

Background: Factors mediating orofacial neuropathic pain are not well defined.
Results: Trigeminal nerve injury-induced calcium channel α2δ1 protein up-regulation in trigeminal ganglia and spinal complex correlated with enhanced spinal presynaptic neurotransmission, excitatory synaptogenesis, and orofacial pain states.
Conclusion: This neuroplasticity may mediate orofacial neuropathic pain states by enhancing dorsal horn excitatory synaptic neurotransmission.
Significance: This reveals a mechanism underlying orofacial neuropathic pain states.
Keywords: Gene Regulation, Molecular Pharmacology, Neuroscience, Pain, Synaptic Plasticity
Abstract
To investigate a potential mechanism underlying trigeminal nerve injury-induced orofacial hypersensitivity, we used a rat model of chronic constriction injury to the infraorbital nerve (CCI-ION) to study whether CCI-ION caused calcium channel α2δ1 (Cavα2δ1) protein dysregulation in trigeminal ganglia and associated spinal subnucleus caudalis and C1/C2 cervical dorsal spinal cord (Vc/C2). Furthermore, we studied whether this neuroplasticity contributed to spinal neuron sensitization and neuropathic pain states. CCI-ION caused orofacial hypersensitivity that correlated with Cavα2δ1 up-regulation in trigeminal ganglion neurons and Vc/C2. Blocking Cavα2δ1 with gabapentin, a ligand for the Cavα2δ1 proteins, or Cavα2δ1 antisense oligodeoxynucleotides led to a reversal of orofacial hypersensitivity, supporting an important role of Cavα2δ1 in orofacial pain processing. Importantly, increased Cavα2δ1 in Vc/C2 superficial dorsal horn was associated with increased excitatory synaptogenesis and increased frequency, but not the amplitude, of miniature excitatory postsynaptic currents in dorsal horn neurons that could be blocked by gabapentin. Thus, CCI-ION-induced Cavα2δ1 up-regulation may contribute to orofacial neuropathic pain states through abnormal excitatory synapse formation and enhanced presynaptic excitatory neurotransmitter release in Vc/C2.
Introduction
Chronic pain is a common clinical syndrome representing a major decrease in the quality of daily life of patients and creates enormous social and economical problems. It has been estimated that about 50 and 10% of people in the United States suffering from localized and widespread chronic pain, respectively (1). Among chronic pain conditions, orofacial pain, which can derive from different etiologies including orofacial inflammation and tissue and nerve injuries, can be the most severe and debilitating. Chronic pain states, such as pain sensations resulting from stimuli that are normally innocuous (allodynia) and increased pain sensations to suprathreshold stimuli (hyperalgesia), tend to be persistent even in the absence of noxious stimuli and in some cases refractory to conventional analgesic treatments. In addition, the use of opioids and other current pain medications is usually limited by problems related to tolerance, addiction, or other nonspecific side effects, which could be life-threatening. Ideally, target-specific therapeutic agents could provide an efficient and safe means of pain relief. Unfortunately, the molecular mechanisms associated with neuropathic orofacial pain are poorly understood (2), and as a consequence, the availability of target-specific, effective pharmacological agents for chronic orofacial pain management is very limited.
Clinical data have shown that neuropathic orofacial pain is sensitive to treatments with gabapentin (2, 3), a drug that binds to the calcium channel α2δ1 (Cavα2δ1)4 and α2δ2 (Cavα2δ2) subunits (4, 5). The Cavα2δ is a structural subunit of the voltage-gated calcium channels important for their functional assembly and expression (6–10). Four Cavα2δ genes have been identified that encode the Cavα2δ1, Cavα2δ2, Cavα2δ3, and Cavα2δ4 subunits, respectively (11–13). These subunits have distinct tissue-specific expression patterns, suggesting their diversified functions in different tissues (5). Findings from a non-orofacial neuropathic pain model have indicated that peripheral nerve injury induces up-regulation of Cavα2δ1, but not Cavα2δ2 (14), in dorsal root ganglia and lumbar dorsal spinal cord that correlates with development of neuropathic pain states sensitive to gabapentin (15–17) without affecting normal nociceptive responses (18–21). However, the detailed mechanism underlying Cavα2δ1 dysregulation in neuropathic pain processing is not clear. In addition, it is not clear whether trigeminal nerve injury causes Cavα2δ1 dysregulation in the trigeminal spinal complex, which differs in structure from that of non-orofacial sensory systems. In this study, we examined whether trigeminal nerve injury induced Cavα2δ1 dysregulation in an orofacial neuropathic pain model and if so what is the underlying role of altered Cavα2δ1 expression in mediating spinal neuron sensitization and development of orofacial neuropathic pain states.
MATERIALS AND METHODS
Experimental Animals
Male adult Sprague-Dawley rats (160–250 g) were used in this study. Animals were housed in groups of three to four in cages with soft bedding with free access to food and water under a 12/12-h light-dark cycle. All animals were acclimated in their cages for 3–4 days before any experiments, which were carried out according to protocols approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Surgery
All surgeries were performed under an operation microscope (MZ95, Leica Microsystems, Wetzlar, Germany). Rats were anesthetized with isoflurane (4% for induction, 2% for maintenance) in O2, shaved above the left eye, and maintained in a prone position. The surgical procedure of chronic constriction injury to the infraorbital nerve (CCI-ION), a branch of the trigeminal nerve, was performed similar to that described by Kernisant et al. (22). Briefly, an anterior-posterior skin incision above the left eye was made following the curve of the frontal bone. The fascia and muscles were then gently pushed laterally from the bone until the contents of the orbit could be gently retracted laterally so that the ION lying on the maxillary bone could be visualized. The ION was then gently freed from surrounding connective tissues. Two loose ligatures (6-0 silk) were placed 3–4 mm apart around the ION using fine forceps and a suture-loaded needle with a bended tip and then loosely ligated. The incision was closed with a 5-0 silk suture, and the rat was recovered on a warm heating pad. In the sham-operated rats, the same procedure was used to expose the left ION, but the ION was not ligated.
For intrathecal catheter implantation, a catheter (PE-10 tubing, 8 cm long) was prepared such that the inserting end of the tubing was blocked with superglue, and holes ∼1.0 cm from the blocked tip were made with a 30-gauge needle. This allowed the insertion of the distal end of the catheter into the subarachnoid space so drugs could be delivered through the holes near the spinal subnucleus caudalis and C1/C2 cervical dorsal spinal cord (Vc/C2) region. Two loose overhand knots, which do not occlude the tubing, were tied and secured by superglue; one was 1.5 cm from the indwelling tip for stopping further insertion of the catheter through the dura mater, and the other was 2.0 cm from the indwelling tip for securing the catheter in place by suturing. A small incision was made on the back of the neck, and the posterior atlantooccipital membrane was exposed after tissues were gently retracted caudally from the occipital bone. A small nick was made in the posterior atlantooccipital membrane (dura mater) using a 25-gauge needle, and a catheter (sterilized PE-10 tubing filled with saline) was inserted gently and caudally (∼0.5 cm) and sutured with muscle ligaments together before closing muscle and skin layers with 5-0 silk sutures. After recovery from anesthesia on a warm pad, rats were returned to their cages and housed individually. Rats were sacrificed at a designated time after behavioral studies, TG and dorsal Vc/C2 were collected, and either used immediately for biochemical studies or kept at −80 °C until use.
Behavioral Tests
Orofacial behavioral tests as described by Vos et al. (23) were performed blindly 1 day before and at designated times after CCI-ION surgeries. For gabapentin treatments, behavioral tests started before and then 30 min and 1, 2, 4, 6, 8, and 24 h after the injection. For daily intrathecal injection of Cavα2δ1 antisense or mismatched oligodeoxynucleotides, the rats were tested before each daily injection and at designated times after the last injection.
The rats were shaved at and near the vibrissal pad under light isoflurane anesthesia 1 day before the testing and placed individually in plastic cages for acclimatization at least 1 h before the testing. During this period, the experimenter reached slowly into the cage to touch the cage wall slightly with a plastic rod similar to the handle of a von Frey filament. The behavioral test started after the rats were calm. When necessary, the acclimatization period was extended.
Mechanical sensitivity was tested with a series of graded von Frey filaments (numbers 3.61, 3.84, 4.08, 4.31, 4.56, 4.74, 4.93, and 5.18 equivalent to 0.4, 0.6, 1.0, 2.0, 4.0, 6.0, 8.0, and 15 g of force, respectively), starting with the 4.31 von Frey filament, and the 50% withdrawal thresholds were determined by the up-down method (24). Briefly, the examiner applied the von Frey filament slowly to the ION territory, near the center of the vibrissal pad, with sufficient force to bend the filament slightly for 2–3 s. A positive response was considered if one or more of the following behaviors occurred: 1) a brisk withdrawal reaction in which a rat pulled briskly backward, 2) escape/attack in which a rat avoided further contact with the filament either by passively moving its body away from the stimulation to assume a crouching position against the cage wall or sometimes with the head buried under the body or attacked the filament actively through biting and grabbing movements, and 3) asymmetric face grooming in which a rat displayed an uninterrupted series of at least three face wash strokes directed to the stimulated facial area often preceded by a brisk withdrawal reaction. Presence of a positive response led to the use of the next filament with a lower stimulating force. Absence of a positive response led to the use of the next filament with a higher stimulating force. This testing paradigm continued until responses to six von Frey stimuli, starting from the one before the first change in response (either negative or positive), were measured. The responses were then converted into a 50% withdrawal threshold value using the following formula: 50% withdrawal threshold = 10(X + kd)/104 where X is the value of the final von Frey hair used in log units, k is the tabular value for the pattern of positive/negative responses from Chaplan et al. (25), and d is the mean difference between stimuli in log units. When consecutive positive or negative responses were observed at the end of a stimulus session, scores of 0.25 or 15 g were assigned, respectively. As reported by other research groups, a small percentage of CCI-ION rats displayed milder bilateral orofacial hypersensitivities postinjury (23, 26). These animals were excluded from the studies.
To determine whether locomotor functions were altered in rats following gabapentin treatments, the Basso, Beattie, Bresnahan Locomotor Rating Scale (27) and the rotarod test were performed blindly in saline or gabapentin (100 mg/kg intraperitoneally)-treated rats. For the Basso, Beattie, Bresnahan Locomotor Rating Scale test, animals were acclimated to their testing environment daily for a week before the testing. The locomotor functions were examined based on a scale focused on evaluating consistent coordination between the front and hind limbs including paw position, toe clearance, trunk stability, and tail position (28). For the rotarod (Economex, Columbus Instruments) test, each rat was placed on the rotarod for seven trials. The first two trials were for training, and then data from the last five trials were averaged. The maximum time for each trial was 120 s. The initial speed of the rotarod was set to 4 rpm, and the acceleration rate was 0.1 rpm.
Western Blots
To measure protein expression levels, frozen TG and dorsal Vc/C2 samples were extracted in 50 mm Tris buffer, pH 8.0 containing 0.5% Triton X-100, 150 mm NaCl, 1 mm EDTA, and protease inhibitors. Cell extracts were subjected to denaturing NuPAGE Tris acetate gel (Invitrogen) electrophoresis and then electrophoretically transferred to PVDF membranes. 5% low fat milk in Tris-buffered saline containing 0.1% Tween 20 was used to block nonspecific binding sites for at least 1 h at room temperature. The Cavα2δ1 monoclonal antibodies (mouse, Sigma-Aldrich; 1:1000) or Cavα2δ2 polyclonal antibodies (rabbit, Alomone Labs Ltd., Israel; 1:200) in phosphate-buffered saline containing 0.1% Tween 20 were incubated with the membranes for 1 h at room temperature or overnight at 4 °C. After washing, the antibody-protein complexes were detected using appropriate secondary antibodies linked to horseradish peroxidase. Antibodies against β-actin (mouse, Novus Biologicals, LLC, Littleton, CO; 1:10,000) were used for loading controls. The antibody-protein complexes were visualized by chemiluminescent reagents. The band densities were quantified by either imaging quantification or densitometry within the linear range of the film sensitivity curve. Cavα2δ protein levels were normalized by taking the ratios of the Cavα2δ band density to that of β-actin within each sample before cross-sample comparisons to calculate the percent changes in Cavα2δ protein levels in the experimental groups compared with that in the control groups. Variations in Cavα2δ band densities in the control groups (contralateral side) were determined by comparing each band density with the mean of the band densities from at least two different control samples run in the same Western blot after taking the ratios of Cavα2δ to β-actin band densities.
Drug Treatments
Gabapentin (Endo Life Sciences, Inc. Farmingdale, NY) was dissolved in sterile saline and intraperitoneally injected (50–100 mg/kg; 1-ml total volume) into CCI-ION rats with allodynia after 3 weeks of CCI-ION. The antisense (AGCCATCTTCGCGATCGAAG) oligodeoxynucleotides against the Cavα2δ1 gene (GenBankTM accession number M86621) and the mismatched control (CGATACCTCGCTGGCTAAAG) were synthesized commercially with phosphorothioate modification on three nucleotides at each end to increase its stability (GeneLink, Hawthorne, NY). The oligodeoxynucleotides were precipitated, washed in 75% ethanol, and dissolved in sterile saline before injections. Cavα2δ1 antisense or mismatched oligodeoxynucleotides were intrathecally injected for 4 days (50 μg/10 μl/rat/day) through the catheter into allodynic rats (started after 3 weeks of CCI-ION) followed by flushing with saline (5 μl to compensate for tubing dead volume). The same volume of saline was injected as a control. A daily von Frey filament test was performed before the daily injection and after the final injection.
Immunohistochemistry
Vc/C2 spinal cord and TG samples were collected from three animals in each group after 1- and 3-week CCI-ION and 3-week sham surgeries. Samples were fixed in 4% paraformaldehyde overnight, cryoprotected in 30% sucrose, then mounted in optimal cutting tissue medium (OCT, Sakura Fintech, Torrance, CA), and sectioned into 10-μm slices by a cryostat (CM1900, Leica Microsystems). The thin sections were pretreated with heat-based antigen retrieval and then incubated for 2 days at 4 °C in Antibody Diluent Solution (Dako, Carpinteria, CA) with combinations of primary antibodies from different species against proteins of interest including Cavα2δ1 (rabbit polyclonal, Thermo Fisher Scientific Inc., Waltham, MA or mouse monoclonal, Sigma-Aldrich), Vglut2 (guinea pig polyclonal, Synaptic Systems, Goettingen, Germany), PSD95 (rabbit polyclonal, Invitrogen), and synaptophysin (SYN; goat polyclonal, Santa Cruz Biotechnology, Dallas, TX). Control staining was done in parallel with omission of the primary antibody. After washing, the sections were incubated for 2 h at room temperature with respective Alexa Fluor 488 or Alexa Fluor 594 secondary antibodies. Isolectin B4 (IB4)-positive staining was detected with Alexa Fluor 488-conjugated IB4 (Invitrogen). Stained sections were then preserved using Vectashield Hard Set Mounting Medium containing DAPI for nucleus staining (Vector Laboratories, Inc., Burlingame, CA) and coverslipped. Images were taken using a Zeiss LSM710 or Zeiss LSM780 confocal microscope (University of California Irvine Optical Biology Core) in stacks of 14 consecutive 0.3-μm Z-sections. Images were then cropped down to three to seven consecutive Z-sections with the best signal and merged (as specified below). Merged images were used for analysis using Volocity (Version 6.0, PerkinElmer Life Sciences).
Sensory neurons in captured images of TG sections (merged stacks of seven consecutive Z-sections of 0.3 μm each, 100 μm apart, and 10 images/side/rat from three rats) were identified based on their morphology and nucleus size and manually selected by drawing the longest line across each cell with a nucleus using the Volocity line function. Background subtraction was performed by subtracting the average fluorescence intensity of the neuron with the lowest fluorescence intensity in each image. Only profiles with Cavα2δ1 immunoreactive intensity at least 1-fold over the baseline average intensity were analyzed. Sizes of immunoreactive cells were quantified using Volocity.
Fluorescent immunoreactivity from rat Vc/C2 sections was analyzed after normalization of background threshold among control (contralateral side) samples from different sets of experiments using the Volocity Percentage Intensity function, and the same background threshold was used for both the contralateral and injury sides for each set of experiments. Cavα2δ1 immunoreactivity was analyzed from merged stacks of seven consecutive Z-sections of 0.3 μm each, 100 μm apart, and three images/side/rat from three rats. Captured images from Vglut2/Cavα2δ1-co-stained samples (merged stacks of three consecutive Z-sections of 0.3 μm each, 100 μm apart, and nine images/side/rat from three rats in each group) were analyzed to determine the number of total Vglut2+, Vglut2+/Cavα2δ1+, and Vglut2+/Cavα2δ1− puncta. SYN/Cavα2δ1-co-stained samples (merged stacks of three consecutive Z-sections of 0.3 μm each, 100 μm apart, and three images/side/rat from three rats) were analyzed for total SYN+, SYN+/Cavα2δ1+, or SYN+/Cavα2δ1− puncta. Vglut2/PSD95-co-stained samples (merged stacks of three consecutive Z-sections of 0.3 μm each, 100 μm apart, and six images/side/rat from three rats) were analyzed for total PSD95+ or Vglut2+/PSD95+ puncta. Data from the ipsilateral side were compared with date from the contralateral side.
Electrophysiological Recording on Vc/C2 Slices
Vc/C2 was collected from animals deeply anesthetized with isoflurane. Transverse slices (300 μm) were cut with a Vibratome (VT1200) in an ice-cold (4 °C) sucrose solution containing 230 mm sucrose, 26 mm NaHCO3, 2.5 mm KCl, 1.25 mm NaH2PO4, 0.5 mm CaCl2, 10 mm MgSO4, and 10 mm glucose, pH 7.4, 290–305 mosmol/liter, equilibrated with 95% O2 and 5% CO2. The slices were incubated for >1 h before recording in an artificial cerebrospinal fluid containing 130 mm NaCl, 3 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 10 mm glucose, pH 7.4, 295–305 mosmol/liter, which was continuously equilibrated with 95% O2 and 5% CO2. Electrodes made from borosilicate glass (World Precision Instruments, Sarasota, FL) with a horizontal electrode puller (P-97, Sutter Instrument Co., Novato, CA) had a resistance of 5–7 megaohms when filled with an intracellular solution containing135 mm potassium gluconate, 5 mm KCl, 5 mm EGTA, 0.5 mm CaCl2, 10 mm HEPES, 2 mm Mg-ATP, and 0.1 mm GTP, pH 7.4, 295–300 mosmol/liter. The flow rate of oxygenated artificial cerebrospinal fluid in the recording chamber was adjusted to 1 ml/min. Neurons in the Vc/C2 dorsal horn were visualized with an infrared differential interference contrast microscope (FN1, Nikon Instruments, Inc., Melville, NY) and a charge-coupled device camera (Andor Clara, Andor Technology, South Windsor, CT).
α-Amino-3-hydroxyl-5-methylisoxazole-4-propionic acid (AMPA) receptor-mediated miniature excitatory postsynaptic currents (mEPSCs) were recorded at a −70-mV membrane holding potential at 32 ± 0.5 °C in the presence of tetrodotoxin (1 μm; Sigma), strychnine (1 μm; Sigma), bicuculline (30 μm; Research Biochemicals, Inc., Natick, MA), and aminophosphonovalerate (50 mm; Research Biochemicals, Inc., Natick, MA) to block tetrodotoxin-sensitive Na+, glycinergic, GABAergic, and N-methyl-d-aspartate (NMDA) currents, respectively. At this holding potential, over 90% of dorsal horn neuron mEPSCs are AMPA receptor-mediated because they are sensitive to blockade by 6-cyano-7-nitroquinoxaline-2,3-dione, an AMPA receptor antagonist (29). NMDA receptor-mediated mEPSCs were not examined because they are not activated at this holding potential because of voltage-dependent Mg2+ blockade (30), and they only represent less than 10% of the mEPSCs even at a depolarizing potential to remove the Mg2+ blockade (29). Series resistance (8–15 megaohms) was monitored periodically during the recording, and cells with >15% changes were excluded from analysis. For experiments involving gabapentin treatments, the same neurons were recorded before treatment, during the 10-min gabapentin treatment, and after 15-min washout. Recording signals were filtered at 2 kHz and digitized at a sampling rate of 5 kHz using a low noise data acquisition system (Digidata 1440A, Molecular Devices).
Statistics
A p value <0.05 indicated significance determined by variance analyses for multigroup comparisons or by two-tailed, unpaired Student's t tests for pairwise comparisons as indicated.
RESULTS
Trigeminal Nerve Injury Induced Up-regulation of the Calcium Channel Cavα2δ1 Subunit Proteins That Correlated with Orofacial Tactile Allodynia
To determine whether trigeminal nerve injury induced up-regulation of the Cavα2δ1 gene and if so whether it correlated with development of orofacial neuropathic pain, we examined Cavα2δ1 expression levels in trigeminal ganglia and Vc/C2 in an orofacial neuropathic pain model derived from CCI-ION (23). As indicated in Fig. 1, unilateral CCI-ION resulted in an initial orofacial hyposensitive state as indicated by an initial increase in orofacial thresholds (to the 15-g cutoff maximal thresholds) to stimuli in the injury side for ∼10 days postinjury. This was followed by an orofacial hypersensitive state shown as increased orofacial sensitivity (or reduced threshold) to von Frey filament stimulation (about 3 weeks postinjury) in the injury side compared with the non-injury side and sham-operated rats. These CCI-ION-induced abnormal, biphasic orofacial sensitivities were also reported in the original study describing this model (23). The allodynic state lasted for about 7–8 weeks followed by a gradual recovery in the injured rats.
FIGURE 1.
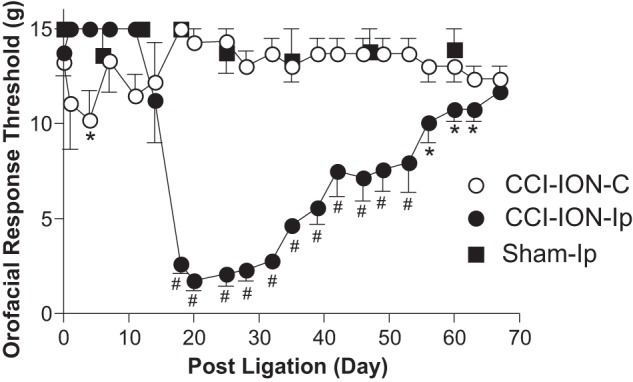
CCI-ION led to development of orofacial tactile allodynia in adult male rats. Orofacial sensitivity to von Frey filament stimulation was tested in the whisker pad area of rats before and at the designated times after CCI-ION or sham operations. Data presented are the means ± S.E. (error bars) from five to seven rats in each group. C, contralateral to injury; Ip, ipsilateral to injury. *, p < 0.05; #, p < 0.001 compared with preinjury level by two-way analysis of variance with Bonferroni post-tests.
If CCI-ION also induced Cavα2δ1 dysregulation that played a role in orofacial pain processing, one would expect to see a correlation between changes in Cavα2δ1 expression in TG and/or Vc/C2 and orofacial allodynia development. To test this hypothesis, we examined the expression level of Cavα2δ1 in TG and Vc/C2 at different stages of orofacial allodynia development: (i) 1 week postinjury but before the onset of allodynia, (ii) 3 weeks postinjury when the injured rats displayed severe allodynia, and (iii) 10 weeks postinjury when the injured animals recovered from allodynia. As shown in Fig. 2 (A and B), CCI-ION induced Cavα2δ1 up-regulation in TG but no change in Vc/C2 before the onset of orofacial allodynia (1 week post-CCI-ION). However, CCI-ION induced Cavα2δ1 up-regulation in both TG and Vc/C2 at the orofacial allodynia stage (3 weeks post-CCI-ION) without altering the levels of Cavα2δ2 (Fig. 2C), which also binds gabapentin (5). When the CCI-ION rats were recovered from the allodynia states (10 weeks post-CCI-ION), injury-induced Cavα2δ1 expression levels returned to a level similar to the levels in the non-injury side. This temporal correlation between injury-induced Cavα2δ1 up-regulation and allodynia suggests that increased Cavα2δ1 may play a critical role in orofacial neuropathic pain processing.
FIGURE 2.

CCI-ION led to increased expression of Cavα2δ1 proteins in TG and Vc/C2. Western blots were used to examine Cavα2δ1 protein levels in TG (A) and dorsal Vc/C2 (B) samples at the designated time points post-CCI-ION as well as Cavα2δ2 protein levels in these samples 3 weeks (wk) post-CCI-ION (C). Representative Western blot data from TG or dorsal Vc/C2 are shown on top of each bar graph summarizing respective Western blot data. For normalizing sample loading, ratios of Cavα2δ over β-actin band densities were taken within each sample group before comparisons were made between the injury side and non-injury side. Data presented are the means ± S.E. (error bars) from four to six rats. *, p < 0.05; **, p < 0.01 compared with the non-injury side by Student's t test. C, contralateral to injury; Ip, ipsilateral to injury.
Localization of Injury-induced Cavα2δ1 in CCI-ION Rats with Orofacial Allodynia
To determine the location of injury-induced Cavα2δ1 expression in TG and Vc/C2 at the orofacial neuropathic pain stage, we examined Cavα2δ1 immunoreactivity profiles in thin sections of TG and Vc/C2 samples from 3-week CCI-ION rats with tactile allodynia in the injury side. Data from confocal immunofluorescent staining indicated that the basal level of Cavα2δ1 immunoreactivity in the non-injury side was relatively high in small and some medium size TG neurons but low in large TG neurons. CCI-ION induced a reduction in the number of the smallest profiles (15 μm in diameter) that were Cavα2δ1-immunoreactive. This could be due to reduced Cavα2δ1 expression or dying back of injured small sensory fibers (31). However, CCI-ION increased the number of Cavα2δ1-immunoreactive profiles over 25 μm in diameter with a trend of more pronounced increases in larger diameter profiles (Fig. 3). This suggests that CCI-ION induced Cavα2δ1 dysregulation in different sizes of sensory neurons.
FIGURE 3.
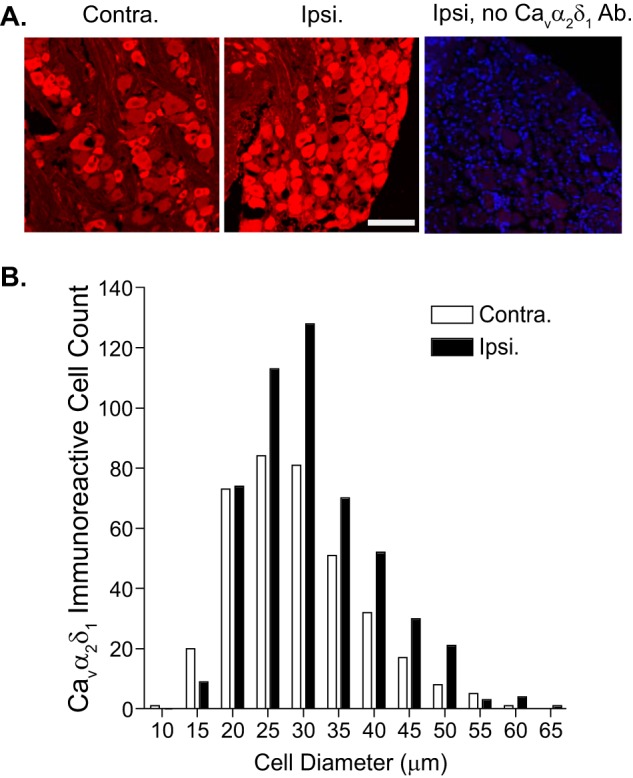
Localization of CCI-ION-induced Cavα2δ1 immunoreactivity in TG. Immunostaining was performed in TG samples collected from 3-week CCI-ION rats with orofacial hypersensitivity in the injury side. A, representative Cavα2δ1 immunoreactive profiles (red) in injury (Ipsi.) and non-injury (Contra.) sides of TG. The right panel shows control staining with omission of the primary antibody (Ab.) in a section from the injury side. Blue, DAPI staining of nuclei. Scale bar, 100 μm for all image panels. B, summarized data quantifying the numbers of Cavα2δ1-immunoreactive profiles of different sizes of neurons from three rats.
Basal level Cavα2δ1 immunoreactivity was detectable in superficial dorsal horn of Vc/C2 (Fig. 4A, Cavα2δ1, Contra.) and the inner portion of which co-localized with non-peptidergic IB4-positive sensory fiber terminals projecting to inner lamina II of the superficial dorsal horn (Fig. 4A, IB4, Cavα2δ1/IB4, Contra.) (32–35). Three weeks after CCI-ION, the intensity of Cavα2δ1 immunoreactivity was increased significantly in the superficial, but not deep, dorsal horn of Vc/C2 in the injury (ipsilateral) side compared with that in the non-injury (contralateral) side (Fig. 4, A and B). The increased Cavα2δ1-immunoreactive intensity in the injury side was mainly due to an increase of surface area of positive Cavα2δ1 immunoreactivity (Fig. 4C) and a small but significant increase in average intensity of Cavα2δ1 immunoreactivity (Fig. 4D). This pattern of Cavα2δ1 dysregulation is similar to that reported in a spinal nerve injury model in which Cavα2δ1 is induced in dorsal root ganglion neurons and then translocated to their central terminals in the superficial dorsal horn of lumbar spinal cord (14, 36). Indeed, CCI-ION-induced Cavα2δ1 up-regulation in TG neurons preceded that in dorsal Vc/C2 (Fig. 2, A and B). Together, it is possible that CCI-ION induces Cavα2δ1 up-regulation in TG neurons followed by its translocation to their central terminals in Vc/C2 superficial dorsal horn.
FIGURE 4.

Localization of CCI-ION-induced Cavα2δ1 immunoreactivity in Vc/C2. Immunostaining was performed in Vc/C2 samples collected from 3-week CCI-ION rats with orofacial hypersensitivity in the injury side. A, representative Cavα2δ1 immunoreactivity (red) in injury (Ipsi.) and non-injury sides (Contra.) of Vc/C2. IB4 reactivity specific to α-d-galactose-terminal glycoconjugates expressed on the cell surface of a subpopulation of non-peptidergic neuronal terminals (33) (green) is shown for anatomical localization of superficial dorsal horn. Scale bar, 140 μm for all image panels. B, summarized data of total intensity of Cavα2δ1 immunoreactivity in superficial (SDH) and deep (DDH) dorsal horn of Vc/C2 samples (AU, artificial units). C, summarized data of the surface area of Cavα2δ1 immunoreactivity in superficial and deep dorsal horn of Vc/C2 samples. D, summarized data of the average intensity of Cavα2δ1 immunoreactivity in superficial and deep dorsal horn of Vc/C2 samples. Data presented are the means ± S.E. (error bars) from nine images in each side from three rats. *, p < 0.05; **, p < 0.01 compared with non-injury side by Student's t test. Contra., contralateral to injury; Ipsi., ipsilateral to injury.
Gabapentin Treatment in CCI-ION Rats Blocked Injury-induced Allodynia
To confirm that up-regulated Cavα2δ1 is involved in the processing of orofacial neuropathic pain, we injected gabapentin, a drug that binds to the Cavα2δ1 proteins (4) and has antiallodynia properties in patients (37–39) and animal models (15–17, 40, 41), intraperitoneally into CCI-ION rats with established allodynia to see whether this treatment could reverse CCI-ION-induced allodynia. Bolus intraperitoneal injection of 50 mg/kg gabapentin had a small and statistically not significant reversal of CCI-ION-induced tactile allodynia. However, increasing the gabapentin dose to 100 mg/kg resulted in a significant reversal of injury-induced orofacial allodynia (Fig. 5A) similar to that observed in other centrally (28) or peripherally induced (15, 41) neuropathic pain models. The gabapentin effects lasted for approximately 6 h, which is similar to the therapeutic duration of the drug. There were no detectable side effects, such as impaired motor functions (Fig. 5, B and C), from the gabapentin treatments. These data support that CCI-ION-induced Cavα2δ1 up-regulation is highly likely to be involved in allodynia processing.
FIGURE 5.
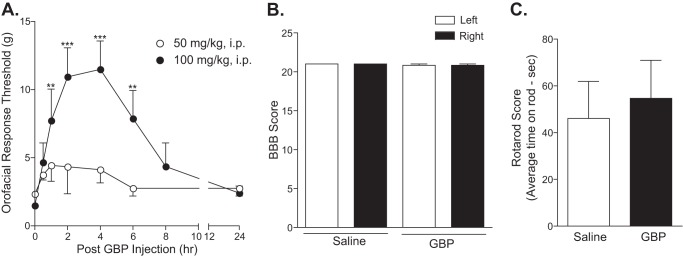
Injury-induced orofacial allodynia could be blocked by gabapentin. Rats with established orofacial allodynia after 3-week CCI-ION were given a bolus intraperitoneal (i.p.) injection of gabapentin with the doses indicated. Behavioral testing was performed in the injury side before the injection and at the indicated time points after the injection (A). Data presented are the means ± S.E. (error bars) from three to four rats. **, p < 0.01; ***, p < 0.001 compared with the pretreatment level by two-way analysis of variance with Bonferroni post-tests. To test whether gabapentin treatment impaired locomotor functions, Basso, Beattie, Bresnahan Locomotor Rating Scale (BBB) (B) and rotarod (C) tests were performed before and 4 h after intraperitoneal gabapentin (100 mg/kg) injection, a time point correlated with maximal reversal of orofacial allodynia in gabapentin-treated CCI-ION rats. Data presented are the means ± S.E. (error bars) from six rats in each group. GBP, gabapentin.
Intrathecal Treatment with Cavα2δ1 Antisense Oligodeoxynucleotides in CCI-ION Rats Resulted in Reversals of Allodynia and Up-regulated Cavα2δ1 Proteins in Vc/C2
To further determine whether CCI-ION-induced Cavα2δ1 up-regulation played a critical role in mediating tactile allodynia, we injected Cavα2δ1 antisense or mismatched oligodeoxynucleotides (50 μg/rat/day) or saline intrathecally through the catheter into the Vc/C2 region of CCI-ION rats for 4 consecutive days, starting after 3 weeks of CCI-ION when the injured rats had developed allodynia, to see whether antisense treatment could block or diminish CCI-ION-induced allodynia by diminishing elevated Cavα2δ1 levels. As indicated in Fig. 6, intrathecal treatments with the antisense, but not mismatched, oligodeoxynucleotides resulted in a time-dependent reversal of tactile allodynia. A similar injection of saline (equal volume) did not cause allodynia reversal (42). The antisense effects had an onset time of 2 days, peaked approximately 5 days after treatment initiation (1 day after the last injection), and lasted for over 2 days after the last injection. To determine whether the antisense effects were due to blockade of CCI-ION-induced Cavα2δ1 up-regulation, we examined Cavα2δ1 protein levels in dorsal Vc/C2 and TG samples collected from CCI-ION rats 1 day after the last treatment with Cavα2δ1 antisense or mismatched oligodeoxynucleotides or saline, which correlated with the peak antiallodynic effects of the antisense oligodeoxynucleotides. Data from Western blot analyses indicated that treatments with antisense oligodeoxynucleotides, but not with mismatched oligodeoxynucleotides or saline, caused a small and statistically not significant reversal of the injury-induced increase of Cavα2δ1 in TG (Fig. 7A). However, similar treatments with antisense oligodeoxynucleotides blocked injury-induced Cavα2δ1 up-regulation in dorsal Vc/C2 (Fig. 7B). This correlation in antisense-mediated blockade between injury-induced allodynia and Vc/C2 Cavα2δ1 up-regulation supports that CCI-ION-induced Cavα2δ1 up-regulation in dorsal Vc/C2 plays a critical role in allodynia development.
FIGURE 6.
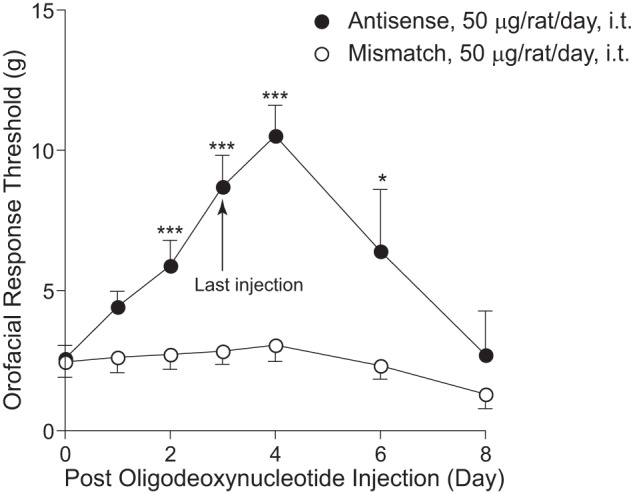
Injury-induced orofacial allodynia could be blocked by intrathecal treatment with Cavα2δ1 antisense, but not mismatched, oligodeoxynucleotides. Rats with established orofacial allodynia after 3-week CCI-ION were given a daily intrathecal (i.t.) injection of Cavα2δ1 antisense (AS) or mismatched (MM) oligodeoxynucleotides (50 μg/rat/day) for 4 consecutive days. Behavioral tests were performed daily in the injury side before the injection and at the designated time points after the last injection. Data presented are the means ± S.E. (error bars) from 12–14 rats up to day 4 after treatment initiation and from three to six rats afterward. *, p < 0.05; ***, p < 0.001 compared with the pretreatment level by two-way analysis of variance with Bonferroni post-tests.
FIGURE 7.

Cavα2δ1 protein levels in TG and Vc/C2 after treatment with antisense and mismatched oligodeoxynucleotides. Cavα2δ1 protein levels in TG and dorsal Vc/C2 samples collected 1 day after the last injection of the 4-day treatment were subjected to Western blot analyses. Representative Western blot data from TG (A) or Vc/C2 (B) are shown on top of each bar graph summarizing respective Western blot data. For normalizing sample loading, ratios of Cavα2δ1 over β-actin band densities were taken within each sample group before comparisons were made between the injury side and non-injury side. Data presented are the means ± S.E. (error bars) from four rats in the saline group and six to seven rats in the treatment groups. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 compared with the non-injury side; $$, p < 0.01 compared with the injury side of the saline-treated group; #, p < 0.05 compared with the injury side of the mismatch group with Student's t test. C, contralateral to injury; Ip, ipsilateral to injury.
Increased Cavα2δ1 in Vc/C2 Superficial Dorsal Horn Correlated with Increased Excitatory Synaptogenesis in CCI-ION Rats with Orofacial Allodynia
Cavα2δ1 proteins have been implicated to play a critical role in mediating aberrant excitatory synaptogenesis in the central nervous system (43). To determine whether CCI-ION led to increased excitatory synaptogenesis associated with Cavα2δ1 up-regulation and orofacial hypersensitivity, we examined the degree of co-localization between Cavα2δ1 immunoreactivity and puncta of an excitatory presynaptic glutamatergic terminal marker, Vglut2, in superficial dorsal horn of Vc/C2 from CCI-ION rats at time points correlated with the absence (1 week postinjury) or presence (3 weeks postinjury) of orofacial allodynia. Vglut2 was selected because knocking out Vglut2, but not Vglut1, from sensory neurons abolished or reduced nerve injury-induced tactile and cold allodynia in peripheral nerve injury models (44–46). Although Vglut3 has been shown to be present in a unique subset of sensory neurons that may play a distinct role in nerve injury-induced behavioral hypersensitivity (47), it was not examined due to the lack of specific antibodies commercially available to us. Our data indicated that the total number of Vglut2-positive (Vglut2+) puncta increased significantly in the injury side of 3-week, but not 1- week, CCI-ION Vc/C2 samples (Fig. 8, A and B). By separating Vglut2+ puncta into Cavα2δ1-positive (Cavα2δ1+) and Cavα2δ1-negative (Cavα2δ1−) groups, we found that all of the increased immunoreactive puncta were both Vglut2+ and Cavα2δ1+. In contrast, the number of Vglut2+ but Cavα2δ1− puncta showed a slight decrease, which might be due to dying back of injured nerves that led to loss of their synaptic terminals. Importantly, the numbers of Vglut2+/Cavα2δ1+ puncta were not changed in Vc/C2 samples taken from 1-week CCI-ION or 3-week sham rats, which did not have orofacial allodynia (Fig. 8, A and B).
FIGURE 8.

CCI-ION-induced Vc/C2 Cavα2δ1 immunoreactivity at excitatory presynaptic terminals correlated with orofacial allodynia development. Co-immunostaining of Cavα2δ1 with excitatory presynaptic marker Vglut2 was performed in Vc/C2 samples collected from CCI-ION rats 1 week (1-WK CCI-ION) or 3 weeks (3-WK CCI-ION) postinjury that correlated with the absence or presence of orofacial hypersensitivity, respectively. A, representative images showing Vglut2 (green) and/or Cavα2δ1 (red) immunoreactivity in superficial dorsal horn of Vc/C2. Arrowheads indicate representative positive immunoreactivities of Vglut2 or Cavα2δ1, respectively, and their co-localization (yellow). Scale bar, 5 μm for all image panels. B, summarized total Vglut2-immunoreactive puncta with (yellow) or without (green) co-localization with Cavα2δ1 immunoreactivity in superficial dorsal horn of Vc/C2 samples collected at the designated time post sham or CCI-ION. Data presented are the means ± S.E. (error bars) collected from 27 images in each side of three rats. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with non-injury (Contra.) side by Student's t test. Contra., contralateral to injury; Ipsi., ipsilateral to injury.
Similarly, the total punctum counts of SYN, another presynaptic marker, were also significantly increased in Vc/C2 superficial dorsal horn of the injury side compared with that from the non-injury side 3 weeks post-CCI-ION (Fig. 9, A and B). These changes were mainly associated with Cavα2δ1 up-regulation because only SYN-positive (SYN+) and Cavα2δ1+ immunoreactive puncta were increased, whereas SYN+ but Cavα2δ1− punctum counts remained unchanged in the injury side compared with those from the non-injury side (Fig. 9, A and B).
FIGURE 9.

CCI-ION-induced Vc/C2 Cavα2δ1 immunoreactivity correlated with excitatory synaptogenesis. Co-immunostaining of Cavα2δ1 with synaptic markers was performed in Vc/C2 samples collected from 3-week (3-WK) CCI-ION rats with orofacial hypersensitivity in the injury side. A, representative images showing SYN (green), a presynaptic marker, and/or Cavα2δ1 (red) immunoreactivity in Vc/C2 superficial dorsal horn. Arrowheads indicate representative positive immunoreactivities of SYN or Cavα2δ1, respectively, and their co-localization (yellow). Scale bar, 5 μm for all image panels. B, summarized total SYN-immunoreactive puncta and those with (yellow) or without (green) co-localization with Cavα2δ1 immunoreactivity in Vc/C2 superficial dorsal horn. Data presented are the means ± S.E. (error bars) collected from nine images in each side of three rats. *, p < 0.05; ***, p < 0.001 compared with non-injury (Contra.) side by Student's t test. C, representative images showing PSD95 (red) and/or Vglut2 (green) immunoreactivity in Vc/C2 superficial dorsal horn. Arrowheads indicate representative positive immunoreactivities of PSD95 or Vglut2, respectively, and their co-localization (yellow). Scale bar, 5 μm for all image panels. D, summarized total PSD95-immunoreactive puncta in Vc/C2 superficial dorsal horn. E, summarized PSD95+ and Vglut2+ immunoreactive puncta in Vc/C2 superficial dorsal horn. For both D and E, data presented are the means ± S.E. (error bars) from 18 images in each side of three rats. *, p < 0.05; **, p < 0.01 compared with non-injury (Contra.) side by Student's t test. Contra., contralateral to injury; Ipsi., ipsilateral to injury.
To determine whether CCI-ION-increased excitatory synapses in superficial dorsal horn of Vc/C2 were functional, which should contain postsynaptic elements, we examined the co-localization of immunoreactivity for Vglut2 and a postsynaptic marker, PSD95, in 3-week CCI-ION Vc/C2 samples. Our data showed that CCI-ION induced a significant increase in the total number of PSD95-immunoreactive puncta (Fig. 9, C and D) that correlated with a similar increase in PSD95-immunoreactive puncta that were also Vglut2-immunoreactive (Fig. 9, C and E). Together, these findings support that CCI-ION-induced Vglut2-positive excitatory synapses are functional as they contain both pre- and postsynaptic elements.
Increased Frequency of Gabapentin-sensitive Excitatory Postsynaptic Currents in Vc/C2 Superficial Dorsal Horn Neurons Correlated with Orofacial Allodynia Development in CCI-ION Rats
To confirm whether CCI-ION-induced Vc/C2 Cavα2δ1 up-regulation contributed to orofacial behavioral hypersensitivity through a central mechanism related to Vc/C2 neuron sensitization, we examined mEPSCs in Vc/C2 superficial dorsal horn neurons from CCI-ION rats either 1 or 3 weeks post-CCI-ION, time points correlating with either the absence or presence of allodynia in injured rats, respectively. Because dorsal horn neuron mEPSCs are induced by presynaptic glutamate release, changes in its frequency or amplitude reflect a presynaptic or postsynaptic mechanism, respectively (29, 48). As indicated in Fig. 10, neither the frequency nor amplitude of mEPSCs differed significantly in Vc/C2 superficial dorsal horn neurons between sham and CCI-ION rats at the 1-week time point (Fig. 10, A–D). However, the frequency, but not the amplitude, of mEPSCs in Vc/C2 superficial dorsal horn neurons was significantly increased in CCI-ION rats compared with that in sham control rats 3 weeks postinjury (Fig. 10, E–H). In addition, the increased mEPSC frequency could be blocked by gabapentin dose-dependently (Fig. 10, I and J). The maximal percent inhibition of mEPSCs by gabapentin was similar to the percent increase of mEPSCs in neurons from injured rats (Fig. 10, G and J). Together, these data suggest that increased Cavα2δ1 in the Vc/C2 region is highly likely to mediate the development of orofacial hypersensitivity by enhancing presynaptic excitatory transmitter release, which can be normalized by gabapentin.
FIGURE 10.
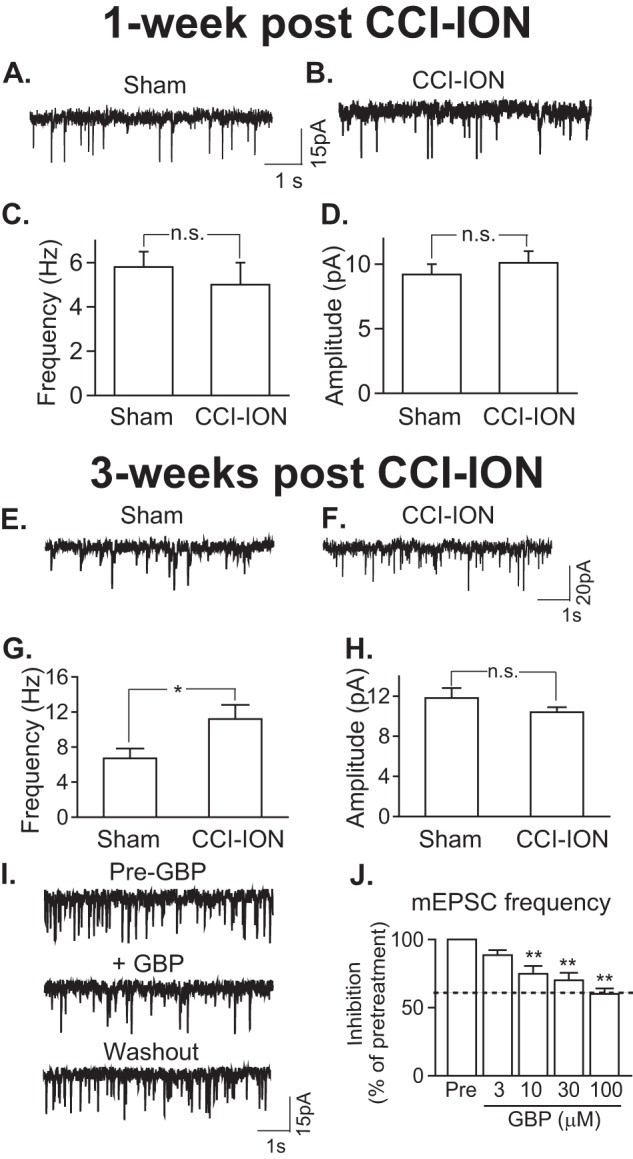
CCI-ION enhanced presynaptic excitatory neurotransmitter release that could be blocked by gabapentin and correlated with orofacial allodynia. A–D, mEPSCs in superficial dorsal horn neurons of Vc/C2 spinal cord slices 1-week post sham or post-CCI-ION. A and B, representative traces of mEPSCs from neurons in the injury side of sham (A) and CCI-ION (B) rats. C and D, summary of mEPSC frequency (C) and amplitude (D) in superficial dorsal horn neurons from the injury side of sham and CCI-ION rats (means ± S.E. (error bars) from 17 neurons of three sham rats and 19 neurons of three CCI-ION rats). n.s., not significant by Student's t test. E–H, mEPSCs in superficial dorsal horn neurons of Vc/C2 spinal cord slices 3-weeks post sham or post-CCI-ION. E and F, representative traces of mEPSCs from neurons in the injury side of sham (E) and CCI-ION (F) rats. G and H, summary of mEPSC frequency (G) and amplitude (H) in superficial dorsal horn neurons from the injury side of sham and CCI-ION rats (means ± S.E. (error bars) from 28 neurons each of four sham rats and four CCI-ION rats, respectively). n.s., not significant; *, p < 0.05 compared with sham control by Student's t test. I, representative mEPSC traces before (upper panel), during (middle panel) 10-min 100 μm gabapentin (GBP) perfusion, and after 15-min washout (lower panel). J, gabapentin inhibition of enhanced mEPSC frequency in Vc/C2 superficial dorsal horn neurons of the injury side from 3-week CCI-ION rats (means ± S.E. (error bars) from three to four neurons in each group). The dotted line represents the baseline level of mEPSC frequency in superficial dorsal horn neurons from sham control rats. **, p < 0.01 compared with the pretreatment level by Student's t test.
DISCUSSION
Orofacial neuropathic pain is a devastating disorder with limited efficacious therapeutic options due to the fact that its underlying mechanisms are not well defined. Findings from this study have indicated that injury to the trigeminal nerve leads to up-regulation of Cavα2δ1, but not Cavα2δ2, in TG and associated dorsal Vc/C2 spinal cord that correlates with Vc/C2 aberrant excitatory synaptogenesis, enhanced presynaptic excitatory inputs, and neuropathic pain state development. Both orofacial allodynia and Cavα2δ1 up-regulation in dorsal Vc/C2 spinal cord can be blocked by treatments with intrathecal Cavα2δ1 antisense, but not mismatched, oligodeoxynucleotides. In addition, gabapentin treatment can normalize CCI-ION-induced Vc/C2 neuron sensitization and reverse orofacial allodynic states. Together, these findings support that trigeminal nerve injury-induced Cavα2δ1 dysregulation may play a critical role in mediating orofacial hypersensitivity through a mechanism involving aberrant excitatory synaptogenesis and elevated presynaptic excitatory neurotransmitter release.
The temporal correlation between Vc/C2 Cavα2δ1 dysregulation (Fig. 2) and orofacial hypersensitivity development (Fig. 1) only in a later stage (3 weeks) postinjury supports that mechanisms underlying acute pathological changes and chronic pain states post-trigeminal nerve injury are distinct. Thus, this model may be useful in studying mechanistic transitions from acute nerve injury to chronic pain development. In addition, the 3-week onset time of allodynia development in this orofacial pain model is much longer than that in the spinal nerve ligation injury model, which develops peak allodynia within the 1st week postinjury (15, 16, 49, 50) including the time required for anterograde transport of elevated Cavα2δ1 from dorsal root ganglion sensory neurons to their presynaptic central axon terminals in lumbar dorsal spinal cord (14, 36). This discrepancy suggests that, in addition to structural differences between the trigeminal and peripheral nervous systems, trigeminal nerve injury-induced pain states may be mediated by a more complex mechanism than simply Cavα2δ1 up-regulation followed by its translocation from TG to Vc/C2. It is possible that the slow onset of allodynia in the CCI-ION model reflects the time required for some pathological changes to occur in Vc/C2 post-CCI-ION, such as dysregulation of factors that are unique to CCI-ION and critical for allodynia development in a later stage. These unique factors may play a critical role in regulating trigeminal nerve injury-induced Cavα2δ1 up-regulation or its translocation from injured trigeminal neurons to their central axon terminals in superficial dorsal horn of Vc/C2. Alternatively, these factors may serve as critical cofactors in Cavα2δ1-mediated orofacial pain states.
Although our data could not exclude any possible contribution of a postsynaptic mechanism to CCI-ION-induced orofacial neuropathic pain states, our behavioral pharmacology, immunostaining, and electrophysiology data support that CCI-ION-induced Cavα2δ1 up-regulation plays an active role in mediating orofacial hypersensitivity mainly through a presynaptic mechanism. This is supported by findings from a non-orofacial neuropathic pain model in which increased Cavα2δ1 expression in dorsal root ganglion sensory neurons leads to spinal neuron sensitization through enhanced presynaptic excitatory neurotransmitter release (29, 51). Although it is technically impossible to ligate the dorsal roots of trigeminal nerves between TG and Vc/C2 to block this pathway, our Western blot data indicate that Cavα2δ1 up-regulation in TG precedes that in Vc/C2 postinjury (Fig. 2), and immunostaining data support that increased Cavα2δ1 in Vc/C2 is co-localized with presynaptic markers (Figs. 8 and 9). In addition, recording data support that CCI-ION-induced Vc/C2 neuron sensitization is driven mainly by enhanced presynaptic excitatory inputs when the CCI-ION rats display behavioral hypersensitivity (Fig. 10, E–H).
In combination with recent findings that Cavα2δ1 is the neuronal receptor for astrocyte-secreted thrombospondins in mediating excitatory synaptogenesis in the central nervous system (43) and that CCI-ION also induces thrombospondin expression in Vc/C2 that contributes to orofacial pain processing (42), our findings are consistent with the notion that elevated presynaptic Cavα2δ1 in the Vc/C2 region may cause aberrant excitatory synaptogenesis (Figs. 8 and 9), leading to spinal neuron sensitization (Fig. 10) and neuropathic pain states (Fig. 1). This is supported by our Western blot data showing that Cavα2δ1 up-regulation is diminished in TG, but abolished in the Vc/C2 region, after Cavα2δ1 antisense, but not mismatched, oligodeoxynucleotide treatments (Fig. 7), which correlates with diminished orofacial allodynia in injured rats (Fig. 6).
Currently, we do not know the underlying mechanism for the transient increase in orofacial sensitivity in the contralateral side within the 1st week of CCI-ION (Fig. 1). It is possible that injury-induced bilateral changes, such as activation of spinal microglia and astrocytes and increased cytokines as reported in other inflammatory and neuropathic pain models (52–56), are responsible for the contralateral effects. These factors, however, failed to elicit behavioral hypersensitivity within the first 2 weeks postinjury in the injury side. This could be due, but not limited, to the following reasons. 1) During this hyposensitive stage, degenerating sensory fibers may contribute to sensory input distortions, such as partial or complete numbness (23). 2) Degenerating and/or regenerating sensory fibers may be actively involved in reorganization of sensory circuits in Vc/C2 but less sensitive to modulations by these factors. 3) A central sensitization state that is required for mediating the behavioral hypersensitivity in the later stage is still under development in this period. These can be supported by our findings that neither abnormal Vc/C2 neuron sensitization (Fig. 10, A–D) nor behavioral hypersensitivity (Fig. 1) in the injury side is detectable in this period.
The fast action of gabapentin in reversing increased mEPSC frequency in Vc/C2 neurons (Fig. 10, I and J) and neuropathic allodynia (Fig. 5A) in this model, however, supports that binding of gabapentin to Cavα2δ1 is critical in normalizing Vc/C2 neuron sensitization and behavioral hypersensitivity. Because the effects of gabapentin are use-dependent (21, 57, 58), it is possible that CCI-ION-induced Cavα2δ1 in Vc/C2 is contributing to the functional integrity of hyperexcitable synapses and formation of abnormal excitatory synapses, both of which are critical in maintaining Vc/C2 neuron sensitization and orofacial behavioral hypersensitivity. However, only about 32% of nerve injury-induced neuropathic pain patients have at least moderate pain relief after gabapentin treatment (59). The reason for the variation in efficacy of gabapentin in neuropathic pain relief is not clear. It may be due, but not limited, to the following possibilities. First, gabapentin efficacy in neuropathic pain relief may correlate with the level of Cavα2δ1 dysregulation among pathological conditions, which could vary significantly. This is supported by data from animal studies (15). Second, other cofactors, such as thrombospondins, that interact with Cavα2δ1 in promoting excitatory CNS synaptogenesis (43) may also play a critical role in neuropathic pain processing (42, 60). Blocking the interactions of Cavα2δ1 with other factors, such as thrombospondins, by gabapentin would be critical in normalizing the activity of pathologically modified synapses in the short term and blocking the progression of abnormal synaptogenesis in the long term (43). Detailed investigations are underway to test these possibilities underlying the role of Cavα2δ1 in promoting neuropathic pain states.
In conclusion, we presented data here to support that CCI-ION leads to Cavα2δ1 up-regulation in dorsal Vc/C2 spinal cord that may contribute to the development of orofacial neuropathic pain states through a central mechanism involving abnormal sensitization of Vc/C2 neurons by enhanced excitatory synaptogenesis and presynaptic release of excitatory neurotransmitters. Identifying and blocking new cofactors of this pathway alone or in combination with gabapentin may provide specific therapeutic benefits for orofacial neuropathic pain management.
Acknowledgment
We thank Ben Vo for technical assistance in some experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants DE014545, DE019298, NS064341, and DE021847 (to Z. D. L.).
- Cavα2δ1
- calcium channel α2δ1
- CCI-ION
- chronic constriction injury to the infraorbital nerve
- TG
- trigeminal ganglia
- Vc/C2
- spinal subnucleus caudalis and C1/C2 cervical dorsal spinal cord
- SYN
- synaptophysin
- IB4
- isolectin B4
- mEPSC
- miniature excitatory postsynaptic current
- Vglut
- vesicular glutamate transporter.
REFERENCES
- 1. Hardt J., Jacobsen C., Goldberg J., Nickel R., Buchwald D. (2008) Prevalence of chronic pain in a representative sample in the United States. Pain Med. 9, 803–812 [DOI] [PubMed] [Google Scholar]
- 2. Clark G. T. (2006) Persistent orodental pain, atypical odontalgia, and phantom tooth pain: when are they neuropathic disorders? J. Calif. Dent. Assoc. 34, 599–609 [PubMed] [Google Scholar]
- 3. Vickers E. R., Cousins M. J. (2000) Neuropathic orofacial pain. Part 2—diagnostic procedures, treatment guidelines and case reports. Aust. Endod. J. 26, 53–63 [DOI] [PubMed] [Google Scholar]
- 4. Gee N. S., Brown J. P., Dissanayake V. U., Offord J., Thurlow R., Woodruff G. N. (1996) The novel anticonvulsant drug, gabapentin (Neurontin), binds to the α2δ subunit of a calcium channel. J. Biol. Chem. 271, 5768–5776 [DOI] [PubMed] [Google Scholar]
- 5. Marais E., Klugbauer N., Hofmann F. (2001) Calcium channel α2δ subunits—structure and gabapentin binding. Mol. Pharmacol. 59, 1243–1248 [DOI] [PubMed] [Google Scholar]
- 6. Mori Y., Friedrich T., Kim M. S., Mikami A., Nakai J., Ruth P., Bosse E., Hofmann F., Flockerzi V., Furuichi T., Mikoshiba K., Imoto K., Tanabe T., Numa S. (1991) Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature 350, 398–402 [DOI] [PubMed] [Google Scholar]
- 7. Gurnett C. A., De Waard M., Campbell K. P. (1996) Dual function of the voltage-dependent Ca2+ channel α2δ subunit in current stimulation and subunit interaction. Neuron 16, 431–440 [DOI] [PubMed] [Google Scholar]
- 8. Williams M. E., Feldman D. H., McCue A. F., Brenner R., Velicelebi G., Ellis S. B., Harpold M. M. (1992) Structure and functional expression of α1, α2, and β subunits of a novel human neuronal calcium channel subtype. Neuron 8, 71–84 [DOI] [PubMed] [Google Scholar]
- 9. Brust P. F., Simerson S., McCue A. F., Deal C. R., Schoonmaker S., Williams M. E., Veliçelebi G., Johnson E. C., Harpold M. M., Ellis S. B. (1993) Human neuronal voltage-dependent calcium channels: studies on subunit structure and role in channel assembly. Neuropharmacology 32, 1089–1102 [DOI] [PubMed] [Google Scholar]
- 10. Kang M. G., Felix R., Campbell K. P. (2002) Long-term regulation of voltage-gated Ca2+ channels by gabapentin. FEBS Lett. 528, 177–182 [DOI] [PubMed] [Google Scholar]
- 11. Qin N., Yagel S., Momplaisir M. L., Codd E. E., D'Andrea M. R. (2002) Molecular cloning and characterization of the human voltage-gated calcium channel α2δ-4 subunit. Mol. Pharmacol. 62, 485–496 [DOI] [PubMed] [Google Scholar]
- 12. Klugbauer N., Lacinová L., Marais E., Hobom M., Hofmann F. (1999) Molecular diversity of the calcium channel α2δ subunit. J. Neurosci. 19, 684–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ellis S. B., Williams M. E., Ways N. R., Brenner R., Sharp A. H., Leung A. T., Campbell K. P., McKenna E., Koch W. J., Hui A., Schwartz A., Harpold M. M. (1988) Sequence and expression of mRNAs encoding the α1 and α2 subunits of a DHP-sensitive calcium channel. Science 241, 1661–1664 [DOI] [PubMed] [Google Scholar]
- 14. Bauer C. S., Nieto-Rostro M., Rahman W., Tran-Van-Minh A., Ferron L., Douglas L., Kadurin I., Sri Ranjan Y., Fernandez-Alacid L., Millar N. S., Dickenson A. H., Lujan R., Dolphin A. C. (2009) The increased trafficking of the calcium channel subunit α2δ-1 to presynaptic terminals in neuropathic pain is inhibited by the α2δ ligand pregabalin. J. Neurosci. 29, 4076–4088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luo Z. D., Calcutt N. A., Higuera E. S., Valder C. R., Song Y. H., Svensson C. I., Myers R. R. (2002) Injury type-specific calcium channel α2δ-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J. Pharmacol. Exp. Ther. 303, 1199–1205 [DOI] [PubMed] [Google Scholar]
- 16. Luo Z. D., Chaplan S. R., Higuera E. S., Sorkin L. S., Stauderman K. A., Williams M. E., Yaksh T. L. (2001) Upregulation of dorsal root ganglion α2δ calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J. Neurosci. 21, 1868–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiao W., Boroujerdi A., Bennett G. J., Luo Z. D. (2007) Chemotherapy-evoked painful peripheral neuropathy: analgesic effects of gabapentin and effects on expression of the α2δ type-1 calcium channel subunit. Neuroscience 144, 714–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Field M. J., Oles R. J., Lewis A. S., McCleary S., Hughes J., Singh L. (1997) Gabapentin (Neurontin) and S-(+)-3-isobutylgaba represent a novel class of selective antihyperalgesic agents. Br. J. Pharmacol. 121, 1513–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hunter J. C., Gogas K. R., Hedley L. R., Jacobson L. O., Kassotakis L., Thompson J., Fontana D. J. (1997) The effect of novel anti-epileptic drugs in rat experimental models of acute and chronic pain. Eur. J. Pharmacol. 324, 153–160 [DOI] [PubMed] [Google Scholar]
- 20. Singh L., Field M. J., Ferris P., Hunter J. C., Oles R. J., Williams R. G., Woodruff G. N. (1996) The antiepileptic agent gabapentin (Neurontin) possesses anxiolytic-like and antinociceptive actions that are reversed by D-serine. Psychopharmacology 127, 1–9 [DOI] [PubMed] [Google Scholar]
- 21. Stanfa L. C., Singh L., Williams R. G., Dickenson A. H. (1997) Gabapentin, ineffective in normal rats, markedly reduces C-fibre evoked responses after inflammation. Neuroreport 8, 587–590 [DOI] [PubMed] [Google Scholar]
- 22. Kernisant M., Gear R. W., Jasmin L., Vit J. P., Ohara P. T. (2008) Chronic constriction injury of the infraorbital nerve in the rat using modified syringe needle. J. Neurosci. Methods 172, 43–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vos B. P., Strassman A. M., Maciewicz R. J. (1994) Behavioral evidence of trigeminal neuropathic pain following chronic constriction injury to the rat's infraorbital nerve. J. Neurosci. 14, 2708–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dixon W. J. (1980) Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 20, 441–462 [DOI] [PubMed] [Google Scholar]
- 25. Chaplan S. R., Bach F. W., Pogrel J. W., Chung J. M., Yaksh T. L. (1994) Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 [DOI] [PubMed] [Google Scholar]
- 26. Iwata K., Imamura Y., Honda K., Shinoda M. (2011) Physiological mechanisms of neuropathic pain: the orofacial region. Int. Rev. Neurobiol. 97, 227–250 [DOI] [PubMed] [Google Scholar]
- 27. Basso D. M., Beattie M. S., Bresnahan J. C. (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J. Neurotrauma 12, 1–21 [DOI] [PubMed] [Google Scholar]
- 28. Boroujerdi A., Zeng J., Sharp K., Kim D., Steward O., Luo Z. D. (2011) Calcium channel α2δ1 protein upregulation in dorsal spinal cord mediates spinal cord injury-induced neuropathic pain states. Pain 152, 649–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nguyen D., Deng P., Matthews E. A., Kim D. S., Feng G., Dickenson A. H., Xu Z. C., Luo Z. D. (2009) Enhanced pre-synaptic glutamate release in deep-dorsal horn contributes to calcium channel α2δ1 protein-mediated spinal sensitization and behavioral hypersensitivity. Mol. Pain 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shimoyama M., Shimoyama N., Hori Y. (2000) Gabapentin affects glutamatergic excitatory neurotransmission in the rat dorsal horn. Pain 85, 405–414 [DOI] [PubMed] [Google Scholar]
- 31. Peyronnard J. M., Charron L., Messier J. P., Lavoie J., Leger C., Faraco-Cantin F. (1989) Changes in lectin binding of lumbar dorsal root ganglia neurons and peripheral axons after sciatic and spinal nerve injury in the rat. Cell Tissue Res. 257, 379–388 [DOI] [PubMed] [Google Scholar]
- 32. Neumann S., Braz J. M., Skinner K., Llewellyn-Smith I. J., Basbaum A. I. (2008) Innocuous, not noxious, input activates PKCγ interneurons of the spinal dorsal horn via myelinated afferent fibers. J. Neurosci. 28, 7936–7944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Silverman J. D., Kruger L. (1990) Selective neuronal glycoconjugate expression in sensory and autonomic ganglia: relation of lectin reactivity to peptide and enzyme markers. J. Neurocytol. 19, 789–801 [DOI] [PubMed] [Google Scholar]
- 34. Snider W. D., McMahon S. B. (1998) Tackling pain at the source: new ideas about nociceptors. Neuron 20, 629–632 [DOI] [PubMed] [Google Scholar]
- 35. Hunt S. P., Mantyh P. W. (2001) The molecular dynamics of pain control. Nat. Rev. Neurosci. 2, 83–91 [DOI] [PubMed] [Google Scholar]
- 36. Li C. Y., Song Y. H., Higuera E. S., Luo Z. D. (2004) Spinal dorsal horn calcium channel α2δ-1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J. Neurosci. 24, 8494–8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Laird M. A., Gidal B. E. (2000) Use of gabapentin in the treatment of neuropathic pain. Ann. Pharmacother. 34, 802–807 [DOI] [PubMed] [Google Scholar]
- 38. Backonja M., Glanzman R. L. (2003) Gabapentin dosing for neuropathic pain: evidence from randomized, placebo-controlled clinical trials. Clin. Ther. 25, 81–104 [DOI] [PubMed] [Google Scholar]
- 39. Dworkin R. H., O'Connor A. B., Backonja M., Farrar J. T., Finnerup N. B., Jensen T. S., Kalso E. A., Loeser J. D., Miaskowski C., Nurmikko T. J., Portenoy R. K., Rice A. S., Stacey B. R., Treede R. D., Turk D. C., Wallace M. S. (2007) Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain 132, 237–251 [DOI] [PubMed] [Google Scholar]
- 40. Field M. J., McCleary S., Hughes J., Singh L. (1999) Gabapentin and pregabalin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by streptozocin in the rat. Pain 80, 391–398 [DOI] [PubMed] [Google Scholar]
- 41. Hwang J. H., Yaksh T. L. (1997) Effect of subarachnoid gabapentin on tactile-evoked allodynia in a surgically induced neuropathic pain model in the rat. Reg. Anesth. 22, 249–256 [DOI] [PubMed] [Google Scholar]
- 42. Li K.-W., Kim D.-S., Zaucke F., Luo Z. D. (2013) Trigeminal nerve injury induced thrombospondin-4 upregulation contributes to orofacial neuropathic pain states in a rat model. Eur. J. Pain 10.1002/j.1532-2149.2013.00396.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eroglu C., Allen N. J., Susman M. W., O'Rourke N. A., Park C. Y., Ozkan E., Chakraborty C., Mulinyawe S. B., Annis D. S., Huberman A. D., Green E. M., Lawler J., Dolmetsch R., Garcia K. C., Smith S. J., Luo Z. D., Rosenthal A., Mosher D. F., Barres B. A. (2009) Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moechars D., Weston M. C., Leo S., Callaerts-Vegh Z., Goris I., Daneels G., Buist A., Cik M., van der Spek P., Kass S., Meert T., D'Hooge R., Rosenmund C., Hampson R. M. (2006) Vesicular glutamate transporter VGLUT2 expression levels control quantal size and neuropathic pain. J. Neurosci. 26, 12055–12066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rogoz K., Lagerström M. C., Dufour S., Kullander K. (2012) VGLUT2-dependent glutamatergic transmission in primary afferents is required for intact nociception in both acute and persistent pain modalities. Pain 153, 1525–1536 [DOI] [PubMed] [Google Scholar]
- 46. Leo S., Moechars D., Callaerts-Vegh Z., D'Hooge R., Meert T. (2009) Impairment of VGLUT2 but not VGLUT1 signaling reduces neuropathy-induced hypersensitivity. Eur. J. Pain 13, 1008–1017 [DOI] [PubMed] [Google Scholar]
- 47. Seal R. P., Wang X., Guan Y., Raja S. N., Woodbury C. J., Basbaum A. I., Edwards R. H. (2009) Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature 462, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brittain J. M., Duarte D. B., Wilson S. M., Zhu W., Ballard C., Johnson P. L., Liu N., Xiong W., Ripsch M. S., Wang Y., Fehrenbacher J. C., Fitz S. D., Khanna M., Park C. K., Schmutzler B. S., Cheon B. M., Due M. R., Brustovetsky T., Ashpole N. M., Hudmon A., Meroueh S. O., Hingtgen C. M., Brustovetsky N., Ji R. R., Hurley J. H., Jin X., Shekhar A., Xu X. M., Oxford G. S., Vasko M. R., White F. A., Khanna R. (2011) Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat. Med. 17, 822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Valder C. R., Liu J. J., Song Y. H., Luo Z. D. (2003) Coupling gene chip analyses and rat genetic variances in identifying potential target genes that may contribute to neuropathic allodynia development. J. Neurochem. 87, 560–573 [DOI] [PubMed] [Google Scholar]
- 50. Kim S. H., Chung J. M. (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50, 355–363 [DOI] [PubMed] [Google Scholar]
- 51. Li C. Y., Zhang X. L., Matthews E. A., Li K. W., Kurwa A., Boroujerdi A., Gross J., Gold M. S., Dickenson A. H., Feng G., Luo Z. D. (2006) Calcium channel α2δ1 subunit mediates spinal hyperexcitability in pain modulation. Pain 125, 20–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sweitzer S. M., Colburn R. W., Rutkowski M., DeLeo J. A. (1999) Acute peripheral inflammation induces moderate glial activation and spinal IL-1β expression that correlates with pain behavior in the rat. Brain Res. 829, 209–221 [DOI] [PubMed] [Google Scholar]
- 53. Clark A. K., Gentry C., Bradbury E. J., McMahon S. B., Malcangio M. (2007) Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur. J. Pain. 11, 223–230 [DOI] [PubMed] [Google Scholar]
- 54. Cao H., Zhang Y. Q. (2008) Spinal glial activation contributes to pathological pain states. Neurosci. Biobehav. Rev. 32, 972–983 [DOI] [PubMed] [Google Scholar]
- 55. Sun S., Cao H., Han M., Li T. T., Zhao Z. Q., Zhang Y. Q. (2008) Evidence for suppression of electroacupuncture on spinal glial activation and behavioral hypersensitivity in a rat model of monoarthritis. Brain Res. Bull. 75, 83–93 [DOI] [PubMed] [Google Scholar]
- 56. Hashizume H., DeLeo J. A., Colburn R. W., Weinstein J. N. (2000) Spinal glial activation and cytokine expression after lumbar root injury in the rat. Spine 25, 1206–1217 [DOI] [PubMed] [Google Scholar]
- 57. Rogawski M. A., Löscher W. (2004) The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat. Med. 10, 685–692 [DOI] [PubMed] [Google Scholar]
- 58. Wamil A. W., McLean M. J. (1994) Limitation by gabapentin of high frequency action potential firing by mouse central neurons in cell culture. Epilepsy Res. 17, 1–11 [DOI] [PubMed] [Google Scholar]
- 59. Gordh T. E., Stubhaug A., Jensen T. S., Arnèr S., Biber B., Boivie J., Mannheimer C., Kalliomäki J., Kalso E. (2008) Gabapentin in traumatic nerve injury pain: a randomized, double-blind, placebo-controlled, cross-over, multi-center study. Pain 138, 255–266 [DOI] [PubMed] [Google Scholar]
- 60. Kim D. S., Li K. W., Boroujerdi A., Peter Yu Y., Zhou C. Y., Deng P., Park J., Zhang X., Lee J., Corpe M., Sharp K., Steward O., Eroglu C., Barres B., Zaucke F., Xu Z. C., Luo Z. D. (2012) Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. J. Neurosci. 32, 8977–8987 [DOI] [PMC free article] [PubMed] [Google Scholar]