

Background: Bcl-3 is an essential negative regulator of cytokine expression.
Results: A mutation in NF-κB p50 that prevents interaction with Bcl-3 abrogates Bcl-3 inhibition of gene expression.
Conclusion: The inhibition of cytokine expression by Bcl-3 requires interaction with NF-κB p50.
Significance: NF-κB p50 is the key target of Bcl-3 anti-inflammatory activity.
Keywords: Cytokine Induction, Inflammation, Mutagenesis, NF-κB transcription factor, Ubiquitination
Abstract
B cell leukemia 3 (Bcl-3) is an essential negative regulator of NF-κB during Toll-like receptor and TNF receptor signaling. Bcl-3 also interacts with a number of transcriptional regulators, including homodimers of the NF-κB p50 subunit. Deletion of Bcl-3 results in increased NF-κB p50 ubiquitination and proteasomal degradation and increased inflammatory gene expression. We employed immobilized peptide array technology to define a region of p50 required for the formation of a Bcl-3·p50 homodimer immunosuppressor complex. Our data demonstrate that amino acids 359–361 and 363 of p50 are critical for interaction with Bcl-3 and essential for Bcl-3-mediated inhibition of inflammatory gene expression. Bcl-3 is unable to interact with p50 when these amino acids are mutated, rendering it incapable of inhibiting the transcriptional activity of NF-κB. Bcl-3 interaction-defective p50 is hyperubiquitinated and has a significantly reduced half-life relative to wild-type p50. Nfkb1−/− cells reconstituted with mutated p50 precursor p105 are hyperresponsive to TNFα stimulation relative to wild-type p105, as measured by inflammatory gene expression. Mutant p105 recapitulates a Bcl3−/− phenotype. This study demonstrates that interaction with p50 is necessary and sufficient for the anti-inflammatory properties of Bcl-3 and further highlights the importance of p50 homodimer stability in the control of NF-κB target gene expression.
Introduction
The members of the NF-κB family of transcription factors are essential regulators of the immune response and control the expression of hundreds of immunomodulatory genes. This transcription factor family is encoded by five genes, resulting in five NF subunits (p65 (RelA), c-Rel, RelB, p50, and p52) that, through the formation of hetero- and homodimers, can potentially generate 15 distinct complexes (1). All subunits of NF-κB share significant sequence homology and are characterized by the Rel homology domain (RHD)2, which is essential for dimerization and DNA binding. Genetic studies in mice have demonstrated that individual NF-κB subunits carry out specific biological roles (2), which may be due to distinct subunit specific cofactors (3) as well as dimer-specific affinities for variable DNA binding sequences (4).
The NF-κB p50 and p52 subunits share a number of properties that distinguish them from other NF-κB subunits. Firstly, they are both generated from the limited proteolytic processing of a precursor: p105 in the case of p50 and p100 in the case of p52. Secondly, p50 and p52 do not contain a transactivation domain and, when complexed as homodimers, they do not possess intrinsic transactivational activity (1). In this regard, p50 homodimers are generally considered as repressors of NF-κB transcription. Interestingly, p50 homodimers have also recently been identified as negative regulators of interferon regulatory factor (IRF)-mediated transcription by binding to interferon-responsive elements in interferon-inducible gene promoters, which act as an NF-κB half-site (5). A significant number of p50 homodimers are present in the nuclei of unstimulated cells, which indicates that p50 plays a critical role in regulating the basal as well as inducible transcription of target genes (5).
In resting cells, NF-κB is sequestered in the cytoplasm through association with members of the IκB protein family, which include IκBα, IκBβ, IκBϵ, as well as p105 and p100. Activating stimuli trigger IκB kinase (IKK)-mediated phosphorylation of cytoplasmic IκB, leading to its ubiquitination and subsequent proteasomal degradation, thereby allowing NF-κB to translocate to the nucleus. There are also a number of atypical IκB proteins, which include Bcl-3, IκBζ, and IκBNS, which reside predominantly in the nucleus and are not degraded following activation of the IKK complex (6). Bcl-3 is perhaps the best studied of the atypical IκB proteins. It was originally identified at the breakpoint junction t(14;19) in B cell chronic lymphocytic leukemia and is classified as an IκB family member by virtue of its seven ankyrin repeat domains that mediate selective interaction with homodimers of NF-κB p50 and p52 (7). Bcl-3 has also been reported to interact with the AP-1 transcription factors c-Jun and c-fos (8); STAT1 (9); peroxisome proliferator-activated receptorγ (PPARγ) (10); CREB-binding protein/p300 (8); HDAC-1, -3, and -5 (11); steroid receptor coactivator 1 (8); TORC3 (12); and the retinoic X receptor (13). Bcl-3 has also been reported to interact with the Src-related kinases Fyn (14) and Lck (15) as well as insulin receptor substrate 3 (IRS3) (16) and Bag-1 (17). Previous data suggest that Bcl-3-mediated stabilization of p50 is required for limiting NF-κB transcriptional activity. In Bcl3−/− macrophages, p50 homodimers undergo increased ubiquitination and proteasomal degradation. Bcl3−/− cells and mice are hyperresponsive to Toll-like receptor and TNF receptor stimulation, as measured by the increased transcription of genes, including TNFα and IL-6 (18). The nature of the interaction between Bcl-3 and p50 has not been explored experimentally, and it is not clear whether Bcl-3 interaction with p50 is essential for the inhibition of cytokine expression or whether the other binding partners of Bcl-3 are also important. This information is critical for our understanding of the regulation of NF-κB activity and cytokine expression by Bcl-3 and p50 homodimers.
Here we employed immobilized peptide array technology to identify the regions of p50 essential for interaction with Bcl-3. We identified amino acids 359–361 and 363 of p50 as critical residues required for the interaction of p50 homodimers with Bcl-3. This allowed us to generate a p50 mutant that is unable to interact with Bcl-3 but that retains normal dimerization and DNA-binding properties. p50 homodimers that are unable to interact with Bcl-3 undergo increased ubiquitination that is not inhibited by overexpression of Bcl-3. Importantly, the Bcl-3 interaction-defective p50 mutant is also defective in its ability to repress NF-κB target gene expression. Overexpression of Bcl-3 is unable to repress NF-κB-mediated gene expression in cells expressing the mutant form of p50. Our study describes the experimental characterization of the interaction between p50 and Bcl-3 and demonstrates, for the first time, that a direct interaction between Bcl-3 and p50 is required for the stability of p50 homodimers and is necessary for the anti-inflammatory function of Bcl-3.
EXPERIMENTAL PROCEDURES
Cell Culture, Plasmids, and Transfection
HEK293T and RAW 264.7 cells were cultured in DMEM containing 10% fetal bovine serum, 2 mm glutamine, and 100 units/ml penicillin/streptomycin. Transfections were performed using Turbofect (Fermentas) or XtremeGENE HP (Roche) according to the instructions of the manufacturer. Nfkb1−/− MEFs were cultured in DMEM containing 10% bovine calf serum, 2 mm glutamine, and 100 units/ml penicillin/streptomycin. Transfection of MEF cells was performed using Attractene (Qiagen) according to the instructions of the manufacturer. For stable transfection, cells were clonally selected by growing in medium containing 0.2 mg/ml zeocin (InvivoGen). Mammalian expression vectors for Bcl-3, p50, and p105 were generated following PCR amplification and ligation of murine cDNAs into the pcDNA3.1-Myc and pRK5-FLAG vectors. The Bcl-3 binding-defective mutants were generated using the QuikChange kit according to the instructions of the manufacturer (Stratagene). Bcl-3 was ligated into pGEX6p1 to produce a GST-Bcl-3 expression vector. The hydrophobicity analysis of p50 was performed using the Bioannotator module of the Vector NTI software suite (Invitrogen) (19).
Bacterial Protein Expression and Purification
For purification of GST and GST-Bcl-3, Escherichia coli BL21 CodonPlus (Stratagene) was transformed with pGEX-6p1 or pGEX-6p1-Bcl-3. Cells were grown to an A600 of 1.0–2.0 at 37 °C and induced with 1.0 mm (GST) or 0.1 mm (GST-Bcl-3) isopropyl 1-thio-β-d-galactopyranoside for 16 h at 20 °C. The bacteria were pelleted; resuspended in a buffer containing 50 mm Tris (pH 8.0), 150 mm NaCl, and 1 mm dithiothreitol; disrupted by sonication; and centrifuged to remove debris. Recombinant proteins were affinity-purified with GSH-agarose (Sigma) and eluted with 10 mm glutathione (Promega) in 50 mm Tris (pH8.5) and 150 mm NaCl.
GST Pulldown Assay
HEK293T cells were transiently transfected with FLAG-p50WT or p50RKR. Whole cell lysates were extracted from cells resuspended in GST lysis and binding buffer (20 mm Tris-Cl (pH 8.0), 200 mm NaCl, 1 mm EDTA (pH 8.0), and 0.5% Nonidet P-40) supplemented with aprotinin, pepstatin, and PMSF. Equal amounts of lysates were precleared in 1 ml of binding buffer with 50 μl of GSH-agarose for 2 h rotating at 4 °C. GST or GST-Bcl-3 were incubated with precleared lysates and affinity-purified with 50 μl of GSH-agarose for 2 h rotating at 4 °C. Following the wash steps, pulldown complexes were eluted from the beads with 2× SDS gel loading buffer (100 mm Tris-Cl (pH 6.8), 4% (w/v) SDS, 0.2% (w/v) bromphenol blue, 20%(w/v) glycerol, and 200 mm β- mercaptoethanol) and analyzed by Western blot analysis.
SPOT Synthesis of Peptides and Overlay Analysis
Peptide libraries of p50 were generated by automatic SPOT synthesis as described previously (20) and synthesized on continuous cellulose membrane supports on Whatman 50 cellulose using Fmoc (N-(9-flurenyl)methoxycarbonyl) chemistry with AutoSpot-Robot ASS 222 (Intavis Bioanalytical Instruments). The interaction of GST and GST-Bcl3 was investigated by overlaying the cellulose membrane with 10 μg/ml of each recombinant protein overnight at 4 °C. Bound protein was detected by immunoblotting with anti-GST and a secondary anti-rabbit antibody coupled with horseradish peroxidase. Specific alanine-scanning substitution arrays were generated using the same synthesis procedure. Bound protein was detected by immunoblotting with anti-GST and an IR-800-conjugated anti-rabbit secondary antibody (Pierce).
Western Blot Analysis and Immunoprecipitation
For Western blot analysis, whole cell lysates were extracted from cells suspended in radioimmune precipitation assay buffer containing 50 mm Tris-HCl (pH 7.4), 1% Nonidet P-40, 0.25% deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF, 1 mm NaF, 1 mm Na3VO4, 2 μg/ml aprotinin, 1 μg/ml pepstatin, and 1 μg/ml leupeptin. Nuclear extracts were obtained using a nuclear extract kit according to the instruction of the manufacturer (Active Motif). Lysates were resolved on SDS-PAGE gels, transferred to nitrocellulose membranes, and immunoblotted with specific antibodies. Phospho- IκBα and IκBα were purchased from Cell Signaling Technology. Anti-FLAG M2, anti-β-actin, and anti-tubulin were purchased from Sigma, and anti-Bcl-3 was purchased from Active Motif. Anti-c-MYC (9E10), anti-HDAC1, and anti-p50 (C-19) were obtained from Santa Cruz Biotechnology. For immunoprecipitation, equal amounts of whole cell extracts were precleared for 30 min at 4 °C with protein G-agarose beads (Millipore) and immunoprecipitated with primary antibody overnight at 4 °C. Pellets were washed three times in radioimmune precipitation assay buffer and resuspended in 2× sample buffer. Equal volumes of resuspended immunoprecipitates were analyzed by Western blot analysis. For ubiquitination assays, cells were incubated with 10 mm N-ethylmaleimide for 30 s and washed in PBS/10 mm N-ethylmaleimide. Cells were lysed in 1% SDS, boiled for 5 min, and sonicated. Cleared lysates were diluted (1:10) in radioimmune precipitation assay buffer supplemented with 20 mm N-ethylmaleimide. Immunoprecipitation was performed as above and analyzed by Western blot analysis using an anti-HA antibody (Roche) to detect ubiquitinated p50 species. For endogenous ubiquitination assays, p50 was immunoprecipitated with anti-NF-κB p50 (Enzo), and Western blots were incubated in 0.5% glutaraldehyde/PBS (pH 7.0) for 20 min prior to Western blotting with anti-ubiquitin (Tebu Bio, catalog no. VU101) to detect ubiquitinated p50 species.
Luciferase Assay
RAW 264.7 and Nfkb1−/− MEFs were transiently transfected with the pLucp19 plasmid for 24 h and cultured with or without 100 ng/ml LPS (Sigma) or 20 ng/ml TNFα (eBiosciences) for an additional 8 h before luciferase activity was measured. Cotransfection of the Renilla luciferase expression vector pRL-TK (Promega) was used as an internal control for all reporter assays. Cells were lysed in 1× passive lysis buffer (Promega), and, for all samples, firefly luciferase activity (25 mm glycylglycine, 15 mm potassium K2PO4 (pH 8), 4 mm EDTA, 15 mm magnesium sulfate, 75 μm luciferin, 1 mm dithiothreitol, 0.1 mm coenzyme A, and 2 mm ATP) was divided by Renilla luciferase activity (1.1 m sodium chloride, 2.2 mm EDTA, 220 mm K2PO4 (pH 5.1), 0.44 mg/ml bovine serum albumin, sodium azide, and 1.43 μm coelenterazine) to normalize for the transfection efficiency, as described previously (21).
EMSA
IR-800 dye-labeled (DY782) NF-κB consensus double-stranded oligonucleotides (5′-AGTTGAGGGGACTTTCCCAGGC-3′ and 3′-TCA ACT CCC CTG AAA GGG TCC G-5′) were purchased from MWG Operon. Binding reactions were prepared using 5 μg of nuclear extract with 10 ng of oligonucleotides in a 30-μl reaction volume containing 10 mm HEPES KOH (pH 7.9), 50 mm KCl, 2.5 mm MgCl2, 1 mm DTT, 10% Ficoll, 1.2 μg of bovine serum albumin, and 3.6 μg of poly[d(I-C)] at room temperature for 15 min. Binding reactions were resolved on a 5% non-denaturing polyacrylamide gel at 300 V and 4 °C in 1× TBE (0.089 m Tris borate, 0.089 m boric acid, and 0.002 m EDTA). Gels were visualized using LI-COR odyssey.
Gene Expression Analysis
For real-time PCR, total RNA was isolated using RNeasy kits (Qiagen), primed with random hexamer oligonucleotides, and reverse-transcribed using a cDyNAmo cDNA synthesis kit (Thermo Scientific). PCR was performed with SensiMix SYBR master mix and Tnf, Il6, Cxcl2, and Ccl2 QuantiTect primers (Qiagen). Data were normalized to 18 S. Gene expression changes were calculated using the 2−ΔΔCT method.
RESULTS
Identification of Regions of p50 Required for Interaction with Bcl-3
The crystal structures of the Bcl-3 and p50 homodimers have been resolved independently, and computational modeling of a Bcl-3·p50 homodimer complex indicates that Bcl-3 makes a number of contacts with amino acids in both subunits of a p50 homodimer (22). To experimentally identify the regions of p50 mediating interaction with Bcl-3, we immobilized p50 using peptide array-based techniques and probed the arrays using purified recombinant GST-Bcl-3 protein. Specifically, a library of overlapping peptides 18 amino acids in length, each shifted by three amino acids and encompassing the entire sequence of p50, was SPOT-synthesized on nitrocellulose membranes to generate p50 arrays. Peptide arrays were overlaid with purified recombinant GST or GST-Bcl-3 protein and, following extensive washing, probed with anti-GST antibody as described previously(23, 24, 25). The suitability of GST-Bcl-3 for use as a p50 peptide library probe was established using a GST pulldown assay in which purified GST-Bcl-3, but not GST, bound to p50 (Fig. 1A). The p50 peptide arrays identified a distinct set of p50 peptides that interacted strongly with GST-Bcl-3 but not with GST (Fig. 1B). These peptides corresponded to residues 337–378 of p50 (murine), which contain the extreme C-terminal region of the RHD of p50.
FIGURE 1.

Analysis of p50 and Bcl-3 interaction using peptide arrays. A, p50 binds specifically to GST-Bcl3 in a GST pulldown assay. Purified bacterial recombinant GST or GST-Bcl-3 was incubated with a whole HEK293 cell lysate overexpressing FLAG-p50 and was affinity-purified with GSH-agarose. Pulldown complexes were immunoblotted with anti-FLAG and anti-GST antibodies. Full-length GST-Bcl-3 is indicated. WB, Western blot. B, peptide arrays of immobilized, overlapping 18-mer peptides, each shifted to the right by three amino acids, encompassing the entire p50 sequence were generated. The arrays were probed with GST or GST-Bcl-3 and detected by immunoblotting with anti-GST antibody. GST-Bcl-3 binding to p50 peptides is shown and is representative of duplicate arrays. Sequences of peptides are shown, with Bcl-3 interacting peptides indicated in gray. C, the 18 amino acids of p50-derived peptide 119 were sequentially substituted with alanine, and a peptide array was probed with GST-Bcl-3. Bcl-3 binding was quantified by densitometry and represented as a percentage of binding of the control parent peptide. Substitution of amino acids Arg-359, Lys-360, Arg-361 and Lys-363 with alanine (shaded in gray) significantly decreased Bcl-3 binding. D, hydrophobicity analysis of p50, with amino acids 359–363 indicated in a region of low hydrophobicity.
To identify the specific amino acids within this region of p50 that are crucial for interaction with Bcl-3, we next employed alanine-scanning arrays containing the amino acid sequence 355–372 of p50 (Fig. 1C). In these arrays, successive amino acids were individually substituted with alanine, differing from the parent peptide by one amino acid, and then incubated with GST-Bcl-3 prior to staining with anti-GST antibody. In each case, the binding of GST-Bcl-3 to the substituted peptide was calculated with respect to the parent peptide contained on the same array. These data demonstrate that alanine substitutions at Arg-359, Lys-360, Arg-361, and Lys-363 significantly decreased GST-Bcl-3 binding when compared with the parent p50 peptide (Fig. 1C). The identified residues lie in the extreme C-terminal region of the RHD of p50 and are not represented in the structures of p50 currently available. However, a hydrophobicity analysis of the primary structure of p50 indicated that these residues are in a region of low hydrophobicity and, therefore, are likely to be available for interaction with Bcl-3 (Fig. 1D).
Arg-359, Lys-360, Arg-361, and Lys-363 of p50 Are Essential for Interaction with Bcl-3
To evaluate the contribution of residues Arg-359, Lys-360, and Arg-361 of p50 to the interaction with Bcl-3, we next performed a GST pulldown assay employing purified GST-Bcl-3 and lysates generated from HEK293T cells expressing wild-type p50 (p50WT) or p50 in which Arg-359, Lys-360, and Arg-361 had been mutated to alanine (p50RKR). As expected, GST-Bcl-3 pulldown readily purified p50WT. However, p50RKR protein was not detectable following GST-Bcl-3 pulldown from p50RKR containing lysates (Fig. 2A). These data confirm our initial findings using peptide microarrays and further validate this method for mapping sites of protein-protein interaction. We further assessed the interaction of the p50RKR mutant with Bcl-3 by cotransfection in HEK293T cells. Similar to our findings using GST pulldown assays, we were unable to detect Bcl-3 in immunoprecipitates of p50RKR, although Bcl-3 was readily detectable in immunoprecipitates of p50WT (Fig. 2B). Similarly, the mutation of Lys-363 abolished the interaction of p50 with Bcl-3 when coexpressed in HEK293T cells (Fig. 2C).
FIGURE 2.
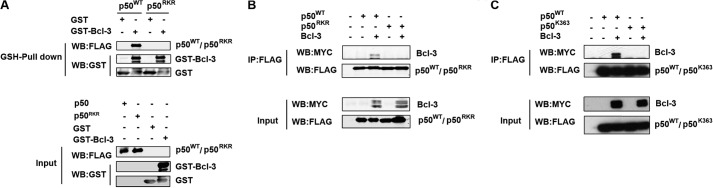
Arg-359, Lys-360, Arg-361, and Lys-363 are essential for interaction with Bcl-3. A, purified bacterial recombinant GST or GST-Bcl3 was incubated with a whole cell lysate overexpressing FLAG-p50 or p50 in which Arg-359, Lys-360, and Arg-361 were mutated to alanine (p50RKR) and was affinity-purified with GSH-agarose. Pulldown complexes were analyzed by Western blot (WB) analysis with anti-FLAG and anti-GST antibodies. Full-length GST-Bcl-3 is indicated. B, HEK293T cells were transfected with p50 or p50RKR with or without Myc-Bcl-3. Whole cell lysates were immunoprecipitated (IP) with anti-FLAG antibody and analyzed by Western blot analysis with the indicated antibodies. C, HEK293T cells were transfected with p50 or p50K363A with Myc-Bcl-3. Whole cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by Western blot analysis with the indicated antibodies. Bcl-3 is a hyperphosphorylated protein and migrates as multiple bands between 48–60 kDa.
To rule out an indirect effect of Arg-359, Lys-360 and Arg-361 mutation on the ability of the p50RKR mutant to interact with Bcl-3, we performed additional characterization of the p50RKR mutant protein. Homodimerization of p50RKR was assessed by cotransfection of HEK293T cells with FLAG-tagged and Myc-tagged p50RKR or FLAG-tagged and Myc-tagged p50WT. Lysates were immunoprecipitated using anti-FLAG antibody and immunoblotted with anti-Myc antibody. This analysis demonstrated no significant differences in the formation of homodimers by p50RKR when compared with p50WT protein (Fig. 3A). We next assessed the heterodimerization of p50RKR using a similar approach. Myc-tagged p50WT or p50RKR was coexpressed with FLAG-tagged NF-κB p65 and lysates immunoprecipitated using anti-FLAG antibody. Immunoblot analysis of FLAG-p65 immunoprecipitates demonstrated no differences in the levels of p50RKR coimmunoprecipitating with p65 relative to p50WT, indicating that the p50RKR mutation has no effect on heterodimerization with p65 (Fig. 3B). IκBα has a high affinity for p65/p50 heterodimers. Therefore, we also assessed the levels of endogenous IκBα in p65 immunoprecipitates from cells expressing p50WT or p50RKR. No significant differences in the amount of IκBα coimmunoprecipitating with p65 was found between cells expressing p50WT or p50RKR (Fig. 3B). Similarly, we observed no difference in the levels of p50RKR interacting with p105 relative to p50WT in cells cotransfected with p105 and p50WT or p50RKR, as assessed by immunoprecipitation (Fig. 3C). Together, these data demonstrate that the p50RKR mutation does not alter p50 dimerization or interaction with IκBα or p105 proteins.
FIGURE 3.
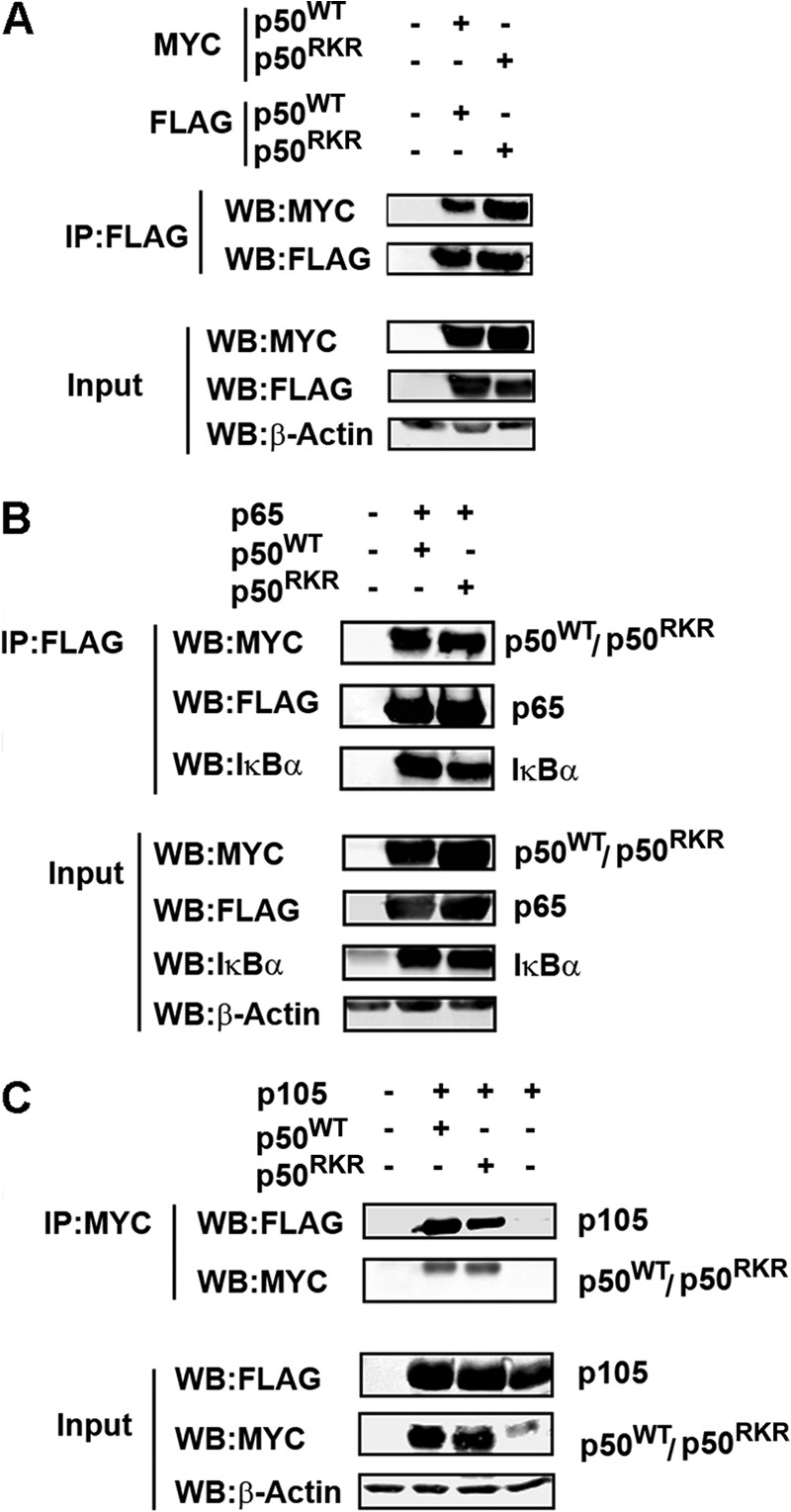
Mutation of Arg-359, Lys-360, and Arg-361 does not affect the dimerization properties of p50. p50RKR can form homodimers (A) and heterodimers with p65 (B) and p105 (C). A and B, HEK293T cells were transfected as indicated. FLAG-p50 or FLAG-p50RKR (A) and FLAG-p65 (B) were immunoprecipitated (IP) from whole cell lysates with anti-FLAG antibody and analyzed by Western blot (WB) analysis with anti c-Myc for Myc -p50 and p50RKR and anti IκBα. C, Myc-p50 or p50RKR was immunoprecipitated from whole cell lysates with anti-c-Myc antibody and analyzed by Western blot analysis with anti-FLAG antibody for p105.
Amino acids 359–361 of p50 have been indicated previously to function in the nuclear translocation of p50 (26). Because Bcl-3 is predominantly nuclear in localization, it was important to rule out the possibility that lack of nuclear p50RKR prevented the interaction with Bcl-3 in cotransfected cells. To address this, we generated nuclear and cytoplasmic fractions of cells transfected with p50RKR or p50WT and analyzed protein levels by immunoblot analysis. As demonstrated in Fig. 4A, subcellular fractionation revealed that p50RKR translocated to the nucleus to similar levels as p50WT, indicating that the mutation of amino acids 359–361 to alanine does not disrupt the nuclear localization of p50. These data were supported by an analysis of the DNA-binding activity of p50RKR using EMSA, which incorporated a double-stranded oligonucleotide probe containing the NF-κB consensus DNA-binding sequence and nuclear extracts from 293T cells transfected with expression vectors for p50WT or p50RKR. No significant differences were seen in the DNA-binding activity of p50RKR compared with p50WT (Fig. 4B). Taken together, these data reveal that the p50RKR mutant is defective in binding to Bcl-3 but retains the p50WT properties of nuclear translocation, dimerization, and DNA binding.
FIGURE 4.
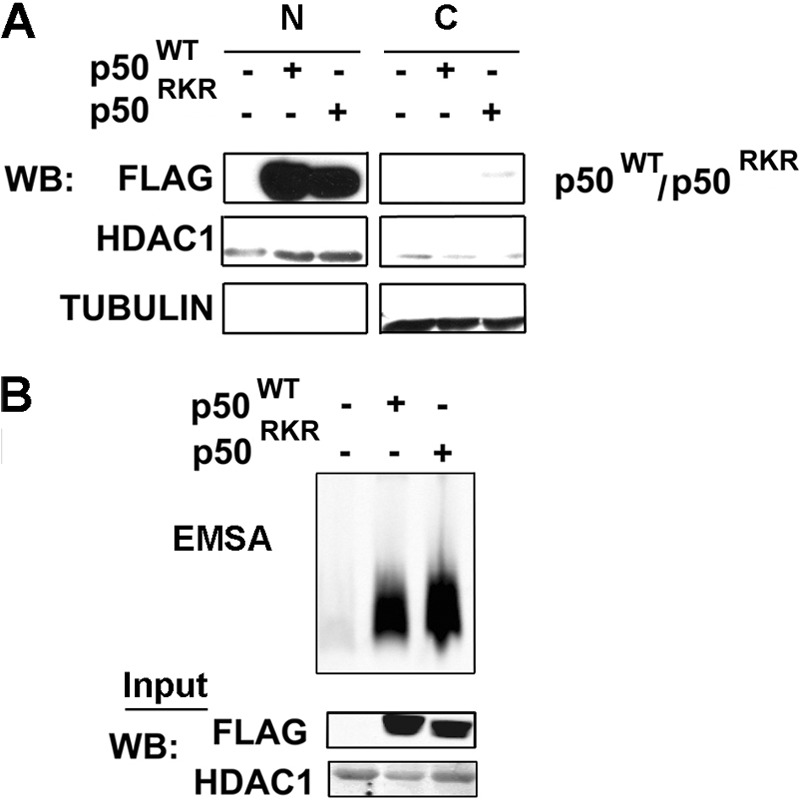
p50RKR can bind DNA and translocate to the nucleus. A, HEK293T cells were transfected with expression plasmids as indicated. Nuclear extracts were prepared from the transfected cells and tested in an EMSA using the consensus NF-κB-binding nucleotide. N, nuclear; C, cytoplasmic; WB, Western blot. B, nuclear and cytoplasmic extracts were prepared from HEK293T cells transfected with expression plasmids as indicated. p50 and p50RKR subcellular localization was analyzed by Western blot analysis with anti-FLAG antibody.
Arg-359, Lys-360, and Arg-361 of p50 Are Critical for Protein Stability
Previous studies of Bcl3−/− cells suggested that Bcl-3 regulates NF-κB-mediated transcription through inhibition of p50 homodimer ubiquitination and proteasomal degradation (18). The ubiquitination of p50 is increased significantly in Bcl3−/− cells, and overexpression of Bcl-3 inhibits p50 ubiquitination (18). To establish whether the inhibition of p50 ubiquitination by Bcl-3 requires interaction with p50, we performed a ubiquitination assay in HEK293T cells transfected with HA-tagged ubiquitin, p50WT, or p50RKR with or without Bcl-3. Following denaturing lysis, p50 was immunoprecipitated and immunoblotted with anti-HA antibody. This analysis revealed significantly increased ubiquitination of p50RKR relative to p50WT (Fig. 5A). Furthermore, in contrast to the complete inhibition of p50WT ubiquitination by Bcl-3, the expression of Bcl-3 failed to inhibit the ubiquitination of p50RKR (Fig. 5A). Similar results were obtained when cells were transfected with p50K363A (Fig. 5B). These data demonstrate that the interaction of Bcl-3 with p50 is required for Bcl-3-mediated inhibition of p50 ubiquitination. p50 ubiquitination leads to degradation by the proteasome and, thereby, controls p50 protein stability. We next assessed the stability of p50RKR by treating p50WT- or p50RKR-expressing cells with the protein synthesis inhibitor cycloheximide and monitoring protein levels over a short time period by immunoblotting. This analysis demonstrated that p50RKR is significantly less stable than p50WT and has a half-life of less than 50% than that of p50WT (Fig. 5C). These data demonstrate that interaction with Bcl-3 is critical for the regulation of p50 ubiquitination and protein stability.
FIGURE 5.

Arg-359, Lys-360, and Arg-361 of p50 are critical for protein stability. A, Bcl-3 cannot block p50RKR ubiquitination. HEK293T cells were transfected as indicated. p50 ubiquitination was determined by immunoprecipitation (IP) from whole cell lysates with anti-FLAG antibody and Western blotting (WB) with anti-HA antibody for HA-ubiquitin (Ub). B, Bcl-3 cannot block p50K363A ubiquitination. HEK293T cells were transfected as indicated. p50 ubiquitination was determined by immunoprecipitation from whole cell lysates with anti-FLAG antibody and Western blotting with anti-HA antibody for HA-ubiquitin. C, reduced half-life of p50RKR. HEK293T cells were transfected with FLAG-p50 or p50RKR. Following transfection, cells were treated with 100 μg/ml cycloheximide (CHX) and harvested at the indicated times. For each sample, p50 protein levels were quantified by densitometry and normalized with β-actin. The half-life of p50 and p50RKR was calculated from three independent experiments and is presented as mean ± S.D.
Arg-359, Lys-360, and Arg-361 of p50 Are Critical for Negative Regulation of NF-κB-dependent Gene Expression
It has been proposed that Bcl-3 regulates NF-κB-mediated transcription through inhibition of p50 homodimer ubiquitination and proteasomal degradation (18). However, additional p50-independent functions of Bcl-3 in regulating transcription cannot be ruled out in studies employing Bcl3−/− cells. Our finding that the interaction of p50 with Bcl-3 is important for regulating the stability of ubiquitination and the stability of p50 led us to examine the regulation of gene transcription by p50RKR. To investigate this, we first employed a luciferase reporter assay incorporating the NF-κB-dependent IL-23p19 gene promoter (27). In agreement with previous reports (28), Bcl-3 expression inhibited the reporter activity in RAW 264.7 macrophage cells following stimulation with LPS. Similarly, overexpression of both p50WT and p50RKR inhibited LPS-induced reporter activity. However, although coexpression of p50WT with Bcl-3 completely abolished LPS-induced reporter activity, the coexpression of p50RKR with Bcl-3 failed to inhibit reporter activity below the level seen when either protein is expressed alone (Fig. 6A). Next, we performed a dose-dependent analysis of p50WT and p50RKR expression on IL-23p19 reporter activity by transfecting increasing but low amounts of the p50WT or p50RKR expression vector in Nfkb1−/− MEFs. Here we observed a dose-dependent inhibition of reporter activity following transfection with p50WT but not with p50RKR (Fig. 6B). Similar results were obtained when Nfkb1−/− MEFs were transfected with an expression vector for wild-type p105 (p105WT) or p105 containing alanine substitutions at positions 359–361 (p105RKR) (Fig. 6C). Together, these data demonstrate that the interaction between Bcl-3 and p50 is critical for the negative regulation of NF-κB target genes by both of these factors.
FIGURE 6.
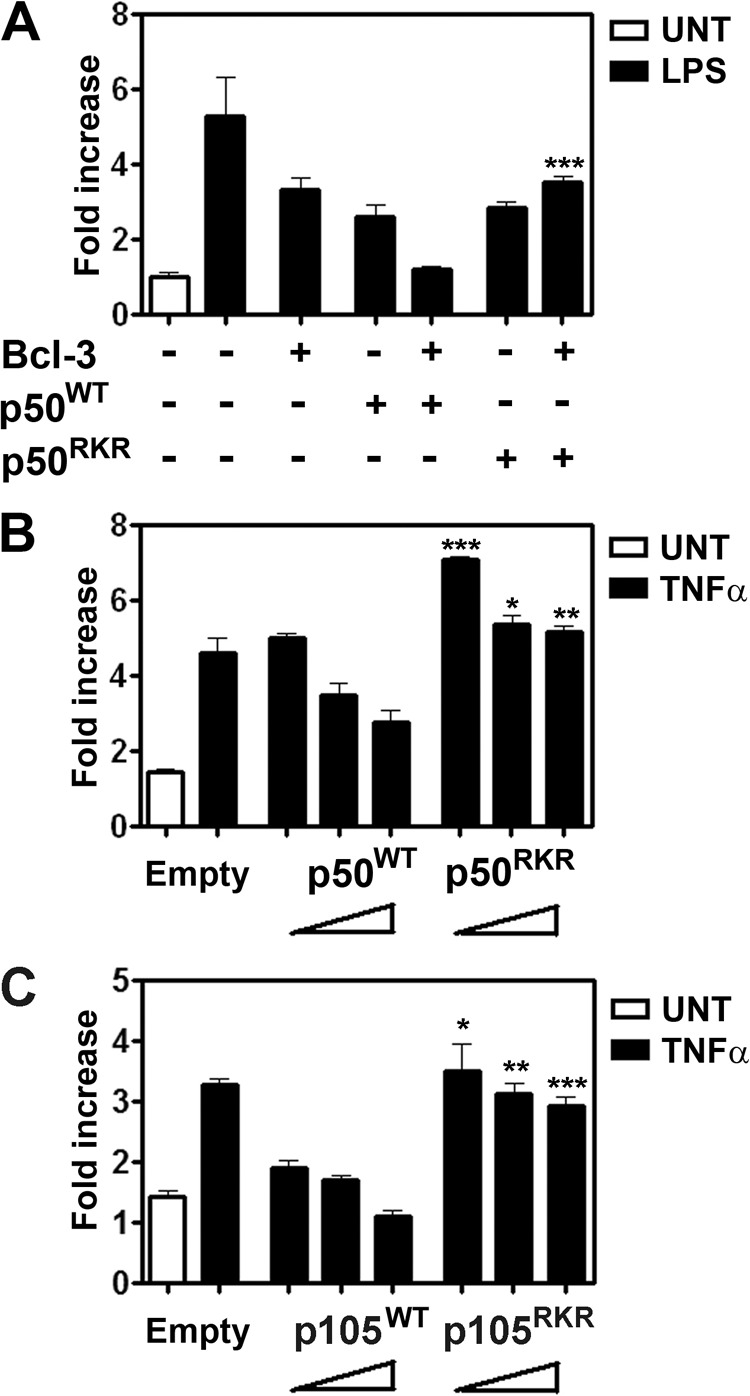
Arg-359, Lys-360, and Arg-361 of p50 are critical for negative regulation of NF-κB-dependent gene expression. A, Bcl-3 is unable to synergize with p50RKR to inhibit LPS-induced activation of IL23 p19 reporter activity. RAW 264.7 cells were transiently transfected with the pLucp19 plasmid and with an empty expression vector or expression vectors containing p50 or p50RKR and Bcl-3 for 24 h and cultured with or without 100 ng/ml LPS for an additional 8 h before luciferase activity was measured. The Renilla luciferase expression vector pRLTK was used as an internal control to normalize the transfection efficiency across all samples. Reporter activity is represented as fold increase over untreated (UNT) cells transfected with the pLucp19 plasmid and empty vector expression. B and C, Nfkb1−/− MEFs were transiently transfected with the pLucp19 plasmid and increasing amounts of expression vectors containing p50 or p50RKR (B) or p105 or p105RKR (C). The total amount of plasmid was kept constant across all samples by adjusting the amount of empty vector used. Cells were transfected for 24 h and cultured with or without 20 ng/ml TNFα for an additional 8 h before luciferase activity was measured. Transfection efficiency was normalized with pRLTK as in A. Data are mean ± S.E. of triplicate samples and are representative of three independent experiments. Statistical significance between corresponding WT and RKR mutant reporter activities was determined by Student's t test. *, p < 0.01; **, p < 0.005; ***, p < 0.0001.
p105RKR Expression Recapitulates the Bcl3−/− Phenotype
To further analyze the lack of p50·Bcl-3 complex formation in the context of an inflammatory signal, we next reconstituted Nfkb1−/− MEFs with expression vectors for p105WT or p105RKR. Stably transfected cells were clonally selected for equivalent expression of p105WT and p105RKR (Fig. 7A). We found no differences in the activation of the NF-κB pathway by TNFα between p105WT and p105RKR cells, as determined by IκBα phosphorylation and degradation (Fig. 7B). TNFα-induced nuclear localization of p50 and p65 was not altered significantly in p105RKR cells relative to p105 WT cells. However, p105RKR cells had significantly reduced p50 protein levels compared with p105WT cells (Fig. 7C), reflecting the reduced stability of p50RKR (Fig. 5C). In agreement with an overexpression analysis (Fig. 2), endogenous Bcl-3 failed to coimmunoprecipitate with p50 RKR in TNFα-stimulated cells, whereas TNF-inducible interaction with wild-type p50 was readily detectable (Fig. 8A). Consequently, endogenous ubiquitination of p50RKR in both untreated and TNFα-stimulated cells was elevated relative to p50WT (Fig. 8B). Real-time PCR analysis of TNFα-induced gene expression revealed that p105RKR cells express significantly higher levels of the NF-κB target genes TNFα, IL-6, CCL2, and CXCL2 when compared with p105WT cells (Fig. 8C). This hyperresponsiveness toward TNFα stimulation in p105RKR cells correlates with the increased ubiquitination and reduced half-life of the p50RKR protein and, importantly, recapitulates the hyperresponsiveness of Bcl3−/− cells (18). Together, these data demonstrate that interaction with Bcl-3 is essential for p50 homodimer repressor function and suggest that repression of transcription by Bcl-3 is dependent on interaction with p50 homodimers.
FIGURE 7.
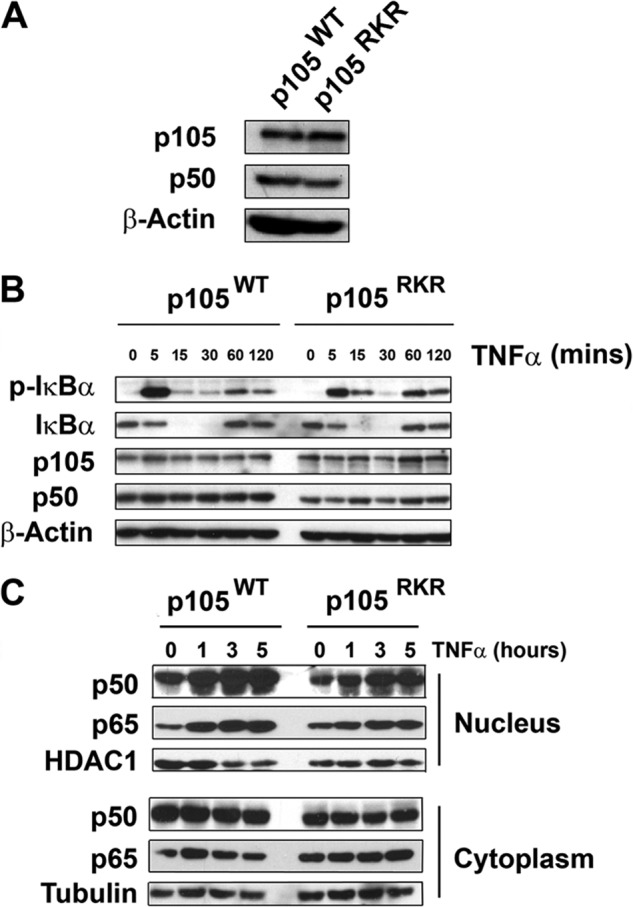
Normal upstream signaling and NF-κB nuclear translocation of p105RKR MEFs. A, Nfkb1−/−MEF cells were stably transfected with expression vectors encoding p105WT or p105RKR. B, p105WT or p105RKR MEFs were stimulated with 20 ng/ml TNFα for the indicated times prior to lysis. Whole cell extracts were analyzed by Western blotting for phosphorylated IκBα and total IκBα. C, p105WT or p105RKR MEFs were stimulated with 20 ng/ml TNFα for the indicated times prior to lysis. Nuclear and cytoplasmic extracts were prepared and analyzed for p50 and p65 proteins by Western blot analysis.
FIGURE 8.
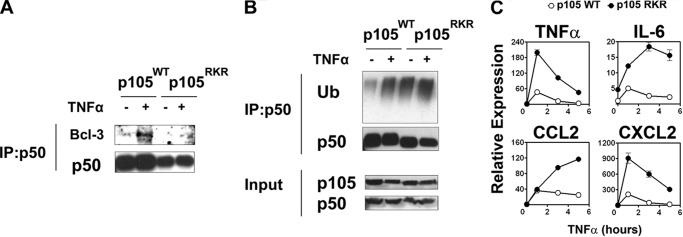
p105RKR MEFs are hyperresponsive to TNF stimulation. A, p105WT or p105RKR MEFs were left untreated (−) or stimulated with 10 ng/ml TNFα (+) for 1 h. Cells were pretreated with MG132 for 30 min prior to harvest. p50WT and p50RKR were immunoprecipitated (IP) from whole cell lysates and analyzed by Western blot analysis with anti-Bcl-3 antibody for endogenous Bcl-3. B, endogenous ubiquitination assay. p105WT or p105RKR MEFs were treated as in A. p50WT and p50RKR were immunoprecipitated from whole cell lysates, and ubiquitination was determined by Western blot analysis with anti-ubiquitin (Ub). C, p105WT or p105RKR MEFs were stimulated with 20 ng/ml TNFα. Gene expression levels were determined by real-time PCR. Data are mean ± S.E. of triplicate samples and are representative of three independent experiments.
DISCUSSION
Bcl-3 is an IκB protein that regulates NF-κB-dependent gene expression through interaction with NF-κB p50 and p52 homodimers (18). In addition to the NF-κB subunits, Bcl-3 interacts with a number of regulators of transcription, including c-Jun and c-fos (8); STAT1 (9); PPARγ (10); CREB-binding protein/p300 (8); HDAC-1, -3, and -5 (11); steroid receptor coactivator 1(8); TORC3 (12); and retinoic X receptor (13); Fyn (14) and Lck (15); insulin receptor substrate 3 (IRS3) (16); and Bag-1 (17). The contribution of these factors to immune regulation by Bcl-3 has not been determined previously. Bcl-3 limits NF-κB-mediated transcription of proinflammatory cytokines and promote LPS tolerance, a state of hyporesponsiveness following repeated or prolonged exposure to LPS (18). Previous studies have demonstrated that Bcl-3 regulates p50 homodimer stability through the inhibition of ubiquitination and subsequent proteasomal-mediated degradation (18). However, studies employing Bcl3−/− cells and mice do not exclude the possibility that Bcl-3 may also function through p50-independent mechanisms to regulate gene expression. Here, by identifying the critical site of p50 necessary for interaction with Bcl-3, we provide the first direct evidence that Bcl-3-mediated repression of NF-κB-mediated gene expression requires interaction with p50. Furthermore, our data demonstrate that p50 that cannot interact with Bcl-3 is inherently unstable and undergoes increased ubiquitination and degradation relative to wild-type p50, even in the presence of overexpressed Bcl-3. Our data also suggest that p50-independent targets of Bcl-3 are not important in the regulation of NF-κB-dependent inflammatory gene expression.
In this study, we employed peptide array techniques to identify residues of p50 necessary for interaction with Bcl-3. This approach, which uses immobilized peptides representing the p50 protein sequence, has distinct advantages over deletional mutagenic approaches. Perhaps chief among these is the avoidance of potential artifacts on the formation of multisubunit complexes, which may result from the deletion of significant portions of a protein. For example, the RHD of p50 is essential for dimerization as well as DNA binding. Even partial deletion of the RHD may have consequences on the formation of a Bcl-3·p50 complex. In this study, the immobilized peptide array approach allowed us to identify a series of overlapping peptides with a strong affinity for purified recombinant Bcl-3 corresponding to amino acids 337–378 at the C-terminal region of p50. The serial substitution of amino acids 355–372 with alanine as immobilized peptide arrays further increased the resolution of the site and identified Arg-359, Lys-360, Arg-361 and Lys-363 as essential residues for interaction with Bcl-3. Subsequently, the mutation of Arg-359, Lys-360, and Arg-361 to alanine in full-length p50 blocked the interaction with purified, recombinant Bcl-3. These data validate peptide array technology as a valuable tool in identifying sites of protein-protein interaction with amino acid level resolution.
The region of p50 containing Arg-359, Lys-360, and Arg-361 is C-terminal to previously identified ankyrin repeat domain interaction sites determined from the crystal structure of a p65/p50 heterodimer complexed with IκBα. Unfortunately, amino acids 359–361 are not represented on the crystal structures of p50-containing complexes, and, therefore, no structural data are available. However, a hydrophobicity plot reveals that these amino acids lie in a region of low hydrophobicity and, thus, are expected to be available for interaction with Bcl-3. Importantly, mutation of these amino acids does not alter the hetero- or homodimer formation properties of p50 or interfere with DNA binding of p50. This allows us to rule out the loss of repressor function of p50RKR homodimers because of a lack of dimerization or DNA binding. Moreover, despite being located in a region reported previously to be important for nuclear localization (26), the mutation of Arg-359, Lys-360, and Arg-361 to alanine had no effect on the nuclear localization of p50, as monitored by subcellular fractionation analysis and DNA binding assays. Our data suggest that additional sequences of p50 are important in its nuclear translocation.
The mutation of Arg-359, Lys-360, and Arg-361 in p50 (p50RKR) functionally recapitulates the phenotype of Bcl3−/− cells described previously. Thus, p50RKR undergoes increased ubiquitination corresponding to a reduced half-life, and cells expressing p50RKR display increased NF-κB transcriptional activity relative to wild-type p50. Critically, Bcl-3 is unable to rescue the increased NF-κB activity in p50RKR-expressing cells. NF-κB transactivation is not inhibited by overexpression of Bcl-3 in cells expressing p50RKR, and Bcl-3 is unable to inhibit p50RKR ubiquitination. The importance of p50 in regulating transcriptional programs during inflammation has been highlighted by two recent studies of Toll-like receptor- (29) and interferon-induced responses (5). Our study further highlights the importance of Bcl-3 and p50 interaction in the regulation of the inflammatory response independently of the interaction of Bcl-3 with other transcriptional regulators.
Acknowledgment
We thank Karen Keeshan for critical evaluation of the manuscript.
This work was supported by Science Foundation Ireland Grant 08/IN.1/B1843.
- RHD
- Rel homology domain
- MEF
- mouse embryonic fibroblast
- CREB
- cAMP response element-binding protein
- IRF
- interferon regulatory factor
- PPARγ
- peroxisome proliferator-activated receptorγ
- IKK
- IκB kinase.
REFERENCES
- 1. Hayden M. S., Ghosh S. (2008) Shared principles in NF-κB signaling. Cell 132, 344–362 [DOI] [PubMed] [Google Scholar]
- 2. Gerondakis S., Grossmann M., Nakamura Y., Pohl T., Grumont R. (1999) Genetic approaches in mice to understand Rel/NF-κB and IκB function. Transgenics and knockouts. Oncogene 18, 6888–6895 [DOI] [PubMed] [Google Scholar]
- 3. Ghosh S., Hayden M. S. (2008) New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 8, 837–848 [DOI] [PubMed] [Google Scholar]
- 4. Siggers T., Chang A. B., Teixeira A., Wong D., Williams K. J., Ahmed B., Ragoussis J., Udalova I. A., Smale S. T., Bulyk M. L. (2012) Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol. 13, 95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheng C. S., Feldman K. E., Lee J., Verma S., Huang D. B., Huynh K., Chang M., Ponomarenko J. V., Sun S. C., Benedict C. A., Ghosh G., Hoffmann A. (2011) The specificity of innate immune responses is enforced by repression of interferon response elements by NF-κB p50. Sci. Signal. 4, ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carmody R. J., Chen Y. H. (2007) Nuclear factor-κB. Activation and regulation during Toll-like receptor signaling. Cell. Mol. Immunol. 4, 31–41 [PubMed] [Google Scholar]
- 7. Palmer S., Chen Y. H. (2008) Bcl-3, a multifaceted modulator of NF-κB-mediated gene transcription. Immunol. Res. 42, 210–218 [DOI] [PubMed] [Google Scholar]
- 8. Na S. Y., Choi J. E., Kim H. J., Jhun B. H., Lee Y. C., Lee J. W. (1999) Bcl3, an IκB protein, stimulates activating protein-1 transactivation and cellular proliferation. J. Biol. Chem. 274, 28491–28496 [DOI] [PubMed] [Google Scholar]
- 9. Jamaluddin M., Choudhary S., Wang S., Casola A., Huda R., Garofalo R. P., Ray S., Brasier A. R. (2005) Respiratory syncytial virus-inducible BCL-3 expression antagonizes the STAT/IRF and NF-κB signaling pathways by inducing histone deacetylase 1 recruitment to the interleukin-8 promoter. J. Virol. 79, 15302–15313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang J., Williams R. S., Kelly D. P. (2009) Bcl3 interacts cooperatively with peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α to coactivate nuclear receptors estrogen-related receptor α and PPARα. Mol. Cell. Biol. 29, 4091–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Viatour P., Dejardin E., Warnier M., Lair F., Claudio E., Bureau F., Marine J. C., Merville M. P., Maurer U., Green D., Piette J., Siebenlist U., Bours V., Chariot A. (2004) GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol. Cell 16, 35–45 [DOI] [PubMed] [Google Scholar]
- 12. Hishiki T., Ohshima T., Ego T., Shimotohno K. (2007) BCL3 acts as a negative regulator of transcription from the human T-cell leukemia virus type 1 long terminal repeat through interactions with TORC3. J. Biol. Chem. 282, 28335–28343 [DOI] [PubMed] [Google Scholar]
- 13. Na S. Y., Choi H. S., Kim J. W., Na D. S., Lee J. W. (1998) Bcl3, an IκB protein, as a novel transcription coactivator of the retinoid X receptor. J. Biol. Chemistry 273, 30933–30938 [DOI] [PubMed] [Google Scholar]
- 14. Weyrich A. S., Dixon D. A., Pabla R., Elstad M. R., McIntyre T. M., Prescott S. M., Zimmerman G. A. (1998) Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc. Natl. Acad. Sci. U.S.A. 95, 5556–5561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao Y., Ramakrishnan A., Kim K. E., Rabson A. B. (2005) Regulation of Bcl-3 through interaction with the Lck tyrosine kinase. Biochem. Biophys. Res. Commun. 335, 865–873 [DOI] [PubMed] [Google Scholar]
- 16. Kabuta T., Hakuno F., Cho Y., Yamanaka D., Chida K., Asano T., Wada K., Takahashi S. (2010) Insulin receptor substrate-3, interacting with Bcl-3, enhances p50 NF-κB activity. Biochem. Biophys. Res. Commun. 394, 697–702 [DOI] [PubMed] [Google Scholar]
- 17. Southern S. L., Collard T. J., Urban B. C., Skeen V. R., Smartt H. J., Hague A., Oakley F., Townsend P. A., Perkins N. D., Paraskeva C., Williams A. C. (2012) BAG-1 interacts with the p50-p50 homodimeric NF-κB complex. Implications for colorectal carcinogenesis. Oncogene 31, 2761–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carmody R. J., Ruan Q., Palmer S., Hilliard B., Chen Y. H. (2007) Negative regulation of Toll-like receptor signaling by NF-κB p50 ubiquitination blockade. Science 317, 675–678 [DOI] [PubMed] [Google Scholar]
- 19. Cowan R., Whittaker R. G. (1990) Hydrophobicity indices for amino acid residues as determined by high-performance liquid chromatography. Pept. Res. 3, 75–80 [PubMed] [Google Scholar]
- 20. Kramer A., Schneider-Mergener J. (1998) Synthesis and screening of peptide libraries on continuous cellulose membrane supports. Methods Mol. Biol. 87, 25–39 [DOI] [PubMed] [Google Scholar]
- 21. Dyer B. W., Ferrer F. A., Klinedinst D. K., Rodriguez R. (2000) A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal. Biochem. 282, 158–161 [DOI] [PubMed] [Google Scholar]
- 22. Michel F., Soler-Lopez M., Petosa C., Cramer P., Siebenlist U., Müller C. W. (2001) Crystal structure of the ankyrin repeat domain of Bcl-3. A unique member of the IκB protein family. EMBO J. 20, 6180–6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neasta J., Kiely P. A., He D. Y., Adams D. R., O'Connor R., Ron D. (2012) Direct interaction between scaffolding proteins RACK1 and 14-3-3ζ regulates brain-derived neurotrophic factor (BDNF) transcription. J. Biol. Chem. 287, 322–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kiely P. A., Baillie G. S., Barrett R., Buckley D. A., Adams D. R., Houslay M. D., O'Connor R. (2009) Phosphorylation of RACK1 on tyrosine 52 by c-Abl is required for insulin-like growth factor I-mediated regulation of focal adhesion kinase. J. Biol. Chem. 284, 20263–20274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Colleran A., Collins P. E., O'Carroll C., Ahmed A., Mao X., McManus B., Kiely P. A., Burstein E., Carmody R. J. (2013) Deubiquitination of NF-κB by ubiquitin-specific protease-7 promotes transcription. Proc. Natl. Acad. Sci. U.S.A. 110, 618–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Latimer M., Ernst M. K., Dunn L. L., Drutskaya M., Rice N. R. (1998) The N-terminal domain of IκB α masks the nuclear localization signal(s) of p50 and c-Rel homodimers. Mol. Cell. Biol. 18, 2640–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carmody R. J., Ruan Q., Liou H. C., Chen Y. H. (2007) Essential roles of c-Rel in Toll-like receptor-induced IL-23 p19 gene expression in dendritic cells. J. Immunol. 178, 186–191 [DOI] [PubMed] [Google Scholar]
- 28. Mühlbauer M., Chilton P. M., Mitchell T. C., Jobin C. (2008) Impaired Bcl3 up-regulation leads to enhanced lipopolysaccharide-induced interleukin (IL)-23P19 gene expression in IL-10−/− mice. J. Biol. Chem. 283, 14182–14189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan Q., Carmody R. J., Qu Z., Ruan Q., Jager J., Mullican S. E., Lazar M. A., Chen Y. H. (2012) Nuclear factor-κB binding motifs specify Toll-like receptor-induced gene repression through an inducible repressosome. Proc. Natl. Acad. Sci. U.S.A. 109, 14140–14145 [DOI] [PMC free article] [PubMed] [Google Scholar]