

Background: Lys-164-monoubiquitinated PCNA is essential for interstrand DNA cross-link (ICL) repair.
Results: A small molecule, T2AA, bi-molecularly binds to PCNA at a PIP-box cavity and close to Lys-164. T2AA inhibited monoubiquitinated PCNA interactions and ICL repair and enhanced DNA double strand breaks.
Conclusion: An inhibitor of monoubiquitinated PCNA inhibits ICL repair.
Significance: Inhibition of monoubiquitinated PCNA could improve chemotherapeutic efficacy.
Keywords: Crystallography, DNA Damage, Small Molecules, Structural Biology, Ubiquitination, PCNA, Interstrand Cross-link
Abstract
Small molecule inhibitors of proliferating cell nuclear antigen (PCNA)/PCNA interacting protein box (PIP-Box) interactions, including T2 amino alcohol (T2AA), inhibit translesion DNA synthesis. The crystal structure of PCNA in complex with T2AA revealed that T2AA bound to the surface adjacent to the subunit interface of the homotrimer of PCNA in addition to the PIP-box binding cavity. Because this site is close to Lys-164, which is monoubiquitinated by RAD18, we postulated that T2AA would affect monoubiquitinated PCNA interactions. Binding of monoubiquitinated PCNA and a purified pol η fragment containing the UBZ and PIP-box was inhibited by T2AA in vitro. T2AA decreased PCNA/pol η and PCNA/REV1 chromatin colocalization but did not inhibit PCNA monoubiquitination, suggesting that T2AA hinders interactions of pol η and REV1 with monoubiquitinated PCNA. Interstrand DNA cross-links (ICLs) are repaired by mechanisms using translesion DNA synthesis that is regulated by monoubiquitinated PCNA. T2AA significantly delayed reactivation of a reporter plasmid containing an ICL. Neutral comet analysis of cells receiving T2AA in addition to cisplatin revealed that T2AA significantly enhanced formation of DNA double strand breaks (DSBs) by cisplatin. T2AA promoted colocalized foci formation of phospho-ATM and 53BP1 and up-regulated phospho-BRCA1 in cisplatin-treated cells, suggesting that T2AA increases DSBs. When cells were treated by cisplatin and T2AA, their clonogenic survival was significantly less than that of those treated by cisplatin only. These findings show that the inhibitors of monoubiquitinated PCNA chemosensitize cells by inhibiting repair of ICLs and DSBs.
Introduction
Proliferating cell nuclear antigen (PCNA)3 is an exceptionally multifunctional DNA clamp crucial for diverse events relevant to DNA replication and DNA damage response. On the replication fork, it is loaded to the border of single-stranded and double-stranded DNA (i.e. the front of DNA replication) as a cyclic homotrimer and recruits numerous components according to ongoing events. The majority of PCNA interaction partners have a characteristic short amino acid sequence called a PIP-box that binds to a cavity of PCNA on its interdomain connecting loop (1). The most important function of PCNA is supporting DNA replication by recruiting DNA polymerases such as pol δ via PIP-box interactions. When the replication fork encounters DNA damage such as a thymidine dimer, the pol δ on PCNA can be switched to another DNA polymerase specialized for translesion DNA synthesis (TLS), such as pol η, to enforce replication beyond the damage (2, 3). Thus, inhibiting the PCNA/PIP-box interaction can be a rational strategy to inhibit TLS (4). Supporting this hypothesis, we previously found that nonpeptide small molecular inhibitors of the PCNA/PIP-box interaction, such as T2AA (5), inhibit both DNA replication and TLS.
ICL is a DNA lesion critical for cytotoxicity of DNA cross-linking agents such as cisplatin and can be repaired by a combination of nucleotide excision repair (NER), TLS, and homologous recombination (HR) (6, 7). Numerous studies have shown the importance of PCNA monoubiquitination at Lys-164 for activating the TLS process by promoting DNA polymerase switching (for instance, see Ref. 3). Therefore, biochemical inhibition of the monoubiquitinated PCNA interaction could result in an ICL repair deficiency. To test this hypothesis, we characterized T2AA by focusing on inhibiting monoubiquitinated PCNA and the consequential effect on ICL repair.
EXPERIMENTAL PROCEDURES
Materials
All chemicals were purchased from Sigma and used as received. All oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). Sources for plasmids, except pGL4.50 and pRL-TK (Promega, Madison, WI), are as indicated in the Acknowledgments. All restriction enzymes and T4 DNA ligase were purchased from New England Biolabs (Ipswich, MA). The following primary antibodies were used according to the manufacturers' recommendations: anti-phospho-BRCA1 (Ser-1524) rabbit IgG, anti-PCNA PC10 mouse IgG, and rabbit anti-RAD18 (D2B8) (Cell Signaling Technology, Danvers, MA); mouse anti-His tag (penta-His) (Qiagen, Hilden, Germany); mouse anti-phospho-Ser-1981 ATM (clone 10H11.E12; Rockland, Gilbertsville, PA); rabbit anti-53BP1 (H-300; Santa Cruz Biotechnology, Dallas, TX), and anti-FANCD2 (clone FL17; Santa Cruz Biotechnology). The secondary antibodies used were as follows: anti-mouse IgG HRP conjugate and anti-rabbit IgG HRP conjugate (Cell Signaling Technology, Danvers, MA); goat anti-mouse IgG conjugated with Alexa Fluor 555, donkey anti-mouse IgG conjugated with Alexa Fluor 488, and donkey anti-rabbit IgG conjugated with Alexa Fluor 555 (Invitrogen). All cells were obtained from American Type Culture Collection (Manassas, VA), except GM04312 cells (Coriell Institute, Camden, NJ), and were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum at 37 °C in a humidified 5% carbon dioxide incubator. All chemical compounds assayed were prepared as 10 mm DMSO solutions, except cisplatin was 5 mm in 0.9% (w/v) aqueous sodium chloride.
Chemical Synthesis
Preparation of all PCNA/PIP-box interaction inhibitors has been described previously (5, 8) except for compound 6, which is described below. A mixture of 4-(2-aminoethyl)phenol (200 mg, 1.46 mmol) in dioxane (3 ml) and water (3 ml) was adjusted to pH ∼10. di-tert-Butyl carbonate (406 μl, 1.75 mmol) was added to the mixture in an ice-water bath, and the solution was stirred for 2 h. The reaction mixture was adjusted to pH ∼3 by HCl (1 m) and extracted with ethyl acetate (3× 20 ml). The combined organic layers were washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude product was purified by flash column chromatography (Biotage SP4, 25+M column, eluting with hexanes/ethyl acetate, 0–50% gradient (v/v)) to isolate tert-butyl 4-hydroxyphenethylcarbamate (compound 7) as a colorless oil (337 mg, 97% yield). 1H NMR (400 MHz, CDCl3), δ 7.03 (d, J = 8.2 Hz, 2H), 6.77 (d, J = 8.4 Hz, 2H), 5.57 (s, 1H), 4.57 (br s, 1H), 3.33 (dd, J = 12.4, 5.9 Hz, 2H), 2.71 (t, J = 7.0 Hz, 2H), 1.44 (s, 9H).
A 30% (w/v) aqueous solution of hydrogen peroxide (430 μl, 4.17 mmol) was added dropwise to a stirring solution of compound 7 (330 mg, 1.39 mmol) and iodine (529 mg, 2.09 mmol) in water (10 ml). The reaction mixture was stirred overnight at 50 °C and an aqueous sodium thiosulfate solution was added (1 m, 10 ml) and extracted with ethyl acetate (3 × 10 ml). The combined organic layers were washed with saturated aqueous NaCl, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude product was purified by flash column chromatography (Biotage SP4, 25+M column, eluting with hexanes/ethyl acetate, 0–30% gradient (v/v)) to isolate tert-butyl 4-hydroxy-3,5-diiodophenethylcarbamate (compound 8) as a white solid (212 mg, 31% yield). 1H NMR (400 MHz, CDCl3), δ 7.50 (s, 2H), 5.65 (s, 1H), 4.53 (br s, 1H), 3.29 (dd, J = 13.4, 6.7 Hz, 2H), 2.67 (t, J = 6.8 Hz, 2H), 1.45 (s, 9H).
A mixture of triethylamine (234 μl, 1.68 mmol) and pyridine (136 μl, 1.68 mmol) was added to a stirring solution of compound 8 (205 mg, 0.419 mmol), (4-methoxyphenyl)boronic acid (96 mg, 0.63 mmol), copper(II) acetate (228 mg, 1.26 mmol), and 4 Å molecular sieve powder in dry dichloromethane (5 ml). The mixture was stirred overnight under a calcium chloride drying tube. Additional (4-methoxyphenyl)boronic acid (63 mg, 0.42 mmol) and copper(II) acetate (150 mg, 0.830 mmol) were added, and the reaction mixture was stirred for an additional 4 h. The solution was diluted with ethyl acetate and filtered through Celite powder. The filtrate was concentrated and purified by flash column chromatography (Biotage SP4, 25+S column, eluting with hexanes/ethyl acetate, 0–30% gradient (v/v)) to isolate tert-butyl-3,5-diiodo-4-(4-methoxyphenoxy)phenethylcarbamate (compound 9) as a white solid (155 mg, 62% yield). 1H NMR (400 MHz, CDCl3), δ 7.68 (s, 2H), 6.86–6.80 (m, 2H), 6.75–6.68 (m, 2H), 4.60 (br s, 1H), 3.77 (s, 3H), 3.36 (q, J = 6.7 Hz, 2H), 2.75 (t, J = 7.0 Hz, 2H), 1.46 (s, 9H).
A 1 m solution of tribromoborane (1.25 ml, 1.25 mmol) in dichloromethane was added dropwise to a solution of compound 9 (149 mg, 0.250 mmol) in dichloromethane (3 ml) at −78 °C. The reaction mixture was stirred overnight under nitrogen and allowed to reach room temperature. The reaction mixture was added to an aqueous sodium hydroxide solution (1 m, 10 ml) and extracted with ethyl acetate (3 × 10 ml). The combined organic layers were washed with saturated aqueous NaCl, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude product was purified by flash column chromatography (Biotage SP4, 25+S KP-NH amine column, eluting with dichloromethane/methanol, 0–10% gradient (v/v)) to isolate 4-(4-(2-aminoethyl)-2,6-diiodophenoxy)phenol (compound 6) (18.8 mg, 16% yield). 1H NMR (400 MHz, DMSO), δ 9.09 (br s, 1H), 7.76 (s, 2H), 6.68 (d, J = 8.9 Hz, 2H), 6.53 (d, J = 8.9 Hz, 2H), 2.77 (t, J = 6.8 Hz, 2H), 2.59 (t, J = 6.9 Hz, 2H), 1.44 (br s, 2H); HRMS (ESI) m/z, [M + H] was calculated for C14H14I2NO2, 481.9114 and was observed at 482.9128.
Protein Crystallization and Structure Determination
Crystals of human PCNA bound to T2AA were obtained by the hanging-drop vapor diffusion method using a buffer (0.1 m sodium citrate, pH 5.5, and 2.0 m ammonium sulfate) as a reservoir solution at 293 K. Prior to x-ray diffraction study, crystals were transferred into a reservoir solution containing 20% (v/v) ethylene glycol for cryo-protection and then cooled in a nitrogen gas stream at 100 K. X-ray diffraction data were collected at a beamline NE-3A in Photon Factory Advance Ring (PF-AR) and processed by programs XDS (9) and SCALA (10). The crystal structure of PCNA bound to the inhibitor molecule was determined by the molecular replacement method with the program MOLREP (11). The structure of PCNA (Protein Data Bank code 2OVX) was used as a model structure. The structure was manually improved with the program COOT (12) and refined by the program REFMAC (13). Structure factors and final coordinates were deposited in the Protein Data Bank Japan (PDBj). Data collection and refinement statistics are shown in Table 1.
TABLE 1.
Data collection and refinement statistics
Values in parentheses are for the highest resolution shell. Ramachandran statistics indicate the fraction of residues in the most favored/allowed/disallowed regions. r.m.s.d., root mean square deviation.
| Data collection | |
| Space group | P4132 |
| Cell dimensions | |
| a = b = c | 192.1 Å |
| Resolution range | 19.82 to 2.80 Å (2.95 o 2.80 Å) |
| Observed reflections | 1,271,545 |
| Unique reflections | 30,376 |
| Rmerge | 0.128 (0.598) |
| Completeness | 99.7% (100%) |
| Mean I/σ(I) | 30.2 (7.4) |
| Refinement | |
| Resolution range | 20.0 to 2.80 Å |
| R/Rfree | 0.183/0.221 |
| r.m.s.d. bond distances/angles | 0.020 Å/2.419° |
| Ramachandran statistics | 91.5/8.5/0% |
| Protein Data Bank ID | 3WGW |
Preparation of Expression Plasmid for His8 pol η(551–713)
The cDNA encoding pol η(551–713) was PCR-amplified from pEGFP-pol η using TATACCATGGATCATCATCATCATCATCATCATCATTCCCCCCAACAAAACCCT and TTGGAATCATTTTTTAAGCCATTAACACATTAGTAGCTCGAGATAT and inserted into the XhoI/NcoI site of pET15b (Novagen) using a Quick Ligation kit (New England Biolabs) and Escherichia coli strain DH5α (Invitrogen) according to the manufacturers' recommendations. The clone was verified by sequencing.
Protein Production
To produce His8-tagged pol η(551–713) protein containing a UBZ domain and a PIP-box, the expression plasmid pET15-pol η(551–713) described above was expressed in E. coli strain BL21(DE3) in Turbo Prime broth (AthenaES) with ampicillin (100 μg/ml). His8-pol η(551–713) expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at an A600 of 8.4, and growth was continued for 16 h at 16 °C. Bacterial cells were harvested by centrifugation for 20 min at 4000 × g in a Beckman JLA8.1 rotor, and the pellets were suspended in lysis buffer (20 mm Tris-HCl, pH 7.9, 500 mm NaCl, and 10% glycerol). Cells were lysed by Microfluidizer, and the soluble fraction was collected by centrifugation at 38,400 × g for 1 h. The lysate was loaded onto a 5-ml charged and pre-equilibrated nickel column (GE Healthcare), and the pol η(551–713) protein was eluted with a linear gradient of imidazole in 20 mm Tris-HCl, pH 7.9, 500 mm NaCl, and 10% (v/v) glycerol on an AKTA explorer. pol η(551–713)-containing fractions were identified by SDS-PAGE, pooled, and dialyzed in 20 mm Tris-HCl, pH 7.9, 500 mm NaCl, and 10% (v/v) glycerol at 4 °C overnight.
Pulldown Assay
A His protein interaction pulldown kit (Thermo Scientific, Waltham, MA) was used, and a 4:1 mixture of TBS and lysis buffer provided in the kit was used as the buffer throughout. For immobilization, 150 μg of His8-pol η(551–713) was incubated with 10 μl of HisPur cobalt resin (Thermo Scientific) in 500 μl of the buffer at 4 °C for 2 h and washed with the buffer four times. To harvest a protein sample containing monoubiquitinated PCNA, GM04312 cells in two culture dishes (15 cm2) were UVC-irradiated (60 J/m2), incubated for 6 h, pre-extracted at 4 °C for 5 min by CSK buffer (100 mm NaCl, 300 mm sucrose, 3 mm MgCl2, and 10 mm PIPES, pH 6.8) containing 0.25% (v/v) Triton X-100 on the dishes, collected as pellets, sonicated at 4 °C in RIPA buffer containing protease inhibitor mixture, and centrifuged to harvest the supernatant lysate. The lysate (100 μg) in 500 μl of the pulldown buffer was mixed with the indicated final concentration of the indicated compounds and the pol η(551–713)-immobilized resin (10 μl settled). After the mixture was incubated at 4 °C for 3 h, the resin was washed with the buffer four times and analyzed by SDS-PAGE, followed by immunoblotting sequentially for PCNA and then for His tag.
Immunoblotting
The cells treated as indicated were washed twice with ice-cold PBS, collected in a spin tube, and lysed with RIPA buffer (approximately twice the volume of the cell pellet and supplemented with Halt Protease Inhibitor Mixture and Halt Phosphatase Inhibitor Mixture (Thermo Scientific)) on ice for 0.5 h. The protein concentration was determined by BCA assay (Thermo Scientific) according to the manufacturer's recommendation. Normalized amounts of samples were loaded onto Novex SDS-PAGE (Invitrogen) as indicated and electrotransferred to a nitrocellulose membrane using an iBlot apparatus (Invitrogen). The membrane was blocked with SuperBlock buffer (Thermo Scientific) and incubated with the indicated primary antibodies in SuperBlock at 4 °C overnight, rinsed with TBS, 0.05% (v/v) Tween 20 three times for 10 min each, incubated with the corresponding secondary IgG-HRP conjugate in SuperBlock at room temperature for 1 h, and rinsed with TBS, 0.05% Tween 20 for 15 min. Proteins probed on the membrane were visualized by chemiluminescence using WestPico reagent (Thermo Scientific) and developed on a BioMax MR film (Eastman Kodak Co.).
Immunofluorescence Imaging
Cells were transfected with a plasmid encoding EGFP-pol η or EGFP-REV1 (20 μg) by electroporation. Cells were propagated in 6-well plates with a coverslip, incubated for 20 h, and treated as indicated. Cells were then rinsed with PBS one time and incubated for another 6 h in fresh medium with or without the indicated compounds (20 μm). Cells were then treated with CSK buffer (10 mm PIPES, pH 6.8, 100 mm NaCl, and 1 mm EGTA) containing 0.2% (v/v) Triton X-100 for 5 min on ice to remove proteins not bound to chromatin, fixed with 4% (w/v) paraformaldehyde in PBS for 15 min at room temperature, washed once with PBS, and kept in cold 100% methanol. Immunofluorescence staining was performed by applying an anti-PCNA monoclonal antibody (1:2000) followed by an anti-mouse IgG conjugated with Alexa Fluor 555 (1:400). The coverslips were mounted with Vectashield (Burlingame, CA) containing DAPI (1 μg/ml) for counterstaining nuclei. The stained cells were examined on a C1Si confocal microscope (Nikon, Tokyo, Japan) with a Cascade 512B photomultiplier (Photometrics, Tucson, AZ). Images were processed using EZC1 (Nikon) and Photoshop 7 (Adobe Systems, San Jose, CA).
Colocalization Analyses
Images were captured using a Marianas System (Intelligent Imaging Innovations, Denver, CO), which incorporates a Zeiss Axioplan microscope, a Yokogawa CSUX spinning disk confocal scan head, and a Photometrics Evolve CCD camera (Photometrics, Tucson, AZ). Excitation was by DPSS lasers at 405, 488, and 561 nm, and the objective used was a 63× 1.4 NA Plan Neofluar. The colocalization was quantified by calculating Pearson's correlation coefficient between the signal intensities of green and red colors in all the pixels on captured confocal images using SlideBook 5.5 software (Intelligent Imaging Innovations, Denver, CO).
Preparation of ICL Repair Reporter Plasmid pGL-ICL
An outline of the preparation is shown in Fig. 3A. The i5OctdU-containing oligonucleotide and iAzideN-containing oligonucleotide (Integrated DNA Technologies, Coralville, IA) were synthesized as 5′-phosphorylated forms. These oligonucleotides contain an EcoRI site and HindIII overhangs with a mutation and were annealed (each 500 μm in water, 40 μl) by heating to 70 °C for 1 min and cooled to 26 °C at −1 °C for 15 s. The mixture was then mixed with aqueous copper(II) sulfate (5 mm, 5 μl) and aqueous sodium ascorbate (5 mm, 5 μl) and incubated at room temperature overnight. The oligonucleotide duplex was isolated by adding sodium acetate (3 m, 6 μl) and acetone (280 μl) and chilling at −20 °C for 1 h. The precipitate was isolated by a centrifuge and purified by excising a single band of the ICL-containing duplex oligonucleotide product from denaturing PAGE (15% TBE/urea).
FIGURE 3.

T2AA inhibits ICL repair. A, reporter assay plasmid pGL-ICL used in this study and outline of the method used to produce it and to verify incorporation of the ICL. An ICL was inserted in between the CMV promoter and luciferase encoding region. The plasmid contains an SV40 origin, which enables it to be replicated in cells expressing SV40 large T antigen. B, chemical structures of compounds. Compound numbers (bold) and their biochemical IC50 for PCNA/PIP-box interaction inhibition (8) (in parenthesis, μm) are shown. C, dose-response of T2AA in the ICL repair inhibition assay. Light and dark gray bars represent lower and higher concentrations indicated in parentheses for each treatment. Error bars represent standard deviation (n = 4). Statistics analyzed by Student's t test: *, p < 0.05. D, inhibitory activity for ICL repair by the compound (20 μm) assayed together. Only two compounds, 2 (T2AA) and 6, were active; regardless, all were active for the PCNA/PIP-box interaction at this concentration. Error bars represent standard deviation (n = 4).
pGL4.50 contains an SV40 origin (residues 3155–3077) and a HindIII site between the CMV promoter region and the luciferase encoding region and was digested by HindIII according to the manufacturer's protocol and purified by agarose electrophoresis. A mixture of the purified linear pGL4.50 (10 μg) and the ICL-containing duplex oligonucleotide (11 μg, 300 eq) was ligated by T4 DNA ligase (1000 units) in 250 μl of T4 DNA ligase buffer at 16 °C for 18 h. The ligation mixture was then diluted to a total of 1500 μl in EcoRI buffer and digested by EcoRI (2500 units) at 37 °C for 20 h. The enzymes were deactivated by standard phenol/chloroform treatment. The mixture was concentrated by butanol extraction and purified by 0.8% (w/v) agarose gel electrophoresis. This EcoRI digestion/purification procedure is for removing any products that do not contain the ICL duplex, thus isolating only linear pGL4.50 in which both 5′ and 3′ ends were ligated by EcoRI-digested fragments of the ICL-containing duplex. The isolated product (i.e. EcoRI-digested pGL-ICL, 4.3 μg) was ligated by T4 DNA ligase (8640 units) in T4 DNA ligase buffer (1440 μl) at 16 °C for 18 h. The ligation mixture was heat-inactivated at 65 °C for 10 min, treated by phenol/chloroform, concentrated by butanol extraction, and purified by ethanol precipitation to isolate the pGL-ICL (3 μg). Small aliquots of the pGL-ICL product were analyzed as follows: 1) treated by HindIII; 2) treated by SalI; 3) SalI and duplex denatured by heating at 95 °C for 5 min and sudden chilling on ice. No digestion in 1) but complete digestion in 2) was observed, which indicated the presence of the ICL duplex insert (data not shown). The same band appearance in 2) and 3) indicated the presence of the ICL, which prohibited strand separation (data not shown). Under this heat and chill condition, the linear pGL4.50 afforded no band in the original location on the gel (data not shown), suggesting complete strand separation and denaturing.
ICL Repair Assay
COS7 cells (8 × 104 cells in DMEM) were seeded into a 24-well plate and allowed to attach for 5 h. The medium was replaced with fresh DMEM containing 1.5 times the final concentration of each indicated compound (200 μl). Immediately, the reporter transfection mixture (pGL4.50 (2.5 ng) or pGL-ICL (10 ng), pRL-TK (1 μg), FuGENE 6 (4 μl, Promega, Madison, WI), and Opti-MEM (96 μl) each per well, according to the manufacturer's recommendation) was added. After 4 h, cells were lifted by trypsin (100 μl), resuspended by adding fresh DMEM containing 1.5 times the final concentration of each indicated compound (200 μl), and replated into a 96-well plate (70 μl per well 4 times). After the cells were cultured for 14 h, the culture medium was removed, and activities of firefly luciferase and Renilla luciferase in the cells were measured by DLR reagent (Promega) in an Envision plate reader (PerkinElmer Life Sciences) according to the manufacturer's recommendation. The ICL repair activity was calculated by dividing the firefly luciferase/Renilla luciferase values from cells transfected by pGL-ICL by those from cells transfected by pGL4.50, which is indicated as 1 for that of cells treated by DMSO.
Neutral Comet Assay
The neutral comet assay was performed using a protocol detailed previously (14). U2OS cells (100,000 per well) were seeded into 6-well plates and allowed to attach overnight. Following exposure to 0 or 30 μm cisplatin for 1 h, the cells were washed with PBS and subsequently exposed to 0 or 15 μm T2AA for 72 h and harvested by trypsin. Cells were mixed with molten low gelling temperature agarose (type VII) and subsequently spread and allowed to set on a glass slide precoated with agarose. Gel-embedded cells were lysed in neutral lysis buffer (0.5% SDS (w/v) and 30 mm EDTA, pH 8.3) for 4 h at 50 °C and then subjected to three washes with TBE buffer (90 mm Tris, 90 mm boric acid, and 2.5 mm EDTA, pH 9.5) overnight. Samples were again submerged in TBE buffer and then subjected to electrophoresis (20 V) for 25 min. Following electrophoresis, samples were rinsed with Milli-Q water and then stained twice with SYBR Green for 5 min. The samples were then destained by rinsing with Milli-Q water for 15 min 3×. Comet tails were visualized using epi-fluorescence microscopy, and the level of DNA damage was measured using the scoring system described previously (15). At least 100 cells were scored per slide.
DR-GFP/I-SceI Recombination Assay
The protocol and reagents used in this assay were developed by Jasin and co-workers (16). HeLa cells stably transfected by a DR-GFP construct (6 × 105 cells in 2 ml of DMEM per well) were seeded into 6-well plates and allowed to attach overnight. Transfection mixture (pCBA-SceI (4 μg) and Lipofectamine 2000 (Invitrogen; used according to the manufacturer's recommendation, 12 μl) in Opti-MEM (total 488 μl)) was added, and the cells were cultured for 6 h. The cells were then trypsin-lifted, resuspended in DMEM, and replated into a 48-well plate (5 × 104 cells in 100 μl of DMEM per well), and 1.5 times the final concentrations of indicated compounds (200 μl) were added, followed by culturing for 72 h. The cells were then trypsin-lifted and filtered through 40-μm nylon mesh prior to running flow cytometry to count at least 10,000 viable single cells. The flow data were acquired using an LSRII flow cytometer equipped with 405, 488, 561, and 640 nm lasers (BD Biosciences). The population of singlet GFP-positive cells was reported as HR%.
Clonogenic Survival Assay
After HeLa (450 cells/well) or U2OS cells (300 cells/well) were seeded into 6-well plates and allowed to attach overnight, each indicated final concentration of cisplatin was added. After 24 h, a 10 μm final concentration of T2AA was added. After another 24 h, the culture medium was replaced with fresh medium, and cells were cultured for another 6 (HeLa) or 7 (U2OS) days. Each treatment was done in triplicate. The cells were gently rinsed twice with PBS, fixed with 3.7% formaldehyde (w/v), and stained by 0.5% crystal violet (w/v). Colonies containing more than ∼40 cells were counted. The survival rate was reported as 1 for the average of colony numbers in wells that received vehicle control (0.9% sodium chloride (w/v) for cisplatin and DMSO for T2AA).
RESULTS
T2AA Binds to PCNA at the PIP-box Cavity and Adjacent to Lys-164
Previously, we determined the crystal structure of PCNA/triiodothyronine (T3, Fig. 3B) in a 1:1 complex and found that T3 specifically binds only to the cavity of PCNA for interacting with PIP-box ligands and generated T2AA (Fig. 3B), a T3 analog that inhibits PCNA/PIP-box interactions (5). PCNA functions are not solely dependent on the PIP-box interaction but also on other protein/protein interactions mediated by post-translational modifications such as ubiquitination (4). Thus, we investigated whether T2AA could interfere with PCNA interactions other than PIP-box interactions by determining the crystal structure of the PCNA-T2AA complex. Unexpectedly, we found that T2AA binds to PCNA in a 2:1 ratio (Fig. 1A and Table 1). The binding mode of T2AA to the PIP-box cavity (Fig. 1B) is almost identical to that of T3, which is consistent with biochemical equipotency of T3 and T2AA for inhibiting PCNA/PIP-box interactions (5). In addition, T2AA binds also to a second site adjacent to Lys-164 on the PCNA homotrimer interface (Fig. 1D), a well known site for RAD18-mediated monoubiquitination upon DNA damage (4). The two aromatic rings of T2AA bind to a cavity on the homotrimer interface, and this was verified by the electron density map (Fig. 1C). One of the two iodine atoms on one aromatic ring of T2AA was accommodated in a small hydrophobic groove composed of the aliphatic portion of the Lys-181 side chain and Val-111 on another PCNA monomer, thereby binding to two subunits of the PCNA homotrimer. Another iodine atom was facing toward Asp-165 next to the Lys-164 monoubiquitination site. Another aromatic ring of T2AA was adopted by a hydrophobic interaction to the aliphatic portions of the Lys-181 and Lys-168 on the same subunit. The amino group of T2AA was close to Glu-109 on another PCNA molecule, which was possibly interacting via a salt bridge. These interactions of T2AA allow it to be located close to the Lys-164 (see Fig. 6). Interestingly, this homotrimer interface cavity is neither deep nor entirely hydrophobic as is the PIP-box cavity; regardless, both of them adopt the two aromatic rings of T2AA in a similar binding mode (Fig. 1D). This suggests that the affinity of T2AA is not optimal for this cavity, and thus a higher concentration of T2AA would be needed to inhibit PCNA interactions around this region than is needed for inhibiting PIP-box interactions.
FIGURE 1.

T2AA binds to PCNA in a bimolecular fashion. A, crystal structure of PCNA-T2AA complex. One T2AA molecule binds to a deep cavity that interacts with the PIP-box to which T3 binds (black box) (5). Another T2AA molecule binds to a shallow cavity on the PCNA homotrimer interface adjacent to Lys-164 (red box). B, close-up view (stereo) of the interaction of T2AA to the PCNA PIP-box adopting cavity. C, T2AA on the PCNA homotrimer interface (stick model) is superimposed to the Fo − Fc electron density map (blue cage) contoured at 3.0 σ. D, close-up view (stereo) of the interaction of T2AA with the PCNA homotrimer interface.
FIGURE 6.
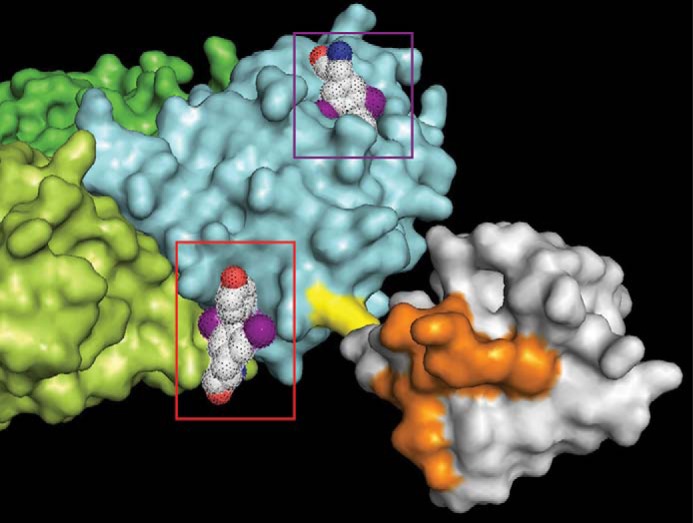
Model of T2AA binding to Lys-164-monoubiquitinated PCNA. The PCNA/T2AA cocrystal structure (Fig. 1) was merged with a crystal structure of Lys-164-monoubiquitinated PCNA (31) by SuperPose 1.0 (50). Cyan, lime, and green, each PCNA monomer; yellow, Lys-164; gray, ubiquitin; orange, binding surface to pol η UBZ domain (32). T2AA bound to the PIP-box adopting cavity is indicated in the violet box, and T2AA bound to the PCNA homotrimer interface is indicated in the red box, each mesh-dotted space-filling model; red, oxygen; blue, nitrogen; violet, iodine. The T2AA on the homotrimer interface is close to the Lys-164 and on the same side of the pol η UBZ-binding surface on the Lys-164-ubiquitin.
T2AA Inhibits Interactions of Monoubiquitinated PCNA but Not PCNA Monoubiquitination
We assayed T2AA for interaction of the monoubiquitinated PCNA with a protein that preferentially binds to monoubiquitinated PCNA over nonubiquitinated PCNA. pol η has been characterized extensively for its bidentate interaction with monoubiquitinated PCNA via its UBZ domain and PIP-box (17). We have produced a truncated pol η protein containing a UBZ domain and a PIP-box that reportedly binds to monoubiquitinated PCNA preferentially (18–20), and we performed a pulldown assay for monoubiquitinated PCNA in the presence of T2AA. T2AA clearly prevented isolation of monoubiquitinated PCNA in the precipitate in a dose-dependent manner, whereas T3 did not (Fig. 2B).
FIGURE 2.

T2AA inhibits interaction of PCNA to pol η or REV1 but does not inhibit RAD18-mediated PCNA monoubiquitination or Fanconi anemia pathway activation upon UV irradiation. A, GM04312 cells induce monoubiquitinated PCNA by UV light in higher level than do U2OS cells. The indicated cells were irradiated by UVC in the indicated dose and cultured for 6 h. The cells were then collected and lysed by sonication in RIPA buffer. The lysates were analyzed by immunoblotting for PCNA. B, pulldown assay using immobilized His8-pol η(551–713) fragment and chromatin fraction of GM04312 cells irradiated by UVC (60 J/m2). The nonubiquitinated PCNA was not significantly isolated with any precipitates after washing (20). His8-pol η(551–713) served as a loading control. Densitometry quantification was performed by dividing band intensity of the monoubiquitinated PCNA by that of His8-pol η(551–713) for each lane, and values normalized to DMSO control are indicated. Chromatin co-foci of PCNA/EGFP-pol η (C) and PCNA/EGFP-REV1 (D) in U2OS cells that were treated by cisplatin (33 μm, 2 h) followed by incubation with DMSO or the indicated compound (Fig. 3B, 20 μm) for 6 h. Immunostaining for PCNA on pre-extracted cells was performed. Confocal images of representative cells treated with or without T2AA are shown. The scale bar is 10 μm. Colocalization of the indicated foci was determined by calculating Pearson's correlation coefficients between red and green signals of all the pixels within a DAPI-positive area for more than 50 cells. The dots in the graph indicate the correlation coefficient of individual cells. Statistics analyzed by Student's t test are as follows: ****, p < 0.0001. The bars in the graph indicate averages. E, recruitment of RAD18 to PCNA is not affected by T2AA. U2OS cells that were not treated (NT) or treated by cisplatin (33 μm, 2 h) were followed by incubation with DMSO or T2AA (20 μm) for 6 h. Immunostaining and colocalization analysis were performed as described above. F, indicated cells were irradiated by UVC (60 J/m2) and incubated with DMSO or T2AA (20 μm) for 8 h. The cells were extensively pre-extracted by cold PBS supplemented with 0.5% Triton X-100 and 5 mm N-ethylmaleimide (deubiquitinase inhibitor) for 5 min to remove nonchromatin proteins (note that monoubiquitinated (mUb) PCNA is exclusively on chromatin). The cells were then collected and lysed by sonication in RIPA buffer. The lysates were analyzed by immunoblotting for PCNA. Recombinant PCNA protein served as a control. G, HeLa cells were irradiated by UVC (60 J/m2) and incubated with indicated agents at 20 μm for 5 h. The cells were collected and lysed by sonication in RIPA buffer. The lysates were analyzed by immunoblotting for FANCD2. The actin band was used as a loading control.
To determine whether T2AA inhibits interactions of monoubiquitinated PCNA on chromatin, we analyzed chromatin-PCNA colocalization of pol η and REV1, TLS polymerases that preferentially bind to and are functionally facilitated by monoubiquitinated PCNA (21, 22). U2OS cells were transfected with either EGFP-pol η or EGFP-REV1, treated with cisplatin to induce nuclear foci localization, and cultured with or without T2AA. Chromatin colocalization with PCNA was imaged and quantified relative to the T2AA treatment. Clearly, the PCNA and the EGFP-pol η or EGFP-REV1 was colocalized in the untreated cells but not in the cells cultured with T2AA (Fig. 2, C and D). This effect was also observed with compound 6, which was active for ICL repair inhibition, but not with compound 1, which was inactive for ICL repair inhibition (Fig. 3, B and D). This suggests that T2AA decreased interactions of pol η and REV1 with PCNA that was likely Lys-164-monoubiquitinated on chromatin upon damage to DNA by cisplatin, which could be a key mechanism of the ICL repair inhibition of T2AA.
To determine whether T2AA affects PCNA monoubiquitination, we exposed cells to UVC to trigger PCNA monoubiquitination and cultured the cells with T2AA. The amount of monoubiquitinated PCNA species in the chromatin-bound protein fractions was not reduced by T2AA, suggesting that T2AA does not inhibit PCNA monoubiquitination (Fig. 2F). RAD18 is an E3 ligase for PCNA monoubiquitination. To verify T2AA's effect of PCNA/RAD18 interaction, their chromatin colocalization was analyzed after cells were treated with cisplatin, T2AA, or both. RAD18 foci were significantly up-regulated by cisplatin, indicating a cisplatin-mediated DNA damage response. PCNA foci were formed modestly by cisplatin and significantly by T2AA, indicating DNA replication stalling. The RAD18 foci and PCNA foci were colocalized in the cells treated with both cisplatin and T2AA (Fig. 2E). This suggests that T2AA does not inhibit RAD18 recruitment to PCNA upon cisplatin treatment. RAD18-mediated PCNA Lys-164 monoubiquitination reportedly activates the Fanconi anemia pathway (23). FANCD2 monoubiquitination was not inhibited by T2AA (Fig. 2G).
T2AA Inhibited ICL Repair
Most cisplatin-induced DNA cross-links consist of intrastrand cross-links that are repaired by NER or tolerated by TLS. A plasmid reactivation assay with a reporter plasmid containing an SV40 origin and nonspecific cisplatin adducts measures NER activity when assayed in cells lacking SV40 large T antigen and TLS activity when assayed in cells expressing T antigen but deficient in NER. Using this assay, we previously found that T2AA inhibited TLS but not NER (5, 8). The more chemotherapeutically crucial cisplatin-induced DNA lesion is the ICL. ICL removal is believed to be a multistep process in which NER, TLS, and HR are sequentially executed (6). Therefore, we anticipated that T2AA would inhibit ICL repair as well. To validate this, we used a reporter plasmid that site-specifically contains an ICL (Fig. 3A). To enable scalable production of the plasmid for assaying numbers of compounds, we used click chemistry to generate the ICL reliably and site-specifically in an oligonucleotide duplex. Then we inserted the ICL duplex between the CMV promoter and luciferase coding regions of the pGL plasmid, which contains an SV40 origin (24) that enables replication driven by large T antigen. The generated pGL-ICL plasmid or corresponding intact pGL plasmid was transfected into COS7 cells (expressing T antigen), and the cells were cultured to allow ICL repair and reporter expression. The luciferase signal from pGL-ICL represents the activity of the ICL repair-dependent reporter expression, and the signal from the intact pGL represents the reporter expression only. Thus, ICL repair activity is indicated by dividing the former by the latter. T2AA inhibited ICL repair in a dose-dependent manner in this assay (Fig. 3C). However, when we assayed several inhibitors of PCNA/PIP-box interaction (8), we observed little inhibitory activity in those compounds except for T2AA (compound 2) and an analog that is structurally close to T2AA (compound 6), regardless of their efficacy for inhibiting PCNA/PIP-box interaction (Fig. 3, B and D). This indicates that the ICL repair in this assay is not entirely dependent on PCNA/PIP-box interaction and that T2AA is unique in its ability to inhibit ICL repair.
T2AA Promoted DSB Formation by Cisplatin
Given that T2AA inhibits ICL repair, it could prevent resolution of DSBs formed as an intermediate of ICL repair (6); thereby, DSBs could persist in the presence of T2AA. To investigate this possibility, we performed a neutral comet assay that is commonly used to detect DSBs in genomic DNA. When U2OS cells were treated by cisplatin only or T2AA only, the comet tail formation was almost unchanged from background level. However, when cells were treated by cisplatin followed by T2AA, comet tail formation was very significant, indicating that T2AA induced collapse of repair of ICL that was induced by the cisplatin (Fig. 4A). Ser-1524-phosphorylated BRCA1 was significantly up-regulated when U2OS cells received T2AA in addition to cisplatin (Fig. 4B), suggesting that T2AA enhances ATM-BRCA1 pathway activity resulting from DSB induction (25). Inhibition of HR by Rad51 depletion reportedly led to colocalization of phospho-ATM and 53BP1 (26), a marker for DSBs more specific than phospho(Ser-139)-histone H2AX. We previously found that T2AA dramatically up-regulates phospho(Ser-139)-histone H2AX induction mediated by cisplatin in HeLa cells (5). To determine whether this response is due to DSB persistence, we analyzed formation of the chromatin colocalized foci for DSBs. In cisplatin-treated HeLa cells, formation of the colocalized foci was further promoted by T2AA (Fig. 4C), implicating promotion of DSB induction in the inhibition of ICL repair by T2AA. PCNA is an essential component of recombination-associated DNA synthesis during extension of the D-loop in the process of DSB repair, which is based on interactions of PCNA with pol δ or pol η via PIP-box interaction (27–29). We assayed T2AA in the DR-GFP/I-SceI recombination assay (16) and observed inhibition activity (Fig. 4D). Taken together, these findings show T2AA's ability to inhibit DSB repair.
FIGURE 4.

T2AA induces DSB response. A, DSB quantification by neutral comet assay. U2OS cells were exposed to cisplatin (30 μm) for 1 h followed by DMSO or T2AA (15 μm) and cultured for 72 h. Cell electrophoresis and comet tail scoring were performed. Representative comet tail images and the comet tail score for each treatment are shown. B, phospho-Ser-1524 BRCA up-regulation in U2OS cells after being treated as indicated for 3 days. Actin served as a loading control. C, chromatin co-foci of phospho-Ser-1981 ATM and 53BP1 in HeLa cells were examined by treating cells with or without cisplatin (15 μm) for 1 h followed by DMSO or T2AA (15 μm) and culturing for 72 h. Cells were immunostained with anti-phospho-Ser-1981-ATM (green) and anti-53BP1 (red) antibodies. Frequency of positive colocalization (percentages of cells exhibiting more than 10 yellow foci in the nucleus (arrows) on confocal images) was 42% for cisplatin/T2AA cotreatment, 0% for other treatments. D, recombination activity assay as described by Jasin and co-workers (16) after cells were cultured with DMSO or T2AA (10 μm) for 72 h.
T2AA Sensitizes Cancer Cells to Cisplatin in Clonogenic Survival
Finally, to validate the chemotherapeutic utility of T2AA for DNA damage enhancement, HeLa and U2OS cells were subjected to a clonogenic survival assay. Cells were first treated with cisplatin followed by T2AA. The colony numbers were counted after the cells were released from the treatment. As T2AA arrests cells in the S-phase (5), the T2AA treatment delayed the growth cycle for the colony, but the plating efficiency was little changed. In contrast, when cells were treated with cisplatin and T2AA, their plating efficiency was significantly less than that of cells treated with cisplatin only (Fig. 5).
FIGURE 5.

T2AA sensitizes cells to cisplatin. Clonogenic survival of HeLa (A) or U2OS (B) cells after cisplatin treatment (2 days) with or without T2AA (10 μm, 1 day). Error bars represent standard deviation (n = 3).
DISCUSSION
Previously, we determined the crystal structure of a PCNA-T3 complex in which the T3 molecules were not found in the homotrimer interface (5). This indicates that the structural difference between T2AA and T3 is a determinant for the interface binding. T3 is an amino acid, and T2AA is an amino alcohol in which the amino group is basic. On the PCNA homotrimer interface, the amino group of T2AA is close to Glu-109, possibly forming a salt bridge interaction, which is impossible for T3 due to an intramolecular zwitterion. The small size of the groups around the amino group could be another signature for this interaction, which possibly reduces the steric effect preventing it. Another possible key interaction is that of the phenol group of T2AA and Lys-167. In the T3 compound, there is a large iodine atom adjacent to the phenol, which could hinder this possible interaction.
Numerous studies have attempted to determine the structural outcome of interactions of the monoubiquitin moiety on PCNA Lys-164, as this interaction is important for divergent DNA damage responses, including TLS and Fanconi anemia pathway activation. Several studies have suggested that the ubiquitin moiety on PCNA is quite flexible, as shown by observations that even a linear artificial PCNA-ubiquitin chimera preferentially binds to REV1 (30). However, RAD18-mediated PCNA monoubiquitination is specific to Lys-164 regardless that there is another lysine nearby (Lys-167); therefore, some structural outcome specific to the Lys-164-monoubiquitin could be crucial for the DNA damage response in the natural endogenous system. Recently, the crystal structure of bona fide Lys-164-monoubiquitinated PCNA was determined (31). In this structure, the monoubiquitin surface with which the pol η UBZ domain interacts (32) is facing toward the side of the PCNA homotrimer interface, where T2AA binds. Therefore, T2AA could hinder interaction of the UBZ domain to the protein surface composed of the Lys-164-monoubiquitin and PCNA (Fig. 6). This hypothesis could be verified when the crystal structure of the Lys-164-monoubiquitinated PCNA/pol η UBZ domain complex is determined.
Previously, we found that PCNA/PIP-box interaction inhibitors inhibit TLS assayed by reactivation of pGL plasmid that was nonspecifically damaged by cisplatin in NER-null cells (8). Most of the DNA damage by cisplatin consists of base monoalkylations or intrastrand cross-links. Thus, most of the response observed in the assay should have been TLS for intrastrand cross-links but not for ICL repair. Recent studies suggested that TLS polymerases for ICL repair are different from those for intrastrand cross-link tolerance (26). Both pol η and REV1 are regarded as TLS polymerases that are functionally supported by Lys-164-monoubiquitinated PCNA, but their importance in ICL repair is quite different. pol η is well known as highly efficient for TLS over thymidine dimer intrastrand cross-links; however, it is not essential for ICL repair in vitro (33). In contrast, REV1 is a deoxycytidyltransferase rather than a DNA polymerase, and it is essential for ICL repair (33), where it presumably engages by coordinating the REV7 subunit of pol ζ (34, 35).
From the viewpoint of PCNA interaction, the largest difference between pol η and REV1 is their PIP-box dependence. pol η has PIP-boxes that are functionally essential (17). REV1 does not have a PIP-box but a BRCT domain that interacts to PCNA at the PIP-box-interacting cavity (36) and two UBM domains and preferentially binds to monoubiquitinated PCNA (30, 37). Therefore, only small molecules that inhibit interactions of monoubiquitinated PCNA, such as T2AA (Fig. 1A), could inhibit REV1 functions and thus inhibit ICL repair. However, we cannot eliminate a possibility that the effects of T2AA could be solely due to inhibition of the PIP-box, although it is unlikely, because T3 failed to efficiently inhibit interaction of monoubiquitinated PCNA to the pol η UBZ domain/PIP-box (Fig. 2A) even though it inhibits the PIP-box interaction (5). We attempted to investigate this possibility by performing a pulldown inhibition assay for monoubiquitinated PCNA by using baits lacking a PIP-box. Unfortunately, neither the pol η UBZ domain/PIP-box, in which the PIP-box was inactivated by mutation (18), nor the UBM1-UBM2 domains of REV1 efficiently precipitated the monoubiquitinated PCNA (data not shown), preventing the assay of compounds directly for PIP-box-independent interactions of the monoubiquitinated PCNA.
T2AA's effect for chromatin PCNA co-foci inhibition was stronger for REV1 than for pol η (Fig. 2, B and C), suggesting that T2AA could have inhibited REV1 functions. It is unknown why T2AA inhibited PCNA co-foci with REV1 more efficiently than with pol η. It could be due to different affinities of pol η and REV1 for the monoubiquitinated PCNA homotrimer. Yeast REV1 reportedly binds to PCNA on the homotrimer region by its PAD domain (38). If this interaction is also valid in human REV1, a small molecule that binds to the PCNA homotrimer region, such as T2AA, could efficiently inhibit REV1 functions.
Studies have shown that ICL repair is a process that requires REV1 and pol ζ (33, 39) or pol κ, which requires Lys-164-monoubiquitinated PCNA to interact with its UBZ domain (40). Given that the REV7 subunit of pol ζ is essential in ICL repair (7), the functions of REV1 and pol ζ, which are facilitated by Lys-164-monoubiquitinated PCNA, could be targeted for efficiently inhibiting ICL repair.
There are several mechanistic possibilities for increasing DSB response by a cisplatin/T2AA combination. Inhibition of ICL repair could promote a transient DSB intermediate by endonucleases (41) such as Mus81-Eme1 (42). In the accepted ICL repair mechanism, the ICL is first unhooked to form a single-stranded DNA with an overhang that was the ICL. Subsequently, possibly REV1 and pol ζ extend the excised strand by TLS (7). Inhibition of REV1 and pol ζ functions thus may trigger a further attempt to remove the overhang by excising it on the single strand, which results in a DSB intermediate. Furthermore, because REV1 and pol ζ also facilitate HR (43), inhibition of their functions could inhibit HR as well, leading to a failure to resolve the DSB by recombination. pol ζ function also could be partly due to a PCNA/PIP-box interaction, because pol ζ needs p66, a subunit of pol δ3 that interacts with PCNA by its PIP-box, to complete the TLS (44–46).
We (5, 8) and other groups (47) have shown that PCNA is a possible drug target for chemosensitization by interfering with its numerous functions for DNA damage response. One of the characteristic features of PCNA is that it is homotrimeric on DNA, and each monomer has at least three functional sites and residues as follows: the PIP-box cavity, Lys-164, and Lys127 (4). We previously showed that inhibiting PCNA/PIP-box interaction was sufficient for inhibiting TLS on a nonspecifically damaged plasmid DNA, in which most DNA damage should consist of base alkylation and intrastrand cross-link. As long as PCNA/PIP-box interaction inhibitors can suppress TLS for such damage, they can chemosensitize cells to alkylation drugs to some extent. However, ICL is a much more lethal form of DNA damage, and ICL repair inhibition should be much more efficient for chemosensitization; therefore, a compound that inhibits the monoubiquitinated PCNA interactions can be more effective. Indeed, we previously observed that T2AA is much more efficacious for sensitizing U2OS cells for cisplatin in cell growth than other PCNA/PIP-box inhibitors regardless of their similar efficacy in TLS inhibition (8). Several studies have shown that, among TLS polymerases, REV1 and pol ζ but not others are needed for ICL repair (26, 33). The role of PCNA Lys-164-monoubiquitination in the functional facilitation of REV1 has been shown (22). It is possible that, after the NER step in the ICL repair process, REV1 and pol ζ are recruited to the monoubiquitinated PCNAs. In this scenario, interaction of the PCNA homotrimer interface should be an attractive target for inhibiting ICL repair.
It would be worthwhile to investigate whether compounds that bind to the Lys-164-monoubiquitination site but not the PIP-box interaction site are effective for chemosensitization and selectively inhibit ICL repair over DNA replication. Although this could be unlikely, given that proteins interacting with Lys-164-monoubiquitinated PCNA that have been discovered to date commonly have both a PIP-box and a ubiquitin-binding domain (48, 49), there may be other proteins that are important to the repair of ICLs and DSBs and that interact with Lys-164-monoubiquitinated PCNA solely by the ubiquitin-binding domain. Generating inhibitors specific to the PCNA homotrimer interface could address this possibility. The structure of the T2AA interaction with the PCNA homotrimer interface suggests that the affinity of this interaction is not optimal, and thus there is some room to increase the interface affinity, for instance, by targeting Asp-165 by changing one of the iodine atoms of T2AA (Fig. 1D).
Acknowledgments
We thank Dr. Scott Perry for flow cytometry support; Dr. Jenifer Peters for light microscopy support; Dr. Maria Jasin (Memorial Sloan-Kettering Cancer Center) for pCBA-SceI plasmid; Dr. Roger Greenberg (University of Pennsylvania) for HeLa (DR-GFP) cells; Dr. Toshiyasu Taniguchi (Fred Hutchinson Cancer Research Center) for helpful discussions; The Sanger DNA Sequencing core for plasmid DNA sequencing; the beamline staff at Photon Factory for the kind support for data collection; and David Galloway for grammatical advice and preparation of this manuscript.
This work was supported by the American Lebanese Syrian Associated Charities and American Cancer Society Research Scholar Grant RSG CDD-120969 (to N. F.), and KAKENHI from MEXT, Japan (to A. H. and H. H.).
The atomic coordinates and structure factors (code 3WGW) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PCNA
- proliferating cell nuclear antigen
- HR
- homologous recombination
- ICL
- interstrand DNA cross-link
- NER
- nucleotide excision repair
- PIP-Box
- PCNA interacting protein box
- T2AA
- T2 amino alcohol
- T3
- 3,3′,5-triiodothyronine
- TLS
- translesion DNA synthesis
- pol
- polymerase
- DSB
- double strand break
- EGFP
- enhanced GFP
- ATM
- ataxia telangiectasia mutated.
REFERENCES
- 1. Moldovan G. L., Pfander B., Jentsch S. (2007) PCNA, the maestro of the replication fork. Cell 129, 665–679 [DOI] [PubMed] [Google Scholar]
- 2. Sale J. E., Lehmann A. R., Woodgate R. (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waters L. S., Minesinger B. K., Wiltrout M. E., D'Souza S., Woodruff R. V., Walker G. C. (2009) Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73, 134–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mailand N., Gibbs-Seymour I., Bekker-Jensen S. (2013) Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 14, 269–282 [DOI] [PubMed] [Google Scholar]
- 5. Punchihewa C., Inoue A., Hishiki A., Fujikawa Y., Connelly M., Evison B., Shao Y., Heath R., Kuraoka I., Rodrigues P., Hashimoto H., Kawanishi M., Sato M., Yagi T., Fujii N. (2012) Identification of small molecule proliferating cell nuclear antigen (PCNA) inhibitor that disrupts interactions with PIP-box proteins and inhibits DNA replication. J. Biol. Chem. 287, 14289–14300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deans A. J., West S. C. (2011) DNA interstrand cross-link repair and cancer. Nat. Rev. Cancer 11, 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Räschle M., Knipscheer P., Knipsheer P., Enoiu M., Angelov T., Sun J., Griffith J. D., Ellenberger T. E., Schärer O. D., Walter J. C. (2008) Mechanism of replication-coupled DNA interstrand cross-link repair. Cell 134, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Actis M., Inoue A., Evison B., Perry S., Punchihewa C., Fujii N. (2013) Small molecule inhibitors of PCNA/PIP-box interaction suppress translesion DNA synthesis. Bioorg. Med. Chem. 21, 1972–1977 [DOI] [PubMed] [Google Scholar]
- 9. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 11. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 12. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 13. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 14. Olive P. L., Wlodek D., Banáth J. P. (1991) DNA double strand breaks measured in individual cells subjected to gel electrophoresis. Cancer Res. 51, 4671–4676 [PubMed] [Google Scholar]
- 15. Collins A. R. (2004) The comet assay for DNA damage and repair: principles, applications, and limitations. Mol. Biotechnol. 26, 249–261 [DOI] [PubMed] [Google Scholar]
- 16. Pierce A. J., Johnson R. D., Thompson L. H., Jasin M. (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13, 2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kannouche P. L., Wing J., Lehmann A. R. (2004) Interaction of human DNA polymerase η with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 14, 491–500 [DOI] [PubMed] [Google Scholar]
- 18. Durando M., Tateishi S., Vaziri C. (2013) A non-catalytic role of DNA polymerase eta in recruiting Rad18 and promoting PCNA monoubiquitination at stalled replication forks. Nucleic Acids Res. 41, 3079–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Despras E., Delrieu N., Garandeau C., Ahmed-Seghir S., Kannouche P. L. (2012) Regulation of the specialized DNA polymerase η: revisiting the biological relevance of its PCNA- and ubiquitin-binding motifs. Environ. Mol. Mutagen. 53, 752–765 [DOI] [PubMed] [Google Scholar]
- 20. Watanabe K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., Yamaizumi M. (2004) Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 23, 3886–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garg P., Burgers P. M. (2005) Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases η and REV1. Proc. Natl. Acad. Sci. U.S.A. 102, 18361–18366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wood A., Garg P., Burgers P. M. (2007) A ubiquitin-binding motif in the translesion DNA polymerase Rev1 mediates its essential functional interaction with ubiquitinated proliferating cell nuclear antigen in response to DNA damage. J. Biol. Chem. 282, 20256–20263 [DOI] [PubMed] [Google Scholar]
- 23. Geng L., Huntoon C. J., Karnitz L. M. (2010) RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J. Cell Biol. 191, 249–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tseng B. Y., Ahlem C. N. (1984) Mouse primase initiation sites in the origin region of simian virus 40. Proc. Natl. Acad. Sci. U.S.A. 81, 2342–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cortez D., Wang Y., Qin J., Elledge S. J. (1999) Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double strand breaks. Science 286, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 26. Hicks J. K., Chute C. L., Paulsen M. T., Ragland R. L., Howlett N. G., Guéranger Q., Glover T. W., Canman C. E. (2010) Differential roles for DNA polymerases η, ζ, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol. Cell. Biol. 30, 1217–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sebesta M., Burkovics P., Juhasz S., Zhang S., Szabo J. E., Lee M. Y., Haracska L., Krejci L. (2013) Role of PCNA and TLS polymerases in D-loop extension during homologous recombination in humans. DNA Repair 12, 691–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J., Holzschu D. L., Sugiyama T. (2013) PCNA is efficiently loaded on the DNA recombination intermediate to modulate polymerase δ, η, and ζ activities. Proc. Natl. Acad. Sci. U.S.A. 110, 7672–7677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X., Stith C. M., Burgers P. M., Heyer W. D. (2009) PCNA is required for initiation of recombination-associated DNA synthesis by DNA polymerase δ. Mol. Cell 36, 704–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo C., Sonoda E., Tang T. S., Parker J. L., Bielen A. B., Takeda S., Ulrich H. D., Friedberg E. C. (2006) REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell 23, 265–271 [DOI] [PubMed] [Google Scholar]
- 31. Zhang Z., Zhang S., Lin S. H., Wang X., Wu L., Lee E. Y., Lee M. Y. (2012) Structure of monoubiquitinated PCNA: implications for DNA polymerase switching and Okazaki fragment maturation. Cell Cycle 11, 2128–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bomar M. G., Pai M. T., Tzeng S. R., Li S. S., Zhou P. (2007) Structure of the ubiquitin-binding zinc finger domain of human DNA Y-polymerase η. EMBO Rep. 8, 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Enoiu M., Jiricny J., Schärer O. D. (2012) Repair of cisplatin-induced DNA interstrand cross-links by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 40, 8953–8964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wojtaszek J., Lee C. J., D'Souza S., Minesinger B., Kim H., D'Andrea A. D., Walker G. C., Zhou P. (2012) Structural basis of Rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polymerase (pol) ζ, and pol κ. J. Biol. Chem. 287, 33836–33846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kikuchi S., Hara K., Shimizu T., Sato M., Hashimoto H. (2012) Structural basis of recruitment of DNA polymerase ζ by interaction between REV1 and REV7 proteins. J. Biol. Chem. 287, 33847–33852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pustovalova Y., Maciejewski M. W., Korzhnev D. M. (2013) NMR mapping of PCNA interaction with translesion synthesis DNA polymerase Rev1 mediated by Rev1-BRCT domain. J. Mol. Biol. 425, 3091–3105 [DOI] [PubMed] [Google Scholar]
- 37. Guo C., Tang T. S., Bienko M., Parker J. L., Bielen A. B., Sonoda E., Takeda S., Ulrich H. D., Dikic I., Friedberg E. C. (2006) Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol. Cell. Biol. 26, 8892–8900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sharma N. M., Kochenova O. V., Shcherbakova P. V. (2011) The non-canonical protein binding site at the monomer-monomer interface of yeast proliferating cell nuclear antigen (PCNA) regulates the Rev1-PCNA interaction and Polzeta/Rev1-dependent translesion DNA synthesis. J. Biol. Chem. 286, 33557–33566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sarkar S., Davies A. A., Ulrich H. D., McHugh P. J. (2006) DNA interstrand cross-link repair during G1 involves nucleotide excision repair and DNA polymerase ζ. EMBO J. 25, 1285–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Williams H. L., Gottesman M. E., Gautier J. (2012) Replication-independent repair of DNA interstrand cross-links. Mol. Cell 47, 140–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sengerová B., Wang A. T., McHugh P. J. (2011) Orchestrating the nucleases involved in DNA interstrand cross-link (ICL) repair. Cell Cycle 10, 3999–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hanada K., Budzowska M., Modesti M., Maas A., Wyman C., Essers J., Kanaar R. (2006) The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA cross-links into double-strands breaks. EMBO J. 25, 4921–4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sharma S., Hicks J. K., Chute C. L., Brennan J. R., Ahn J. Y., Glover T. W., Canman C. E. (2012) REV1 and polymerase ζ facilitate homologous recombination repair. Nucleic Acids Res. 40, 682–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baranovskiy A. G., Lada A. G., Siebler H. M., Zhang Y., Pavlov Y. I., Tahirov T. H. (2012) DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. J. Biol. Chem. 287, 17281–17287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Makarova A. V., Stodola J. L., Burgers P. M. (2012) A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 40, 11618–11626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johnson R. E., Prakash L., Prakash S. (2012) Pol31 and Pol32 subunits of yeast DNA polymerase δ are also essential subunits of DNA polymerase ζ. Proc. Natl. Acad. Sci. U.S.A. 109, 12455–12460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tan Z., Wortman M., Dillehay K. L., Seibel W. L., Evelyn C. R., Smith S. J., Malkas L. H., Zheng Y., Lu S., Dong Z. (2012) Small-molecule targeting of proliferating cell nuclear antigen chromatin association inhibits tumor cell growth. Mol. Pharmacol. 81, 811–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moldovan G. L., Dejsuphong D., Petalcorin M. I., Hofmann K., Takeda S., Boulton S. J., D'Andrea A. D. (2012) Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol. Cell 45, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Centore R. C., Yazinski S. A., Tse A., Zou L. (2012) Spartan/C1orf124, a reader of PCNA ubiquitylation and a regulator of UV-induced DNA damage response. Mol. Cell 46, 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maiti R., Van Domselaar G. H., Zhang H., Wishart D. S. (2004) SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 32, W590–W594 [DOI] [PMC free article] [PubMed] [Google Scholar]