (9) (Fig. 1, C and D), we treated resting memory CD4 T cells with HIV-1. Activation of WAVE2 was also observed, and the phosphorylation was blocked by a CXCR4 antagonist AMD3100 (Fig. 1E). Consistent with these data, pseudotyping HIV with the CD4/CXC4-independent VSV-G (vesicular stomatitis virus G glycoprotein) caused a decrease in WAVE2 activation (Fig. 1, F and G). These results demonstrated that HIV actively engages WAVE2 and the Arp2/3 complex through CXCR4 signaling to modulate actin activity in resting T cells. Actin dynamics are also important for HIV infection of macrophages (29), we therefore treated macrophages with R5 HIV. Both HIV-1YU2 and HIV-1NL4–3(AD8) induced WAVE2 phosphorylation in macrophages (Fig. 1, H and I), implying that HIV-1-induced WAVE2 phosphorylation is conserved in both X4 and R5 infection.
FIGURE 1.
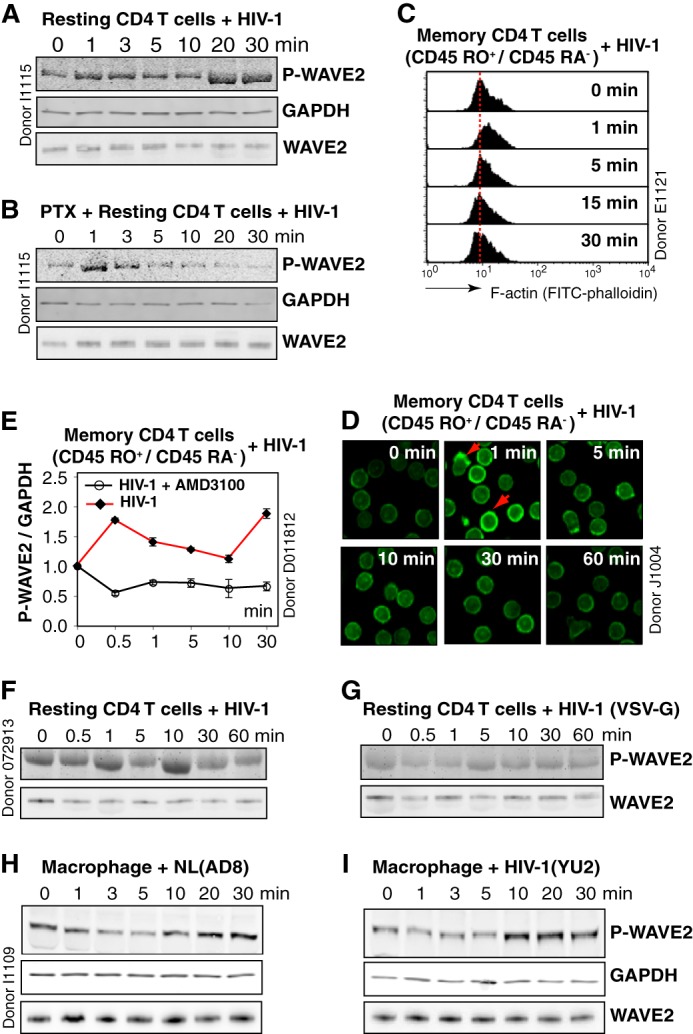
HIV-1 triggers WAVE2 serine 351 phosphorylation. A, resting CD4 T cells were treated with HIV-1NL4–3 over the indicated time course, and WAVE2 serine 351 phosphorylation was detected by an IR fluorophore-conjugated, anti-phospho-S351 WAVE2 antibody. The same blot was also stained with an anti-human GAPDH antibody for loading control. WAVE2 was also stained with an anti-WAVE2 antibody on a blot duplicate. B, resting CD4 T cells were treated with 50 ng/ml PTX, similarly treated with HIV-1, and analyzed for WAVE2 S351 phosphorylation. C and D, resting memory CD4 T cells (CD45RO+/CD45RA−) were treated with HIV-1 for a time course, and F-actin was stained with FITC-phalloidin and analyzed with flow cytometry (C) or confocal microscopy (D). Red arrows indicate localized actin polymerization. E, resting memory CD4 T cells were pre-treated or not with AMD3100 (100 nm), and then infected with HIV-1 for a time course. WAVE2 serine 351 phosphorylation was similarly analyzed and quantified. F and G, resting CD4 T cells were treated with HIV-1NL4–3 or with HIV-1(VSV-G), and WAVE2 serine 351 phosphorylation was measured. H and I, blood monocyte-derived macrophages were treated with HIV-1NL4–3 (AD8) or HIV-1YU2 for a time course, and WAVE2 serine 351 phosphorylation was similarly analyzed.
ShRNA Knockdown of Arp3 Inhibits HIV-1 Nuclear Migration and Infection of CD4 T Cells
WAVE2 activates Arp2/3, leading to actin nucleation and filament branching. To explicate the role of Arp2/3 in HIV-1 infection, we first performed a transient knockdown of Arp3 in CEM-SS cells. Cells were electroporated with 3 different dosages of a lentiviral vector, pLKO.1-Puro, carrying Arp3 shRNA or a non-targeting shRNA (NTC). Knockdown cells were infected with HIV-1 at 3 days post electroporation. As shown in Fig. 2A, we observed significant inhibition of HIV infection (6). To further confirm these results, we performed lentiviral vector-mediated stable shRNA knockdown of Arp3 in CEM-SS T cells, using a similar approach to the knockdown of LIMK1 (8). Individual cells were cloned, and we obtained 5 clones carrying stable shRNA against Arp3 (Fig. 2B). All of these cells showed a decreased capacity to support HIV-1 infection, and 4 of the 5 clones, including shArp-09, shArp-10, shArp-12, and shArp-13, showed a strong impairment in supporting HIV infection (Fig. 2C).
FIGURE 2.
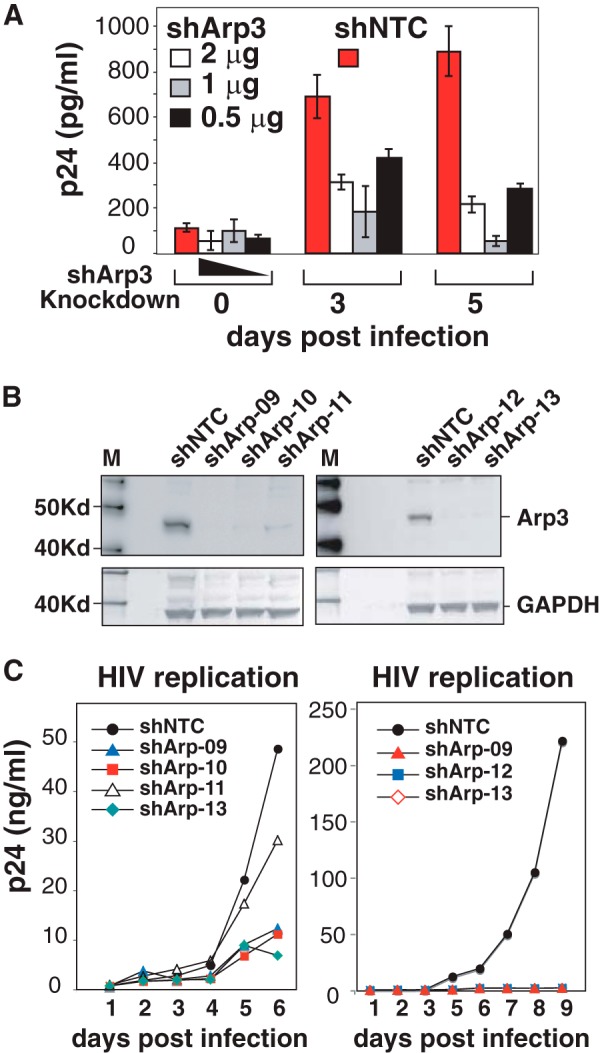
Arp3 knockdown diminishes HIV-1 infection. A, transient shRNA knockdown of Arp3 inhibits HIV replication. CEM-SS cells were electroporated with indicated amounts of vector pLKO.1-Puro carrying Arp3 shRNA (shArp3) or a non-targeting shRNA (shNTC). Knockdown cells were infected with HIV-1 at 3 days post electroporation. Viral replication was measured by p24 release. B, Western blot measurement of cell lines carrying stable Arp3 knockdown. Individual shRNA Arp3 knockdown cell clones and the control shNTC cells were analyzed by Western blot using an antibody again Arp3. The same blot were also probed with an antibody again GAPDH for loading control. C, stable Arp3 knockdown diminishes HIV-1 infection. The Arp3 knockdown clones, shArp-09, shArp-10, shArp-11, shArp-12, shArp-13, and the control shNTC cells, were identically infected with HIVNL4–3 for 2 h. Cells were washed twice and then culture for 6–9 days to monitor viral replication by p24 release.
To further delineate the molecular mechanism of how inhibiting Arp3 affects HIV early steps, we selected shArp-12 and shArp-13 for further characterization. Both clones exhibited diminished Arp3 expression (70 and 80% reduction, respectively) (Fig. 3A). Additionally, shArp-12, but not shArp-13, had decreased surface expression of CD4, while both shArp-12 and shArp-13 had slightly decreased surface levels of CXCR4 (Fig. 3B). These data are consistent with a previous finding, in which actin modulation following LIMK knockdown affected surface expression of CD4 and CXCR4 by altering receptor cycling (8). In addition, shArp-13 grew slightly more slowly than the control shNTC cells (Fig. 3C). Nevertheless, both shArp-12 and shArp-13 have been grown in the laboratory for over a year, and the knockdown phenotypes have been stably maintained.
FIGURE 3.

Arp3 knockdown diminishes HIV-1 infection and viral nuclear migration. A, Arp3 knockdown in shArp-12, shArp-13, and the control shNTC cells was analyzed by Western blot. B, shArp-12, shArp-13, and shNTC cells were stained for surface expression of CD4 and CXCR4. C, shArp-12, shArp-13, and shNTC cells were measured for growth rate. D, shArp-12, shArp-13, and shNTC cells were infected with HIVNL4–3 to monitor viral replication by p24 release. E, these cells were similarly infected with a VSV-G-pseudotyped virus, HIV-1(VSV-G), and monitored for p24 release. F, entry of HIV-1 (left panel) into shArp-12, shArp-13, and shNTC was measured with a Nef-luciferase-based entry assay (left panel). A VSV-G pseudotyped reporter virus was similarly used in the Nef-luciferase-based entry assay (right panel). As a control, cells were also pretreated with 20 mm NH4Cl during infection to inhibit endocytotic entry (right panel). G, viral entry into shArp-12, shArp-13, and shNTC were also measured with a BlaM-Vpr-based entry assay. For control, the cells were also pre-treated for 1 h with T20 peptide (0.1 mg/ml) and then infected. H, shArp-12, shArp-13, and shNTC cells were infected with a single-cycle HIV-1(Env) that was pseudotyped with HIV-1 gp160. Cells were infected for 2 h, washed, and then taken at different time points for PCR quantification of viral DNA. I, viral 2-LTR circles following HIV infection at 48 h were also measured. J, viral mRNA was also extracted following infection at 24 and 48 h, and quantified by reverse-transcriptase PCR for the nef transcripts.
We further tested the ability of shArp-12 and shArp-13 to support HIV infection. Both cells demonstrated a highly diminished ability to support HIV-1 infection, as measured by viral p24 release for ∼10 days (Fig. 3D). Given that HIV-1 fusion and entry are dependent on surface CD4/CXCR4 and the cortical actin (13), we similarly infected both cells with a VSV-G-pseudotyped HIV-1, which enters cells through endocytosis, a process independent of CD4/CXCR4 and having less viral contact with the cortical actin. HIV-1(VSV-G) infection of shArp-12 and shArp-13 led to a 70% reduction of viral p24 release in comparison with shNTC at day 4 (Fig. 3E), likely resulting from the effect of Arp2/3 on endocytosis (30). Indeed, when viral entry was measured, we observed an ∼40% decrease of viral entry in shArp-12 and shArp-13 (Fig. 3F, right panel). These results suggested that knockdown of Arp3 has much more profound inhibition of wild type (wt) HIV replication; this is likely related to gp120-mediated entry, which is dependent on CD4, the chemokine co-receptors, and the cortical actin. Surprisingly, when we measured wt viral entry using a Nef-luciferase-based fusion assay (21), neither shArp-12 nor shArp-13 exhibited a reduction in entry (Fig. 3F). However, when we measured viral entry using highly concentrated, β-lactamase-Vpr-tagged virion particles (20), shArp-13 but not shArp-12 demonstrated a decrease in entry (Fig. 3G). A similar discrepancy in assay results has also been reported in our previous study of the LIMK1 knockdown cells (8). The difference likely resulted from different levels of reporter viruses used. The β-lactamase-Vpr assay requires the use of highly concentrated virus, and may need global actin reorganization for receptor clustering (8), whereas in the Nef-luciferase assay, low-levels of viral entry may depend on the natural association of CD4/CXCR4 (31, 32) without the need of extensive actin reorganization.
We further monitored viral DNA synthesis, and both shArp-12 and shArp-13 exhibited an enhancement of viral DNA synthesis from 2 to 48 h compared with the shNTC control (Fig. 3H). We also measured 2-LTR circles, a correlative of nuclear entry, and found that 2-LTR circles were significantly reduced in shArp-12 and shArp-13 by ∼70 and 95%, respectively, at 48 h post-infection (Fig. 3I). Measurement of the nef transcripts, a correlative of nuclear entry and transcription corroborated these findings, indicating a marked reduction of the nef transcription in shArp-12 and shArp-13 cells at 24 and 48 h (Fig. 3J). Therefore, Arp3 knockdown inhibits viral nuclear translocation and subsequent viral replication without inhibiting initial viral DNA synthesis.
The Arp2/3 Inhibitor CK548 Inhibits HIV-1 Nuclear Migration and Infection of CD4 T Cells
Nolen et al. (15) recently described several novel, small molecule inhibitors of Arp2/3. We used the Arp2/3 inhibitors CK548 and CK636 to confirm the Arp3 knockdown results. Firstly, to determine whether CK548 and CK636 can inhibit actin polymerization mediated by the Arp2/3 complex independent of any possible role in endocytosis or receptor recycling, we tested both inhibitors using an in vitro actin-based motility assay (33, 34). Both CK548 and CK636 reduced the formation of actin comet tails in a dose-dependent manner, with IC50 values of approximately 10 μm (Fig. 4, A–E). The in vivo effects of 50 μm CK548 and CK636 on SDF-1-induced Jurkat T cell chemotaxis were further examined. We found that CK548, but not CK636, inhibited Jurkat chemotaxis toward SDF-1 (2) (Fig. 4F).
FIGURE 4.

Effects of the Arp2/3 inhibitors CK548 and CK636 on actin polymerization in vitro and on T cell chemotaxis. A–D, in vitro effects of CK548 and CK636 on actin polymerization in an in vitro WCA-bead actin polymerization assay as illustrated in D. DMSO, 50 μm CK548, or CK636, along with 8.5 mm actin (10% labeled), 9 mm profilin, 100 nm Arp2/3, and 200 nm capping protein were added for the assay. CT, actin comet tail. E, quantification of TIRF images from the polymerization assay. F, Jurkat T cells were assayed in SDF-1-induced chemotaxis in transwell plates after a 2-h treatment with DMSO, 50 μm CK548, or 50 μm CK636.
To determine the effects of these inhibitors on HIV-1 infection, a highly specific HIV Rev-dependent indicator cell line, Rev-CEM (19), was pre-treated with each inhibitor and subsequently infected with HIVNL4–3. CK548 inhibited HIV-1 replication in a dosage-dependent manner (97–100% at 50–100 μm, respectively), while CK636 only modestly inhibited HIV-1 replication (19% at 100 μm) (Fig. 5A). The lower inhibition of HIV by CK636 is consistent with the lower effectiveness of CK636 in inhibiting SDF-1-mediated chemotaxis of Jurkat T cells, and may reflect a difference in intracellular stability or mechanisms of action between CK548 and CK636 (15) (see discussions below).
FIGURE 5.

CK548 inhibits HIV-1 infection and viral nuclear migration. A, an HIV Rev-dependent indicator (GFP) T cell, Rev-CEM, was pretreated with DMSO, CK548 or CK636 for 1 h, and then infected with HIV-1NL4–3 for 2 h. Cells were washed, and the inhibitors were added back after infection. HIV-dependent GFP expression was measured at 72 h postinfection by flow cytometry. To exclude drug cytotoxicity, PI was added into the cell suspension prior to flow cytometry, and only viable cells (R1 gate) were used for measuring GFP expression. B, CEM-SS T cells were pre-treated with DMSO or CK548 for 1 h, and then infected with HIV-1NL4–3 as above. Viral replication was measured by p24 release. C, drug cytotoxicity was meaured in CK548-treated and infected cells by PI staining and flow cytometry. Shown is the percentage of PI-positive cells. D–F, CEM-SS T cells were pretreated with DMSO or CK548 for 1 h, and then infected with a single-cycle HIV-1(Env) that was pseudotyped with HIV-1 gp160. Cells were infected for 2 h, washed, and cultured in the presence of the drug for 48 h. Total cellular DNA was extracted at 12 and 48 h postinfection and then PCR-amplified to quantify viral DNA (D) and viral 2-LTR-circles (E). The relative ratio of 2-LTR circles to viral total DNA was also plotted (F). ND, not detectable. G, effect of CK548 on viral budding and release was also measured. Cells were infected with HIV-1(Env) for 12 h, washed three times, and then incubated with CK548 at different dosages. Viral p24 release was measured.
CK548 was further tested in HIV infection of CEM-SS T cells and drastically diminished HIV replication at 25 to 50 μm, concentrations at which drug cytotoxicity was not observed (Fig. 5, B and C). Further detailed studies revealed that CK548 did not inhibit HIV-1 DNA synthesis at the lower dosage (12.5 μm), but caused a decrease in viral DNA at higher dosages (25 and 50 μm) early (12 h), likely resulting from lower entry (Fig. 5D). However, at later time points (48 h), CK548 enhanced viral DNA synthesis at all dosages (Fig. 5D). Examination of viral nuclear DNA, the 2-LTR-circles, revealed that 2-LTR-circles were highly diminished and undetectable at early time points (12 h) in CK548-treated cells, whereas at later time points (48 h), 2-LTR-circles were decreased (12.5 and 25 μm CK548) and remained undetectable (50 μm CK548) (Fig. 5E). If normalized by total viral DNA, the 2-LTR-circles were significantly reduced at all CK548 dosages (Fig. 5F). These data suggest that inhibiting Arp2/3 activity enhances viral DNA synthesis; meanwhile, it diminishes viral nuclear migration. This phenotype is consistent with the Arp3 knockdown results (Fig. 3, H–J) and strikingly resembles the phenotype observed in the cofilin knockdown cells (7). In both cases, actin dynamics are inhibited, which may lead to the retention of HIV in the cortical actin layer. Extended retention of HIV PIC in the cortical actin could enhance viral reverse transcription (12, 35) but hinder viral nuclear migration (7).
Recent studies have suggested that actin dynamics may also be involved in viral late steps such as budding (36–41). We tested the effects of inhibiting Arp2/3 on viral release. CEM-SS cells were infected with a single-cycle virus, HIV-1(Env) (pseudotyped with HIV gp160). Infected cells were washed and treated with 3 dosages of CK548 (12.5 to 50 μm). Viral budding was monitored by measuring p24 release. As shown in Fig. 5G, we observed a moderate inhibition of p24 release (a 34% reduction at 50 μm, 120 h postinfection). This result suggests that HIV early steps have a higher dependence than late steps on Arp3 or Arp2/3-mediated acting branching.
We also tested the effect of CK548 on HIV latent infection of blood resting CD4 T cells. Resting T cells were purified from the peripheral blood, pre-treated with CK548 for 1 h, and then infected with HIV-1 for 2 h. Cells were washed, cultured in medium with CK548, and then activated with anti-CD3/CD28 beads. Viral replication was monitored by p24 release. As shown in Fig. 6A, CK548 diminished HIV infection of blood CD4 T cells at all the dosages tested (12.5 to 50 μm). This inhibition was specific to HIV, as CK548 did not inhibit T cell activation at dosages at or below 25 μm (Fig. 6, B and C).
FIGURE 6.
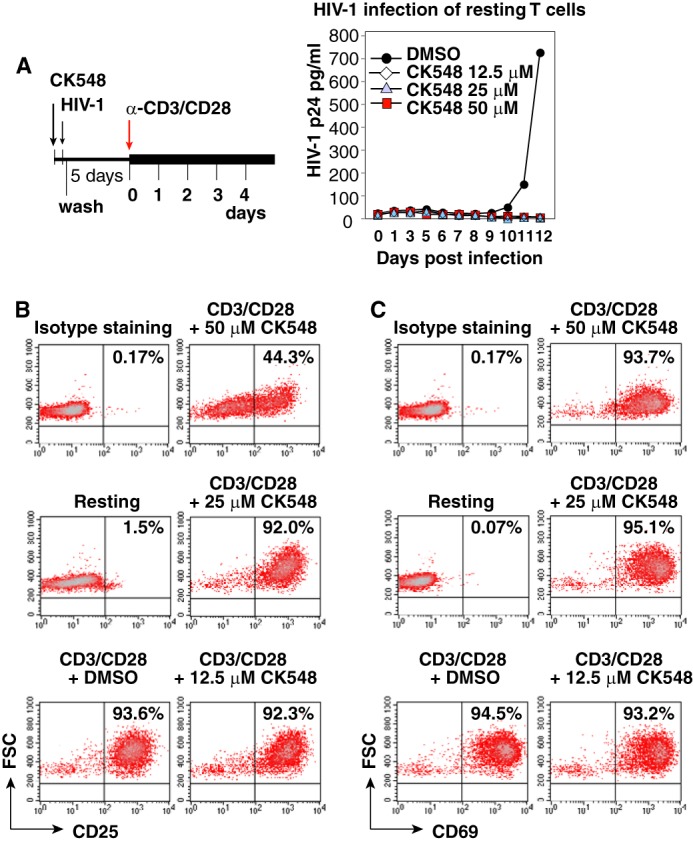
CK548 inhibits HIV-1 latent infection of resting CD4 T cells. A, resting CD4 T cells were pre-treated with CK548 for 1 h, and then infected with HIV-1 for 2 h. Cells were washed, cultured for 5 days in the presence of CK548, and then activated with anti-CD3/CD28 beads (4 beads per cell). Viral replication was monitored by p24 release. B and C, effects of CK548 on T cell activation measured by CD25 and CD69 up-regulation. Resting CD4 T cells were treated with CK548 at different dosages for 1 day, and then stimulated with anti-CD3/CD28 beads (4 beads per cell) for 24 h. Up-regulation of the CD25 and CD69 receptors following stimulation was measured by surface staining with PE-labeled monoclonal antibodies against either CD25 (B) or CD69 (C). Shown are the results from flow cytometry, demonstrating that CK548 did not inhibit T cell activation at dosages at or below 25 μm.
DISCUSSION
Our study presented the first evidence exhibiting a direct protein phosphorylation event occurring in the WAVE2-Arp2/3 actin nucleation and branching pathway, as a result of HIV-1 binding to the chemokine coreceptors. In conjunction with prior studies (6, 7, 42), there is sufficient evidence to indicate that HIV-1 signaling may impinge upon actin filaments from both the barbed and pointed ends: the pointed end is depolymerized as a result of dynamic signaling to LIMK and cofilin (7, 8), whereas at the barbed end, actin polymerization is nucleated by Arp2/3, which is activated by WAVE2. Both LIMK-cofilin and WAVE2-Arp2/3 signaling occur in HIV infection of T cells and macrophages. HIV-mediated actin signals appear to redundantly use multiple pathways, both the Gαi-dependent and -independent, to ensure proper activation of actin dynamics. These results suggest that these signaling events are a conserved feature of early HIV-1 infection.
Although the exact mechanisms by which HIV-1 utilizes actin remain to be elucidated in detail, the inhibition in viral nuclear migration observed in this study implies a specific and direct role of Arp2/3 in HIV intracellular mobility. There are two possible mechanisms (Fig. 7) by which Arp2/3 could directly affect HIV-1 nuclear migration: one, that HIV-1 associates with actin and passively migrates through actin treadmilling mediated by cofilin and Arp2/3 (7, 13); the other, that Arp2/3 anchors directly onto PIC, resulting in the formation of an actin comet tail that drives viral nuclear migration. Notably, Nef (43), nucleocapsid (44–46), integrase (47), and the large subunit of the reverse transcriptase (48), components of PIC, have all been shown to bind actin, lending credence to both models. Furthermore, Nef has been previously shown to dysregulate N-WASP, another Arp2/3-activating NPF, at the immunological synapse in T cells, which may be consistent with the latter model (49).
FIGURE 7.
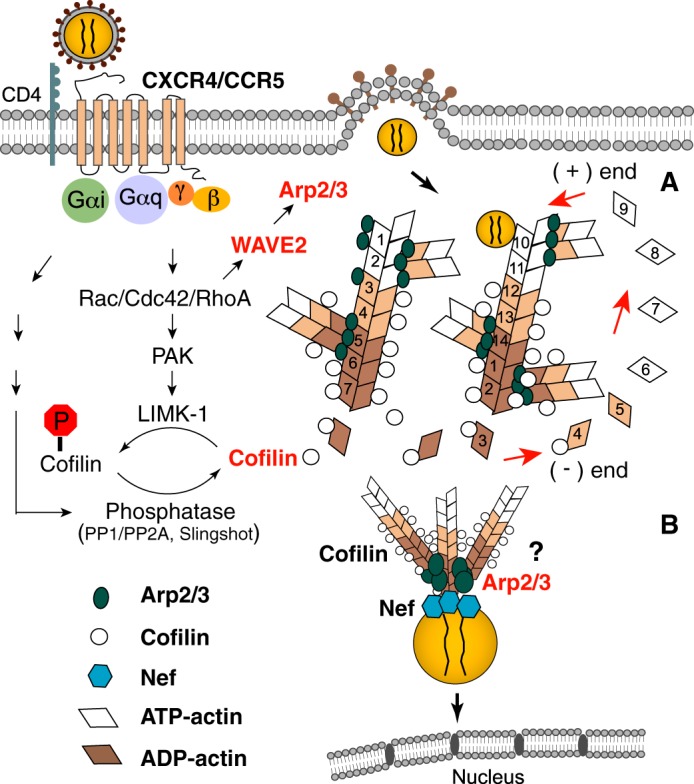
Model of Arp2/3 in HIV intracellular migration. A, binding of HIV gp120 to CXCR4/CCR5 activates G proteins and the downstream Rho family GTPases such as Rac1, leading to the activation of LIMK1 and WAVE2, which modulate cofilin and Arp2/3 activity to promote actin dynamics. Cofilin- and Arp2/3-mediated actin treadmilling promotes intracellular migration of the viral PIC across the actin cortex. B, it is also possible that Arp2/3 anchors directly onto PIC, resulting in the formation of an actin comet tail that drives viral nuclear migration.
Given that the Arp2/3 complex is a fundamental actin modulator, inhibiting Arp3 is expected to have pleiotropic effects on cells. We observed marked phenotypic variations incurred by different CK548 dosages, and between the two Arp3 knockdown cells. However, there is a striking phenotypic consistency in cellular effects on viral post-entry steps when Arp3 was inhibited, either by CK548 or by the shRNA knockdown. In all cases, inhibiting Arp3 enhanced viral DNA synthesis and inhibited viral nuclear migration.
Intriguingly, the Arp2/3 inhibitors, both CK636 and CK548, inhibit the Arp2/3 complex effectively in vitro, but only CK548 effectively inhibits HIV infection. This divergence may reflect a difference in intracellular drug stability or mechanisms of action between CK548 and CK636. Nolen et al. (15) has found that CK636 binds between Arp2 and Arp3, and appears to block them from moving into an active conformation, whereas CK548 inserts into the hydrophobic core of Arp3, blocking its activity. Perhaps, Arp2/3-mediated actin branching may not be an absolute requirement for HIV; it has been known that unbranched actin filaments can still polymerize and treadmill with decreased Arp2/3 activity (2). However, there is a possibility that Arp3 may be present around the docking site of the viral PIC on actin filaments. Thus, CK548 insertion into Arp3 may interfere with the docking of viral PIC onto actin. On the other hand, although CK636 inhibits Arp2 and Arp3 association, it may not impact viral association with actin. Certainly, further detailed mechanistic studies of these two drugs would shed light on how exactly Arp2/3 is involved in HIV PIC interaction with actin.
Actin dynamics have also been suggested to be important for viral assembly and budding (36–41) and HIV cell-cell transmission (50–53). Active actin treadmilling has been suggested to drive viral budding (39). In addition, HIV Gag has been found to bind to actin (44–46) and the actin cross-linking protein filamin A (40), suggesting that these interactions may facilitate the transport and anchorage of Gag to the plasma membrane for viral assembly.
In our study, we observed a moderate inhibition on viral release when Arp3 was inhibited by CK548 (at 50 μm). Interestingly, a recent study has found that depletion of LIMK1 or cofilin impaired HIV-1 particle release (54). In addition, induction of cofilin activity by the cofilin S3 peptide also promoted accumulation of viral particles on the plasma membrane (54). These results suggest that dynamic actin polymerization and depolymerization are required for HIV-1 budding. Perhaps, viral release is more dependent on active actin polymerization and treadmilling than on Arp2/3-mediated actin branching.
In HIV cell-cell transmission, the formation of virological synapase (VS) requires both actin and microtubules for polarized viral budding and viral cell-cell transfer (52, 55, 56). Actin is particularly involved in the clustering and polarization of Env-Gag on the effector cells, and in the stabilization of the CD4/CXCR4 and LFA-1 clusters on target cells (52, 53). In addition, gp120 is shown to induce signal transduction to create an F-actin-depleted zone on target cells, possibly facilitating viral transfer and post transfer events (53). Although the specific role of Arp2/3 in viral cell-cell transmission was not investigated in our study, we expect that Arp2/3 may also be involved in the transmission process. In our study, we mainly focused on addressing the involvement in Arp2/3 in viral early steps using an HIV gp160 pseudotyped single-cycle virus. However, in the multiple round replication of the wild-type HIV-1, Arp2/3-mediated effects on viral assembly and cell-cell transmission may also contribute to the overall levels of viral replication.
The ability of HIV to hijack and modulate the Arp2/3 signaling pathways may also have pathogenic implications. It has been known that genetic defects of Arp2/3 regulators such as WASP cause immunodeficiency (57). HIV-mediated abnormal WAVE2 activity may affect normal T cell function and migration. Finally, the anti-HIV effect of the Arp2/3 inhibitor, as demonstrated in our study, suggest that, pharmacologically, limited targeting of Arp2/3 and upstream regulators could yield novel approaches to prevent HIV transmission. This may offer an advantage in combating viral resistance, given the critical role of these cellular cofactors in the HIV life cycle.
Acknowledgments
We thank the George Mason University Student Health Center for blood donation; the National Institutes of Health AIDS Research and Reference Reagent Program for reagents; W. Ou of FDA for the fusion assay; and J. Guernsey for editorial assistance.
This work was funded by Public Health Service Grants 1R01AI081568 and 1R03AI093157 from NIAID, National Institutes of Health (to Y. W.) and in part by the 2010 NYCDC AIDS Ride (to M. S.).
- NPF
- nucleation-promoting factor
- PIC
- preintegration complex
- PI
- propidium iodide
- PTX
- pertussis toxin.
REFERENCES
- 1. Pollard T. D. (2007) Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 36, 451–477 [DOI] [PubMed] [Google Scholar]
- 2. Wu C., Asokan S. B., Berginski M. E., Haynes E. M., Sharpless N. E., Griffith J. D., Gomez S. M., Bear J. E. (2012) Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 148, 973–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Welch M. D., Iwamatsu A., Mitchison T. J. (1997) Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature 385, 265–269 [DOI] [PubMed] [Google Scholar]
- 4. Cudmore S., Cossart P., Griffiths G., Way M. (1995) Actin-based motility of vaccinia virus. Nature 378, 636–638 [DOI] [PubMed] [Google Scholar]
- 5. Goley E. D., Ohkawa T., Mancuso J., Woodruff J. B., D'Alessio J. A., Cande W. Z., Volkman L. E., Welch M. D. (2006) Dynamic nuclear actin assembly by Arp2/3 complex and a baculovirus WASP-like protein. Science 314, 464–467 [DOI] [PubMed] [Google Scholar]
- 6. Komano J., Miyauchi K., Matsuda Z., Yamamoto N. (2004) Inhibiting the Arp2/3 complex limits infection of both intracellular mature vaccinia virus and primate lentiviruses. Mol. Biol. Cell 15, 5197–5207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoder A., Yu D., Dong L., Iyer S. R., Xu X., Kelly J., Liu J., Wang W., Vorster P. J., Agulto L., Stephany D. A., Cooper J. N., Marsh J. W., Wu Y. (2008) HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 134, 782–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vorster P. J., Guo J., Yoder A., Wang W., Zheng Y., Xu X., Yu D., Spear M., Wu Y. (2011) LIM kinase 1 modulates cortical actin and CXCR4 cycling and is activated by HIV-1 to initiate viral infection. J. Biol. Chem. 286, 12554–12564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang W., Guo J., Yu D., Vorster P. J., Chen W., Wu Y. (2012) A dichotomy in cortical actin and chemotactic actin activity between human memory and naive T cells contributes to their differential susceptibility to HIV-1 infection. J. Biol. Chem. 287, 35455–35469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cameron P. U., Saleh S., Sallmann G., Solomon A., Wightman F., Evans V. A., Boucher G., Haddad E. K., Sekaly R. P., Harman A. N., Anderson J. L., Jones K. L., Mak J., Cunningham A. L., Jaworowski A., Lewin S. R. (2010) Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc. Natl. Acad. Sci. U.S.A. 107, 16934–16939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Campbell G. R., Spector S. A. (2008) CCL2 increases X4-tropic HIV-1 entry into resting CD4+ T cells. J. Biol. Chem. 283, 30745–30753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bukrinskaya A., Brichacek B., Mann A., Stevenson M. (1998) Establishment of a functional human immunodeficiency virus type 1 (HIV-1) reverse transcription complex involves the cytoskeleton. J. Exp. Med. 188, 2113–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spear M., Guo J., Wu Y. (2012) The trinity of the cortical actin in the initiation of HIV-1 infection. Retrovirology 9, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takenawa T., Suetsugu S. (2007) The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 8, 37–48 [DOI] [PubMed] [Google Scholar]
- 15. Nolen B. J., Tomasevic N., Russell A., Pierce D. W., Jia Z., McCormick C. D., Hartman J., Sakowicz R., Pollard T. D. (2009) Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature 460, 1031–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu Y., Marsh J. W. (2001) Selective transcription and modulation of resting T cell activity by preintegrated HIV DNA. Science 293, 1503–1506 [DOI] [PubMed] [Google Scholar]
- 17. Yu D., Wang W., Yoder A., Spear M., Wu Y. (2009) The HIV envelope but not VSV glycoprotein is capable of mediating HIV latent infection of resting CD4 T cells. PLoS Pathog. 5, e1000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu Y., Beddall M. H., Marsh J. W. (2007) Rev-dependent lentiviral expression vector. Retrovirology 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu Y., Beddall M. H., Marsh J. W. (2007) Rev-dependent indicator T cell line. Current HIV Research 5, 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cavrois M., De Noronha C., Greene W. C. (2002) A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 20, 1151–1154 [DOI] [PubMed] [Google Scholar]
- 21. Saeed M. F., Kolokoltsov A. A., Davey R. A. (2006) Novel, rapid assay for measuring entry of diverse enveloped viruses, including HIV and rabies. J. Virol. Methods 135, 143–150 [DOI] [PubMed] [Google Scholar]
- 22. Kelly J., Beddall M. H., Yu D., Iyer S. R., Marsh J. W., Wu Y. (2008) Human macrophages support persistent transcription from unintegrated HIV-1 DNA. Virology 372, 300–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Butler S. L., Hansen M. S., Bushman F. D. (2001) A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7, 631–634 [DOI] [PubMed] [Google Scholar]
- 24. Wu Y., Marsh J. W. (2003) Early transcription from nonintegrated DNA in human immunodeficiency virus infection. J. Virol. 77, 10376–10382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Danson C. M., Pocha S. M., Bloomberg G. B., Cory G. O. (2007) Phosphorylation of WAVE2 by MAP kinases regulates persistent cell migration and polarity. J. Cell Sci. 120, 4144–4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pocha S. M., Cory G. O. (2009) WAVE2 is regulated by multiple phosphorylation events within its VCA domain. Cell Motil. Cytoskeleton 66, 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alfano M., Schmidtmayerova H., Amella C. A., Pushkarsky T., Bukrinsky M. (1999) The B-oligomer of pertussis toxin deactivates CC chemokine receptor 5 and blocks entry of M-tropic HIV-1 strains. J. Exp. Med. 190, 597–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alfano M., Vallanti G., Biswas P., Bovolenta C., Vicenzi E., Mantelli B., Pushkarsky T., Rappuoli R., Lazzarin A., Bukrinsky M., Poli G. (2001) The binding subunit of pertussis toxin inhibits HIV replication in human macrophages and virus expression in chronically infected promonocytic U1 cells. J. Immunol. 166, 1863–1870 [DOI] [PubMed] [Google Scholar]
- 29. Contreras X., Mzoughi O., Gaston F., Peterlin M. B., Bahraoui E. (2012) Protein kinase C-delta regulates HIV-1 replication at an early post-entry step in macrophages. Retrovirology 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Derivery E., Sousa C., Gautier J. J., Lombard B., Loew D., Gautreau A. (2009) The Arp2/3 activator WASH controls the fission of endosomes through a large multiprotein complex. Dev. Cell 17, 712–723 [DOI] [PubMed] [Google Scholar]
- 31. Lapham C. K., Ouyang J., Chandrasekhar B., Nguyen N. Y., Dimitrov D. S., Golding H. (1996) Evidence for cell-surface association between fusin and the CD4-gp120 complex in human cell lines. Science 274, 602–605 [DOI] [PubMed] [Google Scholar]
- 32. Zaitseva M., Romantseva T., Manischewitz J., Wang J., Goucher D., Golding H. (2005) Increased CXCR4-dependent HIV-1 fusion in activated T cells: role of CD4/CXCR4 association. J. Leukoc. Biol. 78, 1306–1317 [DOI] [PubMed] [Google Scholar]
- 33. Kuhn J. R., Pollard T. D. (2005) Real-time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophys. J. 88, 1387–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu X., Kuhn J. R. (2012) Actin filament attachments for sustained motility in vitro are maintained by filament bundling. PLoS One 7, e31385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De B. P., Burdsall A. L., Banerjee A. K. (1993) Role of cellular actin in human parainfluenza virus type 3 genome transcription. J. Biol. Chem. 268, 5703–5710 [PubMed] [Google Scholar]
- 36. Ott D. E., Coren L. V., Kane B. P., Busch L. K., Johnson D. G., Sowder R. C., 2nd, Chertova E. N., Arthur L. O., Henderson L. E. (1996) Cytoskeletal proteins inside human immunodeficiency virus type 1 virions. J. Virol. 70, 7734–7743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ott D. E., Coren L. V., Johnson D. G., Kane B. P., Sowder R. C., 2nd, Kim Y. D., Fisher R. J., Zhou X. Z., Lu K. P., Henderson L. E. (2000) Actin-binding cellular proteins inside human immunodeficiency virus type 1. Virology 266, 42–51 [DOI] [PubMed] [Google Scholar]
- 38. Jolly C., Mitar I., Sattentau Q. J. (2007) Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J. Virol. 81, 5547–5560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gladnikoff M., Shimoni E., Gov N. S., Rousso I. (2009) Retroviral assembly and budding occur through an actin-driven mechanism. Biophys. J. 97, 2419–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cooper J., Liu L., Woodruff E. A., Taylor H. E., Goodwin J. S., D'Aquila R. T., Spearman P., Hildreth J. E., Dong X. (2011) Filamin A protein interacts with human immunodeficiency virus type 1 Gag protein and contributes to productive particle assembly. J. Biol. Chem. 286, 28498–28510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Graziano F., Elia C., Laudanna C., Poli G., Alfano M. (2011) Urokinase plasminogen activator inhibits HIV virion release from macrophage-differentiated chronically infected cells via activation of RhoA and PKCepsilon. PLoS One 6, e23674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harmon B., Campbell N., Ratner L. (2010) Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 6, e1000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Niederman T. M., Hastings W. R., Ratner L. (1993) Myristoylation-enhanced binding of the HIV-1 Nef protein to T cell skeletal matrix. Virology 197, 420–425 [DOI] [PubMed] [Google Scholar]
- 44. Rey O., Canon J., Krogstad P. (1996) HIV-1 Gag protein associates with F-actin present in microfilaments. Virology 220, 530–534 [DOI] [PubMed] [Google Scholar]
- 45. Liu B., Dai R., Tian C. J., Dawson L., Gorelick R., Yu X. F. (1999) Interaction of the human immunodeficiency virus type 1 nucleocapsid with actin. J. Virol. 73, 2901–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wilk T., Gowen B., Fuller S. D. (1999) Actin associates with the nucleocapsid domain of the human immunodeficiency virus Gag polyprotein. J. Virol. 73, 1931–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Turlure F., Devroe E., Silver P. A., Engelman A. (2004) Human cell proteins and human immunodeficiency virus DNA integration. Front Biosci 9, 3187–3208 [DOI] [PubMed] [Google Scholar]
- 48. Hottiger M., Gramatikoff K., Georgiev O., Chaponnier C., Schaffner W., Hübscher U. (1995) The large subunit of HIV-1 reverse transcriptase interacts with β-actin. Nucleic Acids Res. 23, 736–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haller C., Rauch S., Michel N., Hannemann S., Lehmann M. J., Keppler O. T., Fackler O. T. (2006) The HIV-1 pathogenicity factor Nef interferes with maturation of stimulatory T-lymphocyte contacts by modulation of N-Wasp activity. J. Biol. Chem. 281, 19618–19630 [DOI] [PubMed] [Google Scholar]
- 50. Mothes W., Sherer N. M., Jin J., Zhong P. (2010) Virus cell-to-cell transmission. J. Virol. 84, 8360–8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lehmann M., Nikolic D. S., Piguet V. (2011) How HIV-1 takes advantage of the cytoskeleton during replication and cell-to-cell transmission. Viruses 3, 1757–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jolly C., Kashefi K., Hollinshead M., Sattentau Q. J. (2004) HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199, 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vasiliver-Shamis G., Cho M. W., Hioe C. E., Dustin M. L. (2009) Human immunodeficiency virus type 1 envelope gp120-induced partial T-cell receptor signaling creates an F-actin-depleted zone in the virological synapse. J. Virol. 83, 11341–11355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wen X., Ding L., Hunter E., Spearman P. (2013) LIM kinase-mediated actin dynamics modulate retrovirus budding. The 20th Conference on Retroviruses and Opportunistic Infections, March 3–6, Atlanta, Georgia [Google Scholar]
- 55. Jolly C., Sattentau Q. J. (2004) Retroviral spread by induction of virological synapses. Traffic 5, 643–650 [DOI] [PubMed] [Google Scholar]
- 56. Sattentau Q. J. (2010) Cell-to-Cell Spread of Retroviruses. Viruses 2, 1306–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Symons M., Derry J. M., Karlak B., Jiang S., Lemahieu V., McCormick F., Francke U., Abo A. (1996) Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell 84, 723–734 [DOI] [PubMed] [Google Scholar]