

Background: PMCA4 actions in vascular smooth muscle are not understood.
Results: Alternative splicing of PMCA4 changes during vessel injury. Cell cycle arrest and downstream effectors of PMCA4 deletion are rescued by PMCA4a, PMCA4b, and PMCA4b unable to bind PDZ- proteins but not by inactive PMCA4.
Conclusion: Ca2+ efflux activity of PMCA4 regulates G1 progression.
Significance: PMCA4 Ca2+ efflux regulates cell cycle.
Keywords: Alternative Splicing, Calcium ATPase, Cell Cycle, Cell Proliferation, Vascular Smooth Muscle Cells, AP-2β, Decorin, Plasma Membrane Calcium Atpase-4, Rgs16, p15(Ink4b)
Abstract
We explored the role played by plasma membrane calcium ATPase-4 (PMCA4) and its alternative splice variants in the cell cycle of vascular smooth muscle cells (VSMC). A novel variant (PMCA4e) was discovered. Quantitative real-time-PCR-quantified PMCA4 splice variant proportions differed in specific organs. The PMCA4a:4b ratio in uninjured carotid arteries (∼1:1) was significantly reduced by wire denudation injury (to ∼1:3) by modulation of alternative splicing, as confirmed by novel antibodies against PMCA4a/e and PMCA4b. Laser capture microdissection localized this shift to the media and adventitia. Primary carotid VSMC from PMCA4 knock-out (P4KO) mice showed impaired [3H]thymidine incorporation and G1 phase arrest as compared with wild type (P4WT). Electroporation of expression constructs encoding PMCA4a, PMCA4b, and a PMCA4b mutant lacking PDZ binding rescued this phenotype of P4KO cells, whereas a mutant with only 10% of normal Ca2+ efflux activity could not. Microarray of early G1-synchronized VSMC showed 39-fold higher Rgs16 (NFAT (nuclear factor of activated T-cells) target; MAPK inhibitor) and 69-fold higher Decorin (G1 arrest marker) expression in P4KO versus P4WT. Validation by Western blot also revealed decreased levels of Cyclin D1 and NFATc3 in P4KO. Microarrays of P4KO VSMC rescued by PMCA4a or PMCA4b expression showed reversal of perturbed Rgs16, Decorin, and NFATc3 expression levels. However, PMCA4a rescue caused a 44-fold reduction in AP-2β, a known anti-proliferative transcription factor, whereas PMCA4b rescue resulted in a 50-fold reduction in p15 (Cyclin D1/Cdk4 inhibitor). We conclude that Ca2+ efflux activity of PMCA4 underlies G1 progression in VSMC and that PMCA4a and PMCA4b differentially regulate specific downstream mediators.
Introduction
Progression through the cell cycle involves Ca2+ transporters including Ca2+ channels and Ca2+ efflux pumps (for review, see Ref. 1), and sarco-endoplasmic reticulum Ca2+ ATPase-2 (SERCA2) splice variants are differentially expressed at various stages of the cell cycle (for review, see Ref. 2). However, whether and how plasma membrane Ca2+ ATPases (PMCAs),2 a family of low capacity, high affinity plasma membrane-associated Ca2+ efflux pumps encoded by four genes (PMCA1–4) (for review, see Refs. 3 and 4) also participated in this process was not known.
Alternative splicing at the amino (N) and carboxyl (C) terminal sites of PMCA4 pre-mRNA can produce x or z splice variants at site N and a, b, or d splice variants at site C, each leading to the expression of a different protein (4–6). PMCA4 is expressed later in development than PMCA1 but is expressed more or less ubiquitously in adult mammalian organs (4). PMCA gene transcription is controlled by c-Myb, leading to changes in the free intracellular Ca2+ concentration ([Ca2+]i) during the cell cycle progression of vascular smooth muscle cells (VSMC) (7–9).
Our initial observations in VSMC showed that inhibiting c-Myb expression or function prevented cell cycle progression and lowered [Ca2+]i (10). We found these changes to be mediated by increased PMCA1 and PMCA4 expression. Indeed, transient overexpression of PMCA1a in VSMC elevated the Ca2+ efflux rate, decreased the [Ca2+]i, and reduced the rate of cell proliferation by over 2.5-fold (10). Whereas PMCA4b overexpression in mice increased blood pressure and myogenic tone (11), mice with functional knock-out of PMCA4 showed impaired phasic contractions and apoptosis in portal vein smooth muscle in vitro provided the mice were also heterozygous for the PMCA1 gene deletion (12). Together, these findings suggested that PMCA4 plays a role in VSMC biology.
We now present evidence that the relative proportions of PMCA4a and PMCA4b splice variants are regulated after wire denudation injury of carotid arteries in vivo. Quiescent VSMC in vivo have roughly equal proportions of PMCA4a and PMCA4b splice variants until arterial injury leads to a significant decrease in the ratio of PMCA4a to PMCA4b. Mice lacking functional PMCA4 (P4KO) show reduced remodeling after injury in vivo, although primary VSMC isolated from such mice manifest reduced cell proliferation and a G1 phase arrest that can be rescued by either the PMCA4a or PMCA4b splice variant alone. A PMCA4b mutant lacking the carboxyl-terminal PDZ binding domain also rescued the G1 phase arrest of P4KO VSMC, whereas a PMCA4b mutant with only 10% of the Ca2+ efflux activity of the wild-type pump could not. Microarray data show that functional PMCA4 ablation caused strong increases in Decorin (a G1 arrest marker) and regulator of G-protein signaling-16 (Rgs16; mitogen-activated protein kinase (MAPK) inhibitor, nuclear factor of activated T-cells (NFAT) target) expression, although rescue with either PMCA4a or PMCA4b restored normal Decorin and Rgs16 expression levels. Having said this, PMCA4a and PMCA4b rescues did differ in their global outcomes, with PMCA4a causing decreased expression of the transcription factor AP-2β (an inhibitor of c-Myc, AP-2α, and cell cycle), whereas PMCA4b caused decreased expression of p15 (Cdkn2b (cyclin-dependent kinase inhibitor 2b)), an inhibitor of Cyclin D1/Cdk4. Together, these findings lead us to conclude that the Ca2+ efflux activity of PMCA4 mediates G1 phase cell cycle progression in VSMC and that PMCA4a and PMCA4b appear to work through different effector proteins to do so.
MATERIALS AND METHODS
Mice and Cell Culture
C57BL6/J mice were purchased (Charles River Laboratories, Wilmington, MA). Frozen PMCA4 knock-out hearts (lacking exons 2 and 3) were a gift from Ludwig Neyses (Manchester, UK) (13, 14). Because deletion of exons 2–3 disrupts the reading frame of the resulting PMCA4 locus, these mice do not produce any PMCA4 protein (supplemental Fig. S2, panel B). PMCA4 functional knock-out mice distinct from the Neyses laboratory mice and lacking exon-11 (encoding codons 440–520, including aspartate 466, which is the catalytic phosphorylation site; GenBankTM accession number NM_001167949.2) were a kind gift from Gary Shull (12). These exon-11-deficient mice express a mutant PMCA4 mRNA as a consequence of exon-10 splicing to exon-12 (12), which preserves the reading frame and enables these mice to produce functionless mutant PMCA4a and PMCA4b proteins (devoid of a catalytic site). These mice are referred to as P4KO mice, with their wild-type littermates denoted as P4WT. Protocols for mouse tissue harvest and carotid artery wire denudation injury have been described (15) and were approved by the Animal Care Committee of University Health Network. Primary VSMC cultures were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (Wisent; Montreal, Quebec, Canada), 50 ng/ml rat recombinant PDGF-BB (Sigma), and 1% penicillin-streptomycin in humidified air containing 5% CO2 at 37 °C.
Cloning of PMCA4 cDNA Variants
PMCA4a, PMCA4b, and PMCA4e cDNA were cloned from total RNA extracted from mouse bladder, a mouse VSMC cell line (MOVAS; ATCC number CRL-2797), and mouse brain of C57 BL6/J mice, respectively (NucleoSpin RNA II kit; Takara-Clontech, Mountain View, CA) and PCR-cloned using mouse PMCA4-specific primers (see supplement Table S1) targeting either the 5′ or 3′ half of PMCA4 mRNA into pGemT-Easy vector (Promega). Clones containing the 5′ and 3′ halves of PMCA4 were digested with BamHI, gel-eluted, and ligated to generate full-length cDNA clones for each splice variant and sequenced. These cDNA sequences have been deposited in GenBankTM under accession numbers JF683384 (PMCA4a), EU493095 (PMCA4b), and EU493094 (PMCA4e).
Antibody Generation and Validation
Peptides (supplemental Fig. IB) specific for PMCA4b and PMCA4a/e carboxyl termini were designed (Aves Labs; Tigard, OR) and used to inject chickens. Polyclonal chicken antibodies with expected specificity for PMCA4 splice variants were collected and affinity-purified on columns containing the immobilized peptide antigen.
Western Blotting
Protein extracts made from primary VSMC and various mouse organs including uninjured and injured carotid arteries were resolved on mini-PAGE gels (Novex; Invitrogen), blotted, and hybridized to various antibodies and visualized either by HRP-conjugated secondary antibodies (ECL kit; PerkinElmer Life Sciences) or by near infrared dye conjugated secondary antibodies using the Odyssey laser scanner (LiCor, Lincoln, NE).
qRT-PCR
Total RNA was extracted from various organs of C57BL6/J mice with the PicoPure RNA extraction kit (Invitrogen). After DNase treatment and the addition of 1.0 pg of EGFP RNA to each sample (reverse transcription efficiency control), cDNA was synthesized, and qRT-PCR was carried out with primers specific for EGFP (cDNA synthesis efficiency), β2-microglobulin (housekeeping gene), PMCA4a, PMCA4b, and PMCA4e splice variants (see supplemental Table S1) using SYBR GREEN master mix in an ABI HT-7900 (Invitrogen) thermal cycler. qRT-PCR primers for PMCA4 splice variants were first used to gel elute amplified products from each primer pair, clone them into pGemT-Easy vector, and sequence them to confirm the identity of the amplicon for each splice variant. Next, the PMCA4a, PMCA4b, and PMCA4e primer pairs were tested under various amplification conditions to measure amplification efficiency. Amplification conditions were chosen where the individual Ct values for each splice variant differed by <5% from the mean Ct value for all three variants. This ensured that amplification efficiency would not vary significantly among the PCR reactions for the three PMCA4 splice variants. The amplification conditions employed consisted of a hot start of 94 °C for 10 min followed by 50 cycles of 94 °C for 15 s (denaturation), 69 °C for 30 s (annealing), and 72 °C for 30 s (extension). Gel eluted amplicons for EGFP, microglobulin, and the three PMCA4 splice variants were diluted to generate copy number standards containing 1 × 106, 1 × 105, 1 × 104, 1 × 103, 100, and 10 copies/μl. The absolute copy number for each particular mRNA was calculated via the absolute quantification method (Protocol 4304965; Applied Biosystems) that employs Ct values of copy number standards to calculate absolute copy numbers of each template in each sample. The absolute copy numbers of PMCA4 splice variants were then normalized to the copies of EGFP (cDNA synthesis efficiency control) and microglobulin (housekeeping gene control) cDNA in each sample. Finally, normalized copy numbers of each PMCA4 splice variant in each sample were summed, and copy numbers of each splice variant were divided by this sum to obtain the relative proportion of each splice variant in each sample.
Immunohistochemistry (IHC), Immunofluorescent Staining, and Morphometry
Mouse carotid artery cross-sections (paraformaldehyde-fixed, paraffin-embedded) were used for immunostaining with various antibodies and fluorophore-conjugated secondary antibodies (Jackson ImmunoResearch and Invitrogen) and visualized by confocal imaging on the Olympus Fluoview microscope using Fluoview software (Advanced Optical Microscopy Facility, University Health Network). Regions of interest were drawn on the images, and fluorescence intensity was quantified in the regions of interest using Fluoview software. Quantification was done on at least two different regions of interests of both uninjured and injured carotid artery sections embedded in one paraffin section (three uninjured and three injured arteries per section), immunostained together on one slide, and imaged via z-stacks using the same laser intensity for all sections. For carotid artery morphometry, sections from P4KO and P4WT mice (day 9 post injury) were immunostained with anti-PMCA4a/e antibody, and the area of the entire media and entire remodeled adventitia was measured at 40× resolution on the Olympus Fluoview microscope using Fluoview software.
Laser Capture Microdissection
Mouse carotid arteries (uninjured and injured) or mouse bladder fresh-frozen in OCT (Optical Cutting Temperature Compound, Sakura Finetek, Torrance, CA) were sectioned (10 μm thick) and mounted on laser capture microdissection microscope slides. Sections were lightly stained with Histogene (Invitrogen), fixed with 70% ethanol, and taken through a dehydration series ending in xylene and dried. An laser capture microdissection microscope and laser set up (Advanced Optical Microscopy Facility, University Health Network) was used to microdissect and capture medial and adventitial layer regions of carotids (or bladder smooth muscle layer) from the stained and fixed sections. RNA was extracted with the PicoPure kit, and qRT-PCR was carried out as described above.
Tritiated Thymidine Uptake Assays
Primary VSMC were seeded into 4 replicate wells of 6-well plates, allowed to grow overnight, and then serum-starved (0.1% serum; DMEM) for 24 h. Serum-synchronized cells were then stimulated with 10% serum and 50 ng/ml PDGF-BB for 24 h. Cells were given tritiated thymidine for the last 1 h before harvest. Cells were lysed and processed for liquid scintillation counting.
BrdU-PI Flow Cytometry
Primary VSMC were seeded into T-75 flasks, allowed to grow overnight, and then serum-starved (0.1% serum; DMEM) for 24 h. Synchronized cells were then stimulated with 10% serum and 50 ng/ml PDGF-BB for 24 h. Cells were labeled with BrdU for the last 1 h before harvest. Cells were harvested, fixed in 70% ethanol, and processed for anti-BrdU FITC-conjugated antibody staining as well as propidium iodide staining and used for flow cytometry.
Microarray Analysis
Primary VSMC from the carotid arteries of P4KO and P4WT mice were grown in quadruplicate T-75 flasks, serum-starved for 24 h, serum- and PDGF-BB-stimulated for 24 h, Hoechst-stained for the last 1 h before harvest, and then flow-sorted to obtain the G0/G1 subpopulation from all 8 samples (n = 4 for each genotype). Total RNA was extracted from the 8 samples and used for microarray analysis. In a second microarray, cells were starved for 24 h and serum- and PDGF-BB-stimulated for 24 h (G1/S stage), and RNA was extracted from P4WT cells, P4KO cells, PMCA4b-rescued P4KO cells, and vector-rescued P4KO cells. In a third experiment, RNA was extracted from P4KO (n = 4), and P4KO+PMCA4a-rescued (n = 4) cells was used for microarray analysis. RNA was submitted to The Center for Applied Genomics (SickKids Hospital, Toronto, ON) for microarray analyses. Table 1 and supplemental Tables S2–S8 show genes increased or decreased in expression or which are known to be implicated in G1 phase arrest or are part of the NFAT pathway and were seen to be strongly modulated in P4KO cells as compared with P4WT cells. The preprocessing includes three steps: background correction (performed in GenomeStudio software), quantile normalization (16), and log2 transformation of normalized data. The last two steps were performed in lumi R package (17). The Center for Applied Genomics used LIMMA (linear models for microarray data) (18) to identify differentially expressed gene signatures under different conditions. Briefly speaking, it starts by fitting a linear model for each gene in the data, then an empirical Bayes method is used to moderate the standard errors for estimating the moderated t-statistics for each gene, which shrinks the standard errors toward a common value. This test is similar to an analysis of variance method for each gene except that the residual standard deviations are moderated across genes to ensure more stable inference for each gene. The moderated standard deviations are a compromise between the individual gene-wise standard deviations and an overall pooled standard deviation. We evaluated the false discovery rate using the Benjamini multiple testing procedure (19). Our PMCA4 microarray data have been placed in the PubMed microarray database GEO under accession number GSE38320.
TABLE 1.
Summary of microarray results
Cdkn2b, cyclin-dependent kinase inhibitor 2b.
| Comparison | Gene modulated (-fold change; p value) |
|---|---|
| Early G1 P4KO vs. P4WT | Dcn (+69.1; p = 8.95E-09) |
| Rgs16 (+39.4; p = 6.38E-09) | |
| E2f2 (−3.6; p = 4.02E-05) | |
| Late G1/S P4KO vs. P4WT | Dcn (+9.3; p = 3.24E-20) |
| Rgs16 (+15.1; p = 2.00E-18) | |
| PMCA4a-rescued vs. P4KO | Tfap2b (−44.6; p = 2.5E-10) |
| Dcn (−1.27; p = NS) | |
| Rgs16 (+1.1; p = NS) | |
| PMCA4b-rescued vs. P4KO | Cdkn2b (p15) (-50.4; p = 8.51E-24) |
| Dcn (−2.4; p = 1.59E-13) | |
| Rgs16 (−18.5; p = 3.07E-19) |
P4KO Electroporated with PMCA4 cDNA
Electroporation was carried out with the 4D-Nucleofector (Lonza; Mississauga, ON), and transfection efficiencies of ∼70% were regularly achieved (supplemental Figs. S4 and S5). Transfected cells were selected with the appropriate antibiotic (puromycin or G418), and the success of the drug selection was validated by RT-PCR (supplemental Fig. S6). P4KO cells were electroporated with either wild-type mouse PMCA4a, wild-type mouse PMCA4b, human PMCA4b mutant lacking a carboxyl-terminal PDZ binding domain (PMCA4b-ΔPDZ), or human PMCA4b-Asp mutant (catalytic site point mutant with 10% efflux activity compared with wild-type) or with vector where all constructs carried an antibiotic resistance cassette. Electroporated cells were allowed to recover for 24 h and then were subjected to 14 days of antibiotic selection before being used for BrdU-PI flow cytometry, microarray, or Western blots.
Statistics
qRT-PCR data, flow cytometry data, and IHC staining quantification are expressed as the mean ± S.E. and represent results from at least two separate experiments (each having n ≥ 3 samples per group). Statistical analyses were performed using Student's t test. Statistical significance was defined as p ≤ 0.05.
RESULTS
Cloning Mouse PMCA4 Splice Variants
cDNA constructs encoding PMCA4a, PMCA4b, and the novel splice variant termed PMCA4e were cloned from total RNA extracted from mouse bladder, the murine VSMC cell line MOVAS, and mouse brain, respectively. Sequence analysis showed that the PMCA4e cDNA clone contains a novel exon-20 sequence that is 10 nucleotide longer than the evolutionarily conserved 181-nucleotide sequence of other mammalian PMCA4a variants. These additional 10 nucleotide result in a premature stop codon being created in the mature, spliced PMCA4e mRNA. The predicted amino acid sequences at the C terminus of the three cloned splice variants are shown in supplemental Fig. S1A. The sequence of the extra 10 nucleotides included in the mouse PMCA4e splice variant novel exon-20 is conserved in the genomes of man, chimpanzee, monkey, dog, cat, rabbit, mouse, and rat (supplemental Fig. S1C), which suggests a unique functional role for PMCA4e. PHI-BLAST analysis showed that other members of the PMCA family (viz. PMCA1 and PMCA3) also express splice variants similar to the PMCA4e variant we have cloned and share the GSE peptide motif at their carboxyl tails (supplemental Fig. S1C).
PMCA4 Splice Variant-specific Antibodies
The PDZ binding domain lies within the last seven amino acids of the carboxyl terminus of mouse PMCA4b (viz. PSLETPV), which is absent from the mouse PMCA4a tail. Two peptides were designed (Aves Labs); one coding for the PMCA4b carboxyl tail and the other coding for an exon-20-specific epitope expected in both PMCA4a and PMCA4e variants (supplemental Fig. S1B), which corresponds to the carboxyl end of the calmodulin binding motif.
Hearts from homozygous Neyses laboratory-generated PMCA4 knock-out adult mice (PMCA4−/−) and littermate wild-type controls (PMCA4+/+) as well as C57/BL6/J mice were used in genomic PCR (14). This confirmed the genetic deletion of PMCA4 (exons 2–3) in knock-out hearts, with the 816-bp PMCA4 (exon3–4)-specific band identified only in hearts from PMCA4+/+ and C57BL6/J mice (supplemental Fig. S2A). Next, Western blots revealed that the PMCA4b antibody recognized a ∼130-kDa protein from brain, stomach, and heart of C57BL6/J mice but did not bind to protein extracts made from PMCA4−/− heart (supplemental Fig. S2B). Similarly, the PMCA4a/e antibody detected a ∼130-kDa protein in extracts from C57BL6/J brain and stomach but not heart from C57BL6/J mice, presumably because of very low or absent PMCA4a/e expression in the normal mouse heart.
PMCA4 Splice Variants Show Differential mRNA and Protein Expression Levels in Specific Mouse Organs
Having previously demonstrated a functional consequence of PMCA1a and PMCA4b overexpression (10, 11), we were interested in exploring the regulation of alternative splicing at splice site C in the PMCA4 pre-mRNA that generates PMCA4a, -4b, and -4e splice variants. To separate the consequences of transcriptional regulation (via the endogenous PMCA4 promoter) from those of alternative splicing modulation, the relative proportions of each PMCA4 splice variant were calculated from absolute copy number data obtained by quantitative RT-PCR. We reasoned that promoter up- or down-regulation would increase the absolute copy numbers of all alternatively spliced transcripts (so that analysis using absolute copy numbers would tend to reflect the effects of promoter regulation), whereas changes in the relative proportions of each PMCA4 splice variant would accurately reflect regulation of alternative splicing. Others engaged in studies of alternative splicing have also used this strategy (20).
In this manner we determined the relative proportions of PMCA4 splice variants in various mouse organs (Fig. 1A). We found that mouse organs could be grouped into three categories: organs predominantly expressing PMCA4e (brain), organs predominantly expressing PMCA4a (bladder), and organs predominantly expressing PMCA4b (all other organs tested), excluding liver, which showed negligible PMCA4 expression. We then used our PMCA4b and PMCA4a/e antibodies to probe PMCA4 splice variant proteins (Fig. 1B) and found that the organs surveyed also displayed distinct PMCA4a/e and PMCA4b expression profiles.
FIGURE 1.
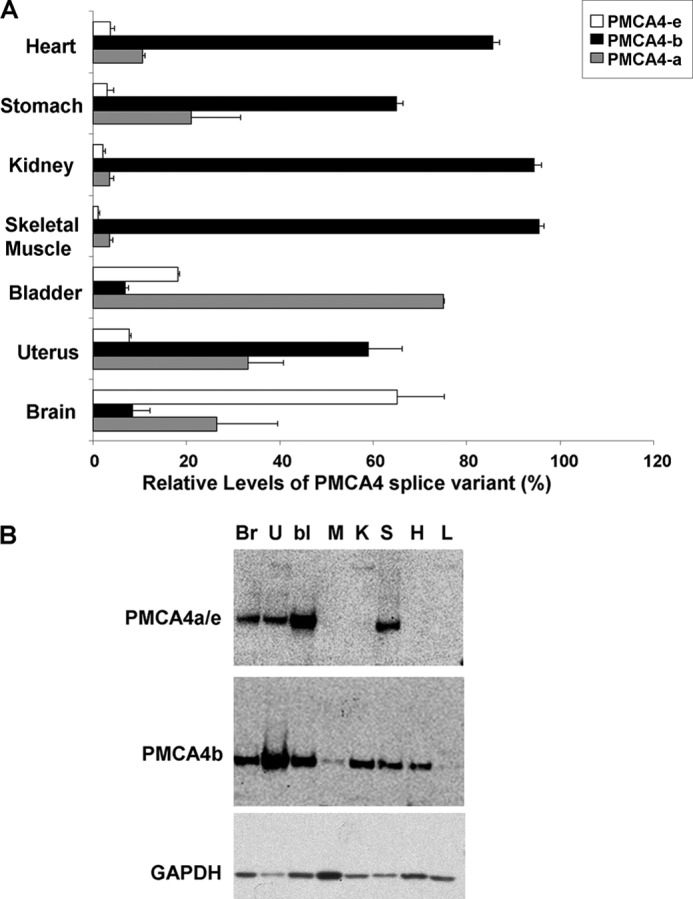
Distinct PMCA4 splice variant expression levels in mouse organs. A, qRT-PCR-determined relative proportions of PMCA4 splice variants in various organs of C57BL6/J mice. Bars represent mean relative proportion for each splice variant mRNA extracted from the organs of three C57BL6/J mice, normalized for cDNA conversion efficiency (EGFP) and loading (β2-microglobulin). Liver gave no PMCA4 splice variant copies upon amplification. B, immunoblots of microsomal protein extracts from various organs of C57BL6/J mice. Microsomal extracts were made from the organs of three C57BL6/J mice and resulted in nine separate immunoblots (three each for PMCA4-a/e, PMCA4b, and GAPDH); representative blots are shown. Br, brain; U, uterus; bl, bladder; M, skeletal muscle; K, kidney; S, stomach; H, heart; L, liver.
Regulation of PMCA4 Alternative Splicing in Proliferating VSMC in Vivo
We next searched for a disease model in which PMCA4 alternative splicing may be modulated. When we examined mouse carotid arteries at day 9 post wire denudation injury, we found that the roughly equal proportion of PMC4a and PMCA4b splice variants observed in uninjured controls was dramatically altered, with preferential expression of PMCA4b at the expense of PMCA4a splice variant in injured arteries (Fig. 2A). Western blot analysis showed that this shift in alternative splicing was also manifest at the protein level (Fig. 2B). Laser capture microdissection coupled with qRT-PCR showed that injured media and remodeled adventitia have a higher proportion of the PMCA4b transcript as compared with uninjured media, whereas a sample from bladder smooth muscle expressed a higher proportion of PMCA4a variant (supplemental Fig. S3).
FIGURE 2.
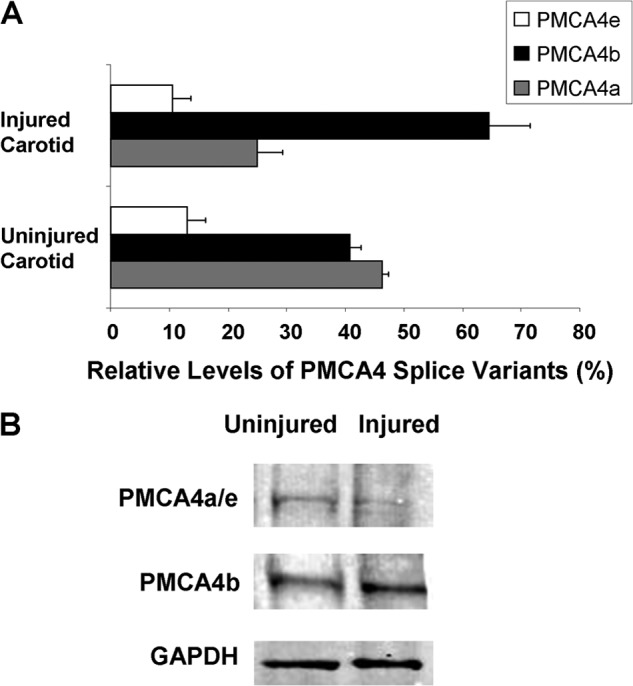
PMCA4 splice variant mRNA and protein expression levels are regulated after arterial injury. A, qRT-PCR-determined relative proportions of PMCA4 spice variants in uninjured and injured carotid arteries from male C57BL6/J mice (n = 4). p values for uninjured versus injured, PMCA4a = 0.001 and PMCA4b = 0.01, by Students' t test. B, immunoblots of protein extracts made from uninjured and injured mouse carotid arteries. Representative blots from two independent experiments are shown.
IHC confocal imaging showed the expression of a smooth muscle marker (sm22α) in uninjured and injured carotid artery cross-sections, whereas trichrome staining showed increased collagen deposition in remodeled adventitia (Fig. 3A). IHC staining with a proliferation marker (proliferating cell nuclear antigen) was only visible in injured arteries. Consistent with qRT-PCR and Western blot, IHC showed a decrease in PMCA4a/e and an increase in PMCA4b staining in injured carotids as compared with uninjured arteries that was further corroborated by quantifying immunofluorescent intensities with PMCA4 splice variant-specific antibodies in injured and uninjured medial layers (Fig. 3B).
FIGURE 3.

Immunofluorescence-based quantification revealed regulated PMCA4 splicing in VSMC of injured carotid arteries. A, confocal images of uninjured and injured carotid artery cross-sections from male C57BL6/J mice stained for PMCA4a/e, PMCA4b, smooth muscle marker (sm22α), proliferation marker (proliferating cell nuclear antigen), and collagen deposition (trichrome). Scale bars in panel A trichrome images = 500 μm, in confocal images = 20 μm, and in panel D confocal images = 50 μm. B, confocal microscopy Z-stack quantification of PMCA4a/e staining. Each bar represents a mean of 3 mouse carotid artery cross-sections in which ∼40–70 cells or cell clusters were used to quantify fluorescent intensity through 20–40 focal planes of 5-μm-thick sections. C, morphometric analysis of injured carotid arteries in PMCA4 wild-type (P4WT) and PMCA4 knock-out (P4KO) mice (n = 4). Carotid artery cross-sections were immunostained for PMCA4a/e with media and adventitia captured as regions of interest for area measurements on FluoView microscope (40× objective) and software (Olympus, Mountain View, CA). The ratio of adventitia:media areas are decreased in injured carotid arteries of P4KO mice (P4WT = 3.1 ± 0.5 versus PMKO = 1.4 ± 0.1; p = 0.01; n = 4). D, representative immunofluorescent micrographs of P4WT and P4KO injured carotid arteries immunostained for PMCA4a/e show reduced medial thickening in P4KO mice.
PMCA4 functional knock-out mice kindly provided by Gary Shull (Cincinnati, OH) lack exon-11, which encodes the central catalytic loop (81 amino acids; residues 440–520; see “Materials and Methods”) (12), resulting in a protein product devoid of any Ca2+ efflux activity (12). In the current manuscript, these mice are referred to as P4KO mice, with their wild-type littermates termed P4WT. Carotid artery cross-sections from P4WT and P4KO mice at day 9 post-injury were used for morphometry (Fig. 3, C and D). There was a 2-fold decrease in the adventitia:media ratio of injured P4KO arteries compared with P4WT arteries. This decreased remodeling in P4KO mice suggested two interrelated possibilities: (i) absence of functional PMCA4 impairs VSMC proliferation in vivo, and (ii) increased proportion of PMCA4b observed in injured arteries may play a physiologically relevant role in VSMC proliferation.
PMCA4 Knock-out Causes G1 Phase Arrest, Which Can Only Be Rescued by PMCA4 with Intact Ca2+ Efflux Activity
To probe the correlation between relative proportions of PMCA4 splice variants and VSMC proliferation, we isolated primary carotid VSMC from P4WT and P4KO mice. [3H]Thymidine uptake assays using cell cycle-synchronized VSMC cultures showed an ∼70% reduction in G1 to S phase cell cycle progression of P4KO VSMC compared with P4WT cells (Fig. 4A). Similarly, BrdU-PI flow cytometry showed an ∼30% increase in G1-stage P4KO cells as compared with P4WT (Fig. 4B), suggesting a G1 stage arrest.
FIGURE 4.
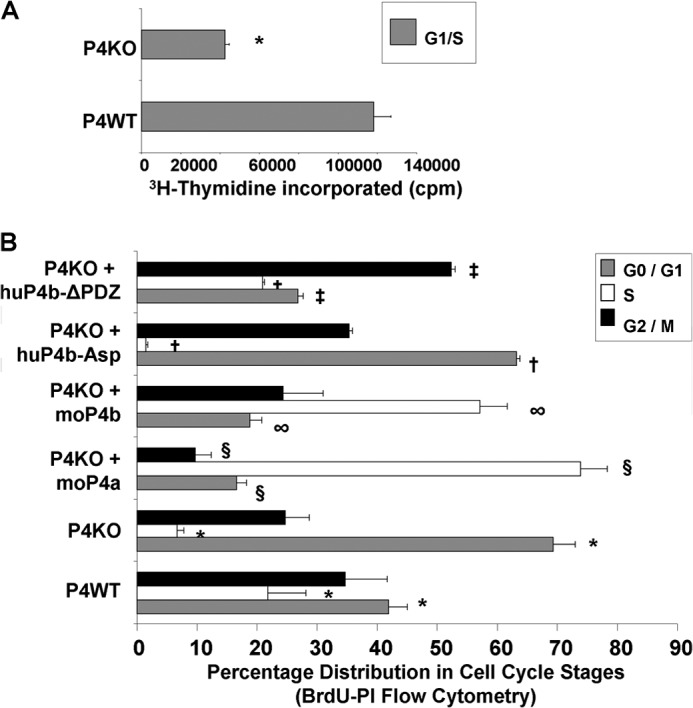
Ca2+ efflux activity of PMCA4 is required to rescue the G1 arrest of functional PMCA4 ablation. A, [3H]thymidine uptake at the late G1/S-stage was assayed in cell cycle-synchronized PMCA4 wild-type (P4WT) and PMCA4 knock-out (P4KO) primary VSMC (n = 3 experiments). P4KO cells show a 64% reduction in thymidine uptake as compared with P4WT cells (*, p = 2.8E-08, Student's t test). B, flow cytometric determination of cell cycle distributions of P4WT and P4KO primary VSMC. P4KO cells were electroporated with expression constructs encoding mouse PMCA4a, mouse PMCA4b, human PMCA4b-Asp mutant (10% Ca2+ efflux activity compared with wild-type), or human PMCA4b-ΔPDZ mutant (lacking PDZ-binding domain) and G0-synchronized by serum starvation. After synchronized serum stimulation for 22 h, the cell cycle distribution of each population was determined by BrdU-PI flow cytometry (n = 3 independent experiments with multiple replicates for all samples). *p = 0.004 for P4WT versus P4KO; §, p = 1.85E-6 for PMCA4a-rescued P4KO versus P4KO; ∞, p = 0.001 for PMCA4b-rescued P4KO versus P4KO; †, p = 2.9E-16 for huP4b-Asp-rescued P4KO versus PMCA4b-rescued P4KO; ‡, p = 0.0001 for huP4b-ΔPDZ-rescued P4KO versus P4KO. Student's t test was used for all comparisons.
Importantly, electroporation of either mouse PMCA4a or PMCA4b expression constructs rescued the cell cycle phenotype of P4KO cells (Fig. 4B), i.e. PMCA4a- or PMCA4b-rescued P4KO VSMC manifest a 40–50% increase in S phase cells and an ∼50% reduction in G1 phase cells. Western blots confirmed (supplemental Fig. S7) that as a result of these electroporations, PMCA4a was overexpressed 1.5-fold in P4KO+4a cells, whereas PMCA4b was overexpressed 1.8-fold in P4KO+4b cells as compared with P4KO cells. Although expression of a PMCA4b mutant lacking the PDZ binding domain was still able to rescue the G1 arrest of P4KO cells, expression of a PMCA4b-Asp mutant (with only 10% of the Ca2+ efflux activity of wild-type PMCA4) failed to rescue the G1 phase arrest of P4KO cells (Fig. 4B).
In a separate series of experiments assessing cell cycle progression at multiple time points after serum stimulation of G0-synchronized P4WT, P4KO, and PMCA4a- or PMCA4b-rescued cell populations, we confirmed that P4KO cells suffer from a G1 phase arrest that is completely alleviated by either PMCA4a or PMCA4b cDNA expression (Fig. 5). Together these data show that whereas specific PDZ binding domain-mediated PMCA4b/nNOS (21) or PMCA4b/CASK interactions (22) are not necessary for PMCA4b to regulate cell cycle, the presence of Ca2+ efflux activity is required for PMCA4 to mediate G1-to-S phase progression in VSMC.
FIGURE 5.
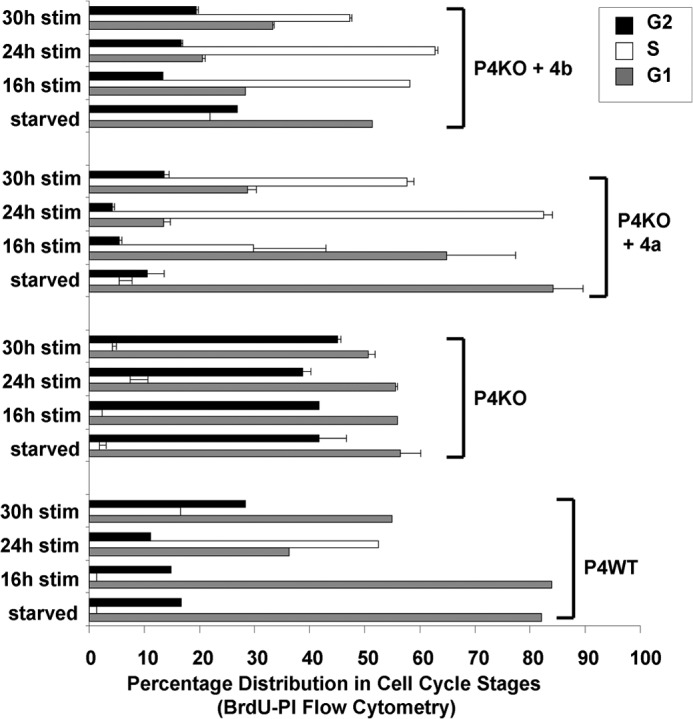
Correction of the impaired cell cycle progression of PMCA4 knock-out VSMC. Cell cycle distributions of P4WT, P4KO, or P4KO VSMC electroporated with mouse PMCA4a or PMCA4b cDNA were determined by BrdU-PI flow cytometry (n = 2 independent experiments with multiple replicates for all samples) at various time points in the cell cycle using serum-synchronized populations.
Microarray Analysis and Validation of Identified Targets
Early G1 stage P4KO and P4WT cells were collected via flow sorting (by gating for G0/G1 phase Hoechst-stained cells) and used for microarray analysis. This unbiased exploration revealed that Decorin (+69.1-fold; p = 8.9E-09) and Rgs16 (+39.4-fold; p = 6.3E-09; Fig. 6A and supplemental Tables 2 and 3) were markedly increased in P4KO cells as compared with P4WT cells. Decorin is a known inhibitor of the VSMC cell cycle and implicated in G1 phase arrest (for review, see Ref. 23). Decorin is known to reduce cyclin D1 expression via the epidermal growth factor receptor (EGFR) or NFAT pathways (24–26), with NFAT activation also known to inhibit Rgs16 expression (27) (see the proposed model, supplemental Fig. S8). This suggested that the high Rgs16 expression observed in P4KO cells (as compared with P4WT cells) may be due to reduced NFAT expression or activation in P4KO cells. Consistent with this, immunoblot analysis showed that Decorin and Rgs16 levels were increased, and Cyclin D1 levels were decreased in P4KO cells as compared with P4WT cells (Fig. 7).
FIGURE 6.
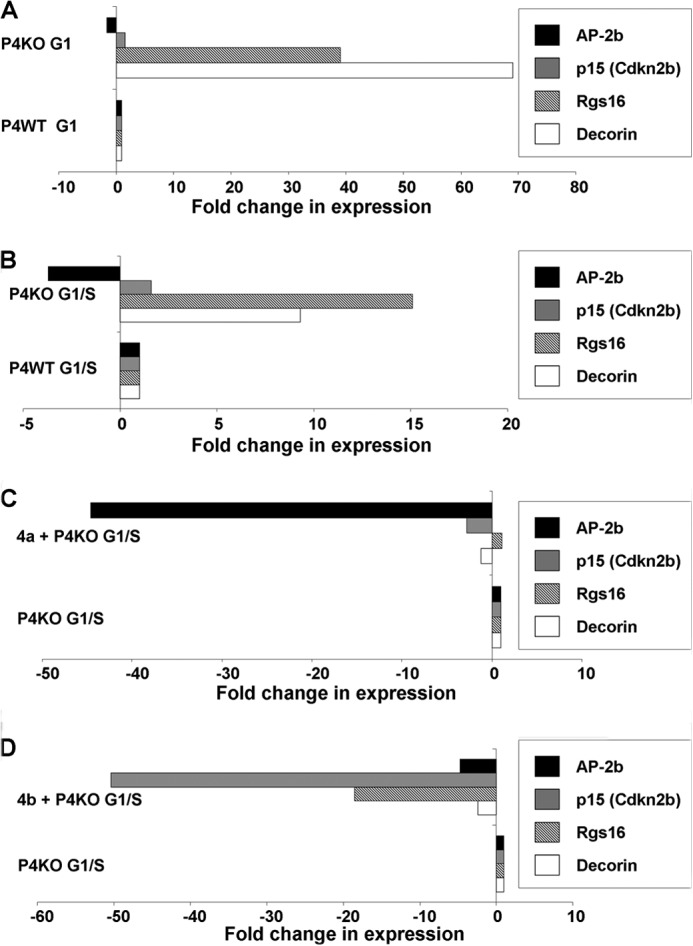
Putative downstream cell cycle mediators of PMCA4 splice variants identified by microarray. RNA isolated from cell cycle-synchronized, Hoechst-stained, and flow-sorted G0/G1 (A) and late G1/S-synchronized (B) populations of P4WT and P4KO VSMC was used for microarray analysis. PMCA4a (C)- and PMCA4b (D)-rescued P4KO VSMC at late G1/S were also subjected to microarray analysis versus P4KO VSMC at the same cell cycle stage. Bar graphs show -fold change in expression in pair-wise comparisons between various cell cycle stage and genotype samples for Decorin, Rgs16, AP2-β, and p15 mRNA levels as determined by microarray analysis.
FIGURE 7.
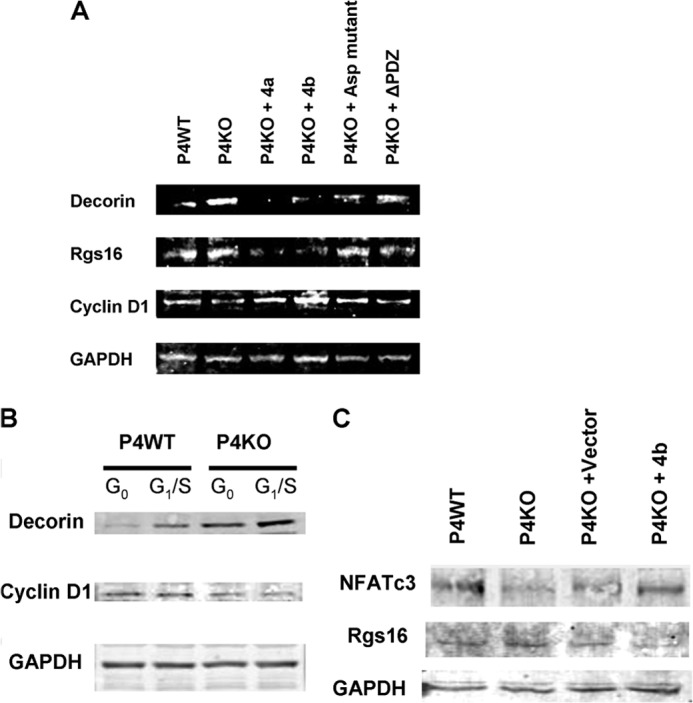
Validation of putative downstream cell cycle mediators of PMCA4 by immunoblot. A, protein extracts were made from late G1/S-synchronized P4WT, P4KO, and P4KO VSMC electroporated with mouse PMCA4a, mouse PMCA4b, human PMCA4b-Asp mutant (10% of normal efflux activity), or human PMCA4b-ΔPDZ mutant (lacking the PDZ binding domain) and probed for Decorin, Rgs16, Cyclin D1, and GAPDH (n = 2 separate experiments). B, P4WT and P4KO primary cells were serum synchronized for 48 h and harvested (G0) or stimulated with serum + PDGF-BB for 24 h (G1/S) for Western blots (n = 2) for Decorin, Cyclin D1, and GAPDH. C, protein extracts from late G1/S-synchronized VSMC (P4WT, P4KO, P4KO+Vector, and P4KO+mouse PMCA4b) were immunoblotted (n = 2) for NFATc3, Rgs16 and GAPDH. Representative immunoblots are shown.
Similar results were observed in vivo. First, Decorin-staining intensity trended 18% lower in medial layers of injured as compared with uninjured carotid arteries of P4WT mice (0.82 ± 0.05 versus 1.00 ± 0.06; n = 3, p = 0.1), suggesting injury-induced decreases in Decorin expression accompanied the reduced PMCA4a:PMCA4b expression ratio. Second, this pattern was lost in P4KO mice, in which carotid artery denudation injury failed to reduce Decorin expression (1.10 ± 0.03 versus 1.00 ± 0.04; n = 3, p = NS), supporting a role for PMCA4 in regulating Decorin expression in vivo. Indeed, similar to the microarray-documented difference in Decorin expression observed in primary VSMC cultures from P4KO versus P4WT mice, levels of Decorin expression were significantly higher in injured carotid arteries of P4KO animals as compared with P4WT (1.10 ± 0.03 versus 0.82 ± 0.05; n = 3, p = 0.01) (data not shown).
Because Decorin and Rgs16 have both been implicated in the NFAT pathway, we next examined mRNA levels of NFAT facilitator proteins Rcan1, Rcan2, and the NFAT target E2F2 in our microarray results (supplemental Table S4). Rcan2 was reduced −6.6-fold (p = 3.8E-08) and E2F2 was decreased −3.6-fold (p = 4.0E-05) in P4KO as compared with P4WT cells, suggesting diminished NFAT action. To examine this more directly, cytoplasmic protein extracts were probed for NFATc3 levels and found to be significantly decreased in P4KO cells as compared with P4WT cells (Fig. 7).
PMCA4a rescue of P4KO cells followed by late G1/S microarray analysis (Fig. 6, B and C) suggested some reversal of the expression levels of Decorin (PMCA4a rescue versus P4KO = −1.27-fold; p = NS) and Rgs16 (PMCA4a rescue versus P4KO = +1.1; p = NS); however, these changes did not reach statistical significance (supplemental Table S5). Rather, PMCA4a-rescued P4KO cells showed a sharp decline in the expression of a known anti-proliferative transcription factor viz. AP-2β (PMCA4a rescue versus P4KO = 44.6; p = 2.5E-10) (supplemental Table S5). AP-2β inhibits the activity of AP-2α (28) and also represses c-Myc transactivation (29). AP-2α is also known to bind to the ErbB2 promoter (EGFR family member) and promote proliferation via the EGFR pathway (30) activated by vessel injury in vivo (31). AP-2β is also implicated in VSMC proliferation in vivo by its role in the closure of the ductus arteriosus (32).
Additional microarray analysis using RNA harvested from late G1/S-stage P4KO, P4WT, and PMCA4b-rescued- versus vector-transfected-P4KO cells showed that Decorin was down-regulated −2.4-fold (p = 1.59E-13) in PMCA4b-rescued versus P4KO cells (down −15.3-fold in PMCA4b-rescued versus control P4KO cells; p = 5.5E-22), whereas it remained up-regulated +9.3-fold (p = 3.24E-20) in P4KO versus P4WT G1/S stage cells (Fig. 6D; and supplemental Tables S6–S8). Rgs16 was also decreased −18.5-fold (p = 3.07E-19) in PMCA4b-rescued versus P4KO cells (−3.9-fold in PMCA4b-rescued versus vector-control P4KO cells; p = 9.2E-14), whereas it continued to be increased +15.1-fold (p = 2.0E-18) in P4KO versus P4WT at the G1/S stage (Fig. 6D; and supplemental Tables S6–S8). Interestingly, Cdkn2b (cyclin-dependent kinase inhibitor 2b; also known as p15 or Ink4b) was markedly decreased in PMCA4b-rescued cells as compared with P4KO cells (PMCA4b versus P4KO = −50.4; p = 8.51E-24). p15 is a cyclin-dependent kinase inhibitor that forms a complex with CDK4 and prevents activation of Cyclin D1 (33).
Immunoblot analysis using late G1/S-stage cytoplasmic protein extracts showed that Rgs16 was increased in P4KO (and control vector-rescued P4KO) cells versus P4WT cells but decreased in PMCA4b-rescued P4KO cells (Fig. 7). Western blots also showed that the decrease in NFATc3 expression observed in P4KO cells was reversed by rescue with PMCA4b (Fig. 7C). Together, these data clearly demonstrate that PMCA4 splice variants rescue common Decorin, Rgs16, and NFATc3 alterations observed in VSMC lacking any PMCA4 expression (P4KO) and also that expression of PMCA4a and PMCA4b result in the regulation of unique downstream mediators.
DISCUSSION
In the current study we present evidence that PMCA4 gene deletion impairs cell cycle progression and causes a G1 phase arrest in VSMC. Furthermore, quiescent VSMC in vivo have roughly equal proportions of PMCA4a and PMCA4b splice variants, whereas proliferating VSMC in vivo appear to have relatively higher levels of PMCA4b than PMCA4a expression. This suggested differential roles for these two PMCA4 splice variants in the VSMC cell cycle. However, when P4KO VSMC were electroporated with either PMCA4a or PMCA4b cDNA, cells are rescued from the G1 phase arrest, and altered expression levels of Decorin and Rgs16 are corrected. Also, our data show that whereas the PDZ binding domain unique to PMCA4b (i.e. absent in PMCA4a) is not needed for this effect, Ca2+ efflux activity is required to rescue the G1 phase arrest of P4KO cells. This observation suggest that the Ca2+ efflux activity of PMCA4 is critical to G1-to-S-phase progression in VSMC but leaves open the possibility that other domains within the PMCA4 protein may also regulate the VSMC cell cycle.
Prior to out current results, a possible mechanism through which PMCA4 alternatively spliced domains could regulate cell cycle included PMCA4b binding to the tumor suppresser RASSF1 (34), which mediates G1 cell cycle arrest via p21 expression (35) or via p53 and p16 accumulation (36). Instead of directly testing the involvement of such a candidate, we used an unbiased, open-ended microarray strategy to discover putative PMCA4 targets. This analysis strongly implicated two proteins (Decorin, Rgs16) previously not known as PMCA4-regulated targets capable of modulating cell cycle. Both of these proteins are markedly increased upon PMCA4 gene deletion, which is reversed by expression of PMCA4a or PMCA4b (but not vector DNA) in P4KO cells. Interestingly, both these PMCA4 target proteins participate in the NFAT pathway. Decorin is an established natural onco-suppressive factor and a novel ligand for EGFR (37, 38), which is increased in VSMC in vivo after vascular injury (31) and triggers a signaling cascade involving Akt and terminating in cyclin D1 induction (39). Decorin can inhibit EGFR, has been implicated in G1 phase arrest (by inducing p21 expression), and is deregulated in many types of human cancers (23, 24, 40). Decorin is also known to inhibit the VSMC cell cycle by triggering Myc degradation (41) and can also inhibit FosL1 (42). Under normal conditions FosL1 and Jun dimerize to form AP-1, which can induce the NFAT family of transcription factors (43), leading to cyclin D1 expression (26). Important confirmation of our in vitro microarray and Western blot results regarding Decorin included the novel finding that the decrease in Decorin expression observed in injured carotid arteries of P4WT mice is lost in P4KO mice, which manifest significantly higher levels of Decorin expression after injury.
Rgs16, the other putative PMCA4 target identified here, is known to block cell cycle progression by inhibiting the MAPK and Akt pathways and is also modulated by the NFAT pathway. Rgs16 overexpression in MCF7 breast cancer cells inhibits EGFR-mediated proliferation and Akt phosphorylation, whereas shRNA-mediated extinction of Rgs16 augments cell growth (44). Transient as well as stable Rgs16 expression in CHO cells can attenuate IP3 production and p38 MAPK activation by platelet-activating factor receptor (45). In T cells, PKC activation induces TNFα, which increases Rgs16 expression. Importantly, the calcineurin/NFAT pathway can repress this effect on Rgs16 (27).
Our results suggest that PMCA4a (supplemental Fig. S8) as well as PMCA4b (supplemental Fig. S9) function to limit Decorin and Rgs16 expression (as well as increasing NFATc3 and cyclin D1 expression) and that PMCA4a may additionally function via inhibiting AP-2β expression (which de-represses c-Myc and induces EGFR expression), whereas PMCA4b may work via decreasing the CDK4 inhibitor p15, allowing cyclin D1 to activate CDK4.
It is indeed surprising that although PMCA4a and PMCA4b proteins show very different expression patterns in quiescent and injured carotid arteries in vivo, both these splice variants rescue the G1 arrest prevalent in proliferating primary VSMC derived from the carotid arteries of P4KO mice. Potential explanations include the following. (i) Our rescue of P4KO cells was with the PMCA4x/a splice variant, whereas the PMCA4a splice variant actually expressed in quiescent carotid artery media may be PMCA4z/a. RNA-Seq of injured and uninjured carotid arteries would be required to explore this possibility as would the cloning and rescue by a PMCA4z/a (and PMCA4z/b) splice variant in vitro. (ii) The P4KO mice we have employed express “non-functional” PMCA4a and PMCA4b. As such, endogenous non-functional PMCA4b proteins may form multimeric complexes with exogenous PMCA4a enabling the non-functional PMCA4b molecules to regain normal Ca2+ efflux activity within complexes functionally defined by the protein-protein interactions of PMCA4b. In this scenario, although P4KO cells were electroporated with PMCA4a, the cells were able to regenerate “functional” PMCA4b pumps associated with a normal Ca2+ efflux activity. (iii) Electroporation of PMCA4a into de-differentiated, proliferating P4KO VSMC in culture may not mimic differentiated, quiescent VSMC in vivo, where a balance between PMCA4a and PMCA4b expression levels might be the critical factor driving the differentiated, non-proliferative state. In this explanation, expressing PMCA4a alone in de-differentiated, proliferating VSMC may not be enough to regain the quiescent, non-proliferative state. To achieve the latter, a fine balance in the expression levels and possibly differential subcellular localization of the two splice variants of PMCA4a and PMCA4b might be what is required.
The eukaryotic cell cycle is known to be regulated by Ca2+ signals at various check points within the cycle (for review, see Ref. 1). Both voltage- and ligand-gated Ca2+ transporters are known to control cell cycle checkpoints by modulating intracellular Ca2+ signals or by hitherto unknown mechanisms involving macromolecular complexes with growth factor receptors, cell adhesion molecules, or cytoplasmic regulatory proteins (1). The latter may or may not depend on the Ca2+ transport function of these transporters but do involve the participation of their non-catalytic domains in signaling cascades that modulate cell cycle checkpoints. For example, the voltage-gated potassium channel subunit hERG1 (Kcnh2 or Kv11.1) regulates proliferation by a mechanism involving its recruitment into multiprotein membrane complexes including integrins and growth factor receptors (46). Other examples where Ca2+ channel domains not involved in the Ca2+ transport function regulate transcription via a protein signaling cascade include Cav1.2 (Cacna1c), the L-type Ca2+ channel 250-kDa α1C subunit that forms the Ca2+ transporting transmembrane pore. An endogenously cleaved 15-kDa carboxyl-terminal fragment of Cav1.2 (calcium channel-associated transcription regulator (or CCAT)) translocates to the nucleus, binds to a nuclear protein, associates with endogenous promoters, and regulates the expression of a wide variety of endogenous genes including the bone morphogenetic protein inhibitor follistatin-like-1 (47, 48) protein and thus de-represses stem cell quiescence (45, 46). In a separate study involving the point mutation of the Cav1.2 calmodulin binding domain, it was shown that Ca2+/calmodulin binding to Cav1.2 enables specific activation of the Ras/MAPK pathway, CREB activation, and CREB-driven transcription (47). Importantly, it was discovered that the ability of Cav1.2 to generate a sustained [Ca2+]i rise was not needed for it to activate the Ras/MAPK/CREB pathway leading to gene transcription (47).
Further studies focused on elucidating the molecular mechanisms that allow PMCA4 to regulate the VSMC cell cycle are needed. The current study supplies the impetus for researchers examining PMCAs in other tissue systems to explore the roles of PMCA4a and PMCA4b in other cellular processes. The only other known example of physiological consequences of a PMCA4a/PMCA4b switch in vivo is the observed increase in relative PMCA4a splice variant expression (and decreased relative PMCA4b levels) during functional sperm maturation (49). In that model of differentiation, there was a steady shift from predominantly PMCA4b expression in the testis, caput, and corpus epididymis to predominantly PMCA4a expression in the cauda epididymis. It is interesting to note that differentiation (associated with lack of cell proliferation) appears to require an increase in the relative level of PMCA4a expression in this model of sperm maturation. This correlates with the observed decrease in relative PMCA4a expression in proliferating VSMC in vivo reported here and is reminiscent of the 2.5-fold reduction in rate of cell proliferation resulting from transient overexpression of PMCA1a splice variant in mouse VSMC (10) as well as the high relative proportion of PMCA4a variant in bladder smooth muscle cells (SMCs) (quiescent SMCs) and the roughly equal relative proportions of PMCA4a and PMCA4b in uninjured carotid artery SMCs (quiescent VSMC).
Supplementary Material
This work was supported by operating grants (to M. H.) from the Canadian Institutes of Health Research (MOP-64352) and the Heart and Stroke Foundation of Ontario (T-5254 and NA6713).

This article contains supplemental Tables S1–S8 and Figs. S1–S9. The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) JF683384, EU493095, and EU493094
PMCA4 microarray data have been placed in the PubMed microarray database GEO under accession number GSE38320.
- PMCA
- plasma membrane Ca2+ ATPases
- qRT
- quantitative real-time
- VSMC
- vascular smooth muscle cells
- EGFP
- enhanced GFP
- IHC
- immunohistochemistry
- NFAT
- nuclear factor of activated T-cells
- CREB
- cAMP-response element-binding protein
- PI
- propidium iodide
- NS
- not significant
- Rgs16
- regulator of G-protein signaling-16
- Dcn
- Decorin.
REFERENCES
- 1. Becchetti A. (2011) Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. Am. J. Physiol. Cell Physiol. 301, C255–C265 [DOI] [PubMed] [Google Scholar]
- 2. Lipskaia L., Lompré A. M. (2004) Alteration in temporal kinetics of Ca2+ signaling and control of growth and proliferation. Biol. Cell 96, 55–68 [DOI] [PubMed] [Google Scholar]
- 3. Carafoli E., Brini M. (2000) Calcium pumps. Structural basis for and mechanism of calcium transmembrane transport. Curr. Opin. Chem. Biol. 4, 152–161 [DOI] [PubMed] [Google Scholar]
- 4. Strehler E. E., Zacharias D. A. (2001) Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 81, 21–50 [DOI] [PubMed] [Google Scholar]
- 5. Keeton T. P., Burk S. E., Shull G. E. (1993) Alternative splicing of exons encoding the calmodulin-binding domains and C termini of plasma membrane Ca2+-ATPase isoforms 1, 2, 3, and 4. J. Biol. Chem. 268, 2740–2748 [PubMed] [Google Scholar]
- 6. Santiago-García J., Mas-Oliva J., Saavedra D., Zarain-Herzberg A. (1996) Analysis of mRNA expression and cloning of a novel plasma membrane Ca2+-ATPase splice variant in human heart. Mol. Cell. Biochem. 155, 173–182 [DOI] [PubMed] [Google Scholar]
- 7. Afroze T., Husain M. (2000) c-Myb-binding sites mediate G1/S-associated repression of the plasma membrane Ca2+-ATPase-1 promoter. J. Biol. Chem. 275, 9062–9069 [DOI] [PubMed] [Google Scholar]
- 8. Afroze T., Yang L. L., Wang C., Gros R., Kalair W., Hoque A. N., Mungrue I. N., Zhu Z., Husain M. (2003) Calcineurin-independent regulation of plasma membrane Ca2+ ATPase-4 in the vascular smooth muscle cell cycle. Am. J. Physiol. Cell Physiol. 285, C88–C95 [DOI] [PubMed] [Google Scholar]
- 9. Husain M., Bein K., Jiang L., Alper S. L., Simons M., Rosenberg R. D. (1997) c-Myb-dependent cell cycle progression and Ca2+ storage in cultured vascular smooth muscle cells. Circ. Res. 80, 617–626 [DOI] [PubMed] [Google Scholar]
- 10. Husain M., Jiang L., See V., Bein K., Simons M., Alper S. L., Rosenberg R. D. (1997) Regulation of vascular smooth muscle cell proliferation by plasma membrane Ca2+-ATPase. Am. J. Physiol. 272, C1947–C1959 [DOI] [PubMed] [Google Scholar]
- 11. Gros R., Afroze T., You X. M., Kabir G., Van Wert R., Kalair W., Hoque A. E., Mungrue I. N., Husain M. (2003) Plasma membrane calcium ATPase overexpression in arterial smooth muscle increases vasomotor responsiveness and blood pressure. Circ. Res. 93, 614–621 [DOI] [PubMed] [Google Scholar]
- 12. Okunade G. W., Miller M. L., Pyne G. J., Sutliff R. L., O'Connor K. T., Neumann J. C., Andringa A., Miller D. A., Prasad V., Doetschman T., Paul R. J., Shull G. E. (2004) Targeted ablation of plasma membrane Ca2+-ATPase (PMCA) 1 and 4 indicates a major housekeeping function for PMCA1 and a critical role in hyperactivated sperm motility and male fertility for PMCA4. J. Biol. Chem. 279, 33742–33750 [DOI] [PubMed] [Google Scholar]
- 13. Hummler E., Cole T. J., Blendy J. A., Ganss R., Aguzzi A., Schmid W., Beermann F., Schütz G. (1994) Targeted mutation of the CREB gene. Compensation within the CREB/ATF family of transcription factors. Proc. Natl. Acad. Sci. U.S.A. 91, 5647–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schuh K., Cartwright E. J., Jankevics E., Bundschu K., Liebermann J., Williams J. C., Armesilla A. L., Emerson M., Oceandy D., Knobeloch K. P., Neyses L. (2004) Plasma membrane Ca2+ ATPase 4 is required for sperm motility and male fertility. J. Biol. Chem. 279, 28220–28226 [DOI] [PubMed] [Google Scholar]
- 15. You X. M., Mungrue I. N., Kalair W., Afroze T., Ravi B., Sadi A. M., Gros R., Husain M. (2003) Conditional expression of a dominant-negative c-Myb in vascular smooth muscle cells inhibits arterial remodeling after injury. Circ. Res. 92, 314–321 [DOI] [PubMed] [Google Scholar]
- 16. Bolstad B. M., Irizarry R. A., Astrand M., Speed T. P. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 [DOI] [PubMed] [Google Scholar]
- 17. Du P., Kibbe W. A., Lin S. M. (2008) lumi. A pipeline for processing Illumina microarray. Bioinformatics 24, 1547–1548 [DOI] [PubMed] [Google Scholar]
- 18. Smyth G. K. (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- 19. Reiner A., Yekutieli D., Benjamini Y. (2003) Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19, 368–375 [DOI] [PubMed] [Google Scholar]
- 20. Xie J., Jan C., Stoilov P., Park J., Black D. L. (2005) A consensus CaMK IV-responsive RNA sequence mediates regulation of alternative exons in neurons. RNA 11, 1825–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oceandy D., Stanley P. J., Cartwright E. J., Neyses L. (2007) The regulatory function of plasma-membrane Ca2+-ATPase (PMCA) in the heart. Biochem. Soc. Trans. 35, 927–930 [DOI] [PubMed] [Google Scholar]
- 22. Schuh K., Uldrijan S., Gambaryan S., Roethlein N., Neyses L. (2003) Interaction of the plasma membrane Ca2+ pump 4b/CI with the Ca2+/calmodulin-dependent membrane-associated kinase CASK. J. Biol. Chem. 278, 9778–9783 [DOI] [PubMed] [Google Scholar]
- 23. Ständer M., Naumann U., Wick W., Weller M. (1999) Transforming growth factor-β and p-21. Multiple molecular targets of decorin-mediated suppression of neoplastic growth. Cell Tissue Res. 296, 221–227 [DOI] [PubMed] [Google Scholar]
- 24. Hu Y., Sun H., Owens R. T., Wu J., Chen Y. Q., Berquin I. M., Perry D., O'Flaherty J. T., Edwards I. J. (2009) Decorin suppresses prostate tumor growth through inhibition of epidermal growth factor and androgen receptor pathways. Neoplasia 11, 1042–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iozzo R. V., Moscatello D. K., McQuillan D. J., Eichstetter I. (1999) Decorin is a biological ligand for the epidermal growth factor receptor. J. Biol. Chem. 274, 4489–4492 [DOI] [PubMed] [Google Scholar]
- 26. Karpurapu M., Wang D., Van Quyen D., Kim T. K., Kundumani-Sridharan V., Pulusani S., Rao G. N. (2010) Cyclin D1 is a bona fide target gene of NFATc1 and is sufficient in the mediation of injury-induced vascular wall remodeling. J. Biol. Chem. 285, 3510–3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fong C. W., Zhang Y., Neo S. Y., Lin S. C. (2000) Specific induction of RGS16 (regulator of G-protein signalling 16) mRNA by protein kinase C in CEM leukaemia cells is mediated via tumour necrosis factor α in a calcium-sensitive manner. Biochem. J. 352, 747–753 [PMC free article] [PubMed] [Google Scholar]
- 28. Buettner R., Kannan P., Imhof A., Bauer R., Yim S. O., Glockshuber R., Van Dyke M. W., Tainsky M. A. (1993) An alternatively spliced mRNA from the AP-2 gene encodes a negative regulator of transcriptional activation by AP-2. Mol. Cell. Biol. 13, 4174–4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaubatz S., Imhof A., Dosch R., Werner O., Mitchell P., Buettner R., Eilers M. (1995) Transcriptional activation by Myc is under negative control by the transcription factor AP-2. EMBO J. 14, 1508–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Han C. Y., Lim S. C., Choi H. S., Kang K. W. (2008) Induction of ErbB2 by ultraviolet A irradiation. Potential role in malignant transformation of keratinocytes. Cancer Sci. 99, 502–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Singh N. K., Wang D., Kundumani-Sridharan V., Van Quyen D., Niu J., Rao G. N. (2011) 15-Lipoxygenase-1-enhanced Src-Janus kinase 2-signal transducer and activator of transcription 3 stimulation and monocyte chemoattractant protein-1 expression require redox-sensitive activation of epidermal growth factor receptor in vascular wall remodeling. J. Biol. Chem. 286, 22478–22488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao F., Bosserhoff A. K., Buettner R., Moser M. (2011) A heart-hand syndrome gene: Tfap2b plays a critical role in the development and remodeling of mouse ductus arteriosus and limb patterning. PLoS ONE 6, e22908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frost S. J., Simpson D. J., Farrell W. E. (2001) Decreased proliferation and cell cycle arrest in neoplastic rat pituitary cells is associated with transforming growth factor-β1-induced expression of p15/INK4B. Mol. Cell. Endocrinol. 176, 29–37 [DOI] [PubMed] [Google Scholar]
- 34. Armesilla A. L., Williams J. C., Buch M. H., Pickard A., Emerson M., Cartwright E. J., Oceandy D., Vos M. D., Gillies S., Clark G. J., Neyses L. (2004) Novel functional interaction between the plasma membrane Ca2+ pump 4b and the proapoptotic tumor suppressor Ras-associated factor 1 (RASSF1). J. Biol. Chem. 279, 31318–31328 [DOI] [PubMed] [Google Scholar]
- 35. Kim D. I., Lee S. J., Lee S. B., Park K., Kim W. J., Moon S. K. (2008) Requirement for Ras/Raf/ERK pathway in naringin-induced G1 cell-cycle arrest via p21WAF1 expression. Carcinogenesis 29, 1701–1709 [DOI] [PubMed] [Google Scholar]
- 36. Serrano M., Lin A. W., McCurrach M. E., Beach D., Lowe S. W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 [DOI] [PubMed] [Google Scholar]
- 37. De Luca A., Santra M., Baldi A., Giordano A., Iozzo R. V. (1996) Decorin-induced growth suppression is associated with up-regulation of p21, an inhibitor of cyclin-dependent kinases. J. Biol. Chem. 271, 18961–18965 [DOI] [PubMed] [Google Scholar]
- 38. Moscatello D. K., Santra M., Mann D. M., McQuillan D. J., Wong A. J., Iozzo R. V. (1998) Decorin suppresses tumor cell growth by activating the epidermal growth factor receptor. J. Clin. Invest. 101, 406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu G., Hitomi H., Hosomi N., Shibayama Y., Nakano D., Kiyomoto H., Ma H., Yamaji Y., Kohno M., Ichihara A., Itoh H., Nishiyama A. (2011) Prorenin induces vascular smooth muscle cell proliferation and hypertrophy via epidermal growth factor receptor-mediated extracellular signal-regulated kinase and Akt activation pathway. J. Hypertens. 29, 696–705 [DOI] [PubMed] [Google Scholar]
- 40. Schönherr E., Levkau B., Schaefer L., Kresse H., Walsh K. (2001) Decorin-mediated signal transduction in endothelial cells. Involvement of Akt/protein kinase B in up-regulation of p21(WAF1/CIP1) but not p27(KIP1). J. Biol. Chem. 276, 40687–40692 [DOI] [PubMed] [Google Scholar]
- 41. Buraschi S., Pal N., Tyler-Rubinstein N., Owens R. T., Neill T., Iozzo R. V. (2010) Decorin antagonizes Met receptor activity and down-regulates β-catenin and Myc levels. J. Biol. Chem. 285, 42075–42085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tharaux P. L., Chatziantoniou C., Fakhouri F., Dussaule J. C. (2000) Angiotensin II activates collagen I gene through a mechanism involving the MAP/ER kinase pathway. Hypertension 36, 330–336 [DOI] [PubMed] [Google Scholar]
- 43. Huang H., Chang E. J., Ryu J., Lee Z. H., Lee Y., Kim H. H. (2006) Induction of c-Fos and NFATc1 during RANKL-stimulated osteoclast differentiation is mediated by the p38 signaling pathway. Biochem. Biophys. Res. Commun. 351, 99–105 [DOI] [PubMed] [Google Scholar]
- 44. Liang G., Bansal G., Xie Z., Druey K. M. (2009) RGS16 inhibits breast cancer cell growth by mitigating phosphatidylinositol 3-kinase signaling. J. Biol. Chem. 284, 21719–21727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y., Neo S. Y., Han J., Yaw L. P., Lin S. C. (1999) RGS16 attenuates Gαq-dependent p38 mitogen-activated protein kinase activation by platelet-activating factor. J. Biol. Chem. 274, 2851–2857 [DOI] [PubMed] [Google Scholar]
- 46. Pillozzi S., Arcangeli A. (2010) Physical and functional interaction between integrins and hERG1 channels in cancer cells. Adv. Exp. Med. Biol. 674, 55–67 [DOI] [PubMed] [Google Scholar]
- 47. Dolmetsch R. E., Pajvani U., Fife K., Spotts J. M., Greenberg M. E. (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333–339 [DOI] [PubMed] [Google Scholar]
- 48. Yucel G., Altindag B., Gomez-Ospina N., Rana A., Panagiotakos G., Lara M. F., Dolmetsch R., Oro A. E. (2013) State-dependent signaling by Cav1.2 regulates hair follicle stem cell function. Genes Dev. 27, 1217–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brandenburger T., Strehler E. E., Filoteo A. G., Caride A. J., Aumüller G., Post H., Schwarz A., Wilhelm B. (2011) Switch of PMCA4 splice variants in bovine epididymis results in altered isoform expression during functional sperm maturation. J. Biol. Chem. 286, 7938–7946 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.