

Abstract
Objective
A high incidence of structural brain abnormalities has been reported in individuals with pyridoxine-dependent epilepsy (PDE). PDE is caused by mutations in ALDH7A1, also known as antiquitin. How antiquitin dysfunction leads to cerebral dysgenesis is unknown. In this study, we analyzed tissue from a child with PDE as well as control human and murine brain to determine the normal distribution of antiquitin, its distribution in PDE, and associated brain malformations.
Methods
Formalin-fixed human brain sections were subjected to histopathology and fluorescence immunohistochemistry studies. Frozen brain tissue was utilized for measurement of PDE-associated metabolites and Western blot analysis. Comparative studies of antiquitin distribution were performed in developing mouse brain sections.
Results
Histologic analysis of PDE cortex revealed areas of abnormal radial neuronal organization consistent with type Ia focal cortical dysplasia. Heterotopic neurons were identified in subcortical white matter, as was cortical astrogliosis, hippocampal sclerosis, and status marmoratus of the basal ganglia. Highly elevated levels of lysine metabolites were present in postmortem PDE cortex. In control human and developing mouse brain, antiquitin immunofluorescence was identified in radial glia, mature astrocytes, ependyma, and choroid plexus epithelium, but not in neurons. In PDE cortex, antiquitin immunofluorescence was greatly attenuated with evidence of perinuclear accumulation in astrocytes.
Interpretation
Antiquitin is expressed within glial cells in the brain, and its dysfunction in PDE is associated with neuronal migration abnormalities and other structural brain defects. These malformations persist despite postnatal pyridoxine supplementation and likely contribute to neurodevelopmental impairments.
Introduction
Pyridoxine-dependent epilepsy (PDE) has long been recognized clinically as an infantile epileptic encephalopathy in which seizures are poorly responsive to traditional antiepileptic drugs, but rapidly disappear upon the administration of pyridoxine (vitamin B6).1 With continued pyridoxine treatment, seizures generally remain at bay, although neurodevelopmental outcomes are still frequently poor.2,3 In 2006, the genetic basis of PDE was reported: homogygous or compound heterozygous mutations of antiquitin, also known as aldehyde dehydrogenase 7A1 (ALDH7A1).4 Homologs of this gene are well conserved throughout evolution, which functions in plants to maintain cellular turgor.5 In mammals, antiquitin is expressed in the cellular cytosol and mitochondria and is involved in the metabolism of the amino acid lysine, resulting in the normal end product of acetyl coenzyme A (CoA).6 With antiquitin dysfunction in PDE, metabolic intermediates including alpha-aminoadipic semialdehyde (α-AASA), δ1-piperideine-6-carboxylate (P6C), and pipecolic acid (PA) accumulate. The pathophysiology of the disease is thought to result from chemical condensation of P6C with pyridoxal phosphate (PLP), the biologically active form of pyridoxine.4 PLP is a cofactor for over 140 enzymatic activities, including glutamic acid decarboxylase (GAD), which is responsible for the conversion of glutamate to the inhibitory neurotransmitter γ-aminobutyric acid (GABA).7 A reduction in GABA synthesis due to reduced PLP availability may be partially responsible for seizure development in PDE.4
Although the activities of antiquitin in mammalian cells are being identified8,9, and the effects of mutations seen in PDE on its function delineated10,11, studies to assess its distribution in the brain and potential roles in brain development have not been reported. Interestingly, multiple brain malformations have been reported in association with PDE, such as hypoplasia of the corpus callosum and white matter, hydrocephalus, posterior fossa abnormalities, and neuronal migration defects, including cortical dysplasia and heterotopias.2,12,13 In the present study, we characterized the normal distribution of antiquitin in human and developing mouse brain and disruption of its expression in the brain of a child with PDE. In addition, we have identified evidence of neuronal migration abnormalities. These findings may contribute to neurodevelopmental impairments in PDE and are unlikely to be substantially impacted by postnatal pyridoxine supplementation.
Methods
Specimens
Full thickness samples of the cerebral cortex from a child with PDE were taken at autopsy and placed at −80° C. The remainder of the brain was fixed in formalin. Postmortem interval prior to freezing/fixation was about 12 hours. The postmortem specimens as well as sections from her previous epilepsy surgery were shipped to Seattle Children's Hospital for evaluation. Additional frozen and formalin-fixed, paraffin-embedded human brain specimens were obtained from the Seattle Children's Hospital pathology lab and from the National Institute of Child Health and Human Development (NICHD) Brain and Tissue Bank for Neurodevelopmental Disorders at the University of Maryland. Wild-type mouse brains at embryonic day 14.5 (E14.5), postnatal day 0.5 (P0.5), and P20 were fixed in 4% paraformaldehyde, cryoprotected with sucrose, and frozen in Optimal Cutting Temperature (OCT) compound before sectioning. These studies were approved by the Seattle Children's Hospital Institutional Review Board (IRB) and the Institutional Animal Care and Use Committee (IACUC).
Measurement of PDE-associated metabolites
Frozen postmortem human brain tissue (43-116 mg) was homogenized in 1 mL of cold 0.4M borate buffer and centrifuged at 17,500×g for 15 min at 4 °C. The supernatant was frozen and stored at −80° C until use. Quantification of α-AASA, P6C, and PA were performed using liquid chromatography-tandem mass spectrometry (LC-MS/MS) as previously detailed.14
Western blot analysis
Rabbit monoclonal ALDH7A1 (antiquitin) antibody (EP1935Y) against the C-terminus of human ALDH7A1 was developed by Epitomics (Burlingame, CA). Rabbit polyclonal glial fibrillary acidic protein (GFAP) antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit monoclonal anti-glutamic acid decarboxylase (GAD) 65 was from Epitomics, mouse monoclonal anti-GAD 67 was from Abcam (Cambridge, MA), and rabbit polyclonal anti-GAD 65/67 was from Millipore (Temecula, CA).
Frozen cortical tissue (100-200 mg) was separated from underlying white matter and homogenized in 5 mM Tris/HCl (pH 7.4) with 0.32 M sucrose. The homogenates were centrifuged at 3000×g for 5 min at 4 °C. The supernatant was then centrifuged at 40,000×g for 1 h at 4 °C to pellet the membrane fraction. Thirty μg of protein from the cytosolic fraction was electrophoretically separated and transferred to an Immobilon-FL PVDF membrane (Millipore). The membrane was blocked with Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE), incubated in primary antibodies overnight at 4 °C, and then incubated in goat anti-rabbit (Alexa Fluor 680, Molecular Probes, Inc., Eugene, OR) or goat anti-mouse (IRDye 800, LI-COR) infrared dye-labeled secondary antibody for 2 h at room temperature. Infrared fluorescence was used for signal detection and quantitation (Odyssey Infrared Imaging System, LI-COR). Binding of mouse monoclonal antibodies against β-tubulin (Novus) was used as a loading control.
Immunohistochemistry
Specimens were deparaffinized by incubation in xylene, followed by rehydration through graded ethanol/water solutions and equilibration in PBS. Antigen retrieval was performed by heating in 0.01M sodium citrate (pH 6.0) in a 90° C water bath for 45 minutes. Specimens were then blocked in PBS with 10% donkey serum, 0.1% Triton X-100 and 2% BSA for 30 minutes and incubated overnight at 4° C in primary antibodies diluted in blocking solution. Primary antibodies used in these studies included rabbit monoclonal ALDH7A1 antibody (Epitomics), goat polyclonal GFAP (Santa Cruz), mouse monoclonal GFAP (Chemicon), mouse monoclonal MAP2 (Millipore), goat polyclonal calcium/calmodulin-dependent protein kinase IIα (CAMKIIα) (Santa Cruz), rabbit polyclonal GAD65/67 (Millipore), goat polyclonal calretinin (Millpore), goat polyclonal SOX2 (Santa Cruz), and goat polyclonal doublecortin (DCX; Santa Cruz). Slides were then incubated in fluorescent secondary antibodies (Alexa Fluor) at room temperature for 2 hours. Following incubation in DAPI nuclear counterstain and Sudan Black to block endogenous fluorescence, sections were examined and photographed using a Zeiss Axio Imager Z1 epifluorescence microscope or a Zeiss LSM 710 laser scanning confocal microscope. Cell counts were performed using NIH ImageJ software.
Results
PDE case history
The patient presented at eight days of age with clinical seizures and encephalopathy characterized by lethargy, irritability, hypotension and acidosis. In retrospect, fetal seizures may have also occurred. Her seizures were not controlled despite treatment with phenobarbital, phenytoin, topiramate, valproic acid, zonisamide and clonazepam, as well as implantation of a vagus nerve stimulator and use of the ketogenic diet. Typically, she would have clusters of two to three seizures per day for several days followed by a two week seizure-free interval. Prolonged seizures only responded to parenteral benzodiazepines, and on several occasions she required treatment with continuous pentobarbital infusions. Brain MRI at 13 months was normal, as was head circumference (78th%). She underwent surgical lobectomy at 15 months of age due to focal findings on SPECT and ictal EEG with resection of the right occipital and posterior parietal lobes. This procedure did not improve her seizure control. A repeat brain MRI at 20 months revealed porencephaly at the site of her surgical resection, thinning of the corpus callosum, signal abnormality of the white matter suggestive of gliosis in the periatrial region and posterior centrum semiovale, delay in myelination, bilateral high signal intensity in the hippocampus, and prominence of the cortical sulci. When the patient was three years of age, a clinical diagnosis of PDE was made in her younger sister, who also presented with neonatal seizures. Pyridoxine supplementation was therefore started in this patient and her seizures were controlled. Her anticonvulsants were sequentially discontinued without seizure recurrence. Once ALDH7A1 was identified as the gene responsible for PDE, her diagnosis was confirmed by the finding of compound heterozygous mutations, c.[750G>A] (splice error) + c.[505C>T] (Pro169Ser).12 Despite excellent seizure control, she had severe neurodevelopmental disabilities including diffuse hypotonia, poor head control, minimal use of her hands and limited communication skills. Her condition was complicated by severe constipation resulting in impaction requiring surgical management. She experienced several postoperative complications and at 9 years of age was placed in hospice care and expired. Clinical information about this patient was also included in the supplemental data from the case series of Mills et al.12.
Measurement of PDE-associated metabolites in human brain
We have reported methods by which the catabolic metabolites of lysine that accumulate in PDE (α-AASA, P6C, and PA)can be measured simultaneously in plasma samples by LC-MS/MS.14 These metabolites have not previously been measured in brain specimens. In the current study we adapted this method to assess levels of α-AASA, P6C, and PA in extracts from frozen postmortem cortex obtained from the PDE patient and two control individuals (8 and 13 yrs old) with non-neurologic causes of death. As shown in Table 1, significant levels of α-AASA, P6C, and PA were found in the PDE specimen, while these compounds were undetectable in the control specimens.
Table 1.
Lysine metabolites are elevated in postmortem PDE cortex.
| α-AASA (pmol/mg) | P6C (pmol/mg) | PA (pmol/mg) | |
|---|---|---|---|
| PDE | 13.02 | 8.14 | 115.58 |
| Control 1 | ND | ND | ND |
| Control 2 | ND | ND | ND |
Measurements are shown for alpha-aminoadipic semialdehyde (α-AASA), δ1-piperideine-6-carboxylate (P6C), and pipecolic acid (PA), and are expressed as pmol per mg of frozen brain tissue. ND = not detected.
Neuropathological abnormalities in PDE
The postmortem brain was small for age and lacked the right parieto-occipital region due to prior surgery, but showed no other obvious macroscopic malformation. Histologic examination of resected (age 15 months) and postmortem (age 9 years) cortex revealed increased variability of the cortical thickness, overall mild to moderate atrophy, and variable laminar disorganization (Fig 1A). Dyslamination was most severe in the surgically-resected right parieto-occipital cortex, the region associated with abnormal signal on preoperative SPECT scan, where radial microcolumns were seen consistent with focal cortical dysplasia type Ia (FCD-Ia) (Fig 1B). Accordingly, neurons were stacked in microcolumns of >8 neurons15, and indeed some microcolumns included >20 neurons. Patchy cortical gliosis was also seen (Supplementary Fig 1). Increased numbers of interstitial neurons were observed in the white matter, and formed microscopic heterotopia in the insular regions (Fig 1C). The hippocampi were bilaterally sclerotic, with severe loss of neurons in CA1, CA3, and dentate gyrus (Fig 1D).
Figure 1. Neuropathologic abnormalities in PDE brain.
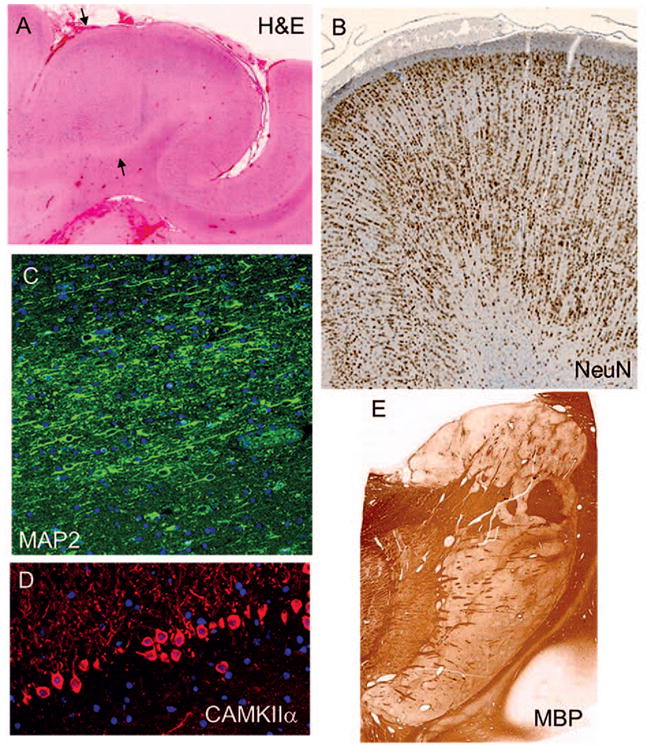
(A,B) Epilepsy surgery specimen, parieto-occipital lobe:focal cortical thickening (arrows, A) with laminar disorganization and microcolumnar neuronal organization (B, NeuN), similar to focal cortical dysplasia type Ia. (C) Postmortem specimen, temporal lobe:cluster of heterotopic neurons in white matter demonstrated by MAP2 immunohistochemistry (IHC). (D) Postmortem hippocampus: severe neuronal loss in dentate gyrus, CAMKIIα IHC. (E) Postmortem basal ganglia: marbled appearance (status marmoratus), highlighted by myelin basic protein (MBP) stain.
The hemispheric white matter was pale, atrophic, and gliotic, with scattered microcalcifications, most abundant around the ventricles. MBP immunohistochemistry demonstrated no myelin deficiency. The ependyma of the lateral ventricles was focally eroded and gliotic. The basal ganglia showed abnormal myelinated fiber organization with gliosis and formation of gray nodules, characteristic of status marmoratus16 (Fig 1E). Ferruginated neurons were observed in the basal ganglia and thalamus, which was also gliotic. The cerebellar cortex displayed variable patchy loss of Purkinje cells with Bergmann gliosis. The cerebellar deep nuclei were gliotic, and the dentate nuclei were broken into discontinuous clusters rather than smooth ribbons. The pontine nuclei and inferior olives were mildly atrophic. The pyramidal tracts were relatively intact.
Together, the neuropathologic findings were consistent with a combination of primary cortical malformation (FCD-Ia), early hypoxic-ischemic injury, and secondary effects of a seizure disorder.
Antiquitin expression in PDE and control human brain
Western blot analysis was performed to assess the levels of antiquitin protein expression in frozen PDE and control postmortem brain specimens. As shown in Figure 2A, the rabbit monoclonal antiquitin antibody recognizes a single band at the expected molecular weight of human antiquitin, ∼58 kDa. Expression of antiquitin was high in the cytosolic fractions of the control human cortex specimens, but very low in the PDE specimen. In immunohistochemistry studies of control human cortex (n = 3, age 31 wk EGA - 16 years, postmortem intervals 11-31 hrs), we found that antiquitin immunofluoresence was present in star-shaped cells and their processes throughout the cortex (Fig 2B). Colocalization of antiquitin with GFAP confirmed its astrocytic location (Fig 3A,B). Antiquitin immunofluorescence was also identified near the pial membrane and its invaginations (Fig 2B), as well as surrounding cortical blood vessels, likely within astrocyte end-feet (Fig 3A). In the PDE specimen astrocytic antiquitin expression was greatly reduced (Fig 2C). At higher magnifications antiquitin was found to be present in small amounts in the astrocyte cell soma adjacent to the nucleus, at times showing perinuclear accumulation, but was not present in astrocyte processes (Fig 3B). Interestingly, in the PDE specimen antiquitin immunofluorescence was still present near the pial membrane and its invaginations, but not surrounding deeper blood vessels (Fig 2C). Antiquitin immunofluorescence was never present in cells with neuronal morphology, and no colocalization was identified between antiquitin and the neuronal marker MAP2 (Supplementary Fig 2).
Figure 2. Reduced antiquitin protein expression in PDE brain.
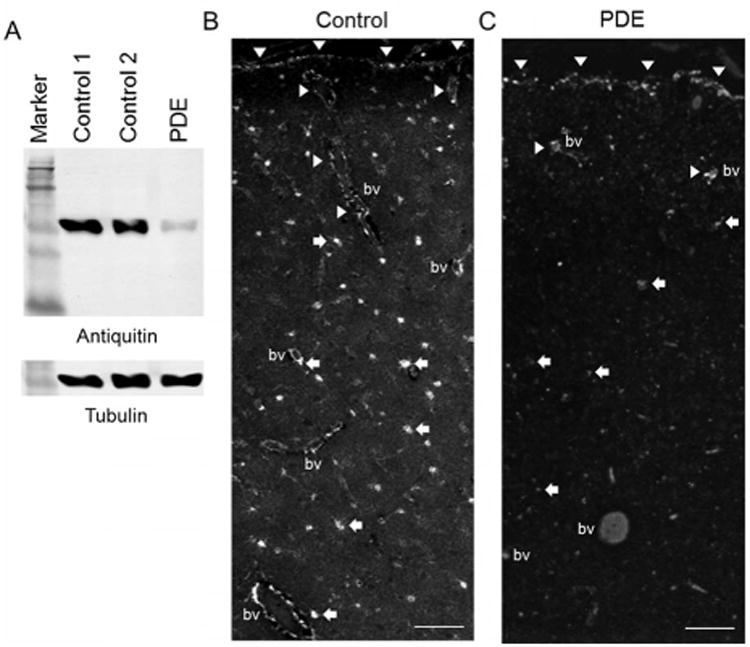
(A) Western blot showing specificity of monoclonal antiquitin antibody and substantially reduced protein expression in PDE cortex. Tubulin binding is shown as a loading control. (B,C) Antiquitin IHC in control (B) and PDE (C) cortex. Arrowheads: pia and its invaginations, arrows: antiquitin-labeled astrocytes (much reduced in PDE), BV: blood vessel. Scale bars = 100 μM.
Figure 3. Colocalization of antiquitin with the astrocyte marker GFAP.
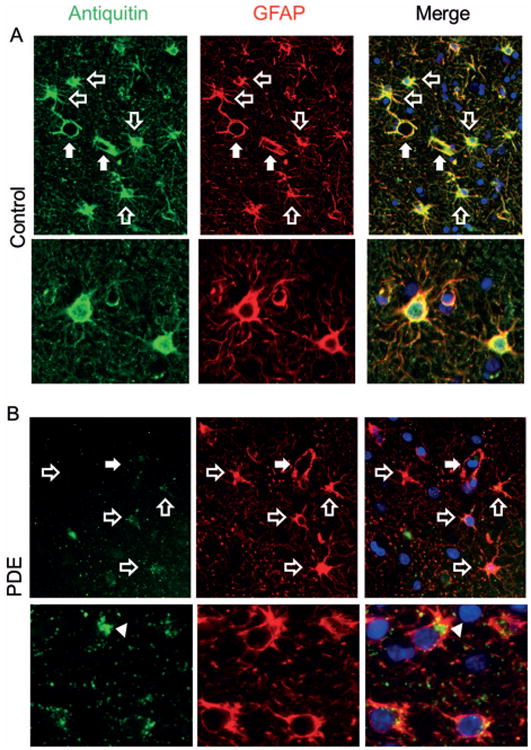
Antiquitin (green) and GFAP (red) immunofluorescence in Control (A) and PDE (B) cortex. In control brain, the distribution of antiquitin and GFAP overlaps. In PDE cortex, antiquitin immunofluorescence is faint and in some astrocytes is accumulated near the nucleus (arrowhead). Open arrows indicate astrocytes; closed arrows indicate small cortical blood vessels, which are surrounded by strong antiquitin immunoreactivity in control, but not PDE, cortex. Blue = nuclear counterstain DAPI.
Because of similarities between the histology of the PDE brain and FCD type Ia, we assessed antiquitin expression in three FCD specimens surgically resected due to intractable epilepsy. These specimens were of the FCD-Ib (abnormal tangential cortical lamination) and FCD-Ic types (abnormal radial and tangential cortical lamination)15; no isolated FCD type Ia (abnormal radial cortical lamination) specimens were available for examination. Antiquitin expression in these samples was found to be normal (Supplementary Fig 3). This finding indicates that reduction in antiquitin expression in PDE is not caused by repeated seizures, and that deficient antiquitin expression is not a common feature of FCD types Ib or Ic.
Antiquitin expression in developing mouse brain
We next assessed the distribution of antiquitin immunofluorescence in mouse brain at E14.5, P0, and P20. At the earliest developmental stage (E14.5), cytosolic antiquitin labeling was seen in cells of the choroid plexus and surrounding the cerebral ventricles (Fig 4A,B). At P0, strong antiquitin immunofluorescence was present in the choroid plexus and ventricular zone, as well as in radial glial processes of the forebrain (Fig 4C,D) and Bergmann glia of the cerebellum (Fig 4E). In P0 hippocampus, antiquitin was co-expressed with GFAP in the supragranular region of the dentate gyrus (Fig 4F). Confirmation of the identity of strongly antiquitin-immunofluorescent cells at P0 as radial/Bergmann glia was provided by demonstration of expression in cells labeled by the nuclear radial glial marker SOX2 (Fig 5A,B). No colocalization was found with DCX, a marker of immature neurons (Fig 5C,D). At P20, antiquitin was identified in mature GFAP-positive astrocytes, as well as choroid plexus and ependymal cells, without colocalization with the neuronal marker CAMKIIα (Fig 4G-J).
Figure 4. Antiquitin distribution in developing mouse brain.
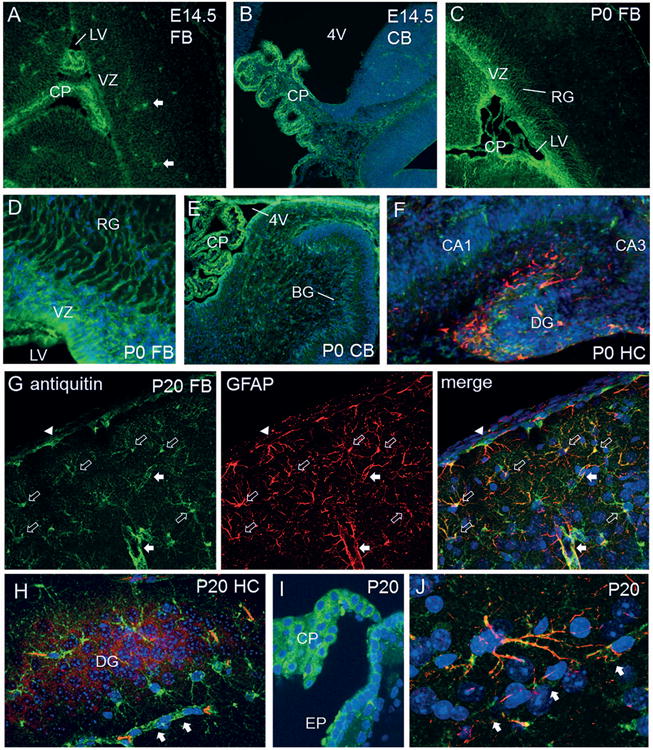
(A) In E14.5 forebrain (FB), antiquitin immunofluorescence (green) is identified in the choroid plexus (CP) of the lateral ventricle (LV) and cells of the ventricular zone (VZ). Filled arrows = non-specifically fluorescent blood vessels. (B) Antiquitin expression in CP of the fourth ventricle (4V) in E14.5 cerebellum (CB). Blue = DAPI. (C) At P0, antiquitin continues to be present in the CP and VZ, and is also visualized in radial glial (RG) processes. (D) Higher magnification of antiquitin expression in RG processes at P0. (E) Antiquitin immunofluorescence in Bergmann glia (BG) of the cerebellum (CB) at P0. (F) Coexpression of antiquitin (green) with GFAP (red) in the supragranular region of P0 mouse hippocampal dentate gyrus (DG). (G-J) P20 mouse brain sections. Antiquitin (green) is colocalized with GFAP (red) in astrocytes and their processes (G). Open arrows: astrocytes, arrowhead: pial astrocyte, closed arrows: blood vessels. (H) In P20 HC, antiquitin (green) is present in astrocytes, but not in DG neurons labeled with CAMKIIα (red). (I) Strong antiquitin immunofluorescence is identified in the CP and ependyma (EP) of the third ventricle. (J) High power image of cortical astrocyte coexpressing antiquitin and GFAP, with its processes enveloping a nearby capillary (arrows).
Figure 5. Antiquitin is expressed SOX2-positive cells.
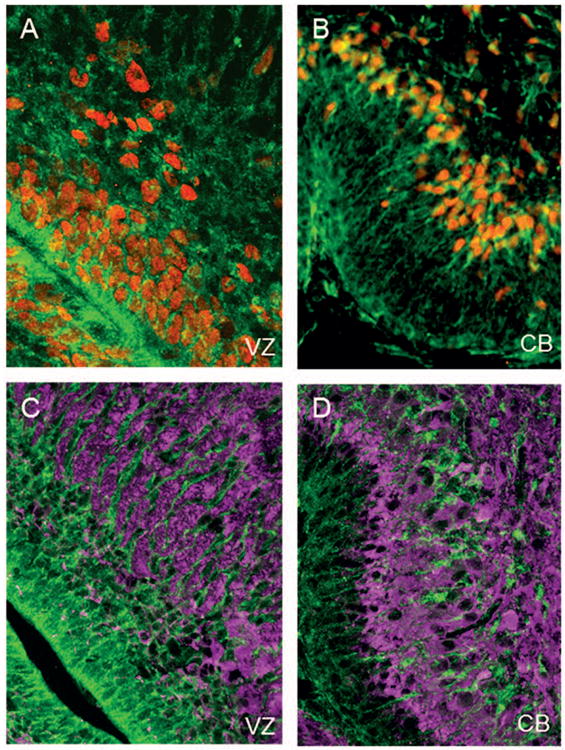
IHC images from the lateral ventricle and cerebellum of a P0 mouse are shown. (A) Antiquitin immunofluorescence (green) is present in ventricular zone (VZ) radial glia labeled with the nuclear marker SOX2 (red). (B) Antiquitin co-expression with SOX2 in Bergmann glia of the cerebellum (CB). (C,D) Antiquitin (green) does not colocalize with DCX (magenta) in immature neurons of the subventricular zone (C) or cerebellum (D).
Markers of inhibitory neurotransmission in PDE
The intractable seizures of PDE have been hypothesized to be largely due to reduced function of the GABA-synthesizing enzyme GAD, which requires PLP as a cofactor.4 Previous studies have found reduced GABA levels in CSF and brain in some individuals with PDE.17,18 We therefore studied components of inhibitory GABAergic neurotransmission in brain specimens from our PDE patient. In contrast to the abnormal laminar organization of pyramidal cells in PDE cortex, the number and distribution of interneurons, as determined by calretinin (Fig 6A-C)and GAD 65/67 (Supplementary Fig 4) immunohistochemistry, were unchanged in PDE cortex. However, total expression levels of GAD 65/67 were lower, suggesting a possible reduction in GABA synthesis capabilities. This was due to reduced expression of the 67 kD GAD isoform (GAD 67) but not the 65 kD one (GAD 65, Fig 6D,E). GAD 67 is thought to mediate the majority of basal GABA synthesis, while GAD 65 is responsible for increases in GABA synthesis during times of increased neuronal activity.19,20 A similar pattern of GAD reduction was seen in cortex resected from children with intractable epilepsy due to FCD (Fig 6D,E), suggesting that this finding may be due to repeated seizures rather than antiquitin dysfunction or pyridoxine sequestration.
Figure 6. Markers of inhibitory neurotransmission in PDE cortex.
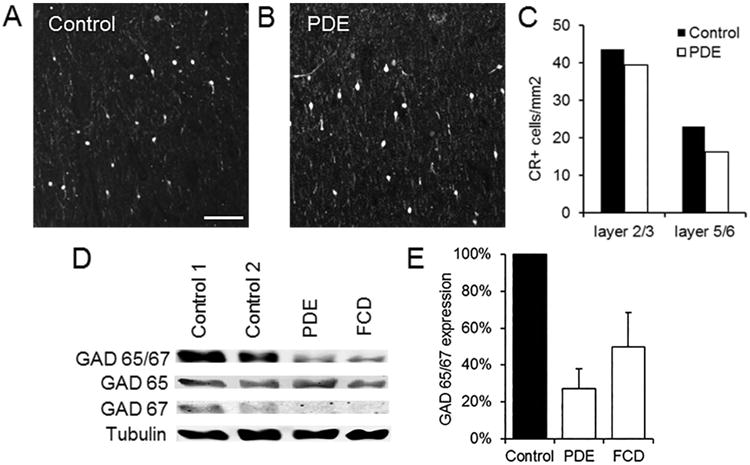
(A,B) Interneuron density in PDE cortex is similar to that in controls. Calretinin immunofluorescence in layer 2/3 of control (A) and PDE (B) cortex. Scale bar: 100 μM. (C) Quantitation of calretinin (CR) positive interneurons in layers 2/3 and 5/6 of control and PDE cortex. (D,E) Total GAD expression is reduced in PDE cortex, due primarily to a decrease in GAD 67, similar to that seen in intractable epilepsy due to FCD. Shown are a Western blot of GAD 65/67, GAD 65, and GAD 67 expression (D) and quantification of GAD 65/67 band intensities in PDE and FCD (n=3) cortex (E).
Discussion
In this study we have for the first time identified antiquitin (ALDH7A1) as a predominantly glial protein that is first expressed during embryonic brain development. This finding provides insight into the pathogenesis of structural brain abnormalities in PDE and the frequent persistence of neurodevelopmental impairments despite control of seizures with postnatal pyridoxine treatment.2,3,12,13 All of the cell types in which antiquitin expression was detected in the mature brain, including astrocytes, ependyma, and choroid plexus epithelium, are within the glial lineage. In the developing brain, antiquitin is also expressed at high levels in radial glia and Bergmann glia, and therefore may play roles in neurogenesis and neuronal migration.21 Our results using immunohistochemistry with a monoclonal ALDH7A1 antibody are supported by in situ hybridization data of E14.5 mouse brain from www.genepaint.org (GenePaint Set IDs EB2198 and MH1649) and the Allen Brain Atlas Prenatal LMD Microarray of human brain at 15, 16, and 21 post conceptual weeks (http://www.brainspan.org; Supplementary Fig 5). Both of these sources demonstrate prominent ALDH7A1 mRNA expression in the ventricular (VZ) and subventricular zones (SVZ), the locations of radial glia and their processes.
Disruption of radial and Bergmann glial function in PDE, either through accumulation of toxic metabolites, dysfunction of PLP-dependent enzymes, or absence of another critical activity of antiquitin, may be responsible for the appearance of cortical dysplasia, neuronal heterotopia, and cerebellar malformations. Our finding of normal distribution of interneurons in PDE is consistent with this hypothesis, since as opposed to pyramidal cell migration, interneuron migration during development is largely tangential rather than radial.22 The specific finding of FCD type Ia in PDE likely indicates disruption of migrational processes around midgestation, a time when the developing human brain begins transitioning from radial columnar architecture to mature horizontal cortical lamination.23 We hypothesize that these neuronal migration abnormalities may at least partially account for neurodevelopmental impairments in children with PDE. In a series of 18 children with FCD type I who underwent surgical resection due to intractable epilepsy, none had intelligence testing within the normal range.24 In addition, individuals with subcortical heterotopia, as seen in our PDE patient, were more likely to exhibit developmental delays than those with subependymal heterotopias.25
Antiquitin is also expressed in the choroid plexus and ependyma, where it may be involved in formation, absorption, or circulation of cerebrospinal fluid. Deficiency of antiquitin function in these cells may contribute to hydrocephalus, which is seen at increased frequency in PDE.12,13 Indeed, in the garden pea the highly conserved antiquitin protein is involved in the regulation of osmotic pressure, and human antiquitin has been shown to provide protection against osmotic stress through the generation of organic osmolytes such as betaine.26 The ependyma may also influence neurogenesis in the SVZ through secretion of trophic factors and provision of metabolic support.27,28 Antiquitin dysfunction and accumulation of toxic metabolites in mature astrocytes may prevent normal regulation of neurotransmission, such as impaired glutamate, GABA, and potassium uptake from the extracellular space, which could contribute to neuronal injury.29 Antiquitin expression in perivascular astrocytic end-feet and the pial glia limitans may be important in both blood-brain barrier function and neuronal migration. In the cobblestone lissencephalies, disruption of the structural integrity of the glia limitans is thought to allow overmigration of neurons into the subarachnoid space.30
The cortical dyslamination and microcolumnar organization identified in our patient were fairly subtle on analysis by routine histologic stains, and only became prominent with the application of the neuronal marker NeuN. The subcortical heterotopias we identified were small and not visible on gross examination. There are 12 previously reported autopsy studies of cases of suspected PDE, all prior to 1978 and therefore without genetic confirmation. Seven of those patients expired before one week of age; in 3 cerebral edema was present while in another 3 there were small degrees of intracerebral hemorrhage. These studies revealed gliosis, neuronal loss in the thalamus, decreased white matter volume with thinning of the corpus callosum, and irregular lobulation of the cerebellum, but no apparent cortical abnormalities17,31,32 and Baxter, P.S. personal communication. However, histologic stains to specifically evaluate neuronal lamination were not performed. In the case series of Mills et al., 2/19 patients had cortical dysplasia or heterotopias evident on neonatal brain MRI.12 In contrast, MRI did not reveal cortical abnormalities in our patient, consistent with reports that a large proportion of pathologically-proven type I FCD lesions are not visible on routine MRI scans.33 Based on our findings, we hypothesize that the prevalence of neuronal migration abnormalities in PDE is likely much higher than appreciated.
Other types of structural brain malformations are even more prominent in PDE. In the combined case series of Mills et al. 2010, Bok et al. 2012, and Peréz et al. 2013 (n=30), 37% had posterior fossa abnormalities (including mega cisterna magna and cerebellar hypoplasia), 30% had corpus callosum dysgenesis or agenesis, and 30% had ventriculomegaly or hydrocephalus. These lesions were present on MRIs obtained both in the neonatal period and later in life, and were even seen in children who had begun pyridoxine supplementation prior to birth.2,12,13 These findings, combined with our current results, provide very strong support for the hypothesis that antiquitin function is required for normal brain development.
Some of the neuropathologic findings in our PDE patient were likely secondary to her history of uncontrolled epilepsy in infancy rather than a direct consequence of antiquitin deficiency, and highlight the importance of early institution of pyridoxine therapy in this disease. She had marked hippocampal sclerosis with neuronal loss in CA1 and the dentate gyrus, a probable consequence of repeated and prolonged seizures.34 The status marmoratus of the basal ganglia may have been secondary to hypoxic-ischemic injury sustained during her septic shock-like presentation in the neonatal period.16 Development of an animal model of PDE is necessary to definitively separate the neuropathologic sequelae of antiquitin deficiency from those due to seizures and other comorbidities.
In a large postnatal mouse brain transcriptome database, ALDH7A1 mRNA was found to be highly enriched in astrocytes as compared with neurons or oligodendroglial cells.35 Interestingly, in that study, prominent astrocytic expression of several other members of the aldehyde dehydrogenase family that have been implicated in human neurodevelopmental disorders was also detected: ALDH3A2 (fatty aldehyde dehydrogenase, associated with Sjögren–Larsson syndrome, a neurocutaneous disorder which includes mental retardation, spastic tetraplegia and cortical laminar disorganization36), ALDH4A1 (pyrroline-5-carboxylate (P5C) dehydrogenase, mutated in type II hyperprolinemia, with neurologic symptoms resulting from P5C-mediated deactivation of PLP similar to the mechanism of PDE; no neuropathologic reports available), ALDH5A1 (succinic semialdehyde dehydrogenase, involved in catabolism of GABA, mutation of which causes human γ-hydroxybutyric aciduria with epilepsy, ataxia, mental retardation, cerebellar atrophy and abnormal MRI signal in the globus pallidi37), ALDH6A1 (acetyl CoA-dependent methylmalonate semialdehyde dehydrogenase, mutation of which is a cause of 3-hydroxyisobutyryic aciduria associated with developmental delay, microcephaly, migrational disorder, and agenesis of the corpus callosum38–40) and ALDH18A1 (Delta1-pyrroline-5-carboxylate synthase (P5CS), deficiency of which causes developmental delay, choreoathetosis, and microcephaly41). These findings demonstrate that the functions of astrocytic aldehyde dehydrogenases are critical for normal neuronal development and activity.
In our PDE case, protein levels of antiquitin were very low (< 10% of control) as determined by western blotting using a monoclonal antibody that recognizes the C terminus of the protein. One of the mutations in our patient, c.750G>A, has been found to induce abnormal mRNA splicing which likely leads to mRNA degradation or production of a markedly truncated protein.42 The other allele harbors the missense mutation P169S, which has not been characterized, although other mutations in this region have been hypothesized to affect substrate binding.11 Based on our findings of very low antiquitin immunoreactivity and evidence of accumulation of protein in the astrocyte soma adjacent to the nucleus, we suggest that this mutation may result in a change in antiquitin structure such that it is degraded or unable to undergo normal intracellular trafficking. The reason for the observed relative persistence of antiquitin immunoreactivity near the pial membrane and its invaginations as compared with other structures is unknown. The effects of other pathogenic antiquitin mutations (>50 reported)11–13 on protein expression levels in the brain and subcellular distribution have not yet been investigated.
While multiple authors have published data demonstrating that α-AASA, P6C and PA are elevated in blood and urine in patients with PDE, and that elevations of α-AASA and P6C (but not always PA) persist despite pyridoxine treatment,11–14,43 our study is the first to demonstrate that 1) α-AASA, P6C and PA are measurable in brain tissue, 2) α-AASA, P6C and PA levels can be determined in frozen postmortem brain tissue, providing another avenue for retrospective PDE diagnosis, and 3) treatment with pyridoxine does not prevent accumulation of α-AASA, P6C and PA in the brain. These are reactive compounds that may directly cause cellular injury and therefore further contribute to the neurodevelopmental phenotype of PDE.43,44 Of note, the postmortem interval prior to brain freezing was longer in the PDE case (12 hrs) than in the control specimens (5 hrs each), and metabolite levels were likely even higher prior to death due to the instability of α-AASA and P6C at room temperature or 4° C.14 In contrast, PA is fairly stable, consistent with our finding of higher levels of this metabolite in postmortem brain as compared with α-AASA or P6C (Table 1).
In conclusion, based on the results of our studies, we hypothesize that postnatal pyridoxine treatment is likely not sufficient to prevent neurodevelopmental impairments in PDE. Although seizures are frequently controlled, pyridoxine supplementation alone would not be expected to ameliorate structural brain abnormalities or glial accumulation of toxic metabolites due lack of ALDH7A1 function. The institution of a lysine-restricted diet may partially address this issue, and trials of this therapy in PDE are planned.45 Prenatal pyridoxine supplementation2,43 and maternal dietary restrictions may also be beneficial. However, as in most metabolic disorders, prenatal restoration of the normal function of the mutated protein would represent the ultimate cure.
Supplementary Material
Supplementary Figure 1. PDE cortex exhibits substantial gliosis. A) Western blot demonstrating markedly increased GFAP expression in PDE as compared with control cortex. Tubulin is shown as a loading control. B) GFAP IHC in control and PDE cortex. PDE cortex shows subpial gliosis, often seen in intractable epilepsy, and astrocyte proliferation in cortical layer I.
Supplementary Figure 2. Antiquitin is not expressed in cortical neurons. IHC of control human cortex showing absence of colocalization between antiquitin (red) and the neuronal marker MAP2 (green).
Supplementary Figure 3. Antiquitin immunofluorescence is normal in intractable epilepsy due to FCD type I. (A) Antiquitin western blot showing normal levels of expression in FCD Ib cortex as compared with controls. (B) Antiquitin immunofluorescence is normally distributed in a different child with FCD type Ib. (C) Normal antiquitin expression in a FCD Ic region demonstrating microcolumnar neuronal organization. Red = antiquitin, green = MAP2.
Supplementary Figure 4. Interneuron density is normal in PDE cortex. (A,B) GAD 65/67 immunofluorescence (red, arrows) in layer 2/3 of control (A) and PDE (B) cortex. Scale bar: 100 μM. (C) Quantitation of GAD positive interneurons in layers 2/3 and 5/6 of control and PDE cortex.
Supplementary Figure 5. ALDH7A1 mRNA expression in prenatal human brain. Graph generated from data downloaded from the Allen Brain Atlas Prenatal LMD Microarray (www.brainspan.org) of ALDH7A1 mRNA expression (probe A_23_P70231) as determined by microarray analysis of laser microdissected regions of human brain at 15, 16, and 21 post conceptual weeks (pcw). Expression is much higher in the subventricular zone (SVZ) and ventricular zone (VZ) than in the marginal zone (MZ), cortical plate (CP), subplate (CP), or intermediate zone (IZ).
Acknowledgments
This work was supported by research endowments of the Division of Neurology, Seattle Children's Hospital (S.M.G.) and by NIH K02 NS072162 (L.A.J.). We thank Dr. J. Siebert for assistance with pathologic examinations; R. Daza and Dr. R. Kahoud for assistance with immunohistochemistry studies; and Drs. K. Millen, E. Turner, R. Hodge, and B. Nelsen for valuable discussions. A special thank you goes to the family that contributed to this study.
References
- 1.Hunt AD, Jr, Stokes J, Jr, McCrory WW, Stroud HH. Pyridoxine dependency: report of a case of intractable convulsions in an infant controlled by pyridoxine. Pediatrics. 1954;13(2):140–145. [PubMed] [Google Scholar]
- 2.Bok LA, Halbertsma FJ, Houterman S, et al. Long-term outcome in pyridoxine-dependent epilepsy. Dev Med Child Neurol. 2012;54(9):849–854. doi: 10.1111/j.1469-8749.2012.04347.x. [DOI] [PubMed] [Google Scholar]
- 3.Basura GJ, Hagland SP, Wiltse AM, Gospe SM., Jr Clinical features and the management of pyridoxine-dependent and pyridoxine-responsive seizures: review of 63 North American cases submitted to a patient registry. Eur J Pediatr. 2009;168(6):697–704. doi: 10.1007/s00431-008-0823-x. [DOI] [PubMed] [Google Scholar]
- 4.Mills PB, Struys E, Jakobs C, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12(3):307–309. doi: 10.1038/nm1366. [DOI] [PubMed] [Google Scholar]
- 5.Lee P, Kuhl W, Gelbart T, et al. Homology between a human protein and a protein of the green garden pea. Genomics. 1994;21(2):371–378. doi: 10.1006/geno.1994.1279. [DOI] [PubMed] [Google Scholar]
- 6.Wong JWY, Chan CL, Tang WK, et al. Is antiquitin a mitochondrial Enzyme? J Cell Biochem. 2009;109(1):74–81. doi: 10.1002/jcb.22381. [DOI] [PubMed] [Google Scholar]
- 7.Percudani R, Peracchi A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003;4(9):850–854. doi: 10.1038/sj.embor.embor914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brocker C, Cantore M, Failli P, Vasiliou V. Aldehyde dehydrogenase 7A1 (ALDH7A1) attenuates reactive aldehyde and oxidative stress induced cytotoxicity. Chem Biol Interact. 2011;191(1-3):269–277. doi: 10.1016/j.cbi.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan CL, Wong JWY, Wong CP, et al. Human antiquitin: structural and functional studies. Chem Biol Interact. 2011;191(1-3):165–170. doi: 10.1016/j.cbi.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 10.Coulter-Mackie MB, Li A, Lian Q, et al. Overexpression of human antiquitin in E. coli: Enzymatic characterization of twelve ALDH7A1 missense mutations associated with pyridoxine-dependent epilepsy. Mol Genet Metab. 2012;106(4):478–481. doi: 10.1016/j.ymgme.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Scharer G, Brocker C, Vasiliou V, et al. The genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy due to mutations in ALDH7A1. J Inherit Metab Dis. 2010;33(5):571–581. doi: 10.1007/s10545-010-9187-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mills PB, Footitt EJ, Mills KA, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency) Brain. 2010;133(7):2148–2159. doi: 10.1093/brain/awq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pérez B, Gutiérrez-Solana LG, Verdú A, et al. Clinical, biochemical, and molecular studies in pyridoxine-dependent epilepsy. Antisense therapy as possible new therapeutic option. Epilepsia. 2013;54(2):239–248. doi: 10.1111/epi.12083. [DOI] [PubMed] [Google Scholar]
- 14.Sadilkova K, Gospe SM, Hahn SH. Simultaneous determination of alpha-aminoadipic semialdehyde, piperideine-6-carboxylate and pipecolic acid by LC–MS/MS for pyridoxine-dependent seizures and folinic acid-responsive seizures. J Neurosci Methods. 2009;184(1):136–141. doi: 10.1016/j.jneumeth.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 15.Blumcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52:158–74. doi: 10.1111/j.1528-1167.2010.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folkerth RD. Neuropathologic substrate of cerebral palsy. J Child Neurol. 2005;20(12):940–949. doi: 10.1177/08830738050200120301. [DOI] [PubMed] [Google Scholar]
- 17.Lott IT, Coulombe T, Di Paolo RV, et al. Vitamin B6-dependent seizures: pathology and chemical findings in brain. Neurology. 1978;28(1):47–54. doi: 10.1212/wnl.28.1.47. [DOI] [PubMed] [Google Scholar]
- 18.Kurlemann G, Ziegler R, Grüneberg M, et al. Disturbance of GABA metabolism in pyridoxine-dependent seizures. Neuropediatrics. 1992;23(5):257–259. doi: 10.1055/s-2008-1071353. [DOI] [PubMed] [Google Scholar]
- 19.Patel AB, de Graaf RA, Martin DL, et al. Evidence that GAD65 mediates increased GABA synthesis during intense neuronal activity in vivo. J Neurochem. 2006;97(2):385–396. doi: 10.1111/j.1471-4159.2006.03741.x. [DOI] [PubMed] [Google Scholar]
- 20.Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19(12):500–505. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- 21.Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Z. Molecular regulation of neuronal migration during neocortical development. Mol Cell Neurosci. 2009;42(1):11–22. doi: 10.1016/j.mcn.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Sarnat HB, Flores-Sarnat L. Radial Microcolumnar Cortical Architecture: Maturational Arrest or Cortical Dysplasia? Pediatr Neurol. 2013;48(4):259–270. doi: 10.1016/j.pediatrneurol.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Blümcke I, Pieper T, Pauli E, et al. A distinct variant of focal cortical dysplasia type I characterised by magnetic resonance imaging and neuropathological examination in children with severe epilepsies. Epileptic Disord. 2010;12(3):172–180. doi: 10.1684/epd.2010.0321. [DOI] [PubMed] [Google Scholar]
- 25.Barkovich AJ, Kjos BO. Gray matter heterotopias: MR characteristics and correlation with developmental and neurologic manifestations. Radiology. 1992;182(2):493–499. doi: 10.1148/radiology.182.2.1732969. [DOI] [PubMed] [Google Scholar]
- 26.Brocker C, Lassen N, Estey T, et al. Aldehyde Dehydrogenase 7A1 (ALDH7A1) Is a Novel Enzyme Involved in Cellular Defense against Hyperosmotic Stress. J Biol Chem. 2010;285(24):18452–18463. doi: 10.1074/jbc.M109.077925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Del Bigio MR. Ependymal cells: biology and pathology. Acta Neuropathol. 2009;119(1):55–73. doi: 10.1007/s00401-009-0624-y. [DOI] [PubMed] [Google Scholar]
- 28.Spassky N. Adult Ependymal Cells Are Postmitotic and Are Derived from Radial Glial Cells during Embryogenesis. J Neurosci. 2005;25(1):10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.David Y, Cacheaux LP, Ivens S, et al. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009;29(34):10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Devisme L, Bouchet C, Gonzalès M, et al. Cobblestone lissencephaly: neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain. 2012;135(Pt 2):469–482. doi: 10.1093/brain/awr357. [DOI] [PubMed] [Google Scholar]
- 31.Baxter P. Pyridoxine dependent and pyridoxine responsive seizures. In: Baxter P, editor. Vitamin Responsive Conditions in Paediatric Neurology. London: MacKeith Press; 2001. pp. 109–165. [Google Scholar]
- 32.Miyasaki K, Matsumoto J, Murao S, et al. Infantile convulsion suspected of pyridoxine responsive seizures. Acta Pathol Jpn. 1978;28(5):741–749. doi: 10.1111/j.1440-1827.1978.tb00913.x. [DOI] [PubMed] [Google Scholar]
- 33.Hauptman JS, Mathern GW. Surgical treatment of epilepsy associated with cortical dysplasia: 2012 update. Epilepsia. 2012;53(4):98–104. doi: 10.1111/j.1528-1167.2012.03619.x. [DOI] [PubMed] [Google Scholar]
- 34.Lado FA, Laureta EC, Moshé SL. Seizure-induced hippocampal damage in the mature and immature brain. Epileptic Disord. 2002;4(2):83–97. [PubMed] [Google Scholar]
- 35.Cahoy JD, Emery B, Kaushal A, et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J Neurosci. 2008;28(1):264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamaguchi K, Handa T. Sjögren-Larsson syndrome: postmortem brain abnormalities. Pediatr Neurol. 1998;18(4):338–341. doi: 10.1016/s0887-8994(97)00192-6. [DOI] [PubMed] [Google Scholar]
- 37.Acosta MT, Munasinghe J, Pearl PL, et al. Cerebellar atrophy in human and murine succinic semialdehyde dehydrogenase deficiency. J Child Neurol. 2010;25(12):1457–1461. doi: 10.1177/0883073810368137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sass JO, Walter M, Shield JPH, et al. 3-Hydroxyisobutyrate aciduria and mutations in the ALDH6A1 gene coding for methylmalonate semialdehyde dehydrogenase. J Inherit Metab Dis. 2011;35(3):437–442. doi: 10.1007/s10545-011-9381-x. [DOI] [PubMed] [Google Scholar]
- 39.Chitayat D, Meagher-Villemure K, Mamer OA, et al. Brain dysgenesis and congenital intracerebral calcification associated with 3-hydroxyisobutyric aciduria. J Pediatr. 1992;121(1):86–89. doi: 10.1016/s0022-3476(05)82549-1. [DOI] [PubMed] [Google Scholar]
- 40.Song X, Anderson V, Guzman M, Rao C. Neuropathology of 3-Hydroxyisobutyric Aciduria, an Autopsy Case Report. Can J Neurol Sci. 2009;36(4):483–486. doi: 10.1017/s0317167100007836. [DOI] [PubMed] [Google Scholar]
- 41.Bicknell LS, Pitt J, Aftimos S, et al. A missense mutation in ALDH18A1, encoding Delta1-pyrroline-5-carboxylate synthase (P5CS), causes an autosomal recessive neurocutaneous syndrome. Eur J Hum Genet. 2008;16(10):1176–1186. doi: 10.1038/ejhg.2008.91. [DOI] [PubMed] [Google Scholar]
- 42.Salomons GS, Bok LA, Struys EA, et al. An intriguing “silent” mutation and a founder effect in antiquitin (ALDH7A1) Ann Neurol. 2007;62(4):414–418. doi: 10.1002/ana.21206. [DOI] [PubMed] [Google Scholar]
- 43.Stockler S, Plecko B, Gospe SM, Jr, et al. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab. 2011;104(1-2):48–60. doi: 10.1016/j.ymgme.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 44.Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. 2008;4(6):697–720. doi: 10.1517/17425250802102627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Karnebeek CDM, Hartmann H, Jaggumantri S, et al. Lysine restricted diet for pyridoxine-dependent epilepsy: First evidence and future trials. Mol Genet Metab. 2012;107(3):335–344. doi: 10.1016/j.ymgme.2012.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. PDE cortex exhibits substantial gliosis. A) Western blot demonstrating markedly increased GFAP expression in PDE as compared with control cortex. Tubulin is shown as a loading control. B) GFAP IHC in control and PDE cortex. PDE cortex shows subpial gliosis, often seen in intractable epilepsy, and astrocyte proliferation in cortical layer I.
Supplementary Figure 2. Antiquitin is not expressed in cortical neurons. IHC of control human cortex showing absence of colocalization between antiquitin (red) and the neuronal marker MAP2 (green).
Supplementary Figure 3. Antiquitin immunofluorescence is normal in intractable epilepsy due to FCD type I. (A) Antiquitin western blot showing normal levels of expression in FCD Ib cortex as compared with controls. (B) Antiquitin immunofluorescence is normally distributed in a different child with FCD type Ib. (C) Normal antiquitin expression in a FCD Ic region demonstrating microcolumnar neuronal organization. Red = antiquitin, green = MAP2.
Supplementary Figure 4. Interneuron density is normal in PDE cortex. (A,B) GAD 65/67 immunofluorescence (red, arrows) in layer 2/3 of control (A) and PDE (B) cortex. Scale bar: 100 μM. (C) Quantitation of GAD positive interneurons in layers 2/3 and 5/6 of control and PDE cortex.
Supplementary Figure 5. ALDH7A1 mRNA expression in prenatal human brain. Graph generated from data downloaded from the Allen Brain Atlas Prenatal LMD Microarray (www.brainspan.org) of ALDH7A1 mRNA expression (probe A_23_P70231) as determined by microarray analysis of laser microdissected regions of human brain at 15, 16, and 21 post conceptual weeks (pcw). Expression is much higher in the subventricular zone (SVZ) and ventricular zone (VZ) than in the marginal zone (MZ), cortical plate (CP), subplate (CP), or intermediate zone (IZ).