

Background: The bacterial second messenger cyclic di-GMP (c-di-GMP) is degraded by EAL phosphodiesterases.
Results: The isolated EAL domain is active only as a homodimer. Substrate binding is coupled with EAL dimerization.
Conclusion: Activity of many full-length EAL phosphodiesterases may be regulated by catalytic domain dimerization.
Significance: A generic mechanism for the regulation of a central node of c-di-GMP signaling is provided.
Keywords: Allosteric Regulation, Bacterial Signal Transduction, Chromatography, Crystal Structure, Phosphodiesterases, c-di-GMP Signaling, Second Messenger
Abstract
The universal second messenger cyclic di-GMP (cdG) is involved in the regulation of a diverse range of cellular processes in bacteria. The intracellular concentration of the dinucleotide is determined by the opposing actions of diguanylate cyclases and cdG-specific phosphodiesterases (PDEs). Whereas most PDEs have accessory domains that are involved in the regulation of their activity, the regulatory mechanism of this class of enzymes has remained unclear. Here, we use biophysical and functional analyses to show that the isolated EAL domain of a PDE from Escherichia coli (YahA) is in a fast thermodynamic monomer-dimer equilibrium, and that the domain is active only in its dimeric state. Furthermore, our data indicate thermodynamic coupling between substrate binding and EAL dimerization with the dimerization affinity being increased about 100-fold upon substrate binding. Crystal structures of the YahA-EAL domain determined under various conditions (apo, Mg2+, cdG·Ca2+ complex) confirm structural coupling between the dimer interface and the catalytic center. The built-in regulatory properties of the EAL domain probably facilitate its modular, functional combination with the diverse repertoire of accessory domains.
Introduction
Most bacteria can exist in two fundamentally different life styles, as motile single cells and as sessile, surface-grown communities called biofilms. The switch between these two cellular states is mediated by the global second messenger cyclic di-GMP (cdG)3 (1). Whereas low levels of cdG promote planktonic behavior, increasing cdG concentrations curb motility, block the expression of toxins and virulence factors, and promote the expression of adhesion factors and a range of different sugar polymers forming the biofilm matrix (2).
CdG synthesis is catalyzed by the GGDEF domains of diguanylate cyclases (DGCs), whereas its degradation is mediated by phosphodiesterases (PDEs) that contain EAL or HD-GYP domains. These abundant and widespread catalytic domains are typically part of multidomain proteins that also carry regulatory domains involved in signal input (PAS, BLUF, Rec etc.). In this way, various environmental or endogenous input cues can control cellular cdG levels. The regulation of DGCs has been studied in detail for PleD from Caulobacter crescentus (3, 4) and for WspR from Pseudomonas aeruginosa (5). Based on these experiments, an “activation by dimerization” mechanism was proposed (reviewed in Ref. 6). More recently, a study on the DGC DgcZ from Escherichia coli has expanded this view by proposing an additional mechanism in which zinc ion-mediated rigidification of the input domain allosterically impedes enzyme activity (7).
EAL-type PDEs hydrolyze cdG in the presence of Mg2+ or Mn2+ to yield the linear pGpG dinucleotide (8, 9). A catalytic mechanism for this class of enzymes was first proposed by Liang and co-workers (10) based on mutagenesis in RocR and the crystal structure of the isolated EAL domain (TdEAL) of a PDE from Thiobacillus denitrificans (PDB code 2R6O). Later, the crystal structure of the YkuI·cdG·Ca2+ complex revealed the spacious substrate-binding site at the C-terminal end of the central β-barrel (11). Finally, based on the substrate·Mn2+ complex structure of the active BLUF-EAL phosphodiesterase BlrP1 from Klebsiella pneumoniae, a two-metal assisted catalytic mechanism was proposed with each of the two manganese ions coordinating the hydrolytic water molecule positioned in-line with the scissile phosphodiester bond (12). This was later confirmed by the TdEAL·Mg2+ complex structure (13). The substrate complex structure of BlrP1 in the presence of calcium revealed a somewhat distinct coordination geometry with only one of the ions coordinated by the hydrolytic water (12) and, thus, rationalized the observation that calcium is inhibiting the enzyme (8).
However, not all EAL domains are catalytically active. Some, such as YkuI, show degenerated active sites, but are still able to bind cdG. Apparently, they have adopted a role as highly specific cdG receptors that are thought to generate an output via their associated domain(s) upon second messenger binding to the EAL domain (14, 15).
YahA from E. coli is a PDE (9) with an N-terminal LuxR-like DNA binding domain. In vivo, it has been demonstrated that YahA overexpression confers a PDE phenotype (16). As a first step toward the understanding of the regulatory node represented by YahA, we embarked on a detailed and quantitative structure-function study of its catalytic EAL domain. The study revealed that the YahA-EAL domain has intrinsic regulatory properties that are likely of general relevance. Although the substrate-binding site is completely contained within the monomer, (i) PDE activity is dependent on the quarternary state of the EAL domain and (ii) cdG binding stabilizes the dimeric state. We propose that these are general properties of multidomain EAL proteins.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification
Two constructs of the EAL domain of YahA from E. coli were generated by cloning the coding sequence for residues 96–362 and 101–362 into pET21b and pET28a vectors (Novagen), respectively, between the NdeI and NotI restriction sites. This yielded constructs YahA96 and YahA101 with a C- and N-terminal His6 tag, respectively. In addition, two single residue mutants (D263N and S298W) were also constructed in the YahA96 and YahA101 backgrounds, respectively. All protein variants were overexpressed and purified using the following protocol.
Protein was expressed in BL21(DE3) host cells in LB media in the presence of 50 μg/ml of ampicillin or kanamycin. At A600 ∼ 0.6, the incubation temperature was shifted to 30 °C and the growing culture was induced with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside. Cells were finally harvested 4 h post-induction by spinning the cultures at 10,000 rpm (Sorvall SLA3000 rotor) for 10 min. The cell pellet was resuspended in buffer A (50 mm Tris-HCl, pH 8.0, 250 mm NaCl, 20 mm imidazole) with protease inhibitor mixture supplied by Roche. Cells were lysed using a French press in two cycles and the lysate was centrifuged for 30 min at 20,000 × g (Sorvall SS34 rotor) and 4 °C to remove any suspended particles. Clear supernatant was collected and loaded on a Ni-NTA affinity column. The column was washed with plenty of buffer A until the baseline was reached and the bound protein was eluted with a 0 to 50% linear gradient of buffer B (50 mm Tris-HCl, pH 8.0, 250 mm NaCl, 1.0 m imidazole) in buffer A.
Fractions containing the desired protein were pooled and concentrated to a final volume of 4–5 ml. The concentrated protein was filtered using a 0.22-μm membrane and then loaded on a Superdex S200–26/60 gel filtration column in SEC buffer (50 mm Tris-HCl, pH 8.0, 250 mm NaCl). Eluted protein peak was collected and pooled. The protein was quantified by recording absorbance at 280 nm and stored at −20 °C at ∼0.5 mg/ml concentration.
Preparation of cdG
CdG was produced enzymatically using a previously reported method (17) by employing a product-inhibition-deficient mutant of diguanylate cyclase DgcZ (also known as YdeH). The dinucleotide was purified from residual GTP, GDP, and phosphate in reversed-phase column chromatography with a Resource-15 RPC column (3 ml, GE Healthcare) with triethylammonium bicarbonate as the starting buffer and ethanol as the eluent. After removing the volatile buffer and the eluent by freeze-drying, the powder was dissolved in water. The concentration of the nucleotide was calculated by recording absorbance at 253 nm and using an absorption coefficient of 28′590 m−1 cm−1 (18). The purity of the product was checked by mass spectrometry.
Crystallization and Data Collection
High resolution crystal structures were determined of WT YahA-EAL in its apo state, as well as of the binary Mg2+ complex and the ternary cdG·Ca2+ complex. Table 1 provides details of crystallization conditions and ligands/cofactors present in the crystallization drops. In its apo form, the protein was crystallized at 8 mg/ml. To grow complex crystals, the protein was concentrated in the presence of 2 mm CaCl2 (or MgCl2). cdG was then added to the concentrated protein to yield a final concentration of 8–10 mg/ml protein with a 1:5 molar ratio of protein to cdG.
TABLE 1.
Crystallization, crystallographic data collection, and structure refinement statistics
Values in parentheses refer to the highest resolution bin.
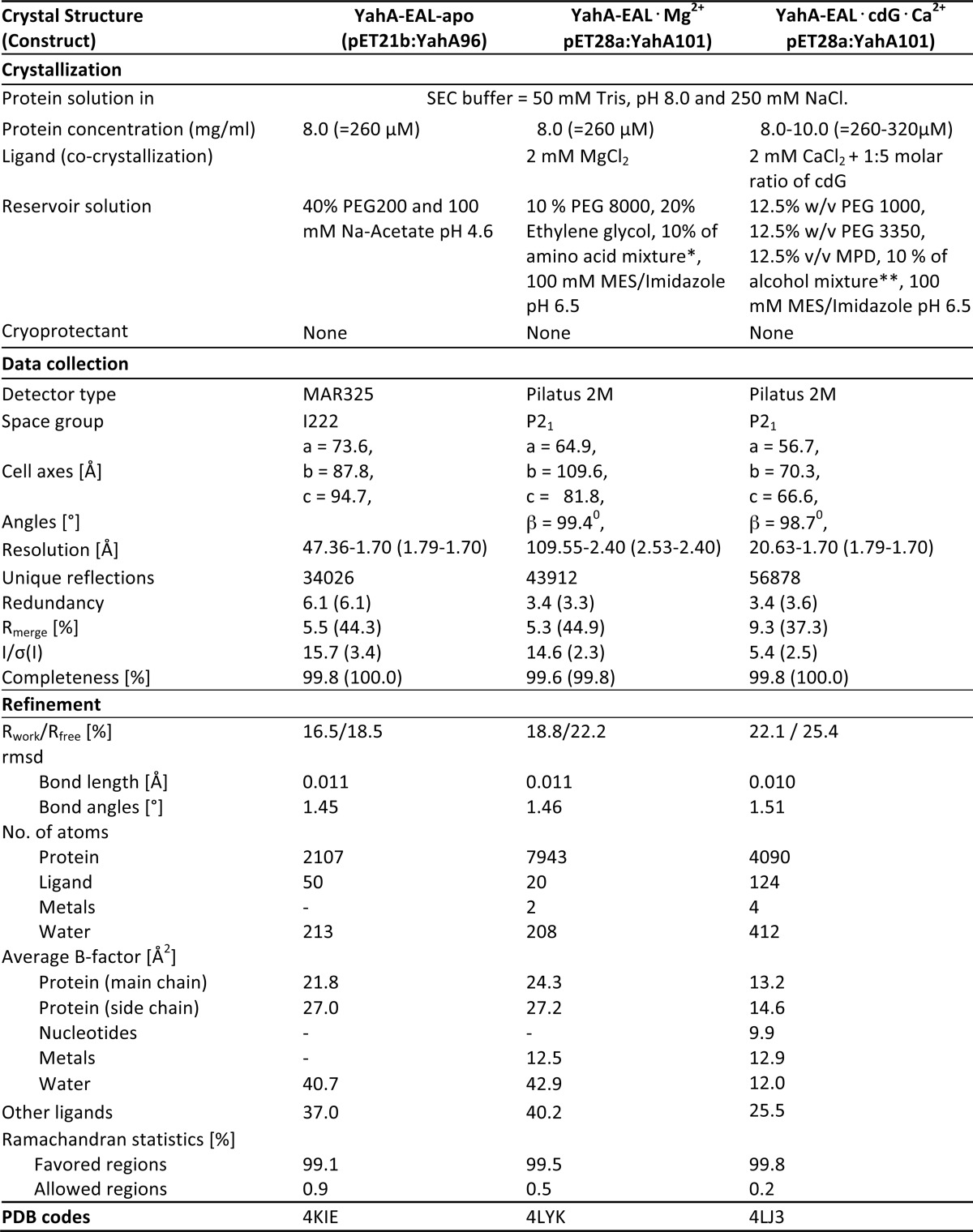
* Amino acid mixture = 200 mm of each of sodium-l-glutamate, dl-alanine, glycine, and l-lysine HCl, 0.2 m dl-serine. ** Alcohol mixture = 200 mm of each of 1,6-hexanediol, 1-butanol, (RS)-1,2-propanediol, 2-propanol, 1,4-butanediol, and 1,3-propanediol.
Crystallization of all the proteins was carried out using sitting drop vapor diffusion method at 20 °C by mixing the protein with the reservoir solution in 1:1 ratio. All single crystal x-ray diffraction datasets were collected at PXIII beam line of Swiss Light source, Villigen, Switzerland. For the YahA-EAL-apo crystal, a total of 150 diffraction images (Δϕ = 10, t = 1 s) were collected. 800 diffraction images (Δϕ = 0.25, t = 0.25 s) were collected for each of the complex crystals.
Structure Solution and Refinement
Diffraction datasets were processed either with MOSFLM (19) or XDS (20) and the resulting intensities were scaled using SCALA (21) from CCP4i suite. At this step, a set of 5% reflections (Rfree set) was taken out for cross-validation (22).
All three structures were solved by molecular replacement using program Phaser (23). For determination of the YahA-EAL apo structure, a YahA-EAL homology model based on TdEAL (PDB code 2R6O) was used. For solving the complex structures, the refined apo YahA-EAL structure was used as search model.
Further refinement of structures was carried out using REFMAC5 (24). Model building was performed using COOT (25). Side chains for some residues were omitted based on the 2Fo − Fc map. Ligand molecules and metal ions were modeled in the Fo − Fc difference electron density map. Finally, water molecules were placed where the Fo − Fc map exceeded 3σ and potential hydrogen bonds could be formed. Model validation was carried out with PROCHECK (26). All structure figures were produced by using DINO.
Microscale Thermophoresis Binding Assay
cdG affinity to the EAL domain was measured by microscale thermophoresis (27) in competition experiments with fluorescein-labeled cdG (fl-cdG) (2′-Fluo-AHC-c-diGMP, Biolog, Bremen, Germany). The experiments were conducted in a Monolith NT.115 device using standard treated capillaries (NanoTemper Technologies). Changes in fluorescence intensity due to thermophoresis were recorded using the blue channel optics of the instrument (λex = 470 ± 15 nm, λem = 520 ± 10 nm), during a 30-s period of infrared laser heating at 70% of maximum laser power followed by a 5-s cooling period. Measurements were performed in buffer containing 50 mm Tris-HCl, pH 8.0, 250 mm NaCl, and 0.1% Tween 20. 2 mm CaCl2 or MgCl2 were added to the buffer when required.
For each protein and each set of conditions, a binding titration and a competition titration were conducted. For the binding titration, a varying concentration of protein (a 16-point 1:1 serial dilution series) was titrated against a fixed concentration of fl-cdG (60 nm). In this experiment, the relative change in fluorescence intensity due to thermophoresis at different concentrations of protein were reported on the binding of fl-cdG to the protein. For the competition titration, a varying concentration of unlabeled cdG was titrated against the same fixed concentration of fl-cdG (60 nm) and protein (at a concentration determined from the first titration, see Table 2). In this experiment, the relative change in fluorescence intensity due to thermophoresis at different concentrations of protein was reported on the displacement of fl-cdG from the protein by unlabeled cdG.
TABLE 2.
Ligand dissociation constants (nm)
CdG and fl-cdG dissociation constants in the absence and presence of 2 mm CaCl2 measured by microscale thermophoresis. [fl-cdG] = 60 nm, [protein] = 2 μm and 90 nm for the cdG competition experiments in the absence and presence of CaCl2, respectively. Values are rounded to 2 digits, p = 0.95 confidence intervals are given in parentheses assuming a 5% error in the individual measurements.
| fl-cdG |
cdG |
|||
|---|---|---|---|---|
| −Ca2+ | +Ca2+ | −Ca2+ | +Ca2+ | |
| YahA-EAL | 489 (281…876) | 33 (12…80) | 22 (6…107) | <1 |
| YahA-EAL(S298W) | 525 (310…883) | 48 (21…90) | 126 (15…526) | <1 |
For evaluation, data were fitted (program ProFit, Quansoft, Zurich, Switzerland) to a ligand competition model with ligands L1 and L2 competing for the same binding site on a protein P. The concentration of the three protein states (P, PL1, and PL2) were calculated from the total concentrations ([P0], [L10], [L20]) according to the analytical solution of the problem (28). As parameters, the model contains the dissociation constants of labeled and unlabeled ligand, Kd1 = [L1]·[P]/[PL1] and Kd2 = [L2]·[P]/[PL2], respectively, and two scale factors that relate the fluorescence signal I of free and bound fl-cdG to their concentrations (Ifree = sc1·[L1], Icompl = sc2·[PL1]). Additionally, the factor sc3 was introduced to correct for errors in protein concentration due to protein loss in the capillary and uncertainty in the protein extinction coefficient that was used for concentration determination. This factor was only refined when the competition data revealed clearly that the fl-cdG was already fully competed out at a [cdG] < [protein] and adopted values in the range between 0.45 and 0.78. Confidence intervals were estimated by a Monte-Carlo method implemented in ProFit, assuming a fixed 5% error on each data point.
Size Exclusion Chromatography (SEC) Coupled with Multiangle Light Scattering (MALS)
A Wyatt silica SEC column (4.6 × 300 mm, 5 μm bead, 300 Å pore) on an Agilent 1100 series HPLC was employed for the determination of the oligomeric state of YahA-EAL under different conditions. The instrument was coupled with a Wyatt miniDawn TriStar multiangle light scattering detector and a Wyatt Optilab rRex refractive index detector. The column was equilibrated for 3 h to obtain stable baseline signals from the detectors before data collection. The inter-detector delay volumes and band broadening, the light-scattering detector normalization, and the instrumental calibration coefficient were calibrated using a standard 2 mg/ml of BSA solution (Thermo Pierce) run in the same buffer, on the same day, according to standard Wyatt protocols. The absolute refractive index of the buffer was measured using the refractive index detector.
The protein sample (20 μl) was loaded on the column in the absence or presence of substrate cdG (5:1 molar ligand/protein ratio) and/or divalent cations (2 mm CaCl2 or MgCl2). All experiments were performed at 6 °C at a flow rate of 0.4 ml/min in SEC buffer (see the purification section). Where appropriate, the SEC buffer contained in addition 2 mm CaCl2 or MgCl2, but no cdG was added to any running buffer.
The molecular weight and mass distribution of the samples were then determined using the ASTRA 5 software (Wyatt Technology). The occupancy of the EAL·cdG complexes at elution was calculated from the UV peak areas of the apo and the complexed protein. No loss of sample on the column and an unchanged extinction coefficient of cdG upon binding were assumed. The extinction coefficient of cdG (ϵcdG(280) = 17,160 m−1 cm−1) was calculated from the published ϵcdG(253) = 28.6 × 103 m−1 cm−1 (18) and the measured cdG absorption spectrum. For the protein, an ϵYahA-EAL(280) of 27,180 m−1 cm−1 was obtained from the EXPASY server.
SEC-MALS derived apparent mass values (Fig. 6C) were used to derive the dimerization affinity of YahA-EAL under various conditions, assuming a fast monomer-dimer equilibrium. According to the mass action law, the monomeric molar fraction, xm = [M]/[M0], is given, by,
with [M0] = [M] + 2 · [MM], total protein concentration at elution; [M], monomer concentration; [MM], dimer concentration.
FIGURE 6.
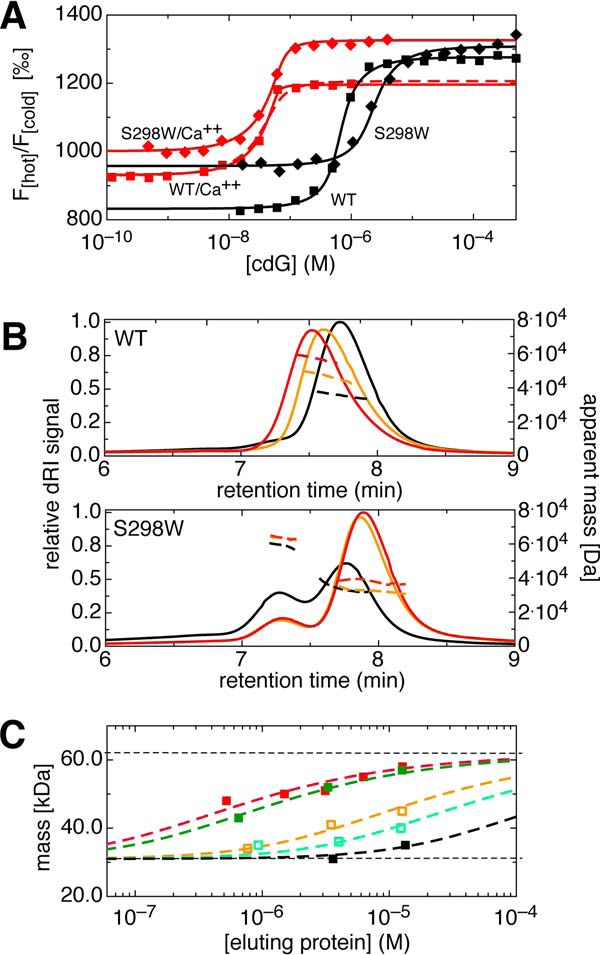
EAL/substrate and EAL/EAL affinity. Concentration of divalent cations was 2 mm, where applicable. A, titration of cdG to YahA-EAL wild-type (squares) and S298W mutant (diamonds) resulting in displacement of fl-cdG (60 nm) from the protein as measured by microscale thermophoresis. Data have been acquired in the absence (black) or presence of Ca2+ (red). The data were fitted to a ligand competition model (solid lines) (28) yielding the cdG dissociation constants given in Table 2. For the wild-type, fit curves for Kd = 0.1 (solid line) and 1.0 nm (broken line) are shown. B, SEC-MALS chromatograms (loading concentration 180 μm) for Yaha-EAL wild-type (top) and the S298W mutant (bottom). The proteins were analyzed in the absence of divalent cations (black), in the presence of Ca2+ (orange), and in the presence of Ca2+ and 540 μm cdG (red). Continuous lines represent the dRI signal (left axis), broken lines the MALS derived apparent mass values (right axis). C, compilation of MALS data (weight-average molecular mass) acquired at various loading concentrations for wild-type YahA-EAL. Data shown are for the apo protein (black), for the binary complexes with Mg2+ (light green) or Ca2+ (orange), and for the ternary complexes (molar cdG:protein ratio, 3:1) with cdG/Mg2+ (green) or cdG·Ca2+ (red). The data were fitted with a dynamic monomer-dimer model in fast exchange with the values of the pure species set to their nominal values (horizontal lines). The derived Kd values are listed in Table 3.
This yields for the weight-averaged apparent mass (see also (29)).
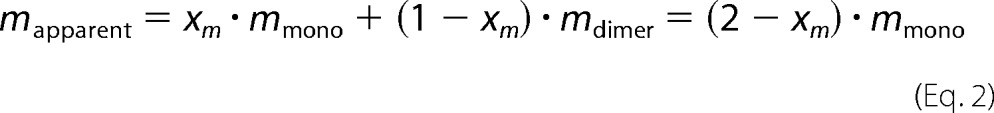 |
The experimental data (Fig. 6C) were fitted (program ProFit) to Equation 2 with a fixed mmono of 31 kDa to yield the dimerization Kd.
Analytical Ultracentrifugation
Sedimentation equilibrium runs were conducted at 8 °C using An-50Ti rotor in a Beckman Coulter XL-I analytical ultracentrifuge. The YahA-EAL protein in SEC buffer supplemented with 2 mm CaCl2 was subjected to ultracentrifugation in the presence of cdG (1.25: 1 m cdG/protein ratio). For each protein concentration (1 μm, 2 μm), three runs (9,700, 16,500, and 28,000 rpm) were performed. The radial absorption profiles (range r = 7.01 to 7.17 cm) were fitted globally (Fig. 7) to a monomer-dimer self-association model (Equation 10 in Ref. 30) to yield the dimerization Kd. The contribution of free cdG ligand was neglected and subsumed in the base line.
FIGURE 7.
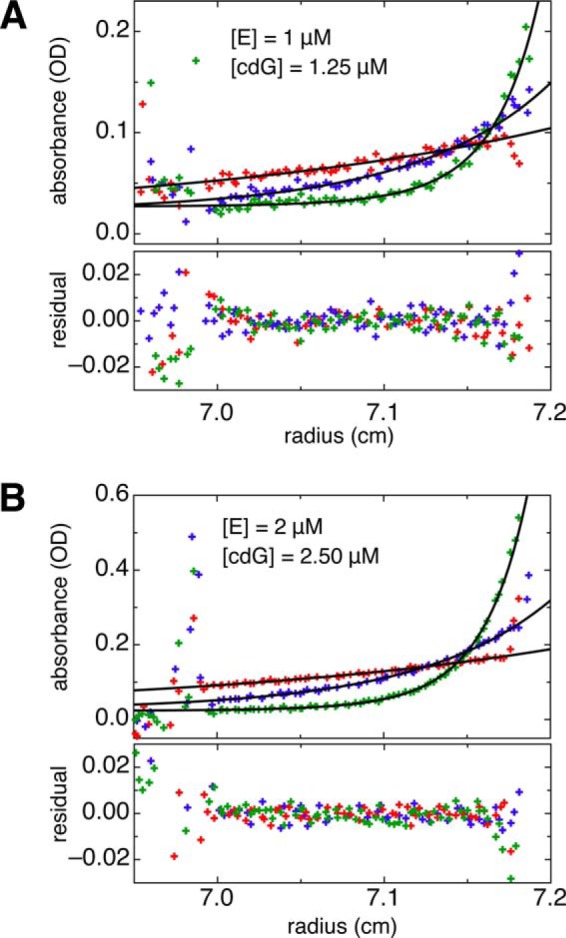
AUC-SE data for YahA-EAL in the presence of 2 mm CaCl2. Protein and substrate concentrations are indicated. For each sample, data are shown for three speeds (9,700, 16,500, and 28,000 rpm). All six runs were fitted globally to a dynamic monomer-dimer self-association model with the monomer mass set to 31 kDa. The fit yielded a dimerization Kd of 0.5 μm (Table 3).
Enzymatic Assay
Enzymatic activity was assayed off-line by FPLC-based steady-state nucleotide quantification following incubation for varying durations. Enzymatic reactions were carried out at room temperature in 50 mm Tris-HCl, pH 9.35, 50 mm NaCl, 5 mm MgCl2, 0.5 mm EDTA, and 50 μm thiamine pyrophosphate as FPLC standard. In all experiments, the saturating substrate concentration (20 μm) was used. The reaction was started by addition of enzyme (×100 concentration) to a total reaction volume of 600 μl. Samples volumes of 100 μl were withdrawn and the reaction was stopped at different time points by addition of 10 μl of 100 mm CaCl2.
The samples were then analyzed using ion-exchange chromatography (1-ml Resource-Q column) after addition of 890 μl of 5 mm ammonium bicarbonate (NH4CO3) to increase the volume to 1 ml. 500 μl of this was then loaded onto the column. The column was washed thoroughly and the bound nucleotides were eluted with a linear NH4CO3 gradient (5 mm to 1 m) over 17 column volumes. The identity of the eluting species was verified by mass spectroscopy.
The amount of pGpG product was determined by integration of the corresponding absorption (253 nm) peak after normalization of the data with respect to the internal thiamine pyrophosphate standard. For each ([M0], [S]) combination of concentrations, the progress curve was fitted to a simple Michaelis-Menten kinetics model (Fig. 8B) yielding the specific activity kcat,meas. Assuming a fast monomer-dimer equilibrium, the apparent specific activity is given by,
with kcat,mono, kcat,dimer: specific activity (turnover number) of monomer and dimer, respectively, and xm given by Equation 1.
FIGURE 8.
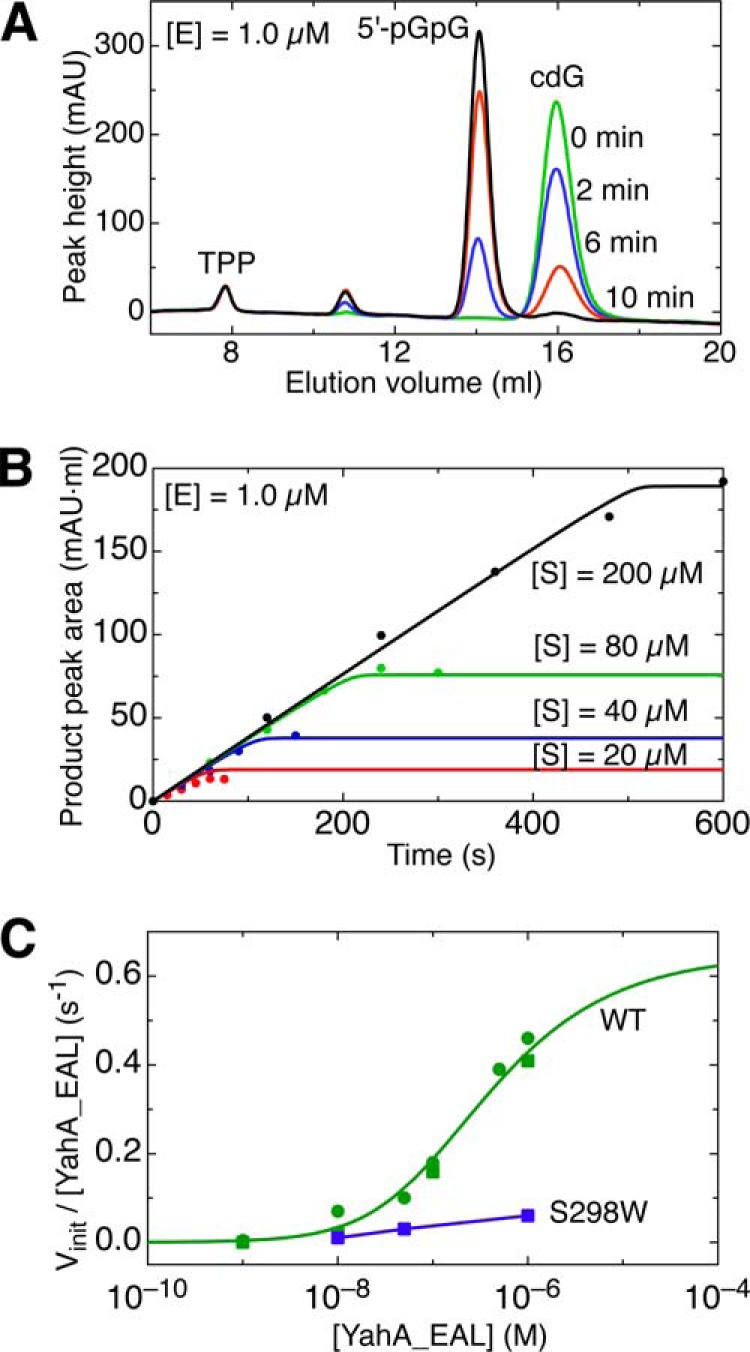
Catalytic activity of the YahA-EAL domain. A, FPLC chromatograms showing time-dependent conversion of cdG to pGpG. CdG (200 μm) was incubated for the indicated time spans with 1 μm YahA-EAL. B, progress curves of pGpG production with the initial cdG substrate concentrations indicated. Data were fitted to a simple Michaelis-Menten kinetics model. C, specific activity vinit/[YahA-EAL] as a function of [YahA-EAL] concentration, acquired at saturating substrate concentration. For the wild-type protein (green), data points are shown from two separate experiments. The data were fitted (continuous line) to a simple monomer-dimer equilibrium model and indicate that the enzyme is inactive as monomer. The dimer interface mutant S298W (blue) is virtually inactive.
Thermodynamic Model of Ligand-induced Dimerization
For the derivation of the thermodynamic coupling between ligand binding and dimerization consider the scheme shown in Fig. 9. Monomer (M) and dimer (MM) are in thermodynamic equilibrium. A ligand L binds to the monomer to form a monomeric ligand complex (LM). This can associate to form a dimeric ligand complex (LMML). In addition, a singly occupied dimer (LMM) has to be considered. The indicated second-order association constants refer to dimerization (K00, K11) or ligand binding (Ks1, Ks10, Ks11) and are defined as follows.
In addition, a third-order association constant K11′ is defined, which also related to species LM and LMML
with
we get
Because the scheme shown in Fig. 9 represents a thermodynamic cycle, the LMML state can be reached via two routes both starting from state M. Therefore, not all association constants are independent, but they are coupled by the following relation.
Assuming no cooperativity in ligand binding to the dimer (Ks10 = Ks11), i.e.
yields the following.
Finally, introducing dissociation constants
FIGURE 9.

A, thermodynamic scheme showing five YahA-EAL states that are in fast thermodynamic equilibrium: monomeric YahA-EAL in the uncomplexed (0) and cdG (ring symbol) complexed (1) state as well as dimeric YahA-EAL in the uncomplexed (00) and singly (10) or doubly (11) occupied dimeric state. Magnesium ions in sites M1 and M2 are indicated. The second-order ligand (Ks1, Ks10, and Ks11) and dimer (K00, K11) association constants and turnover numbers (kcat,10 and kcat,11) are indicated. Assuming no cooperativity, Ks10 = Ks11 and kcat,10 = kcat,11. Monomeric YahA-EAL is inactive, due to the postulated absence of a divalent cation in site M2. Dimeric YahA-EAL hydrolyzes cdG to yield the linear pGpG dinucleotide (open ring symbol). B, generic regulatory mechanism for a full-length EAL phosphodiesterase with associated regulatory domain (R). Wavey lines indicate interfaces that undergo structural changes. The protein is monomeric (top), but dimerizes via the R domains upon signal perception (bottom left). This promotes dimerization of the EAL domains, due to the increase of their local concentration (bottom left). Finally, structural changes in the EAL/EAL interface induced by dimerization are coupled to structural changes in the active site that would affect substrate affinity and/or catalytic activity.
Kd,11 = 1/K11, Kd,00 = 1/K00, Kd,s10 = 1/Ks10, Kd,s1 = 1/Ks1, we get the equivalent relationship.
Thus, any difference in the dimerization propensity of the liganded versus unliganded state is coupled to a difference in ligand affinity to the dimeric versus monomeric state.
RESULTS
Structural Basis
High resolution crystal structures of the EAL domain of YahA from E. coli (YahA-EAL) were determined under various conditions, namely in the uncomplexed state, as a Mg2+ complex and as a ternary YahA-EAL·cdG·Ca2+ substrate complex. Data processing, structure refinement, and validation statistics are shown in Table 1. The modified triose-phosphate isomerase (TIM)-barrel-fold (αβ(βα)7) of YahA-EAL (Fig. 1A) shows close similarity to other EAL structures such as YkuI (11), BlrP1 (12), or TdEAL (13). The primary structure of YahA-EAL with assigned secondary structural elements and conservation logo is shown in Fig. 2. All residues that are important for catalysis (13, 31) are conserved.
FIGURE 1.

Crystal structures of the YahA-EAL domain. Schematic representation with secondary structure elements and chain termini labeled. The β5-α5 loop is highlighted in magenta. A, structure of the monomer of YahA-EAL in complex with Mg2+ (M1). Mutation site Ser-298 is shown in full and labeled. B, structure of the monomer of YahA-EAL in complex with substrate (cdG) and Ca2+ (M1 and M2). C, superimposition of YahA-EAL-apo (brown) with YahA-EAL·Mg2+ (steel blue) and YahA-EAL·cdG·Ca2+ (light green). D, subunit arrangement in YahA-EAL·Mg2+ (canonical EAL dimer). The asymmetric unit contains two dimers, both are virtually identical. E, subunit arrangement in YahA-EAL·cdG·Ca2+ (closed EAL dimer). The asymmetric unit contains one dimer with virtually identical subunit structure. In panels D and E the same color code is used as in panels A and B, but with symmetry mates shown in gray.
FIGURE 2.
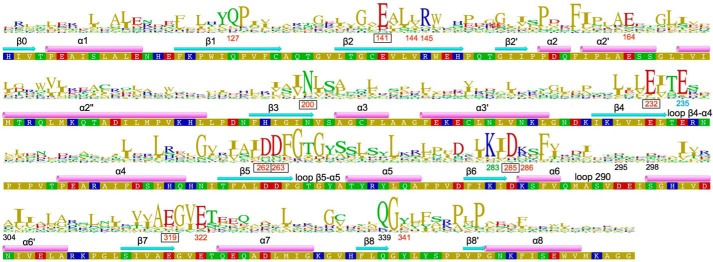
Sequence of the EAL domain of YahA from E. coli (UNIPROT accession name P21514) with experimentally determined secondary structural elements and HMM logo based on a non-redundant set of 62 EAL sequences. Important residues are labeled with their number. cdG binding residues are shown in red. Metal coordinating residues are represented in boxes. The anchoring glutamate is shown in blue, whereas the general base lysine is in green. Other important residues are shown in black.
In the presence of Mg2+, YahA-EAL forms a 2-fold symmetric dimer (Figs. 1D and 3, A and C) with the β5-α5 loop, and the α5 and α6′ helices forming isologous contacts with their counterparts. The residues involved in the interaction are well conserved with a buried surface area of 1100 Å2. Although such dimers have been observed in almost all known EAL crystal structures (for a survey, see Ref. 32), the details of the interactions and the relative orientation of the two domains vary somewhat. Because, in solution, YahA-EAL·Mg2+ is monomeric up to a concentration of 10 μm (see below), dimers were probably stabilized by the high protein concentration (260 μm) used for crystallization or have been incorporated selectively into this crystal form.
FIGURE 3.

YahA-EAL dimer interfaces. A and C, canonical YahA-EAL/Mg2+ dimer. B and D, closed YahA-EAL·cdG·Ca2+ dimer. For each structure, two orthogonal views (front and top) are shown. Hydrogen bonds are represented as broken brown lines, divalent cations as spheres.
Fig. 4A shows that at the C-terminal end of the central β-barrel a magnesium cation (M1) is coordinated by Glu-141 (of the EAL motif), Asn-200, Asp-262, and Glu-232, analogous to the situation in the binary TdEAL·Mg2+ complex (13). Because the metal site is fully occupied and the structure was obtained at a physiological magnesium concentration (2 mm) (33), it is likely that in vivo YahA-EAL is constitutively complexed with magnesium. Adjacent to the M1 site, there is an unusual cluster of acidic side chains, with Asp-263 (the second aspartate of the conserved DD motif (Fig. 2) at the end of β5) in H-bonding distance to the conserved residues Glu-235 and Glu-319. Thus, some of the residues should be protonated, irrespective of the close to neutral crystallization and protein buffer conditions (pH 6.5 and 8.0, respectively). In addition, the conserved residue Asp-285 is at close distance (3.2 Å).
FIGURE 4.

EAL active site structures in the absence and presence of cdG substrate. Divalent cations are colored in green (Mg2+), yellow (Ca2+), or magenta (Mn2+). A, YahA-EAL·Mg2+, B, YahA-EAL·cdG·Ca2+, and C, BlrP1·cdG/Mn2+ (PBD code 3GG0) (12). Water molecules are represented as red spheres, cation coordination bonds by black broken lines, and loop β5-α5 is highlighted in magenta. In A, Asp-263 is H-bonded (orange broken lines) to surrounding carboxylic side chains. In B and C, the hydrolytic water (W) is in-line with the scissile O3′-P bond of the cdG substrate (shown in full with cyan carbons). Note that the location of the “anchoring” glutamate (Glu-235 in YahA, Glu-275 in BlrP1) is distinct in the binary and ternary complexes. D, stereoview of the superposition of YahA-EAL in complex with Mg2+ (ribbon and carbon atoms in gray) and cdG·Ca2+ (ribbon and carbons atom in light green).
When the protein was crystallized in the absence of divalent cations, a different crystal form was obtained (Table 1) in which YahA-EAL forms a disulfide-linked dimer employing Cys-131 and its symmetry mate Cys-131*. This is probably a crystallization artifact, considering the reducing environment under physiological conditions, the residue variability at this position (Fig. 2), and the fact that in solution only EAL monomers were observed even in the absence of reducing agent (see further below). The overall-fold is virtually identical to that of the YahA-EAL·Mg2+ complex with the notable exception of changes in the β5-α5 loop (Fig. 1C), which makes few 2-fold symmetric contacts with the β5-α5 loop of a symmetry-related molecule. The conformation of the loop, which shows high B-factors, is incompatible with the tight canonical YahA-EAL·Mg2+ dimer (Fig. 1D). This re-enforces the notion that EAL dimerization is coupled with β5-α5 loop conformation (31).
Substrate Complex Structure
The structure of the enzyme in complex with the substrate was determined in the presence of 2 mm calcium to prevent substrate turnover (9). The structure is virtually identical to the apo and magnesium complex structures (Figs. 1, A–C). Unexpectedly, the ternary YahA-EAL·cdG·Ca2+ complex showed a non-canonical dimeric assembly (Figs. 1, E and G, and 3, B and D), which was also observed in two other, low resolution crystal forms grown under different crystallization conditions. The dimer interface (1400 Å2) overlaps largely with the canonical β5-α5 loop, α5, α6′ interface, but in addition, involves the α6-α6′ loop (290 loop). Because this loop is not conserved among EAL homologs (Fig. 2), the physiological relevance of this interaction and thus of this dimeric arrangement is unclear, but might fulfill a YahA specific task.
Note that the dimerization loop (β5-α5 loop) adopts distinct conformations dependent on the ligation state (magenta loop in Figs. 4, A and B, and 5). The relevance of this loop (“loop 6”) for catalysis has been recognized before by mutagenesis in RocR (10) and evidence for its flexibility has been obtained by deuterium-hydrogen exchange experiments (31). The conformation of the β5-α5 loop is correlated with distinct side chain orientations of Glu-235 from the adjacent β4-α4 loop (Fig. 5). This highly conserved “anchoring glutamate” interacts with the β5-α5 loop and its mutation to alanine in RocR (E268A) rendered the enzyme inactive (10).
FIGURE 5.
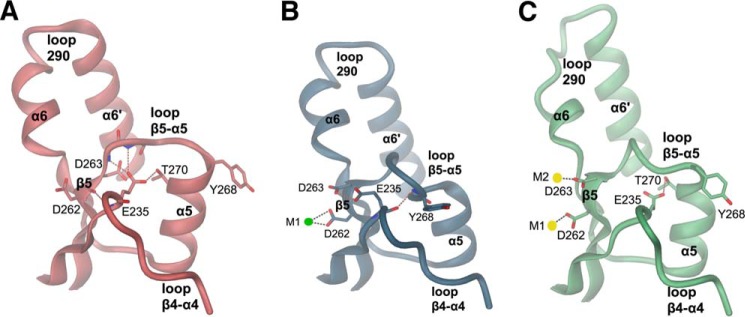
Loop β4-α4, β5-α5, and loop 290 conformations of apo YahA-EAL (A), YahA-EAL·Mg2+ (B), and YahA-EAL·cdG·Ca2+ (C). The distinct interaction patterns between the anchoring glutamate (Glu-235) and loop β5-α5 are represented by broken lines in brown.
As observed for other EAL domains (see Ref. 32 and references therein), the substrate is bound to an extended shallow groove at the C-terminal end of the β-barrel (Fig. 4B). There are two calcium ions, one bound to the M1 site (with identical coordination as in the Mg2+ complex), and the other (M2) coordinated by Asp-263, Glu-285, and Glu-319, i.e. residues that form the acidic cluster in the Mg2+ complex (Fig. 4A). Each of the non-bridging oxygens of the proximal phosphate is within coordinating distance to one of the Ca2+ ions. A water molecule (W) is positioned in-line with the scissile O3′-P bond, but is coordinated only with M1 (Fig. 4B).
A very similar arrangement has been described for the ternary cdG·Ca2+ complex of BlrP1 (12), where substitution of Ca2+ against the active cofactor Mn2+ resulted in a significant shift of M2 and brought this cation within coordinating distance of the catalytic water (Fig. 4C). Correspondingly, Barends et al. (12) proposed a two-metal assisted catalytic mechanism with both cations activating the water and defining the precise relative orientation of the water (W) and the proximal phosphate moiety. The same constellation was observed later for TdEAL·cdG·Mg2+ (13) and can thus be predicted to represent the competent metal constellation for YahA-EAL as well.
In the cdG·Ca2+ and the Mg2+ complex, an unknown small molecule, tentatively assigned as trans-4-(hydroxymethyl) cyclohexanol, was found on the barrel axis at the center of the domain. In the apo structure, a PEG200 molecule was identified at the same position. Although the presence of these molecules is probably artifactual, they identify a large hydrophobic cavity close to the active site that may be occupied in vivo by a bioactive factor.
Substrate Affinity
The affinity of YahA-EAL for cdG was measured by microscale thermophoresis in a competition assay using fluorescein-labeled cdG as a reporter (fl-cdG; Fig. 6A, Table 2). Due to substrate turnover it was not possible to determine fl-cdG and cdG affinity in the presence of magnesium. With calcium as the cofactor, YahA-EAL binds the substrate extremely tightly (Kd ≤ 1 nm), whereas in the absence of divalent cations, the substrate affinity is still considerable (Kd about 20 nm, Table 2). This can be understood by the multitude of interactions the substrate undergoes with the binding site. cdG affinity has been measured before only for the degenerated EAL domain of the FimX receptor (14). Although the protein lacks the conserved cation binding sites, it again showed tight cdG binding (Kd about 100 nm), similar to that of YahA-EAL in the absence of cations.
Fast cdG-dependent EAL Monomer-Dimer Equilibrium
The oligomeric state of YahA-EAL was probed by SEC coupled with MALS (Fig. 6, B and C) and analytical ultracentrifugation-sedimentation equilibrium (AUC-SE, Fig. 7). The SEC-MALS data indicated that the retention time and apparent mass values are dependent on the YahA-EAL concentration (Fig. 6C), which is indicative of a self-association equilibrium that is fast on the time scale of the experiment. The weight-averaged mass values were fitted to the appropriate monomer-dimer equilibrium model (see “Experimental Procedures”) and the results are summarized in Table 3. The protein is largely monomeric up to the maximal loading concentration (180 μm), both in the absence or presence of magnesium. Magnesium and calcium shift the equilibrium somewhat (by a factor of 5 and 14, respectively) toward the dimeric state. However, upon addition of cdG to YahA-EAL·Mg2+ (the probable constitutive form of the protein present in vivo) the dissociation constant is appreciably reduced (40-fold) to a value of 0.6 μm as derived from the MALS data. The potential relevance of this cdG-induced dimerization effect for the mechanism of degenerate EAL effectors (receptors) is discussed further below.
TABLE 3.
Dimer dissociation constants in the absence and presence of cdG (in μm)
The molar cdG:protein ratio was 3:1 and divalent cation concentration 2 mm, where applicable.
| Method |
Kd,00 = K00−1 |
Kd,11 = K11−1 |
|||
|---|---|---|---|---|---|
| −cdG, −M2+a | −cdG, +Mg2+ | −cdG, +Ca2+ | +cdG, +Mg2+ | +cdG, +Ca2+ | |
| MALS (Fig. 6C) | 188 (80…1600) | 36 (21…65) | 13 (0.8…25) | 1.1 (0.6…2.3)b | 0.6 (0.3…1.1)b |
| AUC (Fig. 7) | 0.4 (0.2…0.9) | ||||
| Activity (Fig. 8C) | 0.5 (0.47…0.54) | ||||
a Without divalent cations.
b Sample not fully complexed upon elution; ligand occupancy ∼0.7 as derived from combined UV/RI measurement. p = 0.95 confidence intervals are given in parentheses assuming a 5% error in the individual measurements.
For the substrate complexes, the fit of the MALS data yields only apparent dimer dissociation constants, because cdG was not present in the running buffer. In fact, at elution, the protein showed a substoichiometric amount of bound nucleotide (about 70% as calculated from peak absorption and refractive index values, see “Experimental Procedures”). A similar Kd(app) was obtained with magnesium as divalent cation, suggesting that conversion of the cdG substrate to the pGpG product does not affect the oligomeric state of the enzyme significantly. Finally, the proper equilibrium dimerization Kd of the ternary complex was determined by AUC-SE analysis (Fig. 7). Global fitting of the absorption profiles, acquired at three speeds and two protein concentrations to a self-associating monomer-dimer system (30), yielded a dimerization Kd of 0.4 μm (Table 3). Unfortunately, evaluation of the AUC-SE data for the apo-samples was not possible due to long-term aggregation of the samples.
For further functional studies, a mutant was generated with the aim to prevent dimer formation (both of the canonical and the closed dimer). A residue in the monomer-monomer interface, far away from the active site, was chosen (Ser-298, see Fig. 1A) and replaced by the bulky tryptophan. As indicated by the SEC-MALS chromatogram (Fig. 6B, bottom), the S298W protein is in slow monomer-dimer exchange with the equilibrium indeed largely on the monomeric side, in particular in the presence of substrate. Similar affinities, both of fl-cdG and cdG, were found with respect to the dimer interface mutant S298W (Table 2), demonstrating an intact binding site.
Catalytic Activity Is Dependent on EAL Concentration
It had been suggested before by us (11) and others (12, 31, 34, 35) that EAL phosphodiesterase activity may depend on the quarternary state or arrangement of the catalytic domains, which in turn would be controlled by associated regulatory domains. We tested this hypothesis in a reductionistic approach by altering the monomer-dimer equilibrium of the isolated YahA-EAL domain through concentration variation or site-directed mutagenesis and concomitant measurement of the catalytic activity by FPLC-based nucleotide quantification (Fig. 8, A and B).
Fig. 8C shows that the specific catalytic activity of YahA-EAL indeed strongly depends on the enzyme concentration, converging to 0 at low nanomolar concentrations. The entire profile can be well fitted according to the law of mass action with monomers being inactive and dimers turning over the substrate with a kcat of 0.72 ± 0.06 s−1. This value is in the same range as those observed for PdeA (8), RocR (10), BlrP1 (12), and the isolated EcDos(EAL) domain (9, 32) at micromolar enzyme concentrations. Variation of substrate concentration did not affect the rate of reaction (Fig. 8B) down to the lowest substrate concentration tested (5 μm), suggesting that the Km lies below this value. For unknown reasons, considerably higher specific activity and Km values (kcat = 17 s−1, Km = 35 μm) have been reported before for the isolated EAL domain of YahA (9). The YahA-EAL dimerization Kd, the remaining parameter of the kinetic model, is 0.5 μm. This is in good agreement with the values measured by SEC-MALS and AUC-SE (Table 3).
The mutant S298W shows drastically reduced activity (Fig. 8C) as anticipated from its predominantly monomeric state (Fig. 6B, bottom). The site of the mutation is both solvent-exposed and distant from the catalytic center, so any direct effect of the mutation on that catalytic center appears unlikely. Because activity has been assayed at high substrate concentrations and, in the presence of calcium, substrate binding is virtually unaffected by the mutation (Fig. 6A, Table 2), the reduced activity most probably reflects a drastically reduced kcat.
DISCUSSION
This study focused on some remarkable functional and structural properties of a phosphodiesterase EAL domain in isolation. The structures reported here show YahA-EAL in various ligand-dependent conformational states and qualify it as a prototypic catalytic EAL domain with dimerization propensity. Intriguingly, apart from the canonical dimeric arrangement (11–13) found for YahA-EAL·Mg2+, the ternary substrate complex YahA-EAL·cdG·Ca2+ shows a different subunit organization, although utilizing most of the canonical interface. Whether one or both dimeric configurations can be formed in full-length YahA must await structure investigation. The “closed” dimer reported for FimX (14, 36), although similar in shape, uses a completely different interface.
The EAL domain is in a fast ligand-dependent monomer-dimer equilibrium. A corresponding thermodynamic model including substrate binding to the two states is shown in Fig. 9A. Because the model constitutes a thermodynamic cycle, the dimer association constants for the uncomplexed and complexed protein (K00 and K11, respectively) are related to the ligand binding affinities of monomer and dimer (Ks1 and Ks10, respectively) by the following relationship.
Hereby, lack of cooperativity is assumed, i.e. Ks10 = Ks11 (see “Experimental Procedures”). Accordingly, the dissociation constants are related by the following analogous relationship.
In the presence of Mg2+, the Kd,11/Kd,00 ratio is 0.4/36 = 1/90, which yields a Kd,s10/Kd,s1 ratio of 0.11, i.e. about 10-fold tighter binding of the substrate to the (catalytically competent) dimer than to the monomer. Most probably, not this modest difference in cdG affinity, but the observed drastic change in kcat, with only the dimer being catalytically active, is crucial for the regulation of the enzyme in vivo. The inactive state of the monomer may be largely due to a compromised M2 site that prevents binding of the second Mg2+ ion, which has been shown to be catalytically indispensable in RocR (10) and in TdEAL (13). Indeed, we found for YahA-EAL that the D263N mutation, which affects a residue that coordinates M2, renders the enzyme inactive, although it does not compromise substrate affinity. Unfortunately, the crystal structure of an EAL·substrate·Mg2+ complex in the monomeric state (state 1 in Fig. 9A) is not known and its determination may indeed be difficult, because at the protein concentrations required for crystallization, cdG stabilizes the dimer.
What are the structural features that correspond to the observed thermodynamic coupling between dimerization and cdG binding? The YahA-EAL structures (Fig. 1) probed under various conditions show that the β4-α4, β5-α5, and 290 loops are exquisitely sensitive to the quarternary and the ligation state of the domain. Thus, these structural elements most likely represent the conduit that transmits structural information between the substrate binding pocket and the dimer interface. Indeed, involvement of the β5-α5 loop and the interface in EAL regulation has been proposed before for YkuI, BlrP1, and RocR (11, 12, 35).
We hypothesize that for rendering the enzyme active loop β5-α5, the anchoring glutamate Glu-235 from loop β4-α4 (see Figs. 4 and 5) have to adopt conformations that allows Asp-263 at the end of strand β5 to engage in M2 coordination. The proximal phosphate group of the substrate would then complete the cation coordination shell, and catalysis would proceed.
In full-length YahA, the EAL domain is linked to a LuxR-like DNA binding domain. How the inherent properties of the isolated YahA-EAL are utilized in the full-length protein is subject to further research. In particular, it will be interesting to see whether YahA is a DNA-dependent PDE or a cdG-dependent transcription factor.
Nonetheless, the findings reported here may well be relevant for the understanding of a large variety of EAL domain proteins that process input via various accessory domains including PAS, BLUF, or REC (6). It appears particularly relevant that all known multidomain EAL protein structures (YkuI, BlrP1, RocR, LapD, and FimX) are dimeric or tetrameric and show canonical EAL/EAL interactions. However, no structural information is available for any of these proteins in both the activated and inactivated state.
In the simplest case, the signal sensed by the input domain would induce its homodimerization and synergistically drive the EAL/EAL equilibrium to the dimeric state and, thus, activate the enzyme (Fig. 9B). This is reminiscent of Rec-HTH transcription factor activation by phosphorylation-induced Rec domain dimerization (37). For the Rec-EAL protein RocR, however, the situation seems to be more complex in that a non-symmetric tetramer structure with partially occluded active sites has been observed (35). Alternatively, in the context of a constitutive dimer such as YkuI or BlrP1, the signal may induce a rearrangement of the input domains that, in turn, would affect the EAL/EAL interface and hence PDE activity and/or substrate binding. For EcDOS, a scissor-like movement of the subunits in the isolated PAS dimer upon heme cofactor reduction has been reported (34). This motion may be propagated to the EAL domain that has been observed to exist as a canonical dimer in isolation (32).
Catalytically inactive EAL protein can serve as c-di-GMP effectors, for reviews see Refs. 2 and 6. In this case, the signaling direction is inverted compared with EAL phosphodiesterases in that the (cdG) signal is recognized by the EAL domain and transmitted to various accessory domains. Obviously, thermodynamic coupling between cdG binding and EAL dimerization appears well suited for such receptors. cdG-induced EAL dimerization would constitute a massive signal that could promote dimerization of accessory domain(s) or induce a domain rearrangement within a constitutive dimer as has been proposed for LapD based on structural information (15). Although for FimX, the other EAL effector with a known structure, is a different signaling mechanism, namely competition between cdG and a protein domain for EAL binding, and appears to be operational (14).
To conclude, a regulatory mechanism inherent in the catalytic domain of an enzyme as reported here for EAL may have a strong evolutionary advantage. There would be no need to evolve a separate mechanism of signal transfer from the input to the catalytic domain for each distinct full-length EAL enzyme. Rather, any domain from the large repertoire of regulatory/sensory domain that dimerizes in a signal-dependent manner dimerization may be utilized to regulate EAL activity (Fig. 9B). In this way, various input signals would be linked easily to a catalytic output. The same seems to hold true for diguanylate cyclase, an enzyme that again is active only in the dimeric state (6, 7).
Acknowledgments
We thank Nisha Vinayak for initial investigations and Timm Maier, Shogo Ozaki, and Alberto Reinders for critical reading of the manuscript.
This work was supported by Swiss National Science Foundation Grants 31003A_138414 (to T. Sch.) and 31003A_130469 (to U. J.).
The atomic coordinates and structure factors (codes 4KIE, 4LYK, and 4LJ3) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- cdG
- cyclic di-GMP
- PDE
- phosphodiesterase
- DGC
- diguanylate cyclase
- MALS
- multiangle light-scattering
- AUC-SE
- analytical ultracentrifugation sedimentation equilibrium
- PDB
- Protein Data Bank
- fl-cdG
- fluorescein-labeled cdG
- SEC
- size exclusion chromatography.
REFERENCES
- 1. Hengge R. (2009) Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 7, 263–273 [DOI] [PubMed] [Google Scholar]
- 2. Römling U., Galperin M. Y., Gomelsky M. (2013) Cyclic di-GMP/ The first 25 years of a universal bacterial second messenger. Microbiol. Mol. Biol. Rev. 77, 1–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wassmann P., Chan C., Paul R., Beck A., Heerklotz H., Jenal U., Schirmer T. (2007) Structure of BeF3-modified response regulator PleD. Implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure 15, 915–927 [DOI] [PubMed] [Google Scholar]
- 4. Paul R., Abel S., Wassmann P., Beck A., Heerklotz H., Jenal U. (2007) Activation of the diguanylate cyclase PleD by phosphorylation-mediated dimerization. J. Biol. Chem. 282, 29170–29177 [DOI] [PubMed] [Google Scholar]
- 5. De N., Pirruccello M., Krasteva P. V., Bae N., Raghavan R. V., Sondermann H. (2008) Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol. 6, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schirmer T., Jenal U. (2009) Structural and mechanistic determinants of c-di-GMP signalling. Nat. Rev. Microbiol. 7, 724–735 [DOI] [PubMed] [Google Scholar]
- 7. Zähringer F., Lacanna E., Jenal U., Schirmer T., Boehm A. (2013) Structure and signaling mechanism of a zinc-sensory diguanylate cyclase. Structure 21, 1149–1157 [DOI] [PubMed] [Google Scholar]
- 8. Christen M., Christen B., Folcher M., Schauerte A., Jenal U. (2005) Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J. Biol. Chem. 280, 30829–30837 [DOI] [PubMed] [Google Scholar]
- 9. Schmidt A. J., Ryjenkov D. A., Gomelsky M. (2005) The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase. Enzymatically active and inactive EAL domains. J. Bacteriol. 187, 4774–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rao F., Yang Y., Qi Y., Liang Z.-X. (2008) Catalytic mechanism of cyclic di-GMP-specific phosphodiesterase. A study of the EAL domain-containing RocR from Pseudomonas aeruginosa. J. Bacteriol. 190, 3622–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Minasov G., Padavattan S., Shuvalova L., Brunzelle J. S., Miller D. J., Baslé A., Massa C., Collart F. R., Schirmer T., Anderson W. F. (2009) Crystal structures of YkuI and its complex with second messenger cyclic di-GMP suggest catalytic mechanism of phosphodiester bond cleavage by EAL domains. J. Biol. Chem. 284, 13174–13184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barends T. R., Hartmann E., Griese J. J., Beitlich T., Kirienko N. V., Ryjenkov D. A., Reinstein J., Shoeman R. L., Gomelsky M., Schlichting I. (2009) Structure and mechanism of a bacterial light-regulated cyclic nucleotide phosphodiesterase. Nature 459, 1015–1018 [DOI] [PubMed] [Google Scholar]
- 13. Tchigvintsev A., Xu X., Singer A., Chang C., Brown G., Proudfoot M., Cui H., Flick R., Anderson W. F., Joachimiak A., Galperin M. Y., Savchenko A., Yakunin A. F. (2010) Structural insight into the mechanism of c-di-GMP hydrolysis by EAL domain phosphodiesterases. J. Mol. Biol. 402, 524–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Navarro M. V., De N., Bae N., Wang Q., Sondermann H. (2009) Structural analysis of the GGDEF-EAL domain-containing c-di-GMP receptor FimX. Structure 17, 1104–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Navarro M. V., Newell P. D., Krasteva P. V., Chatterjee D., Madden D. R., O'Toole G. A., Sondermann H. (2011) Structural basis for c-di-GMP-mediated inside-out signaling controlling periplasmic proteolysis. PLoS Biol. 9, e1000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Claret L., Miquel S., Vieille N., Ryjenkov D. A., Gomelsky M., Darfeuille-Michaud A. (2007) The flagellar σ factor FliA regulates adhesion and invasion of Crohn disease-associated Escherichia coli via a cyclic dimeric GMP-dependent pathway. J. Biol. Chem. 282, 33275–33283 [DOI] [PubMed] [Google Scholar]
- 17. Zähringer F., Massa C., Schirmer T. (2011) Efficient enzymatic production of the bacterial second messenger c-di-GMP by the diguanylate cyclase YdeH from E. coli. Appl. Biochem. Biotechnol. 163, 71–79 [DOI] [PubMed] [Google Scholar]
- 18. Gentner M., Allan M. G., Zaehringer F., Schirmer T., Grzesiek S. (2012) Oligomer formation of the bacterial second messenger c-di-GMP. Reaction rates and equilibrium constants indicate a monomeric state at physiological concentrations. J. Am. Chem. Soc. 134, 1019–1029 [DOI] [PubMed] [Google Scholar]
- 19. Leslie A. G. (2006) The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 20. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 22. Brünger A. (1992) Free R-value. A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 23. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 25. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laskowski R. A., Moss D. S., Thornton J. M. (1993) Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 231, 1049–1067 [DOI] [PubMed] [Google Scholar]
- 27. Seidel S. A., Dijkman P. M., Lea W. A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J. S., Srinivasan P., Baaske P., Simeonov A., Katritch I., Melo F. A., Ladbury J. E., Schreiber G., Watts A., Braun D., Duhr S. (2013) Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Z. X. (1995) An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 360, 111–114 [DOI] [PubMed] [Google Scholar]
- 29. Benfield C. T., Mansur D. S., McCoy L. E., Ferguson B. J., Bahar M. W., Oldring A. P., Grimes J. M., Stuart D. I., Graham S. C., Smith G. L. (2011) Mapping the IκB kinase β (IKKβ)-binding interface of the B14 protein, a vaccinia virus inhibitor of IKKβ-mediated activation of nuclear factor κB. J. Biol. Chem. 286, 20727–20735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lebowitz J., Lewis M. S., Schuck P. (2002) Modern analytical ultracentrifugation in protein science. A tutorial review. Protein Sci. 11, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rao F., Qi Y., Chong H. S., Kotaka M., Li B., Li J., Lescar J., Tang K., Liang Z.-X. (2009) The functional role of a conserved loop in EAL domain-based cyclic di-GMP-specific phosphodiesterase. J. Bacteriol. 191, 4722–4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tarnawski M., Barends T. R., Hartmann E., Schlichting I. (2013) Structures of the catalytic EAL domain of the Escherichia coli direct oxygen sensor. Acta Crystallogr. D Biol. Crystallogr. 69, 1045–1053 [DOI] [PubMed] [Google Scholar]
- 33. Walker G. M. (1994) The roles of magnesium in biotechnology. Crit. Rev. Biotechnol. 14, 311–354 [DOI] [PubMed] [Google Scholar]
- 34. Kurokawa H., Lee D.-S., Watanabe M., Sagami I., Mikami B., Raman C. S., Shimizu T. (2004) A redox-controlled molecular switch revealed by the crystal structure of a bacterial heme PAS sensor. J. Biol. Chem. 279, 20186–20193 [DOI] [PubMed] [Google Scholar]
- 35. Chen M. W., Kotaka M., Vonrhein C., Bricogne G., Rao F., Chuah M. L., Svergun D., Schneider G., Liang Z.-X., Lescar J. (2012) Structural insights into the regulatory mechanism of the response regulator RocR from Pseudomonas aeruginosa in cyclic di-GMP signaling. J. Bacteriol. 194, 4837–4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robert-Paganin J., Nonin-Lecomte S., Réty S. (2012) Crystal structure of an EAL domain in complex with reaction product 5`-pGpG. PLoS ONE 7, e52424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gao R., Stock A. M. (2010) Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr. Opin. Microbiol. 13, 160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]