

Abstract
Objective
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system (CNS), characterized by a global increasing incidence driven by relapsing-remitting disease in females. p38 MAP kinase (MAPK) has been described as a key regulator of inflammatory responses in autoimmunity, but its role in the sexual dimorphism in MS or MS models remains unexplored.
Methods
Toward this end, we used experimental autoimmune encephalomyelitis (EAE), the principal animal model of MS, combined with pharmacologic and genetic inhibition of p38 MAPK activity and transcriptomic analyses.
Results
Pharmacologic inhibition of p38 MAPK selectively ameliorated EAE in female mice. Conditional deletion studies demonstrated that p38α signaling in macrophages/myeloid cells, but not T cells or dendritic cells, recapitulated this sexual dimorphism. Analysis of CNS inflammatory infiltrates showed that female, but not male mice lacking p38α in myeloid cells exhibited reduced immune cell activation compared with controls, while peripheral T cell priming was unaffected in both sexes. Transcriptomic analyses of myeloid cells revealed differences in p38α-controlled transcripts comprising female- and male-specific gene modules, with greater p38α dependence of pro-inflammatory gene expression in females.
Interpretation
Our findings demonstrate a key role for p38α in myeloid cells in CNS autoimmunity and uncover important molecular mechanisms underlying sex differences in disease pathogenesis. Taken together, our results suggest that the p38 MAPK signaling pathway represents a novel target for much needed disease modifying therapies for MS.
Introduction
Multiple sclerosis (MS), the most common disabling neurologic disease of young adults, is considered a classical T cell-mediated disease and is characterized by demyelination, axonal damage, and progressive neurological dysfunction1, 2. Recent genetic studies further confirmed the role of cell-mediated immunity in MS, with an emphasis on T helper cell function3. Despite these insights, the etiopathogenesis of this devastating disease is poorly understood and current disease-modifying therapies (DMTs) have limited efficacy. Importantly, like many other autoimmune diseases, MS is characterized by a female bias. Epidemiological studies have demonstrated a significant increase in the incidence of relapsing-remitting MS in females over the last 50 years4. This rate of change is suggestive of environmental factors acting specifically in females at the population level. Despite the fact that such sexual dimorphisms in autoimmunity are well-documented, the mechanistic knowledge for the development of sex-specific DMTs is lacking.
The p38 mitogen-activated kinase (MAPK) pathway plays a prominent role in innate and adaptive immunity 5. p38 MAPK was identified as the target of a series of small molecules that inhibited toll-like receptor (TLR)-induced inflammatory cytokine production by macrophages6. As a key regulator of pro-inflammatory cytokine production, this molecule was expected to be a promising drug target in autoimmune inflammatory disorders where these cytokines were overproduced. Indeed, animal studies have shown efficacy of p38 MAPK inhibitors in models of rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and type 1 diabetes (T1D)7–9, although these compounds have not yet had success in the clinic10, 11. Until recently, this pathway has not been evaluated in MS or its models, despite the fact that MS shares many etiopathogenic features with these autoimmune diseases, such as activation of self-reactive T cells and augmented production of proinflammatory cytokines by innate cells12.
Early evidence for the involvement of p38 MAPK in autoimmune neuroinflammation came from studies showing increased phosphorylation of this kinase in inflammatory cells and glia in the central nervous system (CNS) during the course of experimental autoimmune encephalomyelitis (EAE), the principal model of MS13. Moreover, mRNA for MAPK14 (encoding p38α) was found to be overexpressed in CNS lesions of MS patients14. Subsequently, several recent studies have documented a functional requirement for p38 MAPK signaling in EAE progression. Treatment with pharmacological inhibitors of p38 MAPK inhibited clinical signs of EAE, which correlated with inhibition of pathogenic IL-17 producing T helper cell (Th17) responses15–17. Genetic inhibition of p38α, the predominant p38 MAPK isoform in immune cells, also potently ameliorated EAE, suggesting that p38α is the primary target underlying pharmacologic inhibition of disease17, 18. EAE severity was also reduced by inhibition of p38 MAPK signaling specifically in T cells, either by expression of dominant negative p38 transgene in T cells, or by the mutation of a residue required for T cell-specific activation of p38α/β16, 19. Accordingly, augmentation of p38 MAPK signaling by expression of a constitutively active MKK6 transgene in T cells enhanced EAE severity16. In contrast, Huang et al showed that genetic ablation of p38α in dendritic cells (DCs), but not in T cells or macrophages, inhibited EAE and led to impaired Th17 responses18. Moreover, deletion of apoptosis signal-regulating kinase 1 (ASK1), a TLR-controlled kinase that is known to activate p38 MAPK, attenuated p38 MAPK activation, EAE severity, and production of pro-inflammatory cytokines by astrocytes and microglia, without affecting peripheral T cell responses, suggesting that ASK1-dependent activation of p38 MAPK in glial cells may promote EAE pathogenesis 20. Here, we show sex-specific effects of p38 MAPK signaling in myeloid cells in EAE pathogenesis, suggesting that this signaling pathway may yield novel sex- and cell type-specific targets for treatment of MS.
Materials and Methods
Mice
Lysm-Cre mice (B6.129P2-Lyz2tm1(cre)Ifo/J) 21, Cd11c-Cre mice (B6.Cg-Tg(Itgax-cre)1-1Reiz/J) 22, p38α floxed mice (Mapk14tm1.2Otsu) 23 have been described previously and were obtained from Jackson Laboratories (USA) or RIKEN BioResource Center (Japan). Lck-Cre mice (B6.Cg-Tg(Lck-cre)1Cwi N9) 24 were obtained from Taconic (USA). Wild type C57BL/6J mice were purchased from Jackson Laboratories (USA) and were rested at the animal facility at UVM for at least 2 weeks prior to any experimentation. The experimental procedures used in this study were approved by the Animal Care and Use Committee of the University of Vermont.
EAE induction and scoring
EAE was induced essentially as described previously 25. Briefly, mice were injected s.c. with an emulsion containing 100 μg of MOG35-55 peptide (MEVGWYRSPFSRVVHLYRNGK) (Anaspec, USA) and complete Freund’s adjuvant (CFA) (Sigma-Aldrich, St. Louis, MO) supplemented with 200 μg of Mycobacterium tuberculosis H37Ra (Difco Laboratories, Detroit, MI) in the posterior right and left flanks. One week later all mice were similarly injected at two sites on the right and left flank anterior of the initial injection sites (2×MOG35-55/CFA). Alternatively, mice were immunized with 200 μg of MOG35-55 in CFA, followed by i.v. administration of 200 ng pertussis toxin (List Biological) (1× MOG35-55/CFA/PTX). In some experiments (as indicated), mice received 5 mg/kg/day of SB203580 dihydrochloride (Tocris, Ellisville, MO) by i.p. injection in a total volume of 200 μl or an equal volume of carrier daily starting on the the day of immunization. Mice were scored daily starting at day 10 post-injection as previously described 25. Passive EAE was induced as follows. WT male and female mice were immunized with 2×MOG35-55/CFA. On day 10, effector cells from LN and spleen were harvested and restimulated ex vivo with 10 μg/ml MOG35-55 and 0.5 ng/ml IL-12 for 72 hr. 20×106 sex-matched effector cells were transferred to female and male recipients.
Cytokine quantification
For the detection of cytokines in the cell culture supernatants, ELISAs were performed as described previously 25, using the primary capture mAbs: anti-IFNγ, anti-IL-17A, anti-TNFα, and anti-IL-6 and their corresponding biotinylated detection mAbs (BD Pharmingen, San Diego, CA). Other ELISA reagents included: HRP-conjugated avidin D (Vector Laboratories, Burlingame, CA), TMB microwell peroxidase substrate and stop solution (Kirkegaard and Perry Laboratories, Gaithersburg, MD). rIFNγ, rIL-17A, rGM-CSF (Biolegend, USA), and rTNFα and rIL-6 (BD Pharmingen, San Diego, CA) were used as standards.
For analysis of antigen-specific cytokine production by lymphocytes from mice immunized with 2× MOG35-55/CFA, spleen and draining lymph nodes (DLN) were harvested on day 10 post-immunization, single cell suspensions were prepared at 1×106 cells/ml in RPMI medium with 5% FBS, and stimulated with 50 μg/ml of MOG35-55. Cell culture supernatants were collected at 72 hours and cytokine levels were measured by ELISA as described above.
Isolation and stimulation of thioglycolate-elicited macrophages
Mice were immunized using the 2×MOG35-55/CFA protocol. On day 6 post-immunization, mice were injected with 1ml of a 4% solution of thioglycolate broth (Sigma-Adrich, USA) i.p. 96 hours later mice were sacrificed and the peritoneal cavity was flushed with 15 ml of cold PBS. Cells were washed and cultured overnight in RPMI + 5% FBS, then washed to remove non-adherent cells. The remaining adherent cells were stimulated with purified LPS (Sigma) or heat-killed Mycobacterium tuberculosis H37Ra (Difco, USA).
Flow cytometry
For intracellular cytokine staining ex vivo, mice were immunized with 2×MOG35-55/CFA, spleen and DLN were harvested on day 10 post-immunization, and cells were stimulated with 5 ng/ml of PMA, 250 ng/ml of ionomycin (Sigma-Aldrich) and Golgi Plug reagent (BD Biosciences) for 4 hours. Cells were then stained with the LIVE/DEAD fixable stain (Invitrogen) and then surface stained for the following markers: CD11b, CD4, CD8, TCRγδ, and TCRβ. Cells were then fixed with 1% paraformaldehyde (Sigma-Aldrich), permeabilized with buffer containing 0.2% saponin and stained with anti-IL-17A, anti-IFNγ, and anti-GM-CSF (Biolegend).
For surface marker analysis, unstimulated isolated cells were stained directly ex vivo with the LIVE/DEAD fixable stain (Invitrogen) and then surface labeled for different combinations of following markers: CD11b, CD11c, MHCII, CD80, CD86, Ly6C, Ly6G, MHCII, CD4, CD8, TCRγδ, and TCRβ (Biolegend, USA) and fixed with 1% paraformaldehyde. All antibodies used for flow cytometry were directly conjugated to fluorophores.
Cells were analyzed using an LSR II cytometer (BD Biosciences). Compensation was calculated using appropriate single color controls. Data were analyzed using FlowJo software (Tree Star Inc, Ashland, OR).
CNS-infiltrating mononuclear cell isolation
Animals were perfused with saline and brains and spinal cords were removed. A single cell suspension was obtained and passed through a 70 μm strainer. Mononuclear cells were obtained by Percoll gradient (37%/70%) centrifugation and collected from the interphase. For mRNA analysis, cells were lysed and total RNA was isolated using the RNEasy kit (Qiagen). For intracellular cytokine analysis, cells were washed and stimulated with 50 μg/ml of MOG35-55 for 4 hours in the presence of Golgi Plug reagent (BD Bioscience). Cells were labeled with LIVE/DEAD UV-Blue dye (Invitrogen) followed by surface staining (anti-CD45 from Invitrogen and anti- CD11b, CD11c, Ly6C, Ly6G, MHCII, CD4, CD8, TCRγδ, and TCRβ from Biolegend). Afterwards, cells were fixed, permeabilized and stained for intracellular IL-17A, IFNγ, and GM-CSF (Biolegend) as described above. For surface marker analysis, unstimulated isolated cells were stained directly ex vivo for the following markers: CD45, CD11b, CD11c, MHCII, CD80, CD86, Ly6C, Ly6G, MHCII, CD4, CD8, TCRγδ, and TCRβ.
Cell lysates and immunoblot analysis
Whole-cell lysates were prepared by lysing adherent macrophages directly in Triton lysis buffer, separated by SDS-PAGE, and transferred to PVDF membranes as described previously 25. Primary antibodies used for western blot analysis included anti-phospho-p38, anti-p38α, and anti-GAPDH (Cell Signaling Technologies, Danvers, MA). Anti-mouse and anti-rabbit secondary antibodies were conjugated to DyLight680 and DyLight800, respectively (Jackson ImmunoResearch Laboratories, West Grove, PA). Membranes were imaged using fluorescent detection on the Odyssey CLx instrument (Li-Cor Biosciences, USA), and images were processed using the Image Studio program (Li-Cor Biosciences, USA).
RNA isolation and quantitative real-time PCR (qRT-PCR)
RNA was extracted using the RNEasy kit (Invitrogen) according to manufacturer’s instructions. cDNA was reverse transcribed using the Taqman Gold RT-PCR kit using the oligo-dT method (Applied Biosciences, USA). qRT-PCR was performed using the DyNAmo Colorflash SYBR green qPCR kit (Thermofisher) and the following primer sets: Il10, GAAGCTGAAGACCCTCAGGA and TTTTCACAGGGGAGAAATCG; Tnfa, GAACTGGCAGAAGAGGCACT and AGGGTCTGGGCCATAGAACT; Il6, CCGGAGAGGAGACTTCACAG and GAGCATTGGAAATTGGGGTA; Il1b, AGGCCACAGGTATTTTGTCG and GCCCATCCTCTGTGACTCAT; B2m, CATGGCTCGCTCGGTGACC and AATGTGAGGCGGGTGGAACTG. B2m was used as a reference gene and relative mRNA levels were calculated using the comparative delta-delta CT method, normalizing first by the expression of the reference gene, then normalizing to the mean WT female expression level for each gene of interest.
Microarray sample preparation and hybridization
RNA was isolated using the RNEasy kit (Qiagen) according to manufacturer’s instructions. Microarray was performed on 3 biological replicates for each condition (e.g. female KO). To create each replicate, equal amounts of RNA from 2–3 different mice were pooled.
An RNA input of 25ng was used to generate cDNA through the First Strand and Second Strand synthesis reactions of the Ovation® Pico WTA System V2 from NuGEN. The cDNA samples were then purified using an Agencourt® RNAClean® XP magnetic bead protocol. Following purification, samples were amplified using SPIA reagents from the Ovation® Pico WTA System V2 from NuGEN. A final cDNA purification is performed using an Agencourt® RNAClean® XP magnetic bead protocol. Sample concentrations were determined using a 33ug/mL/A260 constant on a Nanodrop 1000 Spectrophotometer. Approximately 4ug of cDNA generated using the Ovation® Pico WTA System V2 was fragmented and labeled using the Encore® Biotin Module from NuGEN. Efficiency of the biotin labeling reaction was verified using NeutrAvidin (10mg/mL) with a gel-shift assay. Samples were injected into arrays and placed in the Affymetrix Genechip® Hybridization Oven 640 at 45° C and 60 RPM for 16–18 hours overnight. Arrays were stained using the Affymetrix Genechip® Fluidics Station 450 and scanned with the Affymetrix Genechip® Scanner 3000. Mouse Gene 2.0 ST arrays were used (Affymetrix).
Microarray analysis - calculation of probe set statistics
Raw GeneChip data (one DAT file for each chip) includes a collection of images, one for each probe and chip. Each image was summarized by Affymetrix GCOS software using one probe intensity (in CEL files, one per chip). Information from multiple probes can be combined to obtain a single measure of expression for each probe set and sample. Probe-level intensities were calculated using the Robust Multichip Average (RMA) algorithm, including background-correction, normalization (quantile), and summarization (median polish), for each probe set and sample, as is implemented in Partek Genomic Suites®, version 6.6 (Copyright © 2009, Partek Inc., St. Louis, MO, USA). Sample quality was assessed based on the 3′:5′ ratio (3′ arrays only), relative log expression (RLE), and normalized unscaled standard error (NUSE).
Microarray analysis - identification of differential expression and alternative splicing
Univariate linear modeling of sample groups is performed using ANOVA as implemented in Partek Genomic Suites. The magnitude of the response (fold change calculated using the least square mean) and the p-value associated with each probe set and binary comparison are calculated, as well a “step-up,” adjusted p-value for the purpose of controlling the false discovery rate (FDR) 26. For identification of differentially expressed genes between WT and KO in females and males, a binary filter of |FC|>1.5 and p<0.05 was used. ANOVA was also performed to detect alternative splicing, using exon-specific probe sets (at least one probe per exon). A filter of FDR < 0.05 was used as a cut off for alternative splicing analysis.
Bioinformatic identification of biological processes associated with differentially expressed genes
Data were analyzed through the use of Ingenuity Pathways Analysis (IPA; Ingenuity® Systems, www.ingenuity.com). Lists of differentially expressed genes in females and males were uploaded and analyzed using IPA Core Analysis. The Upstream Analysis function was used to identify predicted upstream regulators. The overlap p value generated by Upstream Analysis indicates the significance of the number of genes in the data set regulated by a given upstream regulator. The Z-score indicates the predicted direction of change for a given upstream regulator, with the sign indicating repression (negative) or upregulation (positive). Estrogen (beta-estradiol) and testosterone (dihydrotestosterone) we both in the top ten upstream regulators with the lowest Z-score (indicative of inactivation in p38α-deficient cells) for female and male data sets, respectively.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software Inc, San Diego, CA). The significance of differences in cytokine production and flow cytometry data were determined using 2-way ANOVA. The significance of differences observed in clinical course of EAE was determined by 2-way ANOVA and post-hoc analysis using Fisher’s LSD test for individual time points (significance indicated by “*” above each time point). Nonlinear regression analyses of the mean daily clinical disease scores indicated that the disease course amongst all strain and treatment combinations was best fit by a variable slope dose response curve, which together with daily mean score was used to represent the change in clinical disease over time. EAE data from replicate experiments were analyzed by heterogeneity testing. No significant experiment-to-experiment variation was observed, and therefore the data were pooled accordingly.
Results
Pharmacologic inhibition of p38 MAPK signaling ameliorates EAE in a sex-specific manner
We have previously shown that pharmacologic inhibition of p38 MAPK prevented EAE in female C57BL/6J (B6) mice 16. Since EAE, like MS, can often exhibit sexual dimorphisms27, we tested whether male mice showed a similar therapeutic response. EAE was induced in female and male B6 mice using the 2×MOG35-55/CFA protocol, followed by daily treatment with SB203580, a small molecule inhibitor of p38α and β. Surprisingly, while disease was robustly ameliorated in female mice as previously described (Fig. 1A), SB203580 treatment showed no therapeutic efficacy in male mice; in fact, EAE onset was accelerated (Fig. 1B). Thus, the therapeutic response to p38 MAPK inhibition is sexually dimorphic.
Figure 1. Sex-specific modulation of EAE susceptibility by p38 MAPK.
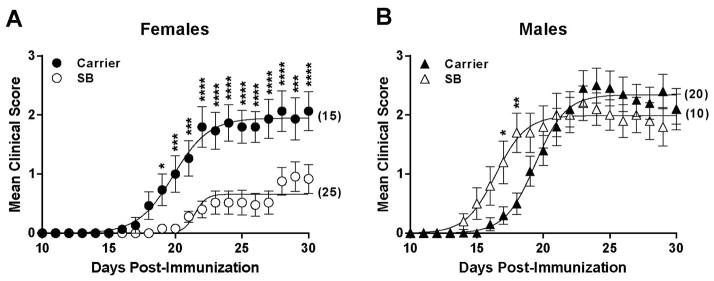
Female (A) and male (B) B6 mice were immunized using the 2×MOG35-55/CFA protocol, followed by daily injections of SB203580 or carrier. Data represent 2 independent experiments, pooled. Data were analyzed as indicated in Materials and Methods. A significant difference in EAE course was observed in females [treatment, p<0.0001; time, p<0.0001; time-by-treatment interaction, p<0.0001], and in males [treatment, p=0.71; time, p<0.0001; time-by-treatment interaction, p<0.0001]. For individual time points, * ≤ 0.05; ** ≤ 0.01, *** ≤ 0.001, **** ≤ 0.0001. Numbers in parentheses indicate the total number of animals studied.
p38α signaling in myeloid cells underlies the sexually dimorphic therapeutic efficacy of p38 MAPK inhibition in EAE
To delineate the contribution of p38 MAPK signaling in different cell types to the sexually dimorphic response to SB203580 treatment, as well as to rule out any sex-specific pharmacokinetic differences, we used cell type-specific genetic ablation of p38α, the predominant isoform expressed in immune cells. Autoreactive Th1 and Th17 cells are thought to initiate disease in both MS and EAE28, and p38 MAPK signaling in Th cells plays an important role in the generation and function of both Th1 and Th17 cells15–17, 29. Moreover, conventional dendritic cells (DCs) are important in the activation of pathogenic Th cells not only in the lymphoid organs, but also in the CNS during disease progression30, and p38α signaling in DCs has been recently shown to promote the generation of encephalitogenic Th17 cells18. Lastly, myeloid cells such as macrophages, microglia, and neutrophils are important mediators of tissue destruction and inflammation in the CNS during EAE and MS31, and p38α is thought to control the release of many pro-inflammatory mediators from these cells5. Therefore, we hypothesized that the sex-specific therapeutic efficacy of SB203580 (Fig. 1) is mediated by inhibition of p38 MAPK signaling in one or more of these cell types. To address this hypothesis, B6 mice expressing a floxed allele of p38α (p38αfl/fl)23 were crossed to mice expressing Cre recombinase under the control of the Lck, Cd11c, or Lysm/Lyz2 promoters21, 22, 24. This selectively ablates p38α in T cells (p38CKOLck), conventional DCs (p38CKOCd11c), or myeloid cells (p38CKOLysm), respectively.
EAE was induced in the resulting mice using the 2×MOG35-55/CFA protocol. Surprisingly, p38CKOLck mice exhibited a disease course similar to littermate p38αfl/fl Cre-negative controls (these are designated as WT throughout), suggesting that p38α in T cells is not essential for EAE in B6 mice (Fig. 2A and B). Deletion of p38α in DCs ameliorated disease, as recently reported18, but in a non sex-specific manner (Fig. 2C and D). However, deletion of p38α in myeloid cells resulted in diminished disease in females, but not males (Fig. 2E and F). EAE in males was in fact augmented by deletion of p38α in myeloid cells (Fig. 2F), as seen with SB203580 treatment (Fig. 1B). Expression of any of the above Cre alleles in p38αwt/fl or p38αwt/wt mice did not affect EAE (data not shown), thereby excluding any non-specific effects of Cre expression. Furthermore, p38α was deleted with equal efficiency in myeloid cells from female and male p38CKOLysm mice (Supplementary Fig. 1). Taken together, these results suggest that inhibition of p38α in myeloid cells underlies the sexual dimorphism observed with pharmacologic inhibition of p38 MAPK (Fig. 1A).
Figure 2. Differential control of EAE by p38α signaling in T cells, DCs, and myeloid cells.

Female (left panels) and male (right panels) WT and p38CKOLck (A and B), WT and p38CKOCd11c (C and D), and WT and p38CKOLysm (E and F) mice were immunized with 2×MOG35-55/CFA. Data represent two pooled independent experiments for each strain combination, and were analyzed as in Fig. 1. No significant effect of KO on EAE course was found in (A and B), females [strain, p=1.0; time, p<0.0001; time-by-treatment interaction, p=0.4], males [strain, p=0.3; time, p<0.0001; time-by-treatment interaction, p=0.6]. In (C and D), a significant effect of KO on EAE course was found in both females [strain, p=0.006; time, p<0.0001; time-by-treatment interaction, p<0.0001], and males [strain, p=0.3; time, p<0.0001; time-by-treatment interaction, p<0.0001]. In (E and F), a significant effect of KO on EAE course was found in both females [strain, p=0.008; time, p<0.0001; time-by-treatment interaction, p<0.0001], and males [strain, p=0.2; time, p<0.0001; time-by-treatment interaction, p=0.02]. For individual time points, * ≤ 0.05; ** ≤ 0.01, *** ≤ 0.001, **** ≤ 0.0001. Numbers in parentheses indicate the total number of animals studied.
p38α signaling in myeloid cells promotes CNS inflammation
Based on the results above, we hypothesized that deletion of p38α in myeloid cells affected their proinflammatory functions selectively in females. Myeloid cells such as macrophages are potent antigen-presenting cells (APCs) that can influence T cell priming and effector responses by presenting antigen and regulating the cytokine milieu30. Thus, we first determined whether deletion of p38α in myeloid cells affected the priming of myelin-specific Th1 and Th17 cells in secondary lymphoid organs. p38CKOLysm mice and WT control littermates were immunized with 2×MOG35-55/CFA, and 10 days later Th1 and Th17 responses in lymph nodes (LN) and spleen were assessed using ex vivo cytokine staining or by measuring MOG35-55-stimulated cytokine production by ELISA. No significant effect of p38α deletion on the production of IFNγ, IL-17, or GM-CSF was found in female or male mice (Fig. 3). Moreover, normal numbers and percentages of myeloid cells were found in lymphoid tissues of p38CKOLysm mice, and the expression of MHC Class II and co-stimulatory molecules CD80 and CD86 on these cells was not affected (Supplementary Fig. 2). Similar results were obtained in thioglycolate-elicited peritoneal macrophages (data not shown). Taken together, these results suggest that p38α in myeloid cells is dispensable for normal myeloid cell homeostasis and for efficient priming of peripheral Th1 and Th17 responses.
Figure 3. Ex vivo peripheral recall responses in p38CKOLysm mice.

Female and male littermate WT and p38CKOLysm mice were immunized with 2×MOG35-55/CFA. On D10, LN and spleen cells were isolated, restimulated with 50 μg/ml MOG35-55, and production of the following cytokines was determined by ELISA: IFNγ (A), IL-17 (B), and GM-CSF (C). Alternatively, LN and spleen cells were stimulated with PMA/Ionomycin in the presence of brefeldin A for 4 hours, then stained and analyzed by intracellular cytokine staining and flow cytometry (D–G). Percentage of TCRβ+CD4+ cells positive for IFNγ (D) or IL-17 (E) is shown. Representative flow cytometry plots are shown for female WT (F) and p38CKOLysm (G) mice. Data are representative of two independent experiments. WT female (n=7), p38CKOLysm female (n=10), WT male (n=10), p38CKOLysm male (n=10). No significant effect of KO was detected.
We next assessed whether deletion of p38α in myeloid cells affected the inflammatory response in the CNS, since peripheral immune responses may not fully represent what occurs in the target organ. p38CKOLysm mice and WT control littermates were immunized with 2×MOG35-55/CFA, and infiltrating mononuclear cells were isolated from the CNS at the peak clinical disease (day 19). Total mononuclear cell numbers were significantly reduced in p38CKOLysm female mice compared to WT females (Fig. 4A), suggesting reduced infiltration of immune cells into the CNS. CD11b expression on macrophages and microglia is upregulated by activation of these cells during neuroinflammation32. There was a reduction in the percentage of activated CD11bhi myeloid cells, with corresponding increase in the CD11bint population in female p38CKOLysm mice (Fig. 4B–E). Furthermore, production of IFNγ, IL-17, and GM-CSF by CD4 T cells was reduced in p38CKOLysm female mice compared to WT females (Fig. 4F–L). None of these changes were seen in male p38CKOLysm mice compared to WT males; in fact, GM-CSF production was significantly increased (Fig. 4H). Taken together, these results indicate that in females p38α in myeloid cells promotes CNS inflammation and indirectly promotes CNS T cell responses, whereas in males it may play an opposing role. To further verify that myeloid cell-specific deletion of p38α impacted the effector phase of EAE, we induced passive EAE in WT or p38CKOLysm mice by adoptive transfer of effector T cells harvested from WT sex-matched donors. EAE severity in p38CKOLysm female mice was significantly reduced compared to WT females (Supplementary Fig. 3A), while in males no significant difference was observed (Supplementary Fig. 3B).
Figure 4. Dampened CNS inflammatory response in female p38CKOLysM mice.

Female and male WT and p38CKOLysm mice were immunized using the 2×MOG35-55/CFA protocol. On day 19, mononuclear cells were isolated from the CNS using a Percoll gradient, counted (A), and surface stained for the indicated markers. Cells were gated on the CD45+TCRβ− population and expression of CD11b (CD11bhi vs. CD11bint) was analyzed (B and C; representative flow cytometry plots shown in D and E). Alternatively, mononuclear cells were stimulated with MOG35-55 for 4 hr in the presence of brefeldin A, then analyzed by intracellular cytokine staining and flow cytometry (F–L). Percentage of TCRβ+CD4+ cells positive for IFNγ (F), IL-17 (G), or GM-CSF (H) is shown. Representative flow cytometry plots are shown in (I–L). WT female (n=6), p38CKOLysm female (n=10), WT male (n=6), p38CKOLysm male (n=6).
p38α signaling in myeloid cells is not required for TLR-induced TNFα and IL-6 production
Based on the results above, we hypothesized that p38α controls a subset of pro-inflammatory mediators in female myeloid cells. p38 MAPK has long been known to control the production of proinflammatory cytokines by macrophages in response to TLR stimulation. Therefore we tested the effect of p38α ablation on macrophage responses to TLR agonists. Thioglycolate-elicited macrophages were isolated from WT and p38CKOLysm mice immunized with 2×MOG35-55/CFA in order to more closely mimic the in vivo environment to which myeloid cells are exposed during EAE. Macrophages were stimulated ex vivo by LPS, a TLR4 agonist, or heat-killed Mycobacterium tuberculosis H37Ra (MTB), the primary adjuvant used to induce EAE which contains several TLR ligands33). These stimuli resulted in increased phosphorylation of p38 MAPK, which was strongly reduced in p38CKOLysm mice (Fig. 5A and B). However, the production of TNFα and IL-6, two TLR-induced cytokines that have been previously shown to be controlled by p38 MAPK5, was not reduced in macrophages from female or male p38CKOLysm mice (Fig. 5C–F). Production of these two cytokines was in fact modestly enhanced in p38CKOLysm macrophages relative to WT at several of the time points assayed, but this was not sex-specific. Similarly, no sex-specific differences in the production of these cytokines were observed using bone marrow-derived macrophages (data not shown). These results suggest that production of cytokines classically associated with p38 MAPK signaling is not reduced in p38CKOLysm macrophages, and hence does not explain the sex-specific effects of p38α deletion on EAE susceptibility.
Figure 5. Macrophage responses to TLR stimulation in p38CKOLysm mice.
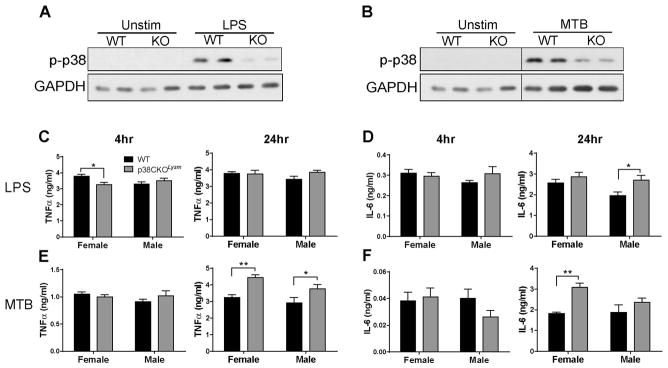
Female and male WT and p38CKOLysm mice were immunized using the 2×MOG35-55/CFA protocol. On day 6 thioglycolate was injected i.p., and elicited peritoneal macrophages were isolated on day 10 post-immunization. Adherent macrophages were stimulated for 30 min with 100 ng/ml LPS (A) or 50 μg/ml heat killed MTB (B), lysed, and analyzed by immunoblot for phosphorylation of p38 MAPK (p-p38). GAPDH is shown as a loading control. Panel (B) is a composite image of two different parts of the same membrane image, as indicated by the lines, treated identically and shown at the same exposure. Alternatively, adherent macrophages were stimulated for 4 or 24 hrs (as indicated) with LPS (C and D) or MTB (E and F), supernatants were collected and analyzed by ELISA for the presence of TNFα or IL-6. Data are representative of 3 independent experiments. * ≤ 0.05; ** ≤ 0.01. WT female (n=6), p38CKOLysm female (n=9), WT male (n=8), p38CKOLysm male (n=8).
p38α signaling in myeloid cells controls unique sex-specific gene expression modules
Since cytokines classically associated with p38 MAPK signaling in macrophages were not altered by p38α deletion in a sex-specific manner, we undertook a genome-wide transcriptomic approach to identify sex-specific p38α-regulated transcripts in macrophages. Thioglycolate-elicited macrophages were isolated from WT and p38CKOLysm female and male mice immunized using the 2×MOG35-55/CFA protocol, and restimulated ex vivo with MTB for 4 hours, at which point RNA was extracted and subjected to microarray analysis. Differential expression analysis revealed three unique modules of p38α-dependent genes in macrophages: non-sex-specific (i.e. controlled by p38α in both sexes), female-specific, and male-specific (Tables 1–3, Fig. 6A and B). The number p38α-dependent transcripts was higher in females than in males, with limited overlap (70 vs. 44 genes; 8 genes in common), suggesting a greater dependence on p38α in females.
Table 1. Non-sex-specific p38α-dependent transcripts in macrophages.
Thioglycolate-elicited macrophages were isolated from female and male p38CKOLysm and WT mice, and stimulated with 50 μg/ml heat-killed MTB in vitro for 4hrs. mRNA was isolated and subjected to microarray analysis to identify differentially expressed genes between p38CKOLysm and WT in both females and males. The criteria for differential expression was set at p<0.05 and signed fold change (|FC|) >1.5. Fold change indicates the change in expression in female p38CKOLysm relative to WT. Non-annotated genes are not shown.
| gene | female | male | ||
|---|---|---|---|---|
| p val | FC | p val | FC | |
| U90926 | 0.00 | −2.51 | 0.02 | −1.69 |
| Mmp13 | 0.00 | −2.18 | 0.00 | −1.78 |
| Il1f9 | 0.01 | −2.02 | 0.01 | −1.91 |
| Serpinb2 | 0.00 | −1.88 | 0.00 | −2.30 |
| Ccr5 | 0.01 | −1.80 | 0.00 | −1.87 |
| Lox | 0.01 | −1.57 | 0.02 | −1.51 |
| Mirlet7e | 0.04 | −1.57 | 0.05 | −1.54 |
| Ch25h | 0.00 | 1.52 | 0.00 | 1.51 |
Table 3. Male-specific p38α-dependent transcripts in macrophages.
Microarray analysis was performed on samples collected as described for Table 1 to identify differentially expressed genes between p38CKOLysm and WT that were unique to males. The criteria for differential expression was set at p<0.05 and |FC|>1.5. Fold change indicates the change in expression in male p38CKOLysm relative to male WT. Non-annotated genes are not shown. Genes known to play a role in EAE/MS pathogenesis (discussed in the text) are italicized and bolded.
| gene | p val | FC |
|---|---|---|
| Mir669a-3 | 0.05 | −1.73 |
| Mup12 | 0.02 | −1.71 |
| Fpr1 | 0.03 | −1.69 |
| Fabp7 | 0.00 | −1.69 |
| Hp | 0.00 | −1.64 |
| Cd5l | 0.04 | −1.63 |
| Rrs1 | 0.02 | −1.61 |
| Il10 | 0.04 | −1.56 |
| Mir7-2 | 0.01 | −1.53 |
| Cav1 | 0.00 | −1.52 |
| Vmn2r26 | 0.02 | −1.52 |
| Olfr1269 | 0.02 | −1.51 |
| Acpp | 0.01 | −1.51 |
| Vmn2r113 | 0.03 | −1.51 |
| Olfr566 | 0.05 | −1.51 |
| Cdc14a | 0.00 | 1.52 |
| Mir505 | 0.04 | 1.53 |
| Atp5s | 0.03 | 1.53 |
| Kansl2 | 0.01 | 1.54 |
| Pgcp | 0.01 | 1.54 |
| Calca | 0.01 | 1.55 |
| Zfp945 | 0.02 | 1.56 |
| Il12b | 0.00 | 1.57 |
| Hgf | 0.00 | 1.60 |
| Mir215 | 0.05 | 1.73 |
| Cdr1 | 0.02 | 2.45 |
Figure 6. Sex-specific p38α-dependent transcript modules in macrophages.

Female and male WT and p38CKOLysm macrophages were isolated as described for Fig. 5, stimulated for 4 hrs with 50 μg/ml heat killed MTB, RNA was extracted, reverse transcribed, and cDNA subjected to microarray analysis. (A) A heat map of gene expression across triplicate samples of WT and p38CKOLysM (KO) macrophages from female and male mice. Each triplicate represents a pool of 2–3 different biological replicates. Expression is shown relative to the centered mean of all samples for a given gene (see Materials and Methods). Genes passing the binary filter of p<0.05 and |FC|>1.5 (of KO relative to WT for either sex) are shown, ordered by FC in descending order. “Up” or “down” refers to the direction of change in KO relative to WT. (B) A Venn diagram indicating the overlap in p38α-dependent transcripts between females and males. Both annotated and non-annotated genes were included.
p38α differentially regulates pro- and anti-inflammatory genes in females and males
MS and EAE are polygenic diseases, where small effects of multiple loci contribute to overall disease susceptibility 34, 35. By analogy, we hypothesize that the effect of p38α deletion in EAE is mediated by the combined effects of multiple genes within the p38α-dependent modules, rather than a single key p38α-dependent gene. Within the non-sex-specific module, several genes of interest were downregulated in the absence of p38α (Table 1). Serpinb2 (plasminogen activator inhibitor 2, PAI-2) and Mmp13 (matrix metalloproteinase 13, MMP-13) have both been previously shown to be controlled by p38α in macrophages, where PAI-2 can inhibit apoptotic responses and IL-1β production36–38. Although the role of MMP-13 in EAE/MS is so far unexplored, MMPs are well-known to be involved in the pathogenesis of these diseases39. Il1f9 encodes IL-36γ, a novel cytokine with a potential role in psoriasis40–42, a Th17-driven autoimmune disease. Mirlet7e encodes a microRNA that was recently shown to promote EAE pathogenesis43. In contrast, Ccr5 (chemokine (C-C motif) receptor 5), which was also downregulated in the absence of p38α, is not required for EAE44. It is important to note that while these genes did not exhibit sex-specific p38α-dependence, their altered expression may nevertheless contribute to the observed sex-specific phenotypes in EAE by interacting with genes within the female- or male-specific p38α-controlled modules.
In females, deletion of p38α downregulated several genes that are known to promote EAE or MS pathogenesis (Table 2). The product of Maoa, monoamine oxidase A, has been successfully targeted to treat EAE45. Oas1g (2′-5′ oligoadenylate synthetase 1G) is an ortholog of human OAS1, which was recently found to be associated with MS susceptibility and severity46–48. Fcgr1 is a mouse ortholog of human FCGR1a (encoding the high affinity IgG Fc receptor), which was upregulated in chronic CNS lesions in MS patients, and targeting the Fc gamma receptor pathway ameliorated EAE in mice14. Lastly, Ccr1 (chemokine (C-C motif) receptor 1) has been shown to be critical for EAE pathogenesis44. In contrast, in males, deletion of p38α resulted in downregulation of Il10, an immunosuppressive cytokine well-known to inhibit EAE49). Meanwhile, Il12b, the p40 subunit of IL-12 and IL-23, and known to be required for EAE50, was upregulated.
Table 2. Female-specific p38α-dependent transcripts in macrophages.
Microarray analysis was performed on samples collected as described for Table 1 to identify differentially expressed genes between p38CKOLysm and WT that were unique to females. The criteria for differential expression was set at p<0.05 and |FC|>1.5. Fold change indicates the change in expression in female p38CKOLysm relative to female WT. Non-annotated genes are not shown. Genes known to play a role in EAE/MS pathogenesis (discussed in the text) are italicized and bolded.
| gene | p val | FC |
|---|---|---|
| Retnlg | 0.021 | −2.17 |
| Snord82 | 0.050 | −2.12 |
| Mir20a | 0.005 | −1.93 |
| Scd1 | 0.048 | −1.91 |
| Mir1b | 0.035 | −1.90 |
| Gpr141 | 0.006 | −1.86 |
| Isg15 | 0.028 | −1.80 |
| Trim30c | 0.002 | −1.79 |
| Snord15a | 0.003 | −1.77 |
| Cox7b | 0.038 | −1.76 |
| Trim30d | 0.006 | −1.74 |
| Syne1 | 0.011 | −1.71 |
| Mpzl3 | 0.027 | −1.69 |
| Hist1h2bn | 0.000 | −1.65 |
| Hist2h3c2 | 0.004 | −1.64 |
| Vsig4 | 0.047 | −1.64 |
| Car2 | 0.038 | −1.64 |
| Mid1 | 0.036 | −1.63 |
| Maoa | 0.004 | −1.62 |
| Adm | 0.016 | −1.61 |
| Snord43 | 0.049 | −1.61 |
| Tfrc | 0.008 | −1.61 |
| Flrt3 | 0.001 | −1.61 |
| Scarna17 | 0.011 | −1.59 |
| Oas1g | 0.017 | −1.59 |
| Mir187 | 0.040 | −1.58 |
| Tpsb2 | 0.013 | −1.58 |
| Ctla2a | 0.005 | −1.57 |
| Snord118 | 0.021 | −1.57 |
| Rn5s20 | 0.049 | −1.55 |
| Ccr1 | 0.009 | −1.54 |
| Scg2 | 0.015 | −1.53 |
| Fcgr1 | 0.003 | −1.52 |
| Dio2 | 0.021 | −1.52 |
| Gorab | 0.005 | −1.52 |
| Zfp68 | 0.008 | −1.51 |
| Mreg | 0.014 | −1.51 |
| Pyhin1 | 0.007 | −1.51 |
| Tut1 | 0.021 | −1.51 |
| Syne1 | 0.040 | −1.50 |
| Hdhd3 | 0.015 | 1.52 |
| Zfp862 | 0.006 | 1.53 |
| Snora30 | 0.024 | 1.56 |
| Mir23a | 0.034 | 1.57 |
| Kcnj13 | 0.032 | 1.58 |
| Ptges3l | 0.000 | 1.61 |
| Pdgfb | 0.005 | 1.62 |
| Kprp | 0.002 | 1.71 |
| Clca2 | 0.044 | 1.78 |
| Mir5123 | 0.020 | 2.32 |
As an additional filter to determine the in vivo relevance of genes differentially regulated by p38α in macrophages stimulated in vitro, we analyzed the expression of multiple transcripts in mononuclear cells isolated from the inflamed CNS at peak EAE (day 21). Consistent with microarray results, we found that Il10 mRNA was downregulated in cells from male but not female p38CKOLysm mice (Fig. 7A), while Oas1g mRNA was downregulated specifically in females in the absence of p38α (Fig. 7B). In contrast to the microarray results, Fcgr1, Ccr1 and Ccr5 mRNAs were unchanged in either sex (Fig. 7C–E), suggesting differential regulation in vivo. Several other transcripts of interest were either undetectable, or their expression was highly variable (data not shown), owing likely to the heterogeneity in CNS-infiltrating cells and/or EAE timing/onset, thus precluding their analysis. We also examined the expression of several pro-inflammatory cytokines thought to be controlled by p38 MAPK. Il1b, was upregulated in males in an inverse relationship with Il10 (Fig. 7F). Tnfa expression was not affected by p38α deletion, while Il6 was upregulated in both females and males in the absence of p38α (Fig. 7G and H), similar to the results observed by ELISA in vitro (Fig. 5). Taken together, these results suggest that differential sex-specific regulation of pro- and anti-inflammatory gene expression by p38α underlies the opposing protective and pathogenic effects of p38 MAPK inhibition in EAE in females and males, respectively, and identify Il10 and Oas1g as key p38α-controlled sex-specific genes in the CNS during peak neuroinflammation.
Figure 7. Sex-specific regulation of inflammatory gene expression in the CNS of p38CKOLysM mice.
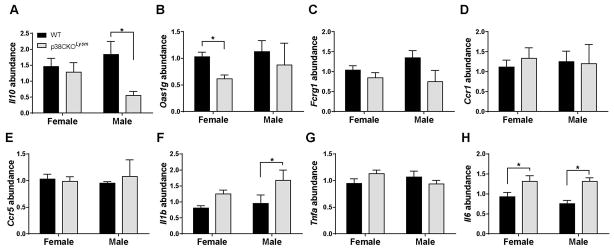
Female and male WT and p38CKOLysm mice were immunized using the 2×MOG35-55/CFA protocol. On day 21, mononuclear cells were isolated from the CNS using a Percoll gradient and RNA was extracted. Relative mRNA abundance of Il10 (A), Oas1g (B), Fcgr1 (C), Ccr1 (D), Ccr5 (E) Il1b (F), Tnfa (G), and Il6 (H) was quantified by qRT-PCR using the delta-delta CT method with B2m as an endogenous control. * ≤ 0.05. WT female (n=8), p38CKOLysm female (n=6), WT male (n=5), p38CKOLysm male (n=4).
Sex hormones contribute to the sexual dimorphism in EAE in p38CKOLysm mice
Bioinformatic analysis of differentially expressed transcript modules in macrophages from female and male p38CKOLysm mice identified estrogen as a highly significant positive upstream regulator of genes within the female-specific module (activation Z score = −2.21; p value of overlap = 2.29E-04), while testosterone was a positive regulator of genes within the male-specific module (activation Z score = −1.47; p value of overlap = 1.32E-06). This suggested that sex hormones may be responsible for differential EAE outcomes in female and male p38CKOLysm mice. To test this hypothesis, we performed gonadectomies (or sham control surgeries) on adult WT and p38CKOLysm mice, followed by EAE induction. Sham surgeries did not affect the sexual dimorphism, as female p38CKOLysm mice were highly resistant to EAE induction compared to WT (Fig. 8A), while EAE in p38CKOLysm males was not significantly different from WT males (Fig. 8B). However, removal of adult sex hormones by gonadectomy completely reversed the sexual dimorphism, as ovariectomized p38CKOLysm females lost EAE resistance (Fig. 8C), and orchiectomized p38CKOLysm males gained it (Fig. 8D). These results demonstrate the role of adult sex hormones in determining the sexual dimorphism in EAE pathogenesis mediated by p38α signaling in myeloid cells.
Figure 8. Sex hormones contribute to the sexual dimorphism in EAE in p38CKOLysm mice.

Female (left panels) and male (right panels) WT and p38CKOLysm mice underwent sham surgeries (Sham) (A and B) or gonadectomies (GndX) (C and D). 3 weeks later, mice we were immunized using the 2×MOG35-55/CFA protocol. Data represent one independent experiment, and were analyzed as in Fig. 1. A significant effect of KO on EAE course was found in (A) [strain, p=0.05; time, p<0.0001; time-by-treatment interaction, p<0.0001] and in (D) [strain, p<0.0001; time, p<0.0001; time-by-treatment interaction, p=0.03]. No significant effect of KO on EAE course was found in (B) [strain, p=0.4; time, p<0.0001; time-by-treatment interaction, p=0.3] and in (C) [strain, p=0.7; time, p<0.0001; time-by-treatment interaction, p=0.7]. For individual time points, * ≤ 0.05; ** ≤ 0.01, *** ≤ 0.001, **** ≤ 0.0001. Numbers in parentheses indicate the total number of animals studied.
Discussion
In this study, we showed that deletion of p38α in T cells did not affect EAE in B6 mice (Fig. 2A and B). This is in contrast to our previous results showing that augmentation of p38 MAPK activity by a constitutively active MKK6 transgene, or its inhibition by a dominant negative p38 transgene, expressed specifically in T cells in B10.BR mice, enhanced or diminished EAE severity, respectively16. This discrepancy may be due to genetic differences between B6 and B10.BR mice, or the fact the transgenic approaches affect all four p38 MAPK isoforms. Furthermore, we found that in transgenic B10.BR mice EAE was modulated in a non-sex specific fashion (data not shown), suggesting a different mechanism from the one described herein.
The finding that SB203580 treatment did not reduce EAE in males (Fig. 1B) is somewhat counter-intuitive given reduced EAE in p38CKOCd11c males (Fig. 2D). However, based on the findings that p38CKOLysm males exhibited augmented EAE (Fig. 2F), we predict that pharmacological inhibition of p38 concurrently in myeloid cells and DCs has opposing effects on EAE in males which effectively cancel each other out. An alternative explanation is the involvement of other cell types targeted by SB203580 besides myeloid cells and DCs. Lastly, pharmacological experiments are difficult to compare to p38α genetic deletion studies due to effects of SB203580 on p38α or potential off-target effects51.
Chi and colleagues recently demonstrated that deletion of p38α in myeloid cells did not affect EAE, although the sex of the animals was not reported18. Another important difference is that Chi and colleagues used pertussis toxin (PTX) as an ancillary adjuvant in the EAE induction protocol, which is absent in the 2×MOG35-55/CFA protocol used in our study (Fig. 2). Because PTX overrides many genetic checkpoints (e.g., see 52–54), we hypothesized that it can override disease protection provided by deletion of p38α in myeloid cells. In agreement with this, we found that WT and p38CKOLysm female and male mice exhibited no difference in EAE disease course when the 1×MOG35-55/CFA/PTX protocol was used (Supplementary Fig. 4). MS exhibits remarkable heterogeneity in disease course and severity, similar to what is seen across in different EAE models elicited with or without PTX.53 It is likely that different adjuvants used in EAE model different infectious/environmental risk factors in MS. Consequently, inhibitors of p38 MAPK-related pathways may have differential therapeutic efficacy depending on disease etiopathogenesis.
Our findings that deletion of p38α in macrophages altered a relatively small subset of transcripts are somewhat surprising, since p38 inhibitors were reported to inhibit wide range of proinflammatory cytokines and mediators. However, our findings are in agreement with previous reports using conditional deletion of p38α, which demonstrated that this kinase controls a limited spectrum of pro-inflammatory genes37, 55. Interestingly, IL-10 production was shown to be dependent on p38α in these reports and in our study. The differences in the results observed between pharmacologic and genetic studies may be due to the poor specificity of inhibitors51. Furthermore, p38 MAPK controls many of its targets, at the post-translational level56, 57, including IL-17 in T cells16, while our current analysis focused mainly on transcriptional control. Nonetheless, while it appears that effects of p38α inhibition in macrophages may be far more limited than previously suggested, it is clear that these subtle effects are sufficient to modulate EAE severity. As such, more specific and/or cell type-targeted inhibitors of p38α could represent an attractive therapeutic approach in MS.
In humans, gender influences immunity, although controversial results regarding gender and/or sex hormone treatment in ex vivo studies are frequently reported58. While p38 MAPK is well-known to control pro-inflammatory functions in human monocytes/macrophages6, to the best of our knowledge, little has been reported on gender differences in this regard. One study examined LPS-induced phosphorylation of p38 in PBMCs from males and females, and found that it was somewhat lower in females, correlating with lower proinflammatory cytokine production59. Hence, it is possible that the p38-dependent sex bias in macrophages observed in our mouse model will translate to humans, but further studies are needed to address this possibility.
The incidence of MS has approximately tripled in the last 50 years, driven by an increase in relapsing-remitting disease in women4. This rate of change strongly suggests the existence of environmental risk factors acting at the population level in females. p38 MAPK is a well-known evolutionarily conserved component of the response to environmental stress stimuli, such as hypertonicity and UV radiation60–62. Recent epidemiological data indicate that the prevalence of MS correlates with decreased UV-radiation exposure more highly in females than it does in males63. The immunomodulatory effects of UV-radiation64 are in part mediated by p38 MAPK signaling62 and UV-irradiation has been shown to influence EAE susceptibility65 independent of vitamin D66. With respect to hypertonicity, two recent reports suggested that dietary sodium may also represent an environmental risk factor for MS, as increased dietary sodium exacerbated EAE, in association with enhanced generation of pathogenic Th17 cells67, 68. p38 MAPK appears to transduce this sodium stress signal, as inhibition of p38 MAPK abrogated sodium-induced upregulation of IL-17 production67. However, the effect of p38 MAPK inhibition on sodium-exacerbated EAE was not reported67. Since our data show that the p38 MAPK signaling pathway is central to EAE pathogenesis in females, it is tempting to speculate that environmental stress signals acting through the p38 MAPK pathway may contribute to the increasing MS risk in females. Consequently, understanding this pathway may reveal mechanistic insight into gene-by-environment interactions in MS etiopathogenesis. Moreover, our results suggest that targeting the p38 MAPK signaling pathway in MS could represent a novel and much needed female-specific DMT.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health Grants AI041747, NS036526, and NS060901 to CT. This work was also supported a postdoctoral fellowship from the National Multiple Sclerosis Society to DNK.
Matt Poynter, John Boyson, Sean Diehl, Laure Case, Roxana del Rio, Naresha Saligrama, Emma Wall and Elizabeth P. Blankenhorn are acknowledged for helpful discussions and advice. Kristiaan Finstaad and Erin Osmanski are acknowledged for technical assistance. Meghann Palermo and Tim Hunter at the VCC facility at UVM are acknowledged for the help with microarrays. Additional thanks to Jeff Bond and the Molecular Bioinformatics Shared Resource of the UVM College of Medicine.
Footnotes
Potential Conflicts of Interest
The authors declare no potential conflicts of interest.
References
- 1.Ramagopalan SV, Sadovnick AD. Epidemiology of multiple sclerosis. Neurol Clin. 2011 May;29(2):207–17. doi: 10.1016/j.ncl.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 2.Greenstein JI. Current concepts of the cellular and molecular pathophysiology of multiple sclerosis. Dev Neurobiol. 2007 Aug;67(9):1248–65. doi: 10.1002/dneu.20387. [DOI] [PubMed] [Google Scholar]
- 3.Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011 Aug 11;476(7359):214–9. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ebers GC. Environmental factors and multiple sclerosis. Lancet Neurol. 2008 Mar;7(3):268–77. doi: 10.1016/S1474-4422(08)70042-5. [DOI] [PubMed] [Google Scholar]
- 5.Rincon M, Davis RJ. Regulation of the immune response by stress-activated protein kinases. Immunol Rev. 2009 Mar;228(1):212–24. doi: 10.1111/j.1600-065X.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- 6.Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994 Dec 22–29;372(6508):739–46. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 7.Liverton NJ, Butcher JW, Claiborne CF, et al. Design and synthesis of potent, selective, and orally bioavailable tetrasubstituted imidazole inhibitors of p38 mitogen-activated protein kinase. J Med Chem. 1999 Jun 17;42(12):2180–90. doi: 10.1021/jm9805236. [DOI] [PubMed] [Google Scholar]
- 8.Hollenbach E, Neumann M, Vieth M, Roessner A, Malfertheiner P, Naumann M. Inhibition of p38 MAP kinase- and RICK/NF-kappaB-signaling suppresses inflammatory bowel disease. FASEB J. 2004 Oct;18(13):1550–2. doi: 10.1096/fj.04-1642fje. [DOI] [PubMed] [Google Scholar]
- 9.Ando H, Kurita S, Takamura T. The specific p38 mitogen-activated protein kinase pathway inhibitor FR167653 keeps insulitis benign in nonobese diabetic mice. Life Sci. 2004 Feb 20;74(14):1817–27. doi: 10.1016/j.lfs.2003.09.045. [DOI] [PubMed] [Google Scholar]
- 10.Genovese MC. Inhibition of p38: has the fat lady sung? Arthritis Rheum. 2009 Feb;60(2):317–20. doi: 10.1002/art.24264. [DOI] [PubMed] [Google Scholar]
- 11.Hammaker D, Firestein GS. “Go upstream, young man”: lessons learned from the p38 saga. Ann Rheum Dis. 2010 Jan;69(Suppl 1):i77–82. doi: 10.1136/ard.2009.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011 Oct 27;365(17):1612–23. doi: 10.1056/NEJMra1100030. [DOI] [PubMed] [Google Scholar]
- 13.Shin T, Ahn M, Jung K, et al. Activation of mitogen-activated protein kinases in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2003 Jul;140(1–2):118–25. doi: 10.1016/s0165-5728(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 14.Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002 May;8(5):500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 15.Lu L, Wang J, Zhang F, et al. Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells. J Immunol. 2010 Apr 15;184(8):4295–306. doi: 10.4049/jimmunol.0903418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noubade R, Krementsov DN, Del Rio R, et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood. 2011 Sep 22;118(12):3290–300. doi: 10.1182/blood-2011-02-336552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Namiki K, Matsunaga H, Yoshioka K, et al. Mechanism for p38alpha-mediated experimental autoimmune encephalomyelitis. J Biol Chem. 2012 Jul 13;287(29):24228–38. doi: 10.1074/jbc.M111.338541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang G, Wang Y, Vogel P, Kanneganti TD, Otsu K, Chi H. Signaling via the kinase p38alpha programs dendritic cells to drive TH17 differentiation and autoimmune inflammation. Nat Immunol. 2012 Feb;13(2):152–61. doi: 10.1038/ni.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jirmanova L, Giardino Torchia ML, Sarma ND, Mittelstadt PR, Ashwell JD. Lack of the T-cell-specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood. 2011 Jun 28; doi: 10.1182/blood-2011-01-333039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo X, Harada C, Namekata K, et al. Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol Med. 2010 Dec;2(12):504–15. doi: 10.1002/emmm.201000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999 Aug;8(4):265–77. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 22.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J Exp Med. 2007 Jul 9;204(7):1653–64. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishida K, Yamaguchi O, Hirotani S, et al. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004 Dec;24(24):10611–20. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee PP, Fitzpatrick DR, Beard C, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001 Nov;15(5):763–74. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 25.Noubade R, Milligan G, Zachary JF, et al. Histamine receptor H1 is required for TCR-mediated p38 MAPK activation and optimal IFN-gamma production in mice. J Clin Invest. 2007 Nov;117(11):3507–18. doi: 10.1172/JCI32792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hochberg YBaY. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57(1):289–300. [Google Scholar]
- 27.Spence RD, Voskuhl RR. Neuroprotective effects of estrogens and androgens in CNS inflammation and neurodegeneration. Front Neuroendocrinol. 2012 Jan;33(1):105–15. doi: 10.1016/j.yfrne.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Segal BM. Th17 cells in autoimmune demyelinating disease. Semin Immunopathol. 2010 Mar;32(1):71–7. doi: 10.1007/s00281-009-0186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rincon M, Enslen H, Raingeaud J, et al. Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. 1998 May 15;17(10):2817–29. doi: 10.1093/emboj/17.10.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chastain EM, Duncan DS, Rodgers JM, Miller SD. The role of antigen presenting cells in multiple sclerosis. Biochim Biophys Acta. 2011 Feb;1812(2):265–74. doi: 10.1016/j.bbadis.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Izikson L, Klein RS, Luster AD, Weiner HL. Targeting monocyte recruitment in CNS autoimmune disease. Clin Immunol. 2002 May;103(2):125–31. doi: 10.1006/clim.2001.5167. [DOI] [PubMed] [Google Scholar]
- 32.Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005 Aug 1;81(3):374–89. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 33.Marta M, Meier UC, Lobell A. Regulation of autoimmune encephalomyelitis by toll-like receptors. Autoimmun Rev. 2009 May;8(6):506–9. doi: 10.1016/j.autrev.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 34.Oksenberg JR, Hauser SL. Decoding multiple sclerosis. Ann Neurol. 2011 Dec;70(6):A5–7. doi: 10.1002/ana.22680. [DOI] [PubMed] [Google Scholar]
- 35.Gourraud PA, Harbo HF, Hauser SL, Baranzini SE. The genetics of multiple sclerosis: an up-to-date review. Immunol Rev. 2012 Jul;248(1):87–103. doi: 10.1111/j.1600-065X.2012.01134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park JM, Greten FR, Wong A, et al. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis--CREB and NF-kappaB as key regulators. Immunity. 2005 Sep;23(3):319–29. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Kim C, Sano Y, Todorova K, et al. The kinase p38 alpha serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol. 2008 Sep;9(9):1019–27. doi: 10.1038/ni.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greten FR, Arkan MC, Bollrath J, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007 Sep 7;130(5):918–31. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002 Sep;39(3):279–91. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- 40.Carrier Y, Ma HL, Ramon HE, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol. 2011 Dec;131(12):2428–37. doi: 10.1038/jid.2011.234. [DOI] [PubMed] [Google Scholar]
- 41.Tortola L, Rosenwald E, Abel B, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. 2012 Nov 1;122(11):3965–76. doi: 10.1172/JCI63451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vigne S, Palmer G, Lamacchia C, et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood. 2011 Nov 24;118(22):5813–23. doi: 10.1182/blood-2011-05-356873. [DOI] [PubMed] [Google Scholar]
- 43.Guan H, Fan D, Mrelashvili D, et al. MicroRNA let-7e is associated with the pathogenesis of experimental autoimmune encephalomyelitis. Eur J Immunol. 2013 Jan;43(1):104–14. doi: 10.1002/eji.201242702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rottman JB, Slavin AJ, Silva R, Weiner HL, Gerard CG, Hancock WW. Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur J Immunol. 2000 Aug;30(8):2372–7. doi: 10.1002/1521-4141(2000)30:8<2372::AID-IMMU2372>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 45.Musgrave T, Benson C, Wong G, et al. The MAO inhibitor phenelzine improves functional outcomes in mice with experimental autoimmune encephalomyelitis (EAE) Brain Behav Immun. 2011 Nov;25(8):1677–88. doi: 10.1016/j.bbi.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 46.O’Brien M, Lonergan R, Costelloe L, et al. OAS1: a multiple sclerosis susceptibility gene that influences disease severity. Neurology. 2010 Aug 3;75(5):411–8. doi: 10.1212/WNL.0b013e3181ebdd2b. [DOI] [PubMed] [Google Scholar]
- 47.Fedetz M, Matesanz F, Caro-Maldonado A, et al. OAS1 gene haplotype confers susceptibility to multiple sclerosis. Tissue Antigens. 2006 Nov;68(5):446–9. doi: 10.1111/j.1399-0039.2006.00694.x. [DOI] [PubMed] [Google Scholar]
- 48.Cagliani R, Fumagalli M, Guerini FR, et al. Identification of a new susceptibility variant for multiple sclerosis in OAS1 by population genetics analysis. Hum Genet. 2012 Jan;131(1):87–97. doi: 10.1007/s00439-011-1053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rott O, Fleischer B, Cash E. Interleukin-10 prevents experimental allergic encephalomyelitis in rats. Eur J Immunol. 1994 Jun;24(6):1434–40. doi: 10.1002/eji.1830240629. [DOI] [PubMed] [Google Scholar]
- 50.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003 Feb 13;421(6924):744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 51.Godl K, Wissing J, Kurtenbach A, et al. An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc Natl Acad Sci U S A. 2003 Dec 23;100(26):15434–9. doi: 10.1073/pnas.2535024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spach KM, Noubade R, McElvany B, Hickey WF, Blankenhorn EP, Teuscher C. A single nucleotide polymorphism in Tyk2 controls susceptibility to experimental allergic encephalomyelitis. J Immunol. 2009 Jun 15;182(12):7776–83. doi: 10.4049/jimmunol.0900142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blankenhorn EP, Butterfield RJ, Rigby R, et al. Genetic analysis of the influence of pertussis toxin on experimental allergic encephalomyelitis susceptibility: an environmental agent can override genetic checkpoints. J Immunol. 2000 Mar 15;164(6):3420–5. doi: 10.4049/jimmunol.164.6.3420. [DOI] [PubMed] [Google Scholar]
- 54.Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y. Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol. 2006 Feb;18(2):399–407. doi: 10.1093/intimm/dxh379. [DOI] [PubMed] [Google Scholar]
- 55.Guma M, Hammaker D, Topolewski K, et al. Antiinflammatory functions of p38 in mouse models of rheumatoid arthritis: advantages of targeting upstream kinases MKK-3 or MKK-6. Arthritis Rheum. 2012 Sep;64(9):2887–95. doi: 10.1002/art.34489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clark AR, Dean JL, Saklatvala J. Post-transcriptional regulation of gene expression by mitogen-activated protein kinase p38. FEBS Lett. 2003 Jul 3;546(1):37–44. doi: 10.1016/s0014-5793(03)00439-3. [DOI] [PubMed] [Google Scholar]
- 57.Schindler JF, Monahan JB, Smith WG. p38 pathway kinases as anti-inflammatory drug targets. J Dent Res. 2007 Sep;86(9):800–11. doi: 10.1177/154405910708600902. [DOI] [PubMed] [Google Scholar]
- 58.Oertelt-Prigione S. The influence of sex and gender on the immune response. Autoimmun Rev. 2012 May;11(6–7):A479–85. doi: 10.1016/j.autrev.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 59.Imahara SD, Jelacic S, Junker CE, O’Keefe GE. The influence of gender on human innate immunity. Surgery. 2005 Aug;138(2):275–82. doi: 10.1016/j.surg.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 60.Sheikh-Hamad D, Gustin MC. MAP kinases and the adaptive response to hypertonicity: functional preservation from yeast to mammals. Am J Physiol Renal Physiol. 2004 Dec;287(6):F1102–10. doi: 10.1152/ajprenal.00225.2004. [DOI] [PubMed] [Google Scholar]
- 61.Cowan KJ, Storey KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol. 2003 Apr;206(Pt 7):1107–15. doi: 10.1242/jeb.00220. [DOI] [PubMed] [Google Scholar]
- 62.Muthusamy V, Piva TJ. The UV response of the skin: a review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch Dermatol Res. 2010 Jan;302(1):5–17. doi: 10.1007/s00403-009-0994-y. [DOI] [PubMed] [Google Scholar]
- 63.Orton SM, Wald L, Confavreux C, et al. Association of UV radiation with multiple sclerosis prevalence and sex ratio in France. Neurology. 2011 Feb 1;76(5):425–31. doi: 10.1212/WNL.0b013e31820a0a9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Norval M, McLoone P, Lesiak A, Narbutt J. The effect of chronic ultraviolet radiation on the human immune system. Photochem Photobiol. 2008 Jan-Feb;84(1):19–28. doi: 10.1111/j.1751-1097.2007.00239.x. [DOI] [PubMed] [Google Scholar]
- 65.Hauser SL, Weiner HL, Che M, Shapiro ME, Gilles F, Letvin NL. Prevention of experimental allergic encephalomyelitis (EAE) in the SJL/J mouse by whole body ultraviolet irradiation. J Immunol. 1984 Mar;132(3):1276–81. [PubMed] [Google Scholar]
- 66.Becklund BR, Severson KS, Vang SV, DeLuca HF. UV radiation suppresses experimental autoimmune encephalomyelitis independent of vitamin D production. Proc Natl Acad Sci U S A. 2010 Apr 6;107(14):6418–23. doi: 10.1073/pnas.1001119107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kleinewietfeld M, Manzel A, Titze J, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic T17 cells. Nature. 2013 Mar 6; doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu C, Yosef N, Thalhamer T, et al. Induction of pathogenic T17 cells by inducible salt-sensing kinase SGK1. Nature. 2013 Mar 6; doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.