

Background: Natriuretic peptide receptor-A (NPRA) lowers blood pressure and blood volume.
Results: Data show that histone deacetylase inhibitors (HDACi) enhance NPRA expression by blocking HDACs and interacting with Sp1, histone acetyltransferase, and acetylated histones.
Conclusion: Results suggest that epigenetic mechanisms regulate NPRA gene transcription.
Significance: Findings will help to identify molecular targets regulating NPRA expression and function in vivo.
Keywords: Chromatin Histone Modification, Gene Transcription, Histone Deacetylase Inhibitors, Natriuretic Peptides, p300, Sp1
Abstract
Atrial natriuretic peptide (ANP) binds guanylyl cyclase-A/natriuretic peptide receptor-A (GC-A/NPRA) and produces the intracellular second messenger, cGMP, which regulates cardiovascular homeostasis. We sought to determine the function of histone deacetylases (HDACs) in regulating Npr1 (coding for GC-A/NPRA) gene transcription, using primary mouse mesangial cells treated with class-specific HDAC inhibitors (HDACi). Trichostatin A, a pan inhibitor, and mocetinostat (MGCD0103), a class I HDAC inhibitor, significantly enhanced Npr1 promoter activity (by 8- and 10-fold, respectively), mRNA levels (4- and 5.3-fold, respectively), and NPRA protein (2.7- and 3.5-fold, respectively). However, MC1568 (class II HDAC inhibitor) had no discernible effect. Overexpression of HDAC1 and HDAC2 significantly attenuated Npr1 promoter activity, whereas HDAC3 and HDAC8 had no effect. HDACi-treated cultured cells in vitro and intact animals in vivo showed significantly reduced binding of HDAC1 and -2 and increased accumulation of acetylated H3-K9/14 and H4-K12 at the Npr1 promoter. Deletional analyses of the Npr1 promoter along with ectopic overexpression and inhibition of Sp1 confirmed that HDACi-induced Npr1 gene transcription is accomplished by Sp1 activation. Furthermore, HDACi attenuated the interaction of Sp1 with HDAC1/2 and promoted Sp1 association with p300 and p300/cAMP-binding protein-associated factor; it also promoted the recruitment of p300 and p300/cAMP-binding protein-associated factor to the Npr1 promoter. Our results demonstrate that trichostatin A and MGCD0103 enhanced Npr1 gene expression through inhibition of HDAC1/2 and increased both acetylation of histones (H3-K9/14, H4-K12) and Sp1 by p300, and their recruitment to Npr1 promoter. Our findings define a novel epigenetic regulatory mechanism that governs Npr1 gene transcription.
Introduction
The binding of atrial natriuretic peptide (ANP)2 and brain natriuretic peptide to their receptor guanylyl cyclase-A/natriuretic peptide receptor A (GC-A/NPRA) produces intracellular second messenger cGMP (1–3). The receptor GC-A/NPRA is a pleiotropic molecule; it stimulates many cellular and molecular responses of ANP in target tissues and cells (4, 5). The physiological actions of ANP/NPRA elicit natriuresis, diuresis, vasorelaxation, endothelial permeability, and antiproliferation, which have critical functions in the maintenance of blood pressure and fluid-volume homeostasis (6–8). Previous studies have indicated the association of allelic variants of the Npr1 (coding for GC-A/NPRA) gene with a family history of hypertension and left ventricular mass in human essential hypertension (9, 10). A limited number of studies have shown that external and internal stimuli regulate Npr1 promoter activity, including hormones such as angiotensin II (11, 12), vitamin D (13), all-trans-retinoic acid (14), cGMP (15, 16), and extracellular osmolality (17). Despite the hallmark significance of NPRA in renal and cardiovascular physiology, the precise mechanism of receptor regulation and activation at the molecular and epigenetic levels still remains elusive.
Histone deacetylases (HDACs) are enzymes having functions that include regulation of chromatin structure, gene expression, and modification of histone and nonhistone proteins (18, 19). Studies have associated HDAC activity with the development and progression of chronic diseases, including kidney disease and cardiac hypertrophy (20–22). HDACs are a family of 18 molecules grouped into 4 classes. Class I comprises four HDAC family members, HDAC1, -2, -3, and -8, which are expressed ubiquitously and display high enzyme activity toward histone substrate (19, 23). Recently, compelling biochemical and genetic evidence has implicated class I HDACs in several pathological conditions, including renal interstitial fibrosis, cardiac hypertrophy, cancer, and pulmonary hypertrophy, in which they act by modulating histone acetylation (24, 25). In contrast, class II HDACs repress cardiac hypertrophic gene expression (22, 26, 27).
The major function of HDACs is to remove acetyl groups from histones, causing condensation of chromatin and a decrease in gene expression (28, 29). Inhibiting the deacetylation of histones results in hyperacetylation and modifies gene expression, either positively or negatively, in a cell type-specific manner (21). Histone deacetylases are the target of several structurally diverse compounds known as HDAC inhibitors (HDACi) (30). Recent studies have shown that HDACi(s) are promising therapeutic agents. Their efficacy appears to be governed by multiple actions on a variety of cell types and pathophysiological processes, including myocyte hypertrophy, fibrosis, inflammation, and renal disease (19, 24, 31). Despite the known significance of NPRA expression and its activity levels in regulating renal pathophysiology, the exact mechanisms of Npr1 gene expression and regulation are not yet clearly understood. The mouse mesangial cells (MMCs), which play an important role in kidney function and express functional GC-A/NPRA, provide a useful model system for elucidating the regulatory mechanisms involved in Npr1 gene transcription and expression (32). In the present study, we examined the effect of pan-HDACi trichostatin A (TSA), class I-selective HDACi mocetinostat (MGCD0103), and class II-selective HDACi MC1568 on Npr1 gene transcription and expression, utilizing cultured MMCs in vitro and intact Npr1 mouse models under physiological conditions in vivo.
EXPERIMENTAL PROCEDURES
Plasmids and Promoter Construct
The murine Npr1 genomic clone was used to design promoter deletion constructs (33). Cloning of the Npr1 proximal promoter-luciferase reporter constructs (−356/+55 bp, −356/+29 bp, and −356/-46 bp) have been described (34, 35). Cloning of the construct −356/−90 bp was done using −356 forward (5′-tacggaacgcgtgagggggggcagcttcctcac-3′) and −90 reverse (5′-acgggaccacaaggcgagccaggcagc-3′) primers. The FLAG-tagged HDAC1, -2, -3, -8, and mutant HDAC2 (pME18S-FLAG-HDAC2-(1–372)) were a gift from Dr. Edward Seto (H. Lee Moffitt Cancer Center & Research Institute, Tampa, FL); pcDNA3.1-p300 WT and pcDNA3.1-p300-(HAT-) were a gift from Dr. Warner C. Greene (Gladstone Institutes, San Francisco, CA). The expression vectors CMV-Sp1 (Addgene plasmid 12097), constructed by Dr. Robert Tijian (University of California, Berkeley, CA) and mutant p180 pCIG-HDAC1 (Addgene plasmid 11053), constructed by Dr. Ramesh Shivdasani (Dana-Farber Cancer Institute, Boston, MA), were obtained from Addgene (Cambridge, MA).
Production of Polyclonal Antibody of NPRA
The peptide sequence ETKAVLEEFDGFE, corresponding to carboxyl terminus residues 1015–1027 in the intracellular region of GC-A/NPRA, was conjugated to keyhole limpet hemocyanin. The keyhole limpet hemocyanin-peptide conjugate (250 μg) was injected intraperitoneally in the presence of complete Freund's adjuant into chickens (GenWay Biotech, Inc., San Diego, CA). Chickens were boosted after 21 days with 100–150 μg of conjugated antigen and incomplete Freund's adjuant. The additional boosts were used every 30 days thereafter in a similar manner and a total of 3 boosts were performed. The eggs were harvested and total IgY was isolated from yolks. The total IgY antibody was evaluated for titer, and IgY was affinity purified using the peptide antigen. The titer of antibody was confirmed by Western blot analysis.
Cell Transfection and Luciferase Assay
MMCs were isolated and cultured in DMEM supplemented with 10% FCS and insulin/transferrin/sodium selenite, as described previously (32). The cells were transfected by Lipofectamine 2000 reagent (Invitrogen) between 4 and 16 passages and luciferase activity was measured as described previously (34). The results were normalized for transfection efficiency as relative to light units per Renilla luciferase activity. In ectopic overexpression experiments, cells were transfected with expression plasmids for HDAC1, -2, -3, 8, HDAC1-mut, HDAC2-mut, Sp1, p300, and p300-mut. Total DNA content was equalized by inclusion of empty vector. Twenty-four hours after transfection, cells were kept for 12 h in DMEM containing 0.1% BSA and treated with HDACi (TSA, MGCD0103, and MC1568) or vehicle (0.1% dimethyl sulfoxide) for 24 h.
Generation of Npr1 Gene-targeted Mice and Treatment with HDAC Inhibitors
Npr1 gene-disrupted heterozygous 1-copy (+/−) and gene-duplicated 4-copy (++/++) mice were produced by homologous recombination in embryonic stem cells as described previously (36–38). The mice were bred and maintained at the Tulane University Health Sciences Center Animal Facility. The mouse colonies were housed under 12-h light/dark cycles at 25 °C and fed regular chow (Purina Laboratory) and tap water ad libitum. All animals were littermate progeny of the C57/BL6 genetic background. Stock solutions of TSA and MGCD0103 were prepared in dimethyl sulfoxide and stored at −70 °C. On the day of injection, TSA and MGCD0103 were thawed, diluted with olive oil to the appropriate concentration, vortexed for 2 min at room temperature, and administered intraperitoneally. Control groups were injected with vehicle (dimethyl sulfoxide and olive oil). Adult male (24–26 weeks old) 1-copy, 2-copy, and 4-copy mice were injected in three groups: Group I (n = 6), vehicle-treated (control); Group II (n = 6), TSA-treated (1 mg/kg); and Group III (n = 6), MGCD0103-treated (5 mg/kg), on alternate days for 2 weeks. All protocols were approved by the Institutional Animal Care and Use Committee at Tulane University Health Sciences Center.
Tissue Collection
Animals were euthanized by administration of a high concentration of CO2. Kidneys were collected, frozen in liquid nitrogen, and stored at −80 °C.
Real-time RT-PCR Assay
Total RNA was extracted from HDACi-treated cells using RNeasy plus Mini Kit (Qiagen, Valencia, CA). First-strand cDNA was synthesized from 1 μg of total RNA using an RT2 First Strand Kit (Qiagen). Real-time RT-PCR was done using the Mx3000P QPCR System; data were analyzed with MxPro software (Agilent Technologies). PCR amplification was done in triplicate in a 25-μl reaction volume using RT2 Real-time SYBR Green/ROX PCR Master Mix and PCR conditions as described previously (14). Primers for amplification of NPRA and β-actin were purchased from Qiagen. β-Actin was amplified from all samples as a housekeeping gene to normalize expression levels of targets between different samples. The reaction mixture without template cDNA was used as a negative control. The Npr1 expression values were normalized to β-actin. Relative expression of the Npr1 gene was determined by the comparative Ct value as described earlier (14).
Whole Cell Lysate and Nuclear Extract Preparation and Immunoblot Assay
Whole cell lysate and nuclear extract were prepared as described earlier (34). The protein concentration of the lysate was measured with a Bradford protein detection kit (Bio-Rad). Immunoblot assay was done as previously described (34). The cell lysate (60 μg of protein) was electrophoresed for 2 h, then transferred to a nylon membrane. The membrane was incubated with primary antibodies of HDAC1, -2, -3, p300, p300/cAMP-binding protein-associated factor (PCAF), Sp1, H3-K9/14ac, H4-K12ac, H3-K9me3, or NPRA and treated with the corresponding secondary anti-rabbit or anti-mouse horseradish peroxidase (HRP)-conjugated antibodies (Table 1). Protein bands were developed using a SuperSignal West Femto Chemiluminescent kit and visualized using an Alpha Innotech detection system from Proteinsimple (Santa Clara, CA). The intensity of protein bands was quantified by Alphaview software.
TABLE 1.
List of the antibodies used in Western blot (WB), immunoprecipitation (IP), and ChIP assay
| Protein | Catalog no. | Source | Assay |
|---|---|---|---|
| HDAC1 | sc-8410 | Santa Cruz Biotechnology Inc. | WB |
| HDAC2 | sc-7899 | Santa Cruz Biotechnology Inc. | WB |
| HDAC3 | sc-11417 | Santa Cruz Biotechnology Inc. | WB |
| H4(K-12)ac | sc-8661-R | Santa Cruz Biotechnology Inc. | WB, ChIP |
| H3(K-9/14)ac | 06-599 | Upstate Biotechnology | WB, ChIP |
| H3(K-9)me3 | A-4036-050 | Epigentek | WB, ChIP |
| p300 | A-4020-100 | Epigentek | WB |
| PCAF | sc-13124 | Santa Cruz Biotechnology Inc. | WB |
| Sp1 | sc-14027 | Santa Cruz Biotechnology Inc. | WB, ChIP, IP |
| Ac-lysine | sc-32268 | Santa Cruz Biotechnology Inc. | WB, IP |
| H3 | sc-10809 | Santa Cruz Biotechnology Inc. | WB |
| β-Actin | A5316 | Sigma | WB |
cGMP Assay
Twenty-four hours after plating, MMCs were made serum-free for 12 h and treated with TSA and MGCD0103 for another 24 h. Cells were stimulated with ANP at 37 °C for 15 min in the presence of 0.2 mm 3-isobutyl-1-methylxanthine, washed three times with phosphate-buffered saline (PBS), and scraped into 0.5 n HCl. Cell suspension was subjected to five cycles of freeze and thaw, then centrifuged at 10,000 × g for 15 min. The supernatant thus collected was used for the cGMP assay. Frozen kidney samples were homogenized in 10 volumes of 0.1 m HCl containing 1% Triton X-100. The homogenate was centrifuged at 1000 × g at 4 °C, and the supernatant collected. Both cell and tissue homogenates were used for cGMP assay, using a direct enzyme-linked immunosorbent assay (ELISA) kit (Enzo Life Sciences, Farmingdale, NY) according to the manufacturer's protocol.
Histone Purification
Total histone was extracted from MMCs using a total histone extraction kit (Epigentek, Farmingdale, NY) according to the manufacturer's protocol. Cells were harvested and suspended in 1× prelysis buffer, kept on ice for 10 min, and centrifuged at 10,000 × g for 1 min at 4 °C. The supernatant was removed and the cell pellet was resuspended in 3 volumes of lysis buffer, incubated on ice for 30 min, then centrifuged at 12,000 × g for 5 min at 4 °C. Balanced dithiothreitol (DTT) buffer (0.3 volumes) was added to the supernatant, which was stored at −80 °C. The protein concentration of the eluted histone was estimated using a Bradford protein detection kit (Bio-Rad) using BSA as a standard.
Quantitative ChIP and Sequential ChIP
Chromatin immunoprecipitation (ChIP) and sequential ChIP were performed using a ChIP-IT Express kit (Active Motif, Carlsbad, CA) as previously described (14). For quantitative ChIP assay, real-time PCR was done with a RT2 Real-TimeTM SYBR Green/ROX PCR Master Mix (Qiagen) according to the supplier's instructions. The Npr1 promoter region, −120 to +73 bp, containing Sp1 and p300 binding sites, as well as a distal promoter region, −1155 to −941 bp (control), was PCR amplified using purified DNA as a template. The primer sequences used to amplify the region −120 to +73 were forward (5′-gaggggaggattcgctgc-3′) and reverse (5′-ctaagaagagcgaggggagc-3′) and region −1155 to −914, forward (5′-ctattgaactatatctccagccccaag-3′) and reverse (5′-ccagttaaatgctgacggcataacgatgaa-3′). The antibodies used in the ChIP assay are listed in Table 1. The reaction conditions were 95 °C for 10 min; 40 cycles at 95 °C for 15 s and 60 °C for 1 min; for the dissociation curve, the preceding was followed by 1 cycle at 95 °C for 1 min, 55 °C for 30 s, and 95 °C for 30 s. For ChIP quantitative PCR, 1% input was used; its value was adjusted to 100% for normalization of the results. Each ChIP DNA threshold cycle number (Ct) was normalized to the input DNA fraction Ct value to account for differences in chromatin sample preparation. Percent input was calculated as 2(−ΔCT(normalized IP) × 100%.
Immunoprecipitation of Acetylated Sp1
Cell lysates (100 μg of protein) from HDACi-treated and untreated MMCs were incubated with 2 μg of polyclonal Sp1 antibody for 2 h at 4 °C. Protein A/G-agarose beads were added and the lysates were incubated overnight at 4 °C. After washing the beads, proteins were eluted by boiling in 2× SDS loading buffer, then electrophoresed. For detection of acetylated Sp1, membranes were incubated with anti-Ac-lys (AKL5C1) antibody (Table 1) and treated with anti-mouse HRP-conjugated secondary antibody. Immunoprecipitate with rabbit IgG was taken as negative control.
Transfection of Small Inhibitory RNA
MMCs were cultured to 80–90% confluence and transfected with HDAC1, HDAC2, and Sp1 small interfering RNA (siRNA; a pool of 3 target-specific 20- to 25-nucleotide sequence siRNAs) purchased from Santa Cruz Biotechnology using Lipofectamine RNAiMAX reagent (Invitrogen). A nontargeting 20–25-nucleotide sequence siRNA was used as a negative control. Four hours after transfection, fresh medium was added to the plates; 24 h later, cells were treated with HDAC inhibitors for another 24 h and lysed to measure firefly and Renilla luciferase activity.
HDAC and Histone Acetyltransferase (HAT) Activity Assay
Total HDAC and HAT activity was measured in nuclear extracts prepared from HDACi-treated cells using colorimetric ELISA kits from, respectively, Active Motif and Epigentek. HDAC enzyme activity was calculated by measuring the amount of HDAC-deacetylated product, which was directly proportional to HDAC enzyme activity. For the HAT enzyme activity assay, the amount of acetylated product was measured. Absorbance was read at 450 nm and results were calculated using a standard curve according to the manufacturer's instructions, and expressed as nanogram/h/mg of protein.
Semi-quantitative Analysis of HDAC1 and HDAC2
The protein content of HDAC1 and HDAC2 was measured using colorimetric EpiQuick HDAC1 and HDAC2 assay kits (Epigentek) according to the manufacturer's protocol. Absorbance was read at 450 nm. Results were calculated using a standard curve and expressed as nanogram/mg of protein.
Quantification of Global Histone H3 and H4 Acetylation
Histone extracts from frozen kidney tissues were prepared using a total histone extraction kit (Epigentek) according to the manufacturer's protocol. Quantification of global levels of lysine-specific histone acetylation was done using EpiQuick global H3-K9/14ac and H4-K12ac colorimetric assay kits (Epigentek) according to the manufacturer's protocol.
Statistical Analysis
Results are expressed as mean ± S.E. of 3–4 independent experiments done in triplicate. Significance was evaluated by one-way analysis of variance, followed by Dunnett's multiple comparison tests using PRISM software (GraphPad Software, San Diego, CA). A p value of <0.05 was considered significant.
RESULTS
The Npr1 proximal promoter region (−356/+55) dose-dependently exhibited 8–10-fold induction in luciferase activity in MMCs treated, respectively, with pan HDACi (TSA) and class I-selective HDACi (MGCD0103) than untreated control cells (Fig. 1A). On the other hand, treating cells with class II-selective HDACi (MC1568) had no effect on Npr1 promoter activity. As compared with untreated controls, there was 4- and 5.3-fold augmentation in Npr1 mRNA levels and a 2.7–3.5-fold increase in NPRA protein expression in cells treated, respectively, with TSA and MGCD0103 (Fig. 1, B and C). Treatment with TSA and MGCD0103 showed 30- and 45-fold stimulation in cGMP levels, respectively, in the presence of ANP as compared with untreated groups (Fig. 1D).
FIGURE 1.
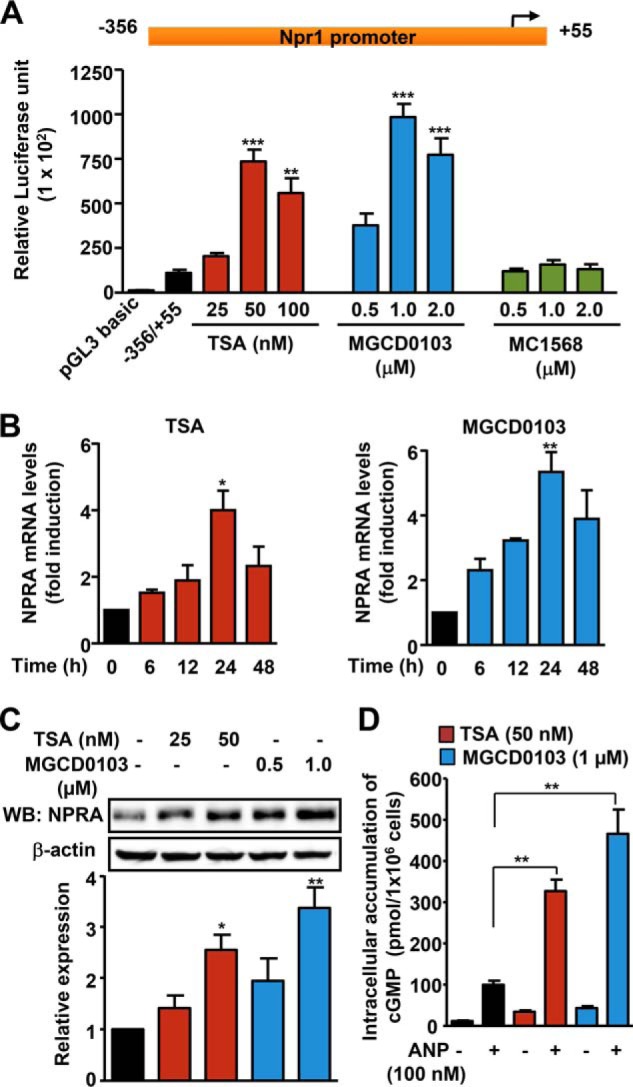
Effect of TSA and MGCD0103 on Npr1 gene transcription and expression. A, luciferase activity of the Npr1 proximal promoter construct −356/+55 in MMCs treated with increasing concentrations of HDACi (TSA, MGCD0103, and MC1568). B, effect of TSA (50 nm) and MGCD0103 (1 μm) on Npr1 mRNA levels in a time-dependent manner as determined by real-time RT-PCR. C, Western blot (WB) analysis of NPRA protein expression in cells treated with HDACi and β-actin expression is shown as the loading control. D, intracellular accumulation of cGMP in MMCs treated with HDACi (TSA, 50 nm; MGCD0103, 1 μm) and ANP. Bar represents the mean ± S.E. of four independent experiments in triplicates. WB, Western blot; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We examined the effect of class I HDACs on Npr1 promoter activity and mRNA expression. Cotransfection of class I HDAC-1, -2, -3, and -8 expression plasmids with the Npr1 promoter construct −356/+55 led, respectively, to 53 and 74% repression of luciferase activity by HDAC1 and -2, whereas HDAC3 and -8 had no effect compared with untransfected control cells (Fig. 2A). Overexpression of HDAC1, -2, and -3 protein levels was observed in transfected MMCs as compared with untransfected cells (Fig. 2B). Knockdown of HDAC1 and -2 protein expression by siRNA released repression of the Npr1 promoter activity seen by luciferase assay (Fig. 2C). Western blot analysis confirmed the knockdown effect of siRNA on endogenous HDAC1 and -2 protein expression in transfected cells compared with control siRNA-transfected cells (Fig. 2D). Ectopic expression of catalytically inactive HDAC1 and HDAC2 mutant plasmid abolished Npr1 promoter repression and markedly induced its activity (Fig. 2E). Western blot analysis confirmed the expression of mutant HDAC1 and -2 proteins in transfected cells compared with control cells (Fig. 2F). There was ∼50–60% attenuation in Npr1 mRNA levels in HDAC1- and HDAC2-transfected cells compared with untransfected cells (Fig. 2G). Overexpression of HDAC1 and -2 in MMCs inhibited NPRA protein expression by 35 and 50%, respectively, compared with control cells (Fig. 2H).
FIGURE 2.
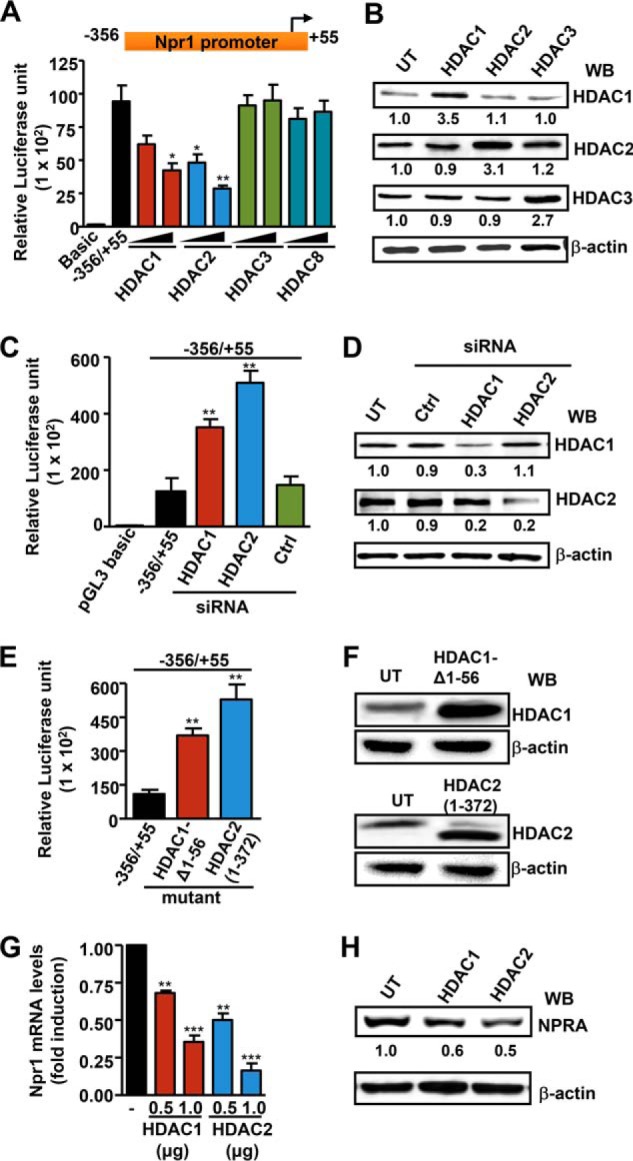
Effect of the overexpression and knockdown of HDAC1 and HDAC2 in transcriptional regulation and expression of the Npr1 gene. A, luciferase activity of Npr1 proximal promoter (−356/+55) in MMCs cotransfected with class I HDACs (HDAC1, -2, -3, and -8) or an empty vector. B, Western blot (WB) analysis of HDAC1, -2, and -3 in transfected cells. β-Actin was used as loading control. C, luciferase activity of the Npr1 promoter cotransfected with HDAC1, HDAC2, and control siRNA (0.1 μm). D, Western blot analysis of siRNA-mediated knockdown of endogenous HDAC1 and HDAC2 proteins in transfected cells with β-actin as loading control. E, luciferase activity of the Npr1 promoter construct −356/+55 cotransfected with catalytically inactive HDAC1 (250 ng) and HDAC2 (250 ng) mutant plasmid. F, Western blot analysis of mutant HDAC1 and -2 proteins in transfected cells with β-actin as loading control. G, Npr1 mRNA levels in cells transfected with HDAC1 and HDAC2 expression plasmid as analyzed by real-time RT-PCR with β-actin as an internal control. H, Western blot analysis of NPRA protein expression in cells transfected with HDAC1 and HDAC2 expression plasmid and β-actin expression is shown as loading control. Data represent mean ± S.E. of four independent experiments. Ctrl, control; UT, untransfected; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Treatment with TSA and MGCD0103, respectively, reduced total HDAC activity by 38 and 66%, no discernible effect occurred in MC1568-treated cells as compared with control cells (Fig. 3A). MMCs treated with MGCD0103 had significantly reduced HDAC1 and -2 protein expression as analyzed by semi-quantitative measurements. In contrast, TSA significantly inhibited only HDAC2 protein expression (Fig. 3B). Treatment of cells with MC1568 had no effect on class I HDAC protein expression. There was significant attenuation of HDAC1 and -2 protein expression in MGCD0103-treated cells and HDAC2 expression in TSA-treated cells, but no change in HDAC3 protein expression was detected as compared with untreated cells (Fig. 3, C and D). TSA and MGCD0103 increased HAT activity by 2- and 2.8-fold, respectively, as compared with untreated cells (Fig. 4A). There was a substantially significant increase in HAT proteins p300 and PCAF in HDACi-treated cells (Fig. 4B). As compared with untreated control cells, both TSA and MGCD0103 significantly augmented global acetylation levels of histone H3-K9/14 (H3-K9/14ac) and H4-K12 (H4-K12ac), but dose-dependently inhibited the trimethylation level of H3-K9 (H3-K9me3) (Fig. 4C). Interestingly, the effect was detected for 24 h (Fig. 4D). Quantitative ChIP assay showed markedly significant accumulation of H3-K9/14ac and H4-K12ac and significantly reduced levels of H3-K9me3 in HDACi-treated cells in Npr1 proximal promoter region −120 to +73, as compared with untreated control cells. In contrast, no binding was observed at the distal promoter region −1155 to −941, taken as a negative control (Fig. 4E).
FIGURE 3.
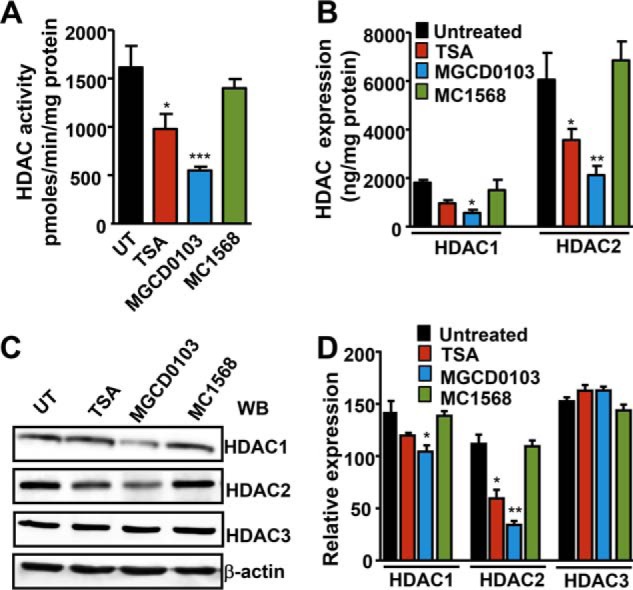
Modulation of total HDAC activity and HDAC1 and HDAC2 protein expression by TSA, MGCD0103, and MC1568. A, total HDAC activity was measured in nuclear extracts of HDACi-treated and untreated MMCs. B, semiquantitative measurement of HDAC1 and HDAC2 protein levels in HDACi-treated cells by colorimetric method. C, Western blot analysis of HDAC1, -2, and -3 protein expressions in HDACi-treated cells with β-actin expression as loading control. D, densitometric analysis for HDAC1, -2, and -3. Bar represents mean ± S.E. of four independent experiments. HDACi (TSA, 50 nm; MGCD0103, 1 μm; MC1568, 1 μm); UT, untreated; WB, Western blot; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 4.

Effect of TSA and MGCD0103 on HAT activity, histone acetylation, and association with the Npr1 promoter. A, quantification of total HAT activity in HDACi-treated MMCs by the colorimetric method. B, Western blot analysis of p300 and PCAF protein expression in HDACi-treated cells. β-Actin was used as loading control. Concentration (C)- and time-dependent (D) effect of HDACi on histones H3-K9/14ac, H4-K12ac, and H3-K9me3 protein levels as analyzed by Western blot. Histone H3 was used as loading control. E, schematic map of the Npr1 promoter showing the −1155/−914 bp and −120/+73 bp regions. Quantitative ChIP assay demonstrating in vivo accumulation of acetylated histones to the Npr1 promoter region. Bar represents mean ± S.E. of three independent experiments. HDACi (TSA, 50 nm; MGCD0103, 1 μm); UT, untreated; Ab, antibody; WB, Western blot; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Luciferase activity of various Npr1 promoter deletion constructs in transfected cells showed that the −356 to −46 construct with two Sp1 binding sites was responsive for TSA- and MGCD0103-mediated Npr1 gene transcription (Fig. 5A). Deletion of Sp1 binding sites in the −365 to −90 construct did not support HDACi-mediated Npr1 promoter activity. To examine the role of Sp1 in TSA- and MGCD0103-mediated Npr1 promoter activation, MMCs were cotransfected with Npr1 promoter and Sp1 expression plasmid. Treatment with TSA and MGCD0103 enhanced Npr1 promoter activity by 14- and 16-fold, respectively, in Sp1-transfected cells as compared with untransfected cells (Fig. 5B). Western blot analysis demonstrated no difference in Sp1 protein expression in HDACi-treated cells. On the other hand, knockdown of Sp1 by siRNA reduced HDACi-mediated Npr1 promoter activity by 76–83% (Fig. 5C). A marked significant decrease in endogenous Sp1 protein expression occurred in Sp1 siRNA-transfected cells compared with untransfected cells (Fig. 5C). The function of Sp1 was further supported by using mithramycin A, a specific inhibitor that interferes with Sp1 binding sites. Mithramycin attenuated HDACi-mediated Npr1 promoter activity by 70–80% (Fig. 5D). Treatment of Sp1-transfected cells with TSA and MGCD, respectively, led to 8- and 11-fold induction in Npr1 mRNA levels as compared with levels in untransfected control cells. In contrast, ablation of endogenous Sp1 protein inhibited Npr1 mRNA levels by 70–75% in HDACi-treated cells as compared with levels in untransfected control cells (Fig. 5E). Quantitative ChIP assay showed that in HDACi-treated cells, the binding of Sp1 to the Npr1 promoter was significantly increased (Fig. 5F). Immunoprecipitation assay showed a significant increase in Sp1 acetylation levels in TSA- and MGCD0103-treated cells as compared with levels in untreated control cells (Fig. 5G).
FIGURE 5.
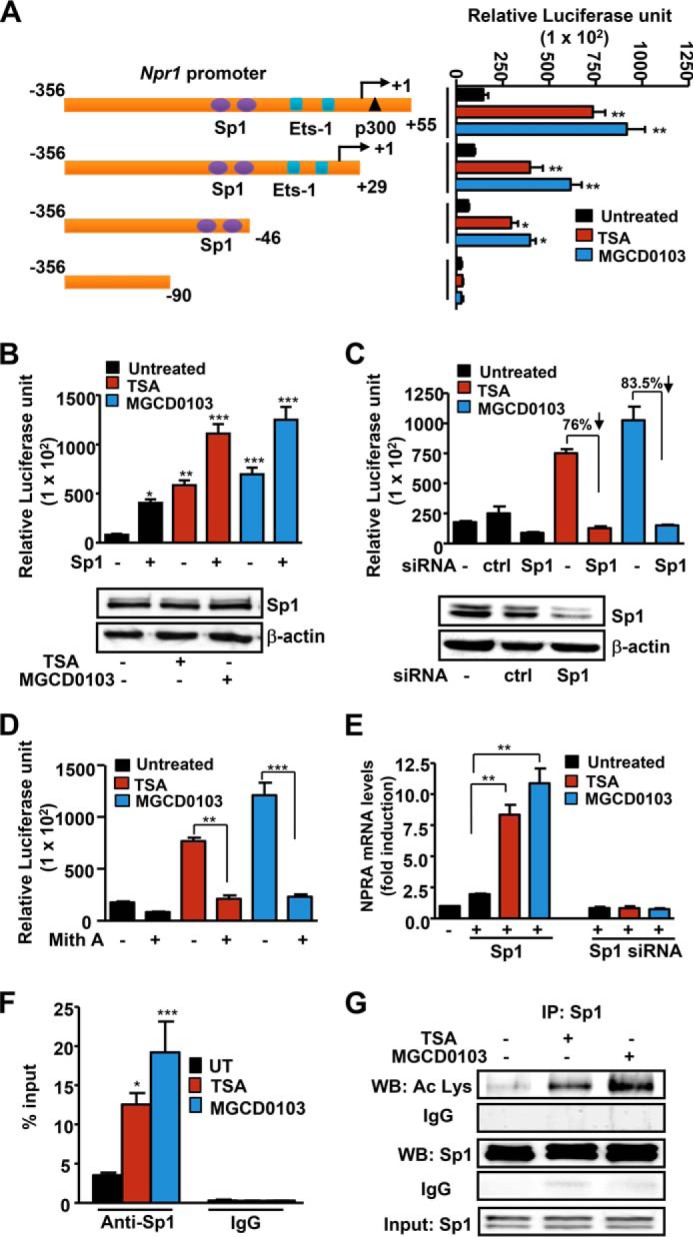
Role of Sp1 in mediating TSA and MGCD0103 effects on Npr1 gene transcription. A, schematic map of the Npr1 promoter deletion construct and their luciferase activity in transfected MMCs treated with HDACi. B, luciferase activity of the Npr1 promoter construct −356/+55 cotransfected with Sp1 plasmid and treated with HDACi. Blots show Western blot analysis of Sp1 protein expression in HDACi-treated cells with β-actin as loading control. C, luciferase activity of the Npr1 promoter cotransfected with Sp1 siRNA and treated with HDACi. Lower panel, Western blot analysis of Sp1 protein expression in Sp1 siRNA-transfected cells and β-actin as loading control. D, luciferase activity of the Npr1 promoter in cells pretreated with mithramycin A and induced with HDACi. E, effect of HDACi on Npr1 mRNA levels in cells transfected with Sp1 expression plasmid or Sp1 siRNA as determined by real-time RT-PCR with β-actin as the internal control. F, quantitative ChIP assay demonstrating recruitment of Sp1 protein on the Npr1 promoter (−120 to +73) in HDACi-stimulated cells as determined by real-time PCR. G, Western blot analysis of acetylated and total Sp1 in the immunoprecipitate from HDACi-treated cells. Input shows Sp1 in lysates as detected by Western blot. Bars represent mean ± S.E. of three independent experiments. HDACi (TSA, 50 nm; MGCD0103, 1 μm); Ctrl, control; UT, untreated; mith A, mithramycin A; WB, Western blot; IP, immunoprecipitation; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
A coimmunoprecipitation assay was done to test the interaction of Sp1 with HDAC1 and -2 proteins in untreated and HDACi-treated cells (Fig. 6A). Western blot analysis of the anti-Sp1-immunoprecipitated fractions from untreated cell lysates showed that Sp1 interacted with HDAC1 and -2 proteins and that this interaction was reduced in the presence of TSA and MGCD0103. A sequential ChIP assay showed enhanced binding of HDAC1 and -2 and Sp1·HDAC complex at the Npr1 promoter in untreated cells (Fig. 6, B and C, upper panel). Treating cells with MGCD0103 significantly reduced HDAC1 and -2 protein recruitment and Sp1·HDAC complex formation at the Npr1 promoter (Fig. 6, B and C, lower panel). We further tested the interaction of Sp1 with p300 in HDACi-mediated Npr1 gene transcription. Simultaneous overexpression of Sp1 and p300 substantially increased Npr1 promoter activity in TSA- and MGCD0103-treated cells as compared with Sp1-overexpressing cells treated with HDACi (Fig. 7A). Interestingly, overexpression of mutant p300 defective in acetyltransferase activity or knockdown of endogenous p300 with siRNA did not support Sp1- and HDACi-mediated Npr1 gene transcription. Overexpression of Sp1 and wild-type and mutant p300, as well as siRNA-mediated knockdown of p300 in transfected cells, was confirmed by Western blot analysis (Fig. 7B). Cotransfection of Sp1 and p300 induced NPRA protein expression, which was further increased by 4.3-fold in MGCD0103-treated cells as compared with untreated control cells (Fig. 7C). Moreover, cells overexpressing Sp1 and mutant p300-HAT did not show HDACi-mediated induction of NPRA protein expression. As compared with untreated controls, cells cotransfected with Sp1 and p300 and treated with MGCD0103 had an almost 60-fold increase in cGMP levels in the presence of ANP (Fig. 7D).
FIGURE 6.

Effect of HDACi on interaction of HDAC1 and -2 with Sp1 on the Npr1 promoter. A, Western blot analysis of HDAC1, HDAC2, or Sp1 complexes isolated by immunoprecipitation with anti-Sp1 antibody from HDACi-treated cells. Sequential ChIP analysis demonstrating in vivo recruitment of HDAC1 (B) and HDAC2 (C) to the Npr1 promoter by Sp1 in MGCD0103-treated and untreated cells. The intensity of DNA bands was quantified by Alpha Innotech analysis software. Data shown represent mean ± S.E. of three independent experiments. MGCD0103 (1 μm); UT, untreated; WB, Western blot; IP, immunoprecipitation.
FIGURE 7.
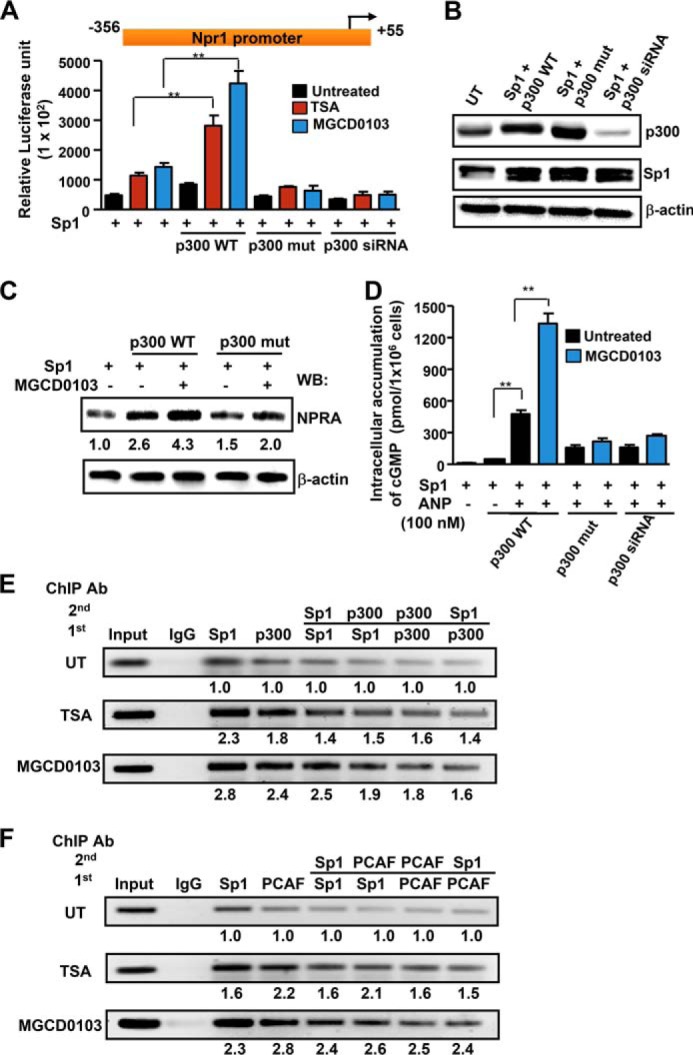
Interaction of p300 and PCAF with Sp1 in the HDACi-mediated effect on Npr1 gene transcription and expression. A, luciferase activity of Npr1 promoter construct −356/+55 cotransfected with Sp1 and wild-type p300, mutant p300-HAT, or p300 siRNA expression plasmid and treated with HDACi. B, Western blot analysis of Sp1, wild-type p300, mutant p300-HAT, and siRNA-mediated knockdown of endogenous p300 in transfected cells. β-Actin was used as loading control. C, Western blot analysis of NPRA protein expression in cells overexpressing Sp1 and wild-type p300 or mutant p300-HAT in the presence of MGCD0103 and β-actin expression is shown as loading control. D, intracellular accumulation of cGMP in cells overexpressing wild-type or mutant p300 and Sp1 protein treated with MGCD0103 and induced with ANP. Sequential ChIP analysis demonstrating in vivo recruitment of p300 (E) and PCAF (F) to the Npr1 promoter by Sp1 in HDACi-treated and untreated cells. The intensity of DNA bands was quantified by Alpha Innotech analysis software. Representative gels from three independent experiments are shown. Data shown represent mean ± S.E. of three independent experiments. HDACi (TSA, 50 nm; MGCD0103, 1 μm); Ctrl, control; UT, untreated; WB, Western blot; IP, immunoprecipitation; **, p < 0.01.
To delineate the interaction of HAT proteins p300 and PCAF with Sp1 on the Npr1 promoter, sequential ChIP assays were used. As shown in Fig. 7E (upper panel), direct Sp1 and p300 binding was observed on the Npr1 promoter and p300 coimmunoprecipitated with anti-Sp1 antibody, indicating that p300 forms a complex with Sp1. Moreover, treatment of cells with TSA and MGCD0103 increased occupancy of Sp1, p300, and Sp1·p300 complex at the Npr1 promoter (Fig. 7E, middle and lower panel). On the other hand, PCAF was also detected in anti-Sp1 immunoprecipitates, confirming that PCAF and Sp1 interact with each other while binding to Npr1 promoter (Fig. 7F, upper panel). Treatment of cells with TSA and MGCD0103 showed higher recruitment of PCAF to the Npr1 promoter; there was significant augmentation of the Sp1·PCAF complex on the Npr1 promoter (Fig. 7F, middle and lower panel).
To further delineate the HDACi-mediated regulation of Npr1 gene expression under physiological conditions in vivo, we have utilized Npr1 gene-disrupted heterozygous 1-copy (+/−), wild-type 2-copy (+/+), and Npr1 gene-duplicated 4-copy (++/++) mice. Total HAT activity in renal tissues of 1-copy, 2-copy, and 4-copy mice treated with HDACi was significantly induced as compared with their vehicle-treated control groups (Fig. 8A). Global acetylation levels of histones H3-K9 and H4-K12 were markedly increased in renal tissues of HDACi-treated mice as compared with vehicle-treated controls groups. Chromatin immunoprecipitation assays were used to elucidate the effect of HDACi on recruitment of acetylated histones, HDAC1 and -2, Sp1, and p300 to the Npr1 promoter in kidney tissues of intact animals in vivo. Both TSA and MGCD0103 augmented the accumulation of acetylated histones H3-K9/14 and H4-K12 at the Npr1 promoter in 1-copy, 2-copy, and 4-copy mice kidneys and significantly prevented the binding of HDAC1 and -2 as compared with untreated control mice (Fig. 8B). On the other hand, there was an increased recruitment of Sp1 and p300 at the Npr1 promoter in Npr1 gene-targeted mice treated with HDACi. Real-time RT-PCR analysis showed that there was a marked significant up-regulation in Npr1 mRNA levels of TSA- and MCGD0103-treated mice kidneys compared with vehicle-treated controls (Fig. 8C). Western blot analysis demonstrated an augmented expression of NPRA protein levels in mice treated with HDACi (TSA- and MGCD0103) compared with their vehicle-treated control groups (Fig. 8D). The treatment with HDACi significantly increased the intracellular accumulation of renal cGMP in the treated mice groups as compared with control mice (Fig. 8E).
FIGURE 8.

HDACi-mediated in vivo regulation of renal NPRA expression and signaling involving HDACs, acetylated histones, Sp1, and p300 in Npr1 gene-targeted mice. A, effect of HDACi on renal HAT activity and global acetylation levels of H3-K9ac and H4-K12ac. B, HDACi-mediated recruitment of acetylated histones, HDAC1 and -2, Sp1, and p300 at the Npr1 promoter (−120 to +73) in treated mice kidneys. Pooled samples from 6 mice in each group were used for ChIP and purified DNA was amplified by conventional PCR. C, Npr1 mRNA levels in TSA- and MGCD0103-treated mice kidneys as analyzed by real-time RT-PCR with β-actin as an internal control. D, Western blot and densitometry analyses of NPRA protein expression in TSA- and MGCD0103-treated Npr1 1-copy, 2-copy, and 4-copy mice kidneys. β-Actin was used as loading control. E, cGMP levels in kidney tissues of HDACi-treated mice as quantitated by ELISA. Data shown represent mean ± S.E. of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (vehicle-treated versus drug-treated same group). Ab, antibody. In each group, six mice (n = 6) were used.
DISCUSSION
The major findings of this study demonstrate that pan-HDACi (TSA) and class I-selective HDACi (MGCD0103) up-regulated Npr1 promoter activity, mRNA expression, protein levels, and intracellular accumulation of cGMP. However, class II-selective HDACi (MC1568) had no effect. Our results show that HDAC inhibitors markedly increased global acetylation levels of histones H3-K9/14 and H4-K12 and their accumulation at the Npr1 proximal promoter. Histone acetylation or decetylation is controlled by two classes of enzymes: the addition of the acetyl group to lysine residues, mediated by HATs, is associated with activation of gene transcription, whereas decreased acetylation levels, mediated by HDACs, is associated with transcription repression (18, 39). Thus, HDACs could repress Npr1 gene transcription via deacetylation of histones, associated with the Npr1 promoter.
Our data provide direct evidence of the involvement of class I HDACs in suppressing Npr1 gene transcription. Indeed, ectopic expression of HDAC1 and -2 noticeably reduced Npr1 promoter activity, mRNA levels, and protein expression, whereas knockdown of endogenous HDAC1 and -2 levels by siRNA markedly induced Npr1 gene expression. These findings suggest that HDAC2 and, to a lesser extent, HDAC1 are the major molecular targets in HDACi-mediated Npr1 gene regulation. The functions of class I HDACs, especially HDAC1 and -2 in transcriptional regulation of genes have been widely studied, as have indications that they may be involved in several pathological conditions via modulation of histone acetylation (22, 24, 25, 40). Several classes of HDACi, including TSA, vorinostat, hydroxamic acid, and sodium butyrate have been shown to attenuate HDAC protein expression and activity and to shift the overall balance in favor of HAT activity, resulting in altered gene expression, cell proliferation, or apoptosis (19, 31, 41, 42). In this study, we found that TSA and MGCD0103 significantly reduced HDAC2 and, to lesser extents, HDAC1 protein expression and total HDAC activity, thereby inducing dynamic changes in chromatin structure within the Npr1 promoter. Previous studies have also shown that HDACi induces protein degradation by regulating the enzymes of the ubiquitin-proteasome pathway. For example, valproic acid has been shown to reduce HDAC2 protein expression by the proteasomal degradative pathway (43, 44).
Studies in humans and mice have shown that functional deletion mutations in the Npr1 promoter seem to be linked with essential hypertension (45). Also, Npr1 promoter polymorphism has been identified as a potent and novel marker of susceptibility to hypertension (46, 47). Our data from Npr1 promoter deletion analysis demonstrated that Sp1 binding sites are essential for TSA- and MGCD0103-mediated up-regulation of Npr1 promoter activity. In addition, overexpression of Sp1 not only significantly induced HDACi-regulated Npr1 gene transcription and knockdown of endogenous Sp1 by siRNA-attenuated Npr1 promoter activity, but also abolished HDACi-induced Npr1 gene transcription. Sp1 belongs to the family of zinc finger-containing Sp1 transcription factors; it can bind to GC-rich motifs in the promoter and regulate multiple housekeeping and growth-related genes (48, 49). Several studies have indicated that Sp1 transcriptional activity can be regulated not only quantitatively, through protein expression, but also qualitatively, by post-translational modifications such as phosphorylation and acetylation (50, 51). There is increasing evidence of nonhistone proteins that are involved in transcription being acetylated at lysine residues by HDACi, then modifying their transcriptional or binding activity to the promoters (28, 52). Our Western blot and immunoprecipitation assays showed that TSA and MGCD0103 did not affect the expression of Sp1, but induced its acetylation in MMCs and also in the kidneys of intact mice. Previous studies have indicated that Sp1 can be acetylated at Lys703 and thus could alter transcriptional activity, protein-protein interactions, and Sp1-containing protein complexes at the gene promoters (51, 52).
Alternatively, HDAC inhibitors may have a direct effect on Sp1, because HDACs can inhibit lysine acetylation by removing acetyl groups from their substrate (29). It has been shown that Sp1 can recruit HDAC1 and -2 to the target gene promoters and that modulation of the interactions between Sp1 and HDAC proteins is crucial for Sp1-dependent gene transcription (52, 53). Our coimmunoprecipitation and sequential ChIP results confirmed that Sp1 is associated with HDAC1 and -2 and that treatment with HDACi decreased levels of HDAC1 and -2 in the multiprotein complex. On the other hand, HDACi has been shown to recruit HAT proteins such as p300/CREB-binding protein and PCAF to sequence-specific transcription factors and has been correlated with the acetylation of transcription factors (54, 55). We found that HDACi-mediated Npr1 promoter activation is dependent on p300-mediated Sp1 acetylation, because ectopic overexpression of mutant p300 defective in HAT activity abolished HDACi effects on Npr1 promoter activity, NPRA protein expression, and intracellular accumulation of cGMP in Sp1 overexpressing cells. Previously, it has been shown that treatment of pancreatic cancer cells with TSA activates the TGF-type II receptor promoter by recruiting p300 and PCAF into a Sp1·NF-Y·HDAC complex (51). Recently, it has been reported that selective inhibition of HDAC1 and -2 leads to transcriptional up-regulation of multidrug resistance protein 1, as well as recruitment of p300, PCAF, and NF-Y by Sp1 acetylation (56). Supporting this notion, our sequential ChIP results showed that HDACi treatment increased the association and recruitment of p300, PCAF, and Sp1 to the Npr1 promoter. A proposed schematic representation of the interaction of HDACs, Sp1, and p300/PCAF to the Npr1 promoter in HDACi-mediated Npr1 gene transcription is shown in Fig. 9. Protein expression of p300 and PCAF was enhanced in the presence of TSA and MGCD0103. Moreover, HDAC inhibitors have been shown to induce expression of HATs such as p300, CREB-binding protein, and PCAF at the mRNA and protein levels and HAT activity (54, 57). Our present results suggest that HDACi(s) up-regulate the renal NPRA/cGMP levels in Npr1 gene-targeted 1-copy, 2-copy, and 4-copy mice in vivo by enhanced recruitment of acetylated histones, Sp1, and p300, with attenuated binding of HDAC1 and -2 to the Npr1 promoter.
FIGURE 9.
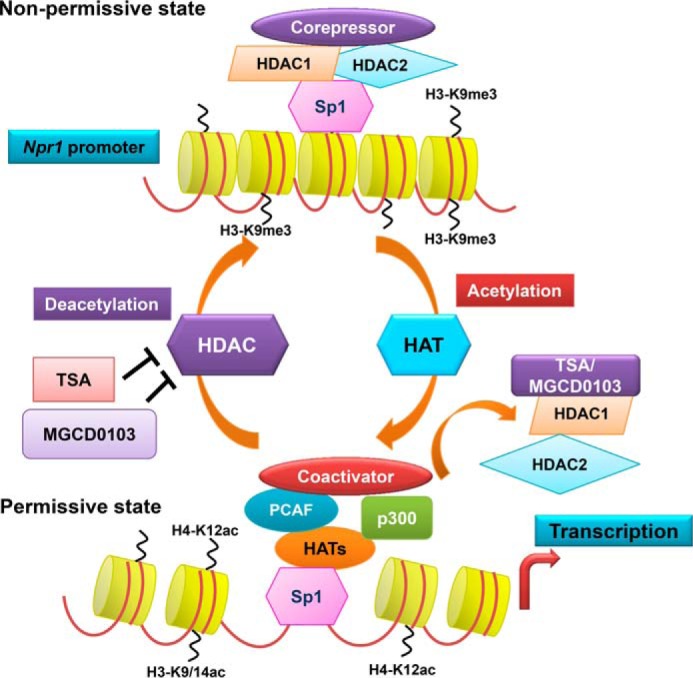
Schematic model showing TSA- and MGCD0103-mediated regulation of Npr1 gene transcription and expression via interactions among regulatory elements and chromatin remodeling. The schematic model depicts that under untreated conditions class I HDACs (HDAC1 and -2) interact with Sp1 and form a corepressor complex and also maintain H3 and H4 in a hypoacetylated state. Under these conditions, transcription factors, including Sp1 has low affinity for promoter. Treatment with HDACi (TSA and MGCD0103) attenuates HDAC activity and induces HAT activity. This allows HAT proteins, p300 and PCAF, whose expression was induced by TSA and MGCD0103, to hyperacetylate H3 at K9/14, H4 at K12, and Sp1, which in association with p300 and PCAF occupies the Npr1 promoter within the newly formed permissive chromatin condition and leads to increased Npr1 gene transcription.
Studies using functional expression and targeted disruption of the Npr1 gene in mice have shown hallmark functional significance of this receptor protein in providing protection against renal and cardiac hypertrophic and fibrotic disorders (58–60). It has recently been shown that glucocorticoids improve renal responsiveness to ANP by up-regulating NPRA expression; in decompensated heart failure, this is accompanied by a remarkable increase in renal cGMP levels (61). The present results of the Npr1 promoter and the identification of transcriptional regulatory factors controlling its activity in cultured cells in vitro and in intact animal models in vivo, will have important implications with respect to identifying new molecular targets for enhancing NPRA/cGMP signaling in the treatment of renal and cardiovascular pathophysiological conditions. Previous studies have also shown that Sp1 regulates corticotrophin-releasing hormone receptor type 2 and p27 gene expression, which are important in both cardioprotection against hypoxia/reoxygenation in cardiomyocytes and smooth muscle cell hypertrophy (48, 62). Sp1 has been shown to regulate the renal protective effect against ischemia/reperfusion injury (63). Accordingly, Sp1 might prove to be an important regulatory factor in Npr1 gene transcription in physiological settings. Moreover, involvement of HDAC1 and -2 in attenuation of Npr1 gene transcription is an important finding in view of the fact that class I HDACs are involved in kidney fibrosis, which should yield new molecular and therapeutic approaches for the treatment high blood pressure and renal diseases.
In conclusion, the present results provide the first evidence that Npr1 gene transcription is modulated by pan-HDACi (TSA) and class I-selective (MGCD0103) by affecting the major molecular targets HDAC1, HDAC2, Sp1, p300, and PCAF. In addition, histone acetylation by TSA and MGCD0103 has a major part in Npr1 gene expression because it precedes HDACi-mediated induction of Npr1 gene transcription and mRNA levels. Our results show that HDACi induces dynamic chromatin changes at the Npr1 promoter by increasing the acetylation of histones H3-K9/14, H4-K12, and Sp1 and their recruitment to the promoter region to facilitate Npr1 expression. Findings from our animal model further demonstrate the involvement of acetylated histones, HDACs, Sp1, and p300 in HDACi-mediated up-regulation of NPRA/cGMP signaling. Collectively, our results provide evidence of a novel epigenetic signaling mechanism that is responsible for regulating Npr1 gene expression and function in cultured cells in vitro and under physiological conditions in the intact animals in vivo. This is an essential step toward understanding NPRA expression and function, which are important regulators of hypertension and cardiovascular homeostasis. Our results demonstrate for the first time that class I-selective HDACi up-regulates Npr1 gene transcription via suppression of HDAC activity and HDAC1 and -2 protein expression, induction of Sp1, and histone acetylation, and recruitment of Sp1, p300, and PCAF complexes to the Npr1 promoter, which may prove useful therapeutic targets in the treatment of hypertension and renal pathophysiological conditions.
Acknowledgments
We thank Alice Y. Yeh and Vickie Nguyen for excellent technical assistance and Kamala Pandey for assistance in the preparation of this manuscript. We sincerely thank Dr. Edward Seto (H. Lee Moffit Cancer Center and Research Institute, Tampa, FL) and Dr. Warner C. Greene (Gladstone Institutes, San Francisco, CA) for the kind gift of expression vectors.
This work was supported, in whole or in part, by National Institutes of Health Grant HL 57531.
- ANP
- atrial natriuretic peptide
- GC-A/NPRA
- guanylyl cyclase/natriuretic peptide receptor-A
- MMCs
- mouse mesangial cells
- HDAC
- histone deacetylase
- HAT
- histone acetyltransferase
- HDACi
- histone deacetylase inhibitor
- TSA
- trichostatin A
- PCAF
- p300-cAMP-binding protein-associated factor
- RT-PCR
- reverse transcription-polymerase chain reaction
- CREB
- cAMP-response element-binding protein.
REFERENCES
- 1. Drewett J. G., Garbers D. L. (1994) The family of guanylyl cyclase receptors and their ligands. Endocr. Rev. 15, 135–162 [DOI] [PubMed] [Google Scholar]
- 2. de Bold A. J. (1985) Atrial natriuretic factor. A hormone produced by the heart. Science 230, 767–770 [DOI] [PubMed] [Google Scholar]
- 3. Pandey K. N., Singh S. (1990) Molecular cloning and expression of murine guanylate cyclase/atrial natriuretic factor receptor cDNA. J. Biol. Chem. 265, 12342–12348 [PubMed] [Google Scholar]
- 4. Levin E. R., Gardner D. G., Samson W. K. (1998) Natriuretic peptides. N. Engl. J. Med. 339, 321–328 [DOI] [PubMed] [Google Scholar]
- 5. Pandey K. N. (2005) Biology of natriuretic peptides and their receptors. Peptides 26, 901–932 [DOI] [PubMed] [Google Scholar]
- 6. Tremblay J., Desjardins R., Hum D., Gutkowska J., Hamet P. (2002) Biochemistry and physiology of the natriuretic peptide receptor guanylyl cyclases. Mol. Cell. Biochem. 230, 31–47 [PubMed] [Google Scholar]
- 7. Pandey K. N. (2011) Guanylyl cyclase/atrial natriuretic peptide receptor-A. Role in the pathophysiology of cardiovascular regulation. Can. J. Physiol. Pharmacol. 89, 557–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Bold A. J., Borenstein H. B., Veress A. T., Sonnenberg H. (1981) A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 28, 89–94 [DOI] [PubMed] [Google Scholar]
- 9. Pitzalis M. V., Sarzani R., Dessì-Fulgheri P., Iacoviello M., Forleo C., Lucarelli K., Pietrucci F., Salvi F., Sorrentino S., Romito R., Guida P., Rappelli A., Rizzon P. (2003) Allelic variants of natriuretic peptide receptor genes are associated with family history of hypertension and cardiovascular phenotype. J. Hypertens. 21, 1491–1496 [DOI] [PubMed] [Google Scholar]
- 10. Rubattu S., Bigatti G., Evangelista A., Lanzani C., Stanzione R., Zagato L., Manunta P., Marchitti S., Venturelli V., Bianchi G., Volpe M., Stella P. (2006) Association of atrial natriuretic peptide and type a natriuretic peptide receptor gene polymorphisms with left ventricular mass in human essential hypertension. J. Am. Coll. Cardiol. 48, 499–505 [DOI] [PubMed] [Google Scholar]
- 11. Garg R., Pandey K. N. (2003) Angiotensin II-mediated negative regulation of Npr1 promoter activity and gene transcription. Hypertension 41, 730–736 [DOI] [PubMed] [Google Scholar]
- 12. Arise K. K., Pandey K. N. (2006) Inhibition and down-regulation of gene transcription and guanylyl cyclase activity of NPRA by angiotensin II involving protein kinase C. Biochem. Biophys. Res. Commun. 349, 131–135 [DOI] [PubMed] [Google Scholar]
- 13. Chen S., Olsen K., Grigsby C., Gardner D. G. (2007) Vitamin D activates type A natriuretic peptide receptor gene transcription in inner medullary collecting duct cells. Kidney Int. 72, 300–306 [DOI] [PubMed] [Google Scholar]
- 14. Kumar P., Garg R., Bolden G., Pandey K. N. (2010) Interactive roles of Ets-1, Sp1, and acetylated histones in the retinoic acid-dependent activation of guanylyl cyclase/atrial natriuretic peptide receptor-A gene transcription. J. Biol. Chem. 285, 37521–37530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hum D., Besnard S., Sanchez R., Devost D., Gossard F., Hamet P., Tremblay J. (2004) Characterization of a cGMP-response element in the guanylyl cyclase/natriuretic peptide receptor A gene promoter. Hypertension 43, 1270–1278 [DOI] [PubMed] [Google Scholar]
- 16. Martel G., Hamet P., Tremblay J. (2010) GREBP, a cGMP-response element-binding protein repressing the transcription of natriuretic peptide receptor 1 (NPR1/GCA). J. Biol. Chem. 285, 20926–20939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen S., Gardner D. G. (2002) Osmoregulation of natriuretic peptide receptor signaling in inner medullary collecting duct. A requirement for p38 MAPK. J. Biol. Chem. 277, 6037–6043 [DOI] [PubMed] [Google Scholar]
- 18. Hassig C. A., Schreiber S. L. (1997) Nuclear histone acetylases and deacetylases and transcriptional regulation. HATs off to HDACs. Curr. Opin. Chem. Biol. 1, 300–308 [DOI] [PubMed] [Google Scholar]
- 19. Yang X. J., Seto E. (2007) HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318 [DOI] [PubMed] [Google Scholar]
- 20. Noh H., Oh E. Y., Seo J. Y., Yu M. R., Kim Y. O., Ha H., Lee H. B. (2009) Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am. J. Physiol. Renal Physiol. 297, F729–739 [DOI] [PubMed] [Google Scholar]
- 21. Villagra A., Sotomayor E. M., Seto E. (2010) Histone deacetylases and the immunological network. Implications in cancer and inflammation. Oncogene 29, 157–173 [DOI] [PubMed] [Google Scholar]
- 22. Trivedi C. M., Luo Y., Yin Z., Zhang M., Zhu W., Wang T., Floss T., Goettlicher M., Noppinger P. R., Wurst W., Ferrari V. A., Abrams C. S., Gruber P. J., Epstein J. A. (2007) Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3β activity. Nat. Med. 13, 324–331 [DOI] [PubMed] [Google Scholar]
- 23. Gregoretti I. V., Lee Y. M., Goodson H. V. (2004) Molecular evolution of the histone deacetylase family. Functional implications of phylogenetic analysis. J. Mol. Biol. 338, 17–31 [DOI] [PubMed] [Google Scholar]
- 24. Bush E. W., McKinsey T. A. (2010) Protein acetylation in the cardiorenal axis. The promise of histone deacetylase inhibitors. Circ. Res. 106, 272–284 [DOI] [PubMed] [Google Scholar]
- 25. Cavasin M. A., Demos-Davies K., Horn T. R., Walker L. A., Lemon D. D., Birdsey N., Weiser-Evans M. C., Harral J., Irwin D. C., Anwar A., Yeager M. E., Li M., Watson P. A., Nemenoff R. A., Buttrick P. M., Stenmark K. R., McKinsey T. A. (2012) Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ. Res. 110, 739–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKinsey T. A., Olson E. N. (2004) Cardiac histone acetylation. Therapeutic opportunities abound. Trends Genet. 20, 206–213 [DOI] [PubMed] [Google Scholar]
- 27. Zhang C. L., McKinsey T. A., Chang S., Antos C. L., Hill J. A., Olson E. N. (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glozak M. A., Sengupta N., Zhang X., Seto E. (2005) Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23 [DOI] [PubMed] [Google Scholar]
- 29. de Ruijter A. J., van Gennip A. H., Caron H. N., Kemp S., van Kuilenburg A. B. (2003) Histone deacetylases (HDACs). Characterization of the classical HDAC family. Biochem. J. 370, 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marks P. A., Richon V. M., Rifkind R. A. (2000) Histone deacetylase inhibitors. Inducers of differentiation or apoptosis of transformed cells. J. Nat. Cancer Inst. 92, 1210–1216 [DOI] [PubMed] [Google Scholar]
- 31. Colussi C., Illi B., Rosati J., Spallotta F., Farsetti A., Grasselli A., Mai A., Capogrossi M. C., Gaetano C. (2010) Histone deacetylase inhibitors. Keeping momentum for neuromuscular and cardiovascular diseases treatment. Pharmacol. Res. 62, 3–10 [DOI] [PubMed] [Google Scholar]
- 32. Pandey K. N., Nguyen H. T., Li M., Boyle J. W. (2000) Natriuretic peptide receptor-A negatively regulates mitogen-activated protein kinase and proliferation of mesangial cells. Role of cGMP-dependent protein kinase. Biochem. Biophys. Res. Commun. 271, 374–379 [DOI] [PubMed] [Google Scholar]
- 33. Garg R., Oliver P. M., Maeda N., Pandey K. N. (2002) Genomic structure, organization, and promoter region analysis of murine guanylyl cyclase/atrial natriuretic peptide receptor-A gene. Gene 291, 123–133 [DOI] [PubMed] [Google Scholar]
- 34. Kumar P., Arise K. K., Pandey K. N. (2006) Transcriptional regulation of guanylyl cyclase/natriuretic peptide receptor-A gene. Peptides 27, 1762–1769 [DOI] [PubMed] [Google Scholar]
- 35. Kumar P., Pandey K. N. (2009) Cooperative activation of Npr1 gene transcription and expression by interaction of Ets-1 and p300. Hypertension 54, 172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oliver P. M., John S. W., Purdy K. E., Kim R., Maeda N., Goy M. F., Smithies O. (1998) Natriuretic peptide receptor 1 expression influences blood pressures of mice in a dose-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 95, 2547–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oliver P. M., Fox J. E., Kim R., Rockman H. A., Kim H. S., Reddick R. L., Pandey K. N., Milgram S. L., Smithies O., Maeda N. (1997) Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc. Natl. Acad. Sci. U.S.A. 94, 14730–14735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pandey K. N., Oliver P. M., Maeda N., Smithies O. (1999) Hypertension associated with decreased testosterone levels in natriuretic peptide receptor-A gene-knockout and gene-duplicated mutant mouse models. Endocrinology 140, 5112–5119 [DOI] [PubMed] [Google Scholar]
- 39. Peserico A., Simone C. (2011) Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gui C. Y., Ngo L., Xu W. S., Richon V. M., Marks P. A. (2004) Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. U.S.A. 101, 1241–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thakur V. S., Gupta K., Gupta S. (2012) Green tea polyphenols causes cell cycle arrest and apoptosis in prostate cancer cells by suppressing class I histone deacetylases. Carcinogenesis 33, 377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pascual M., Do Couto B. R., Alfonso-Loeches S., Aguilar M. A., Rodriguez-Arias M., Guerri C. (2012) Changes in histone acetylation in the prefrontal cortex of ethanol-exposed adolescent rats are associated with ethanol-induced place conditioning. Neuropharmacology 62, 2309–2319 [DOI] [PubMed] [Google Scholar]
- 43. Buchwald M., Krämer O. H., Heinzel T. (2009) HDACi. Targets beyond chromatin. Cancer Lett. 280, 160–167 [DOI] [PubMed] [Google Scholar]
- 44. Krämer O. H., Zhu P., Ostendorff H. P., Golebiewski M., Tiefenbach J., Peters M. A., Brill B., Groner B., Bach I., Heinzel T., Göttlicher M. (2003) The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 22, 3411–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakayama T., Soma M., Takahashi Y., Rehemudula D., Kanmatsuse K., Furuya K. (2000) Functional deletion mutation of the 5`-flanking region of type A human natriuretic peptide receptor gene and its association with essential hypertension and left ventricular hypertrophy in the Japanese. Circ. Res. 86, 841–845 [DOI] [PubMed] [Google Scholar]
- 46. Tremblay J., Hum D. H., Sanchez R., Dumas P., Pravenec M., Krenova D., Kren V., Kunes J., Pausova Z., Gossard F., Hamet P. (2003) TA repeat variation, Npr1 expression, and blood pressure. Impact of the Ace locus. Hypertension 41, 16–24 [DOI] [PubMed] [Google Scholar]
- 47. Usami S., Kishimoto I., Saito Y., Harada M., Kuwahara K., Nakagawa Y., Nakanishi M., Yasuno S., Kangawa K., Nakao K. (2008) Association of CT dinucleotide repeat polymorphism in the 5`-flanking region of the guanylyl cyclase (GC)-A gene with essential hypertension in the Japanese. Hypertens. Res. 31, 89–96 [DOI] [PubMed] [Google Scholar]
- 48. Andrés V., Ureña J., Poch E., Chen D., Goukassian D. (2001) Role of Sp1 in the induction of p27 gene expression in vascular smooth muscle cells in vitro and after balloon angioplasty. Arterioscler. Thromb. Vasc. Biol. 21, 342–347 [DOI] [PubMed] [Google Scholar]
- 49. Wierstra I. (2008) Sp1. Emerging roles–beyond constitutive activation of TATA-less housekeeping genes. Biochem. Biophys. Res. Commun. 372, 1–13 [DOI] [PubMed] [Google Scholar]
- 50. Tan N. Y., Khachigian L. M. (2009) Sp1 phosphorylation and its regulation of gene transcription. Mol. Cell. Biol. 29, 2483–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang W., Zhao S., Ammanamanchi S., Brattain M., Venkatasubbarao K., Freeman J. W. (2005) Trichostatin A induces transforming growth factor β type II receptor promoter activity and acetylation of Sp1 by recruitment of PCAF/p300 to a Sp1·NF-Y complex. J. Biol. Chem. 280, 10047–10054 [DOI] [PubMed] [Google Scholar]
- 52. Swingler T. E., Kevorkian L., Culley K. L., Illman S. A., Young D. A., Parker A. E., Lohi J., Clark I. M. (2010) MMP28 gene expression is regulated by Sp1 transcription factor acetylation. Biochem. J. 427, 391–400 [DOI] [PubMed] [Google Scholar]
- 53. Tsai P. F., Lin S. J., Weng P. L., Tsai S. C., Lin J. H., Chou Y. C., Tsai C. H. (2011) Interplay between PKCδ and Sp1 on histone deacetylase inhibitor-mediated Epstein-Barr virus reactivation. J. Virol. 85, 2373–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gaub P., Tedeschi A., Puttagunta R., Nguyen T., Schmandke A., Di Giovanni S. (2010) HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 17, 1392–1408 [DOI] [PubMed] [Google Scholar]
- 55. Kim J. H., Kim K., Youn B. U., Jin H. M., Kim J. Y., Moon J. B., Ko A., Seo S. B., Lee K. Y., Kim N. (2011) RANKL induces NFATc1 acetylation and stability via histone acetyltransferases during osteoclast differentiation. Biochem. J. 436, 253–262 [DOI] [PubMed] [Google Scholar]
- 56. Xu Y., Jiang Z., Yin P., Li Q., Liu J. (2012) Role for class I histone deacetylases in multidrug resistance. Exp. Cell Res. 318, 177–186 [DOI] [PubMed] [Google Scholar]
- 57. Kim S. H., Kang H. J., Na H., Lee M. O. (2010) Trichostatin A enhances acetylation as well as protein stability of ERα through induction of p300 protein. Breast Cancer Res. 12, R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Das S., Periyasamy R., Pandey K. N. (2012) Activation of IKK/NF-κB provokes renal inflammatory responses in guanylyl cyclase/natriuretic peptide receptor-A gene-knockout mice. Physiol. Genomics 44, 430–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vellaichamy E., Khurana M. L., Fink J., Pandey K. N. (2005) Involvement of the NF-κB/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J. Biol. Chem. 280, 19230–19242 [DOI] [PubMed] [Google Scholar]
- 60. Ellmers L. J., Scott N. J., Piuhola J., Maeda N., Smithies O., Frampton C. M., Richards A. M., Cameron V. A. (2007) Npr1-regulated gene pathways contributing to cardiac hypertrophy and fibrosis. J. Mol. Endocrinol. 38, 245–257 [DOI] [PubMed] [Google Scholar]
- 61. Liu C., Chen Y., Kang Y., Ni Z., Xiu H., Guan J., Liu K. (2011) Glucocorticoids improve renal responsiveness to atrial natriuretic peptide by up-regulating natriuretic peptide receptor-A expression in the renal inner medullary collecting duct in decompensated heart failure. J. Pharmacol. Exp. Ther. 339, 203–209 [DOI] [PubMed] [Google Scholar]
- 62. Cong B., Xu Y., Sheng H., Zhu X., Wang L., Zhao W., Tang Z., Lu J., Ni X. (2014) Cardioprotection of 17β-estradiol against hypoxia/reoxygenation in cardiomyocytes is partly through up-regulation of CRH receptor type 2. Mol. Cell. Endocrinol. 382, 17–25 [DOI] [PubMed] [Google Scholar]
- 63. Wu N., Siow Y. L., O K. (2010) Ischemia/reperfusion reduces transcription factor Sp1-mediated cystathionine β-synthase expression in the kidney. J. Biol. Chem. 285, 18225–18233 [DOI] [PMC free article] [PubMed] [Google Scholar]