

Abstract
Homeostasis of the gastrointestinal epithelium is dependent upon a balance between cell proliferation and apoptosis. Cyclin-dependent kinases (Cdks) are well known for their role in cell proliferation. Previous studies from our group have shown that polyamine-depletion of intestinal epithelial cells (IEC-6) decreases cyclin-dependent kinase 2 (Cdk2) activity, increases p53 and p21Cip1 protein levels, induces G1 arrest, and protects cells from camptothecin (CPT)-induced apoptosis. Although emerging evidence suggests that members of the Cdk family are involved in the regulation of apoptosis, their roles directing apoptosis of IEC-6 cells are not known. In this study, we report that inhibition of Cdk1, 2, and 9 (with the broad range Cdk inhibitor, AZD5438) in proliferating IEC-6 cells triggered DNA damage, activated p53 signaling, inhibited proliferation, and induced apoptosis. By contrast, inhibition of Cdk2 (with NU6140) increased p53 protein and activity, inhibited proliferation, but had no effect on apoptosis. Notably, AZD5438 sensitized, whereas, NU6140 rescued proliferating IEC-6 cells from CPT-induced apoptosis. However, in colon carcinoma (Caco2) cells with mutant p53, treatment with either AZD5438 or NU6140 blocked proliferation, albeit more robustly with AZD5438. Both Cdk inhibitors induced apoptosis in Caco2 cells in a p53-independent manner. In serum starved quiescent IEC-6 cells, both AZD5438 and NU6140 decreased TNF- /CPT-induced activation of p53 and, consequently, rescued cells from apoptosis, indicating that sustained Cdk activity is required for apoptosis of quiescent cells. Furthermore, AZD5438 partially reversed the protective effect of polyamine depletion whereas NU6140 had no effect. Together, these results demonstrate that Cdks possess opposing roles in the control of apoptosis in quiescent and proliferating cells. In addition, Cdk inhibitors uncouple proliferation from apoptosis in a p53-dependent manner.
Keywords: Polyamines, proliferation, Cdk2, Cdk9, Cdk1, p53, H2AX, DNA damage, apoptosis
INTRODUCTION
The intestinal epithelium has one of the most rapid turnover rates with complete renewal of the epithelial mucosa occurring every 3–8 days [1]. Renewal of the gut epithelium is a complex process and depends on a balance between cell proliferation and apoptosis. Proliferation occurs in undifferentiated stem cells located in the crypts of the small intestine. Enterocytes migrate out of the proliferative zone and undergo cell cycle arrest, differentiation, and maturation along the villus surface. Differentiated enterocytes are subsequently removed by anoikis at the villus tip [2]. Spontaneous apoptosis occurs at the base of the crypt and is responsible for the balance between newly proliferating and exfoliating cells [3]. The identification of cellular signaling mechanisms common to both apoptosis and the cell cycle is important to understanding the regulation of the growth of this tissue.
Cell proliferation is controlled by sequential activation and inactivation of a highly conserved family of cyclin-dependent serine threonine protein kinases (Cdks). Binding of regulatory proteins, the cyclins, regulates Cdk activities. Transition through both G1/S and S phase require activation of Cdk2 through association with cyclin E and cyclin A, respectively [4]. During late G2 and early M, cyclin A complexes with Cdk1. Association of Cdk1 with cyclin B regulates mitosis [5]. Cdk9 controls transcriptional elongation, mRNA processing, and histone modification via association with cyclins K and T [6]. Two separate families of Cdk inhibitory proteins are known to regulate Cdk activities. The INK4 family (comprising of p15, p16, p18, and p19) and Cip/Kip family (including p21 and p27) inactivate Cdk-cyclin complexes [7–8] leading to growth arrest. Activation of Cdks triggers phosphorylation of substrate proteins resulting in changes that favor cell cycle progression. A well-known substrate for activated Cdk complexes is retinoblastoma tumor suppressor (Rb). Cdk9 has been shown specifically to phosphorylate the Rb protein [9]. Hyperphosphorylation of Rb occurs during G1-S transition, and hypophosphorylated Rb prevents DNA synthesis [10].
The tumor suppressor p53 is an important coordinator of proliferation and apoptotic signals [11]. We previously reported that p53 plays an obligatory role in apoptosis of intestinal epithelial cells (IEC-6) cells induced by DNA damage [12]. Phosphorylation of H2AX is a nuclear marker of various types of DNA damage [13] and several studies have linked H2AX to p53-dependent apoptosis and Cdk-mediated cell cycle arrest [13–15].
Cdks are master regulators of DNA damage checkpoint and repair pathways [16]. Furthermore, Cdks have putative roles in transcriptional regulation and a controversial role in apoptosis [17]. However, it has not been addressed, whether or not Cdks, traditionally required for gut epithelial proliferation, are also essential for apoptosis. Potential mechanisms related to the regulation of apoptosis by Cdks include numerous upstream and downstream interactions between the Cdk and p53 pathways [14–15, 18]. In proliferating cells, p53 is a direct downstream kinase substrate for Cdks, including cdc2/cyclin B and Cdk2-cyclin A [19]. In addition, indirect regulation of p53 downstream of Cdk4/6 has been demonstrated via phosphorylation of Rb [20]. Furthermore, p53 is a transcriptional activator of the Cdk inhibitor p21Cip1 [21], suggesting that p53 can also act as an upstream regulator of Cdks.
Polyamines are biologically active polycations found in all-eukaryotic cells. Growing lines of evidence implicate polyamines in a number of cellular processes required for proliferation and apoptosis of the intestinal epithelium. Extensive studies from our group have examined the roles of polyamines in gastrointestinal mucosal homeostasis in cultured intestinal epithelial cells (IEC-6) [22–23]. We have consistently shown that inhibition of ornithine decarboxylase (ODC) by alpha-difluromethylornithine (DFMO) and the subsequent depletion of intracellular polyamines inhibits apoptosis induced by genotoxic stress and DNA damage (i.e -radiation and camptothecin) or TNF- /CHX [12, 23–26].
Given the fact that polyamine-depletion decreased Cdk2 activity, increased p53 and p21Cip1 expression [27], and significantly conferred protection from apoptosis induced by DNA-damage [12, 23]; we sought to determine the role of Cdks in apoptosis of intestinal epithelial cells. Data presented in this study demonstrate for the first time the relationship between Cdk-1/2/−9 and key apoptotic regulators including H2AX, p53, p21Cip1, and caspase-3 in the untransformed IEC-6 (expressing wild-type p53) cell line and the p53 mutated Caco2 colon carcinoma cell line. Furthermore, our data demonstrate that Cdks play diverse roles in apoptosis of proliferating and quiescent cells.
MATERIALS AND METHODS
Reagents
Disposable cell culture ware was purchased from Corning Glass works (Corning, NY) and Zellkulture Flaschen (Europe, Switzerland). Media and other cell culture reagents were obtained from Mediatech, Inc (Herndon, VA) and Invitrogen (Long Island, NY). Dialyzed fetal bovine serum (dFBS), insulin, and Camptothecin were purchased from Sigma (St Louis, MO). Recombinant rat TNF- was obtained from BD PharMingen International (San Diego, CA). The Enhanced Chemiluminescence (ECL) Western Blot detection system was purchased from Perkin Elmer (Boston, MA). DFMO was a gift from ILEX Oncology™ Inc, (San Antonio, TX). Phospho-p53 Ser15, total-p53, phospho-H2AX Ser139, phospho-Rb Ser 780, total-Rb, and cleaved active caspase-3 (Asp 175) antibodies were purchased from Cell Signaling (Beverly, MA). p21Cip1 antibody was purchased from BD Biosciences (San Diego, CA). AZD5438 (Cdk1, 2, and 9 inhibitor) and NU6140 (Cdk2 inhibitor) were purchased from Calbiochem, EMD Biosciences (La Jolla, CA). The Cell Death Detection ELISA Plus kit and WST-1 cell proliferation kit were purchased from Roche Diagnostics Corp. (Indianapolis, IN). The IEC-6 cell line (ATCC CRL 1592) and Caco2 (ATCC HTB-37) were obtained from American Type Culture Collection (Rockville, MD). All chemicals were of the highest purity commercially available.
Cell culture
The IEC-6 cell line was derived from normal rat intestine and was developed and characterized by Quaroni et al [28]. IEC-6 cells originate from intestinal crypt cells as judged by immunologic criteria. IEC-6 cells are nontumorigenic and retain the undifferentiated character of epithelial stem cells. Cell stocks were maintained in T-150 flasks in a humidified, 37 °C incubator in an atmosphere of 10% CO2. The medium consisted of Dulbecco’s Modified Eagle Medium (DMEM) with 5% heat inactivated FBS and 10μg insulin and 50μg gentamicin sulfate per ml. The stock flask was passaged weekly, fed 3 times per week, and passages 15–22 were used. To set up experiments, the cells were trypsinized with 0.05% trypsin and 0.53 mM EDTA and counted by a Beckman Coulter Counter (Model Z1). Experimental set-ups with proliferating IEC-6 cells involved overnight attachment followed by addition of AZD5438 (1 M), NU6140 (1 M), and DFMO (5mM) in serum-containing medium for 72h. All experimental set-ups with confluent quiescent IEC-6 cells involved three-days growth in control, 5mM DFMO or DFMO plus 10μM putrescine containing Dulbecco’s modified Eagle medium (DMEM)/5% dFBS. Within 6h of DFMO treatment, putrescine was undetectable and spermidine was absent after 24 hours. The cells were fed on day 2 and serum starved with control, DFMO or DFMO plus putrescine containing medium for 24 hrs (on day 3) to achieve quiescence on day 4. On day 4 only 40% of bound intracellular spermine was detected [29]. Exogenous putrescine (10μM) added to DFMO containing medium acted as a control to indicate that all results were due to the depletion of polyamines and not to DFMO itself.
Caco2 cells were maintained in T-75 flasks in a humidified 37 °C incubator in an atmosphere of 5% CO2. Medium consisted of Modified Eagle’s medium (MEM) with 10% heat-inactivated fetal bovine serum and 50μg gentamicin sulfate per ml. The stock flask was fed every alternate day and passaged weekly. Experimental set-ups with proliferating Caco2 cells involved 2 days growth in control medium followed by addition of AZD5438 (5μM), NU6140 (5μM), and CPT (2μM) in serum-containing medium for 48h.
Apoptosis
The quantitative DNA fragmentation assay was carried out using a cell death detection ELISA kit as described earlier [23–26]. Briefly, floating cells were discarded and the attached cells were washed twice with DPBS. An aliquot of the nuclei-free supernatant was placed in streptavidin-coated wells and incubated with anti-histone-biotin antibody and anti-DNA peroxidase conjugated antibody for 2 hours at room temperature. After incubation, the sample was removed and the wells were washed 3 times with incubation buffer. After the final wash was removed, 100 μl of the substrate, 2,2′-azino-di[3-ethylbenzthiazolin-sulfonate], was placed in the wells for 20 minutes at room temperature. The absorbance was read at 405 nm using a plate reader. Results were expressed as absorbance at 405 nm /min/mg protein.
Proliferation assay
The Cell Proliferation Reagent WST-1 was used to quantify proliferation rates of IEC-6 and Caco2 cells. Briefly, both IEC-6 and Caco2 cells (0.5 × 106 cells per well) were seeded in 48-well plates and proliferating cells were treated with AZD5438, NU6140, and CPT in serum containing medium. After treatment, the cells were washed in DMEM without phenol red and L-glutamine. WST-1 reagent was diluted (1:10 final dilution) in DMEM without phenol red and with L-glutamine. 200μl of diluted tetrazolium WST-1 reagent was added to each well and absorption was measured at 2, 4, and 6h. Diluted WST-1 reagent was used for background control. Absorbance was read at 440nm using an ELISA plate reader.
Western blot analysis
The protocol for western blots has been described earlier [23–26]. Typically, cell monolayers were first washed with ice cold PBS and lysed for 10 min in ice cold extraction buffer containing 20mM Tris-HCl (pH 7.5), 150mM NaCl, 1mM Na2EDTA, 1mM EGTA, 1% Triton-X 100, 2.5mM sodium pyrophosphate, 1mM-glycerophosphate, 1mM Na3VO4 and a protease inhibitor cocktail. Lysates were centrifuged at 10,000 rpm for 10 min at 4 °C. followed by SDS-PAGE. Proteins were transferred to Immobilon-P membranes (Millipore Bedford, MA, USA) and probed with the indicated antibodies overnight at 4 °C in TBS buffer containing 0.1% Tween-20 and 5% non fat dry milk (blotting grade, Biorad). Membranes were subsequently incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h and the immunocomplexes were visualized by the ECL detection system (Perkin Elmer).
Statistical analysis
All data are expressed as mean +/− SE. Experiments were repeated three times, with triplicate samples for each. Representative Western blots from three experiments are shown. ANOVA and appropriate post-hoc testing determined the significance of the differences between means. Values of P<0.05 were regarded as significant.
RESULTS
AZD5438 and NU6140 inhibit proliferation of IEC-6 cells
We first investigated the time-dependent effect of treatment with Cdk inhibitors on the growth of IEC-6 cells. Fig. 1A shows morphological changes in IEC-6 cells treated with DMSO, broad range Cdk 1, 2, and 9 inhibitor (AZD5438), and Cdk2 inhibitor (NU6140) during proliferation. The most conspicuous changes observed in AZD5438-treated cells included cell shrinkage, increased brightness, and detachment from the substratum. These changes became visible after 72h of AZD5438 treatment but were absent in control cells treated with DMSO, suggesting that cells treated with AZD5438 died by apoptosis. By contrast, cells treated with NU6140 for 72h were binucleated and enlarged, indicating failure of cytokinesis and decreased rate of proliferation (Fig. 1A). In order to quantitate proliferation in the presence of Cdk inhibitors, we used the WST-1 colorimetric assay. A time-dependent increase in IEC-6 proliferation was observed in control cells treated with DMSO (Fig. 1B). 1 M AZD5438 prevented proliferation after 24h of treatment. In addition, treatment with AZD5438 (until 72h) significantly reduced proliferation by more than 50%. Since reduction of WST-1 reagent to formazan requires the presence of viable cells with functional mitochondria, these results suggest that the anti-proliferative effect of AZD5438 may be partly due to its ability to induce cell death. Treatment of IEC-6 cells with 1 M NU6140 decreased proliferation at 24h, which continued as long as cells were exposed to the drug (Fig. 1B), suggesting that inhibition of Cdk2 decreased proliferation without causing apoptosis.
Fig. 1. Inhibition of Cdk reduces IEC-6 cell proliferation.

IEC-6 cells were trypsinized and equal numbers of cells were seeded in serum-containing medium. Eighteen hours later the attached cells were treated with DMSO, 1 M AZD5438, and 1 M NU6140 in serum-containing medium. Culture medium was changed everyday containing respective inhibitors and cells were incubated for 72h. (A) Phase contrast-microscopy of cells grown in the presence of DMSO, AZD5438, and NU6140 (B) Proliferating IEC-6 cells were treated with DMSO, 1 M AZD5438, and 1 M NU6140 in the presence of serum for the indicated time periods. Cell proliferation was measured by WST-1 assay as described in Methods. Values expressed are mean SE, n=3. * significantly different compared to cells grown in the presence of DMSO at indicated time points (P< 0.05), ** significantly different compared with the DMSO treated group at 24h.
Induction of DNA damage and apoptosis by inhibition of multiple Cdks in proliferating cells
To assess the functional consequences of Cdk inhibition in proliferating cells, we determined the impact of Cdk inhibition on H2AX activity. Cdk 1, 2, and 9 inhibition by AZD increased the Ser139-phosphorylated form of H2AX suggesting a DNA damage response (Fig. 2A). Phosphorylation of H2AX is required for p53/p21-dependent cell cycle arrest after replication stalling [13]. As expected, levels of phosphorylated p53 and total p53 were markedly higher in AZD5438-treated cells with concomitant increases in p21Cip1 protein (Fig. 2A). Cdk1 protein levels were mostly unaffected by AZD5438 and NU6140, indicating that these inhibitors block Cdk activity without changing protein expression. Increased p53 activation in response to AZD5438 led to caspase-3 activation and caused DNA fragmentation (Fig. 2B). Similar to the effect of AZD5438, NU6140 increased p53 protein and its phosphorylation with a concomitant upregulation of its downstream target, p21Cip1 (Fig. 2A). However, levels of p21Cip1 were higher in cells treated with NU6140 compared to AZD5438-treated cells (Fig. 2A). Interestingly, NU6140 failed to induce H2AX phosphorylation and, consequently, had no effect on caspase-3 activation (Fig. 2A) and DNA fragmentation (Fig. 2B). These results suggest that specific inhibition of Cdk2 promotes p53/p21 signaling-dependent cell cycle arrest without causing apoptosis. Conversely, the primary response of IEC-6 cells to AZD5438 is the concomitant occurrence of growth arrest, DNA damage, and apoptosis.
Fig. 2. AZD5438 induces apoptosis in proliferating IEC-6 cells.

(A) IEC-6 cells were allowed to proliferate for 72h in the presence of DMSO, 1 M AZD5438, and 1 M NU6140. Cell lysates were analyzed by western blot for phospho-H2AX Ser139, phospho-p53 Ser15, Cdk1, total-p53, p21Cip1, and active caspase-3 using specific antibodies. Actin was used as an internal loading control. (B) DNA fragmentation was measured by ELISA as described in methods (mean SE, n=3). *significantly different compared to cells grown in the presence of DMSO (P< 0.05).
Effect of AZD5438 and NU6140 in colon carcinoma cells
Uncontrolled proliferation and aberrations in the Cdk-p53 pathway are common in human cancers [30]. Given that Cdk inhibitors block proliferation and induce apoptosis in IEC-6 cells, which correlate with the activation of p53-mediated pathways (Figs. 1 and 2), we determined the effect of these inhibitors in p53 deficient colon carcinoma Caco-2 cells. Results in Fig. 3A demonstrate that Caco-2 cells grown in the presence of DMSO exhibited normal polygonal cell morphology with broad contours. Proliferating Caco-2 cells exposed to AZD5438 for 48h underwent morphological changes including cell rounding and increased brightness. The appearance of bright rounded cells accompanied by cytosolic vacuolization increased in cells treated with NU6140 for 48h (Fig. 3A). Camptothecin (CPT) treatment during proliferation showed loss of cell-cell contact and detachment of cells from the substratum. Since altered phenotypes are often associated with reduced growth rates, we studied the effect of AZD5438 and NU6140 on the proliferation of Caco-2 cells. DMSO-treated control cells continued proliferating in a time-dependent manner (Fig 3B), as judged by WST-1 assay. We observed that exposure of Caco-2 cells to 5 M AZD5438 or NU6140 for 24h resulted in reduced proliferation. Similarly, treatment of Caco-2 cells with 2 M CPT decreased proliferation at 24h. Caco-2 cells proliferated at a lower rate when treated with NU6140 for 48h. However, AZD5438 treatment for 48h completely blocked growth of Caco-2 cells. Similarly, cell numbers were significantly reduced after 48h of CPT treatment (Fig. 3B).
Fig. 3. Cdk inhibitors regulate proliferation in p53-deficient Caco-2 cells.

(A) Caco-2 cells were trypsinized and equal numbers of cells were seeded in serum-containing medium. Two days later, attached cells were treated with DMSO, 5 M AZD5438, and 5 M NU6140 in serum-containing medium and cells were allowed to proliferate for 48h. Phase-contrast microscopy was used to visualize changes in cell morphology. (B) Proliferating Caco-2 cells were treated with DMSO, 5 M AZD5438, 5 M NU6140 and 2 M CPT in serum-containing medium for the indicated time periods. Cell proliferation was measured by WST-1 assay as described in Methods. Values expressed are mean SE, n=3. * significantly different compared to cells grown in the presence of DMSO at indicated time periods (P< 0.05). (C) Caco-2 cell lysates were analyzed by western blot for Cdk1, phospho-H2AX Ser139, phospho-Rb Ser780, total-p53, p21Cip1, and total-Rb using specific antibodies. Actin was used as an internal loading control.
Western blot analysis of the AZD5438, NU6140, and CPT treated samples showed that both AZD5438 and NU6140 decreased Cdk1 expression, whereas CPT had no effect. Caco-2 cells did not express p53 and p21Cip1 proteins in DMSO treated cells (Fig. 3C), confirming previous observations [31]. In addition, p53 expression did not change in response to AZD5438, NU6140, and CPT treatment in Caco-2 cells. However, treatment of proliferating Caco-2 cells with CPT increased p21Cip1 expression and H2AX phosphorylation at Ser139 suggesting that CPT is capable of inducing a DNA damage response in cells lacking p53. Interestingly, both AZD5438 and NU6140 partially decreased H2AX phosphorylation suggesting that Cdk inhibition fails to trigger DNA damage in cells lacking functional p53. Since Rb is a Cdk substrate, we monitored the effectiveness of these inhibitors by measuring Rb phosphorylation. Decreased retinoblastoma phosphorylation (p-Rb) at Ser780 in response to AZD5438 and NU6140 confirmed the complete inhibition of Cdks. Unphosphorylated total Rb (lower band) increased in cells treated with AZD5438 and NU6140 (Fig. 3C), indicating inhibition of cell cycle progression. Furthermore, levels of phosphorylated Rb were found to be partially lower in CPT treated cells without a concomitant increase in unphosphorylated Rb, indicating that CPT-induced DNA damage occurred concurrent with inhibition of Cdks.
Since Cdk inhibitors and CPT decreased Caco-2 proliferation and induced morphological changes characteristic of the apoptotic process, we investigated caspase-3 activation and DNA fragmentation in these samples. Both AZD5438 and NU6140 failed to activate caspase-3 at 24h as judged by the lack of active fragments (Fig. 4A). AZD5438 had no effect on caspase-3 activation (Fig. 4A) but significantly increased DNA fragmentation at 48h with respect to DMSO-treated cells (Fig. 4B), indicating that AZD5438-induced cell death is caspase-3 independent. CPT partially increased caspase-3 activation at 24h. However, CPT robustly increased the appearance of caspase-3 fragments after 48h (Fig. 4A) and significantly increased DNA fragmentation, indicating cell death (Fig. 4B). NU6140 increased the expression of active caspase-3 and DNA fragmentation at 48h. However, DNA fragmentation in response to NU6140 was lower than the response of AZD5438 (Fig. 4B). Furthermore, the caspase-3 fragment in response to NU6140 had a higher electromobility compared to cells treated with CPT, indicating incomplete proteolytic processing of caspase-3. Interestingly, extent of DNA fragmentation in AZD5438 and NU6140 treated cells was significantly lower compared to cells treated with CPT. These results demonstrate that CPT is a potent inducer of apoptosis compared to Cdk inhibitors in proliferating Caco2 cells.
Fig. 4. Cdk inhibitors induce apoptosis in Caco-2 cells.

(A) Caco-2 cells were treated with DMSO, 5 M AZD5438, 5 M NU6140 and 2 M CPT during proliferation for the indicated time periods. Cell lysates were analyzed for active caspase-3 using specific antibody. Actin was used as an internal loading control. (B) Caco-2 cells were treated with DMSO, 5 M AZD5438, 5 M NU6140 and 2 M CPT during proliferation for 48h. DNA fragmentation was measured by ELISA. Values expressed are mean SE, n=3. *significantly different compared to DMSO treated cells (P<0.05).
Cdks differentially regulate CPT-induced apoptosis in proliferating cells
Consistent with data presented in Fig. 2A, AZD5438 increased p53, p21Cip1 with simultaneous increases in phosphorylation of p53 and H2AX (Fig. 5A). Activation of p53 signaling in response to AZD5438 correlated with caspase-3 activation (Fig. 5A) and DNA fragmentation (Fig. 5B). Similar to the effect of AZD5438, NU6140 increased p53, p21Cip1, and p53 phosphorylation, but had no effect on H2AX phosphorylation. Our earlier data showed that polyamine depletion by DFMO conferred protection from various inducers of apoptosis [12]. We also reported that polyamine depletion decreased Cdk2 activity and induced cell cycle arrest [27]. Thus, we predicted that treatment of proliferating cells with DFMO would inhibit Cdk2 activity in a manner comparable to the inhibition conferred by NU6140. DFMO failed to increase p53 phosphorylation at Ser15, which is consistent with previous observations [12]. Furthermore, treatment of proliferating cells with DFMO marginally increased p21Cip1 without any effect on p53 expression. Both NU6140 and DFMO had no effect on caspase-3 activation.
Fig. 5. NU6140 and DFMO block CPT-induced apoptosis in proliferating IEC-6 cells.
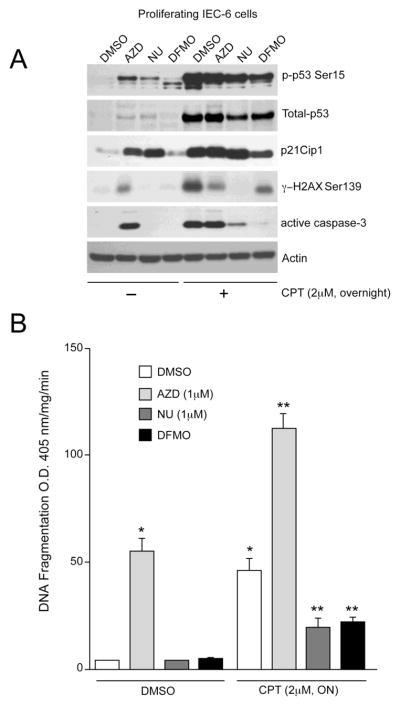
(A) IEC-6 cells were trypsinized from stock flasks and equal numbers were seeded in serum containing medium. The cells were allowed to attach overnight. Attached cells were treated with DMSO, 1 M AZD5438, 1 M NU6140, and 5mM DFMO in the presence of serum. Cells were incubated with these inhibitors for 48h in serum containing medium followed by treatment with or without 2 M CPT for 18h. Cell extracts were analyzed to determine the levels of phospho-p53 Ser15, total-p53, p21Cip1, phospho-H2AX Ser139, and active caspase-3 by western blot analysis. Actin was used as an internal loading control. (B) DNA fragmentation was measured by ELISA as described in methods. Values expressed are mean SE, n=3. *significantly different compared to cells grown in the presence of DMSO (P<0.05), **significantly different compared with cells grown in the presence of DMSO and treated with 2 M CPT (P<0.05).
We have reported earlier that activation of p53 is central during CPT-induced apoptosis in quiescent serum-starved cells [12]. We treated cells overnight with 2 M CPT in the presence of serum. CPT robustly increased H2AX and p53 phosphorylation, which was accompanied by significant increases of p53 and p21Cip1 protein (Fig. 5A). Activation of p53 signaling in response to CPT led to caspase-3 activation and significantly increased DNA fragmentation (Fig. 5B). In addition, inhibition of Cdk1, 2, and 9 during proliferation (AZD5438) significantly augmented CPT-induced DNA fragmentation (Fig. 5B), but had no effect on CPT-induced total p53, phospho-p53, and p21Cip1 levels. Since AZD5438 treated cells contained high levels of phosphorylated H2AX (Fig. 5A), CPT failed to further increase H2AX phosphorylation. Interestingly, higher levels of phospho-p53 and total-p53 were observed in AZD5438+CPT treated cells compared to AZD5438 alone (Fig. 5A). Although AZD5438 potentiated CPT-induced apoptosis, levels of active caspase-3 did not change, indicating extensive proteolytic processing and degradation of active caspase-3. Treatment of proliferating cells with Cdk2 inhibitor (NU6140) completely blocked CPT-mediated H2AX phosphorylation and partially decreased p53 expression and its phosphorylation (Fig. 5A). These results indicate that CPT-induced H2AX phosphorylation is predominantly dependent on Cdk2 activity. NU6140 failed to block CPT-induced p21Cip1 expression indicating the involvement of additional signal transduction pathways in regulating CPT-induced p21Cip1 expression. Furthermore, NU6140 significantly decreased CPT-induced caspase-3 activation and DNA fragmentation, suggesting that inhibition of Cdk2 confers protection from DNA damage-induced apoptosis, and p53 activation does not always correlate with the apoptotic index. Similar to the effect of NU6140, DFMO partially decreased CPT-induced p53 and p21Cip1 protein levels and simultaneously reduced CPT-induced p53 and H2AX phosphorylation. Consequently, DFMO significantly decreased CPT-induced caspase-3 activation (Fig. 5A) and DNA fragmentation (Fig. 5B), further confirming the pro-apoptotic role of Cdk2.
Combined inhibition of Cdk1/Cdk2/Cdk9 sensitizes polyamine-depleted cells to apoptosis
Data presented earlier show that inhibition of Cdk2 activity alone (using NU6140) inhibits proliferation without causing cell death (Figs. 1 and 2). Conversely, concomitant inhibition of Cdk1, Cdk9, and Cdk2 activities (using AZD5438) significantly blocks proliferation and concomitantly induces caspase-3-dependent apoptosis (Fig. 2). In control cells, addition of AZD5438 during proliferation increased levels of active caspase-3 (Fig. 6A) and DNA fragmentation (Fig. 6B), which is consistent with earlier observations presented in figure 2. Polyamine depletion decreased Cdk2 activity [27] and conferred resistance to apoptosis in IEC-6 cells [24–26], and both NU6140 and DFMO inhibited apoptosis in response to CPT (Fig. 5B). To determine the effect of Cdk1, Cdk9, and Cdk2 inhibition in polyamine-depleted cells, we seeded cells in DFMO containing medium followed by the addition of AZD5478 after 24h. Treatment of proliferating cells with AZD5438 in the presence of DFMO induced caspase-3 activation and DNA fragmentation (Fig. 6B), indicating that pro-apoptotic effects of AZD5438 were due to inhibition of Cdk1 and Cdk9 in polyamine depleted cells. However, levels of active caspase-3 in cells grown in the presence of DFMO plus AZD5438 were lower than control cells treated with AZD5438, suggesting that DFMO and AZD5438-mediated inhibition of Cdk2 activity partially contributes to protection in cells with decreased activities of Cdk1 and Cdk9. In addition, levels of active caspase-3 and DNA fragmentation in cells grown in the presence of DFMO plus putrescine plus AZD5438 were similar to those of control cells. To further understand the involvement of Cdk2, cells were seeded in control, DFMO, and DFMO plus putrescine containing medium. After 24h, cells were treated with NU6140 in respective culture medium. NU6140 failed to activate caspase-3 and DNA fragmentation in all three groups (Fig. 6A, 6B). Together, these results indicate that Cdk2 is pro-apoptotic and both Cdk1 and Cdk9 are major anti-apoptotic regulators in proliferating cells.
Fig. 6. AZD5438 reverses the protective effect of DFMO in proliferating cells.

(A) IEC-6 cells were trypsinized from stock flasks and equal numbers were seeded in control, DFMO, and DFMO plus 10 M putrescine containing medium. Eighteen hours later, the attached cells were treated with 1 M AZD5438 and 1 M NU6140 in control, DFMO, and DFMO plus putrescine containing medium. Cells were incubated with these drugs for 72h. Cell lysates were analyzed for active caspase-3 using specific antibody. Actin was used as an internal loading control. (B) DNA fragmentation was measured by ELISA. Values expressed are mean SE, n=3. *significantly different compared to DMSO treated cells (P<0.05).
Role of Cdks in non-proliferating cells
Cdk activity is blocked in contact-inhibited, serum-starved cells. It has been established that in the absence of proliferative signals, normal cells withdraw from the cell cycle into quiescence [32]. To investigate the consequences of Cdk inhibition in serum starved confluent monolayers, we treated confluent monolayers with AZD5438 for 1h followed by treatment with either CPT alone or a combination of CPT and TNF- (CPT+TNF). Activation of Cdks, as determined by phosphorylation of retinoblastoma protein on a Cdk consensus site [33], occurred after 3h of CPT treatment with no change in total Rb protein (Fig. 7A). We next determined whether p53 and Cdks are activated along the same pathway or by independent mechanisms. Both p53 and p21Cip1 protein levels increased in confluent serum-starved cells treated with CPT (Fig. 7A), which is consistent with earlier observations [12]. Concomitant with the increase in p53 signaling, CPT increased caspase-3 activation (Fig. 7A) and DNA fragmentation (Fig. 7B). Treatment of serum-starved confluent cells with 5 M AZD5438 alone had no appreciable effect on p53 and p21Cip1 expression, Rb phosphorylation, and caspase-3 activation (Fig. 7A). However, AZD5438 significantly increased basal DNA fragmentation in serum-starved cells, indicating that AZD5438-induced apoptosis in non-proliferating cells is independent of caspase-3. Pre-treatment of cells with AZD5438, decreased CPT-induced Rb phosphorylation, indicating that AZD5438 prevents CPT-induced Cdk activation. Furthermore, AZD5438 prevented CPT-mediated increases in p53 and p21Cip1 protein levels and, consequently, decreased CPT-induced caspase-3 activation and DNA fragmentation (Fig. 7A, 7B), indicating a pivotal role of Cdks in mediating pro-apoptotic effects of p53 in quiescent cells.
Fig. 7. AZD5438 protects non-proliferating IEC-6 cells from both CPT and CPT+TNF-induced apoptosis.

(A) IEC-6 cells were grown to confluence and serum-starved for 24h. Confluent monolayers were treated with 5 M AZD5438 for 1h followed by 20 M CPT or 20 M CPT+ 10ng/ml TNF- for 3h. Cell lysates were analyzed by western blot for phospho-Rb Ser780, total-pRb, p53, p21Cip1, and caspase-3 using specific antibodies. Actin was used as an internal loading control. (B) DNA fragmentation was measured by ELISA. Values expressed are mean SE, n=3. *significantly different compared to untreated (UT) cells (P<0.05), **significantly different compared to CPT and CPT+TNF treated groups (P<0.05).
In IEC-6 cells, CPT+TNF is a potent inducer of apoptosis [34]. CPT+TNF robustly increased Rb phosphorylation, p53 and p21Cip1 expression, which was accompanied by increased levels of active caspase-3 and DNA fragmentation (Fig. 7B). Pre-treatment with AZD5438, decreased CPT+TNF-induced Rb phosphorylation and simultaneously decreased p53 and p21Cip1 expression leading to a decrease in caspase-3 activation and DNA fragmentation (Figs. 7A, 7B). Taken together, these results indicate that activation of Cdk1, 2, and 9 is essential for both CPT and CPT+TNF-induced apoptosis in non-proliferating cells.
Specific inhibition of Cdk2 rescues various cell types from apoptosis [35], and our data in Fig. 5 indicate that inhibition of Cdk2 (using NU6140) protects proliferating cells from CPT-induced apoptosis. Therefore, we investigated whether inhibition of Cdk2 activity leads to cyto-protective responses in non-proliferating quiescent cells. CPT+TNF increased p53 phosphorylation and protein levels with a concomitant increase of p21Cip1 (Fig. 8A) and DNA fragmentation (Fig. 8B), which is similar to that of the CPT-treated cells. Pre-treatment of cells with increasing doses of NU6140 (1–10 M) slightly increased basal DNA fragmentation. Furthermore, NU6140 prevented CPT+TNF-induced phosphorylation of p53 and total p53 expression in a dose-dependent manner (Fig. 8A). In addition, NU6140 blocked the CPT+TNF-induced increase in p21Cip1 in a dose-dependent manner, which was accompanied by a significant decrease in DNA fragmentation. These results suggest that inhibition of Cdk2 activity confers protection from CPT+TNF-induced apoptosis, which resembles the effects of polyamine-depletion.
Fig. 8. Cdk2 inhibitor NU6140 rescues non-proliferating IEC-6 cells from CPT+TNF-induced apoptosis.

(A) Confluent serum-starved IEC-6 cells were treated with indicated doses of NU6140 for 1h followed by CPT+TNF- treatment for 3h. Cell lysates were analyzed by western blot for phospho-p53 Ser15, total-p53, and p21Cip1 using specific antibodies. Actin was used as a loading control. (B) DNA fragmentation was measured by ELISA. Values expressed are mean SE, n=3. *significantly different compared to DMSO treated cells (P<0.05), **significantly different compared to CPT+TNF treated group (P<0.05).
Since inhibition of Cdks in control cells resulted in protection from apoptosis, we determined whether the inhibition of Cdks in polyamine-depleted cells had a similar effect. In control cells, AZD5438 significantly increased basal DNA fragmentation. Treatment of control cells with CPT+TNF increased DNA fragmentation (Fig. 9A). In control cells treated with CPT+TNF, the inhibition of Cdk1, 2, and 9 (using AZD5438) significantly decreased CPT+TNF-induced DNA fragmentation (Fig. 9A). In cells grown in the presence of DFMO, the level of apoptosis in response to CPT+TNF was significantly less compared with that seen in control cells, which is consistent with previous observations [12, 34]. In response to AZD5438 alone in polyamine-depleted cells, apoptosis significantly increased. The inhibition of Cdks failed to decrease CPT+TNF-induced apoptosis in polyamine-depleted cells. However, the level of apoptosis in the DFMO group in response to CPT+TNF following AZD5438 treatment was lower compared to control cells treated with AZD5438 and CPT+TNF (Fig. 9A). Cells grown in the presence of DFMO plus putrescine had responses identical with control cells, indicating that the effects observed in DFMO-treated cells were due to the depletion of polyamines and not due to the effects of DFMO. Since pre-treatment of confluent, serum-starved, cells with 10 M NU6140 significantly blocked CPT+TNF-induced apoptosis (Fig. 8B), we used 10 M NU6140 to investigate the role of Cdk2 inhibition in polyamine-depleted cells. Similar to previous experimental results (Fig. 8B), NU6140 increased basal DNA fragmentation in control cells (Fig. 9B). Furthermore, pre-treatment of control cells with NU6140 significantly decreased CPT+TNF-induced DNA fragmentation (Fig. 9B). In polyamine-depleted cells, NU6140 did not significantly increase basal DNA fragmentation (Fig. 9B). Polyamine-depletion significantly protected cells from CPT+TNF-induced apoptosis. In addition, NU6140 had no effect on CPT+TNF-induced DNA fragmentation in polyamine-depleted cells. These results suggest that the pro-apoptotic role of Cdk2 is polyamine-dependent, whereas anti-apoptotic activities of Cdk1 and Cdk9 are polyamine-independent.
Fig. 9. Role of Cdk inhibitors in polyamine depleted cells.

(A) Confluent serum-starved IEC-6 cells grown in control, DFMO and DFMO plus putrescine (Put) containing medium were pre-treated with 5 M AZD5438 for 1h followed by CPT+TNF- for 3h. DNA fragmentation was measured by ELISA. Values expressed are mean SE, n=3. *significantly different compared to untreated (UT) cells (P<0.005). (B) Confluent serum-starved IEC-6 cells grown in control and DFMO containing medium were pre-treated with 10 M NU6140 for 1h followed by CPT+TNF- for 3h. DNA fragmentation was measured by ELISA. Values expressed are mean SE, n=3. *significantly different compared to untreated (UT) cells (P<0.05), ** significantly different compared to CPT+TNF treated group (P<0.05), # significantly different compared to control cells treated with CPT+TNF (P<0.05).
DISCUSSION
Apoptosis is required for the regulation of cell number in proliferating tissues. A link between the cell cycle and apoptosis is based on the fact that genes involved in cell cycle progression regulate apoptosis [36]. The key finding of our studies is that Cdks play fundamental roles in proliferation and apoptosis of intestinal epithelial cells. In this study, our data show that inhibition of basal Cdk2 activity in proliferating IEC-6 cells (using NU6140) slows proliferation (Fig. 1B) without inducing H2AX phosphorylation and apoptosis (Fig. 2B). Our previous evidence demonstrated that polyamine depletion reduced Cdk2 activity, induced cell cycle arrest [27], and protected IEC-6 cells from apoptosis [22–24]. Cdk2 orchestrates several cell cycle events in different cell types [37]. Recent studies with animal models have failed to support the concept that Cdks play a pivotal role in driving various phases of the cell cycle. Genetic ablation of Cdk2 does not induce abnormalities in cell proliferation [38], and Cdk2-null mice are viable [39]. Therefore, Cdks have cell-type specific functions, and compensatory roles exist among different Cdk isoforms, which in turn could play pivotal roles in the regulation of proliferation and apoptosis. In IEC-6 cells, concomitant inhibition of Cdk1, 2, and 9 induced more antiproliferative effects than those induced by Cdk2 individually (Fig. 1B). Furthermore, sustained inhibition of Cdk1, 2, and 9 for 72h caused massive DNA damage and apoptosis (Fig. 2). It has been shown that Cdk inhibition during S phase triggers a checkpoint that includes components of the DNA-damage pathway including H2AX, a molecular sensor for double-stranded breaks [40]. Based on comparing signaling events in proliferating IEC-6 cells treated with different Cdk inhibitors, the major alteration was found to be the phosphorylation of H2AX (Fig. 2A). Mechanisms through which Cdk1/2/9 inhibition induces H2AX phosphorylation in proliferating IEC-6 cells are unknown, but our studies implicate that irreversible collapse of replication forks and a decreased capacity for replication result in rapid phosphorylation of H2AX. DNA topoisomerase I (Camptothecin) and II (Etoposide) inhibitors increased phosphorylation of H2AX via Ataxia-telangiectasia mutated and Rad3-related (ATR) kinase signaling at S and G1-phases of the cell cycle, respectively [41] indicating that H2AX phosphorylation is one of the key players in DNA damage response induced by genotoxic stress. Furthermore, both Cdk1 and Cdk2 phosphorylated checkpoint kinase 1 (Chk1) at Ser286 and Ser301 upon stalled replication forks and DNA damage [42] suggesting that cell cycle-dependent processing of DNA damage is dependent on Cdk kinase activity. Zhu et al. demonstrated that treatment of an ovarian cancer cell line with the Cdk2 inhibitor aminothiazole compound-25 increased biochemical features of DNA damage [43]. By contrast, Cdk2 inhibition failed to induce H2AX phosphorylation in exponentially growing cells suggesting a lack of DNA damage (Fig. 2A). Phosphorylation of H2AX is dynamic in nature and is finely tuned by serine/threonine phosphatases. Multiple phosphatases including PP2A, PP4, PP1, and Wip1 have been implicated in negatively regulating H2AX phosphorylation [44]. Furthermore, phosphatases regulate cell cycle progression by removing inhibitory phosphorylations from Cdk1 and Cdk2 [45]. Based on these observations, it is possible to posit that inhibition of Cdk2 during proliferation activates phosphatases, thereby increasing de-phosphorylation of H2AX leading to decreased DNA damage.
Cdks are known to regulate p53 function [19]. Therefore, we investigated whether p53 activation cooperates in regulating the apoptotic response triggered by inhibition of multiple Cdks. Our data show that either pharmacologic inhibition of Cdk2 activity alone or inhibition of multiple Cdks is sufficient to induce p53 activation (Fig. 2A). This is consistent with an earlier observation showing that pharmacologic Cdk inhibitors increased p53-dependent transcription in tumor cells [46]. Based on these observations, we propose that direct regulation of p53 occurs after Cdk inhibition in proliferating gut epithelial cells. The question remains whether p53 activation in proliferating cells treated with different Cdk inhibitors triggers cell cycle arrest or apoptosis. Consistent with the notion of a link between activated p53 and cell cycle arrest, p53 expression and its post-translational modifications dictate cell fate by governing a variety of cellular checkpoints involved in cell cycle arrest, apoptosis, and DNA repair. However, the entire spectrum of p53 actions may be complex and cell type-specific. For instance, p53 has been implicated in both promoting and inhibiting apoptosis in multiple cell types [47–48]. The enhancement of p53 signaling in cells treated with the pharmacologic Cdk2 inhibitor (NU6140) coincided with p21Cip1 accumulation (Fig. 2A), suggesting that Cdk2 inhibition leads to cell-cycle perturbation via the p53-mediated induction of p21Cip1, an endogenous Cdk inhibitor protein. Polyamine depletion delayed cell cycle progression by decreasing Cdk2 activity and increasing the levels of p53-dependent p21Cip1 without causing DNA damage in most cell systems studied [49], suggesting that polyamine depletion mimicked the effects of NU6140. NU6140 completely blocked CPT-induced H2AX phosphorylation, whereas polyamine-depletion had a partial effect (Fig. 5A). Furthermore, both NU6140 and polyamine depletion decreased CPT-induced p53 expression and its phosphorylation. Consequently, both NU6140 and CPT rescued cells from CPT-induced apoptosis (Fig. 5B). These results indicate that either inhibition (via NU6140 treatment) or reduction (via polyamine-depletion) of Cdk2 activity increases the cellular resistance to CPT-induced apoptosis. The increased levels of p21Cip1 in cells treated with NU6140 followed by CPT might be either due to sustained inhibition of Cdk2 or enhanced engagement of the protein in cell-cycle arrest. Co-treatment of proliferating cells with NU6140 and DFMO had no effect on apoptosis (Fig. 6B), indicating that NU6140 and DFMO act in a synergistic manner to prevent cell death. Together, these results unequivocally demonstrate a pro-apoptotic role of Cdk2 and implicate Cdk2 as a master regulator of proliferation via regulation of p53/p21Cip1 signaling.
The multiple effects of Cdk inhibitors upon various members of the Cdk family, may determine the anti- or pro-apoptotic outcome. Chemical inhibitors of Cdks such as roscovitine, purvalanol, and olomoucine block apoptosis in many cell types [50–52]. Under certain conditions, Cdk inhibitors can also induce apoptosis [53]. Although the precise biological mechanism of these inhibitors is not known, it has been demonstrated that inhibition of Cdk1 and Cdk9 may cause apoptosis [54]. In IEC-6 cells, sustained inhibition of Cdk1, Cdk2, and Cdk9 by AZD5438 for 72h resulted in p53 activation, accompanied by increased expression of p21Cip1, phosphorylation of H2AX, and activation of caspase-3 (Figs. 2A, 5A). Since p21Cip1 expression is high in these cells, the assembled cyclin-Cdk1/2/9 complexes are inhibited, which is sufficient to halt cell-cycle progression. As a result, stalled replication induces DNA damage via H2AX phosphorylation. Based on these observations, it is possible to speculate that the critical function of p53 is to prevent proliferation of cells with DNA damage rather than to allow repair to occur. In so doing, p53 uncouples proliferation to the onset of apoptosis in DNA damaged cells. Although AZD5438-treated cells contained higher levels of phosphorylated p53 compared to NU6140, p21Cip1 levels were higher in NU6140 treated cells (Fig. 2A). Caspase-3 mediated cleavage of p21Cip1 has been established as a requisite for apoptotic cell death in various cell lines [55–56]. Consistent with these observations, we have demonstrated caspase-3 induced cleavage of p21Cip1 during apoptosis of IEC-6 cells [25]. Therefore, p21Cip1 cleavage by active caspase-3 and its subsequent degradation can explain the observed decrease in p21Cip1 expression in AZD5438-treated cells. Furthermore, accumulation of p21Cip1 is due to lack of caspase-3 activation in NU6140-treated cells.
Pre-treatment of proliferating cells with AZD5438 followed by CPT potentiated DNA fragmentation and cell death (Fig. 5B). However, AZD5438 did not augment CPT-mediated phosphorylation of p53 and H2AX (Fig. 5B). This could be due the fact that pre-existing DNA damage in cells treated with AZD5438 prevents further potentiation of CPT-induced H2AX phosphorylation and p53 signaling. However, higher levels of phospho-p53 and total-p53 were observed in AZD5438+CPT treated cells compared to AZD5438 alone (Fig. 5A). Therefore, it is likely that a subset of cells with lower levels of DNA damage (in response to AZD5438 treatment) still respond to CPT showing changes in p53/p21Cip1 signaling. Additionally, CPT is capable of activating multiple upstream kinases including MEK1/2, JNK in AZD5438-treated cells, which in turn leads to the phosphorylation of p53. We have shown earlier that MEK1/2 and JNK contribute to CPT-induced p53 phosphorylation in IEC-6 cells [12]. Several studies have shown that phosphorylation of p53 inhibits its degradation by interfering with the p53-Mdm2 interaction, and, thereby, causes accumulation of the protein [57]. Therefore, CPT-induced p53 phosphorylation contributes to the stabilization and accumulation of the protein without changing H2AX phosphorylation in AZD5438-treated cells. Furthermore, these results clearly reveal that Cdk1/2/9 inhibition in dividing cells can selectively modulate p53 function, leading to cell death. Incubation of proliferating cells with AZD5438 in the presence of DFMO reverses the protective effect of polyamine depletion (Fig. 6B) probably due to AZD5438-mediated Cdk1/9 inhibition in polyamine-depleted cells (with reduced Cdk2 activity). Furthermore, AZD5438 sensitized serum-starved, quiescent, polyamine-depleted cells to apoptosis (Fig. 9A). An earlier report showed that polyamine depletion enhances roscovitine-induced apoptosis in colon cancer cells [58]. Thus, the correlation between sustained inhibition of Cdk1/2/9 and increased apoptosis in polyamine-depleted cells indicates an anti-apoptotic role for both Cdk1 and 9.
One interesting facet of this study is that Cdks play a differential role in serum starved, confluent, IEC-6 cells. Treatment of synchronized, quiescent, monolayers with AZD5438 (Fig. 7B) or NU6140 (Fig. 8B) increased basal DNA fragmentation without affecting the level of active caspase-3, suggesting that basal Cdk activities are essential to survival of cells. CPT+TNF increased p53 and p21Cip1 expression and Rb phosphorylation in confluent, serum-starved, monolayers (Figs. 7A, 8A). However, both AZD5438 and NU6140 decreased CPT+TNF-induced p53 and p21Cip1 expression, and, consequently, protected cells from CPT+TNF-induced apoptosis (Figs. 7 and 8). These results indicate that Cdks participate in CPT+TNF-induced apoptosis. Indeed, Knockaert et al. demonstrated that Cdk inhibitors have protective roles in non-proliferating neuronal cells subjected to various chemical insults [53]. The induction of DNA fragmentation by Cdk inhibitors independent of CPT+TNF in non-dividing cells, might explain why these inhibitors could not completely protect cells from CPT+TNF-induced apoptosis (Figs. 9A, 9B).
Because Cdk inhibition appears to regulate p53-dependent proliferation and apoptosis in untransformed IEC-6 cells, we further confirmed the consequences of Cdk inhibition in p53-deficient Caco-2 cells. Proliferating Caco-2 cultures treated with CPT for 48h showed robust activation of caspase-3 (Fig. 4A), which was accompanied by significant reduction in cell numbers (Fig. 3B). However, no significant caspase-3 activity was detected in p53-deficient Caco-2 cultures treated with AZD5438 (Fig. 4A), even at 48h when death occurred as evidenced by increased levels of DNA fragmentation (Fig. 4B). These observations suggest that Caco-2 death after AZD5438 treatment in the absence of p53 is caspase-3-independent. Furthermore, the incomplete processing of active caspase-3 during NU6140 treatment for 48h indicates that caspase-3 activation requires cooperation between the downstream effectors of p53 and Cdk-mediated pathways. The incomplete processing of caspase-3 is fully consistent with several published reports linking processing of caspase-3 to the induction of apoptosis. Bax deficiency results in incomplete processing of caspase-3 and modulates TRAIL-induced apoptosis in cancer cells [59]. We showed earlier that Caco-2 cells lack Bax expression [31], and this could be a contributing factor for the incomplete processing of caspase-3 in cells treated with NU6140. The incomplete processing of caspase-3 was also shown to be responsible for the partial cleavage of XIAP in HL-derived B cell lines [60] and resistance to TRAIL-induced apoptosis in human neuroblastoma cells [61].
Intriguingly, AZD5438 significantly blocked proliferation in the absence of p53 (Fig. 3B). Caco-2 cells proliferated at a lower rate when treated with NU6140. Thus, Cdk inhibitors influence Caco-2 proliferation in a p53-independent manner. In accordance with our findings, Cdk2 inhibitor AT7519 showed anti-proliferative activity in human tumor cell lines that was independent of their p53 status [62]. Furthermore, both AZD5438 and NU6140 equally inhibited Rb phosphorylation in proliferating Caco-2 cells (Fig. 3C), suggesting that p53 deficiency has no effect on Rb phosphorylation. AZD5438 decreased Cdk1 expression confirming inhibition of Cdk1/2/9-cyclin complexes and their activities. However, NU6140 decreased Cdk1 expression, indicating functional redundancy and possible cross talk between Cdk2 and Cdk1-dependent pathways regulating Caco-2 proliferation. Vesely et al. showed that the Cdk inhibitor olomoucine specifically inhibits Cdk2-cyclinE and Cdk2-cyclinA complexes [63]. Since both Cdk inhibitors decreased Cdk1 expression and concomitantly induced apoptosis in cells lacking p53, it is possible that the binding of the inhibitors to Cdks disrupts the Cdk1-cyclinA complexes. As a consequence, Cdk1 undergoes degradation. p21Cip1 was shown to stabilize interactions between Cdks and cyclins [64]. Caco-2 cells lack p21Cip1 expression (Fig. 3C). Therefore, one additional possibility is that disruption of Cdk-cyclin complexes (using pharmacologic Cdk inhibitors) in cells lacking p21Cip1 expression permits destabilization and degradation of Cdk1. CPT robustly increased apoptosis but failed to decrease Cdk1 expression in Caco-2 cells, clearly suggesting that CPT is not capable of disrupting the Cdk1-cyclin regulatory complex.
Exposure of dividing Caco-2 cells to AZD5438 failed to increase H2AX phosphorylation (Fig. 3C), indicating that p53 is necessary for the induction of DNA damage. Conversely, NU6140 decreased H2AX phosphorylation (Fig. 3C) suggesting that inhibition of Cdk2 fails to trigger DNA damage regardless of p53 status. Interestingly, CPT increased H2AX phosphorylation, p21Cip1 expression, and partially decreased Rb phosphorylation in dividing Caco-2 cells (Fig. 3C), suggesting that Caco-2 cells possess an intact apoptotic pathway and that either p73 or E2F1 could regulate CPT-induced H2AX phosphorylation, p21Cip1 expression, and apoptosis in these cells. We showed earlier that p73 and E2F1-mediated expression of PUMA and Siva-1 induces apoptosis in p53-deficient Caco-2 cells [31]. Thus, we reasoned that modulation of p73 signaling using CPT might induce apoptosis without altering Cdk activities in Caco-2 cells.
In conclusion, the results presented show that Cdks differentially modulate apoptosis in proliferating versus quiescent cells (Fig. 10). Therefore, Cdk activities could have important implications in the control of intestinal epithelial homeostasis. Inhibition of Cdk2 protects both proliferating and quiescent cells from CPT-induced apoptosis, confirming that Cdk2 predominantly acts as a pro-apoptotic protein. Furthermore, polyamines regulate IEC-6 survival at least in part by repressing Cdk2 activity. By contrast, Cdk1 and Cdk9 activities appear to be polyamine-independent. However, both Cdk1 and Cdk9 play opposing roles in the regulation of apoptosis. Concomitant inhibition of Cdk1, 2, and 9 activities in proliferating cells triggers DNA damage and p53-dependent apoptosis (Fig. 10). Conversely, inhibition of Cdk1/2/9 in quiescent cells decreases CPT-induced p53 activation and protects cells from apoptosis. In addition, our data show that Cdk inhibitors are capable of blocking proliferation and inducing apoptosis in p53-deficient Caco-2 cells. Thus, our study underlines the therapeutic potential of Cdk inhibitors in colon carcinoma.
Fig. 10. Schematic showing the mechanisms of Cdks involved in apoptosis.

Proliferating cells: Inhibition of Cdk1, Cdk2, and Cdk9 activities (using AZD5438) in proliferating IEC-6 cells triggers p53 phosphorylation and its accumulation. Activated p53 increases p21Cip1 protein, which in turn causes growth arrest. Furthermore, in response to AZD5438, DNA replication fork stalling leads to irreversible DNA damage characterized by increased phosphorylation of histone H2AX at Ser139. AZD5438 failed to induce H2AX phosphorylation in p53 deficient Caco-2 cells suggesting that p53 is necessary for the induction of DNA damage. H2AX phosphorylation coupled with p53 activation induces apoptosis evidenced by increased levels of DNA fragmentation and caspase-3 activation. Campothecin (CPT)-induced DNA damage leads to H2AX phosphorylation Ser139 and p53 activation in proliferating IEC-6 cells. CPT-induced activation of p53 leads to growth arrest (via p21Cip1 expression) and apoptosis. Pre-treatment of cells with AZD5438 potentiated CPT-induced apoptosis.
Conversely, selective inhibition of Cdk2 (using NU6140) during proliferation failed to induce DNA damage and H2AX phosphorylation, and, consequently, blocked caspase-3 activation and apoptosis ( ). Notably, Cdk2 inhibition increased p53/p21 signaling, which led to growth arrest. Inhibition of Cdk2 activity blocked CPT-induced H2AX phosphorylation in proliferating cells and increased the cellular resistance to apoptosis.
Quiescent cells: Pre-treatment of serum-starved quiescent cells with either AZD5438 or NU6140 decreased CPT+TNF-induced p53 activation, p21Cip1 expression, and protected cells from apoptosis.
Acknowledgments
This work was supported by National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) grant DK-16505, and by the Thomas A. Gerwin Endowment. We gratefully acknowledge Mary Jane Viar for technical assistance.
Abbreviations
- IEC-6
intestinal epithelial cells
- p21Cip1
p21Waf1/Cip1
- DFMO
-difluromethylornithine
- TNF-
Tumor necrosis factor-
- CPT
Camptothecin
- ODC
ornithine decarboxylase
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
Fetal bovine serum
- dFBS
dialyzed FBS
- ECL
enhanced chemiluminescence
- DPBS
Dulbecco’s PBS
References
- 1.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. I. Columnar Cell. Am J Anat. 1974;141:461–79. doi: 10.1002/aja.1001410403. [DOI] [PubMed] [Google Scholar]
- 2.Hall PA, Coates PJ, Ansari B, Hopwood D. Regulation of cell number in the mammalian gastrointestinal tract: The importance of apoptosis. J Cell Sci. 1994;107:3569–3577. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- 3.Potten CS, Grant HK. The relationship between ionizing radiation-induced apoptosis and stem cells in the small and large intestine. Br J Cancer. 1998;78:993–1003. doi: 10.1038/bjc.1998.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arellano M, Moreno S. Regulation of CDK/cyclin complexes during the cell cycle. Int J Biochem Cell Biol. 1997;29:559–73. doi: 10.1016/s1357-2725(96)00178-1. [DOI] [PubMed] [Google Scholar]
- 6.Johnsen SA. CDK9 and H2B monoubiquitination: a well-choreographed dance. PLoS Genet. 2012;8:e1002860. doi: 10.1371/journal.pgen.1002860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee MH, Reynisdottir I, Massague J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995;9:639–49. doi: 10.1101/gad.9.6.639. [DOI] [PubMed] [Google Scholar]
- 8.Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E, et al. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simone C, Bagella L, Bellan C, Giordano A. Physical interaction between pRb and cdk9/cyclinT2 complex. Oncogene. 2002;21:4158–65. doi: 10.1038/sj.onc.1205511. [DOI] [PubMed] [Google Scholar]
- 10.Goodrich DW, Wang NP, Qian YW, Lee EY, Lee WH. The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell. 1991;67:293–302. doi: 10.1016/0092-8674(91)90181-w. [DOI] [PubMed] [Google Scholar]
- 11.Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 dynamics control cell fate. Science. 2012;336:1440–4. doi: 10.1126/science.1218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhattacharya S, Ray RM, Johnson LR. Role of polyamines in p53-dependent apoptosis of intestinal epithelial cells. Cell Signal. 2009;21:509–522. doi: 10.1016/j.cellsig.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Fragkos M, Jurvansuu J, Beard P. H2AX is required for cell cycle arrest via the p53/p21 pathway. Mol Cell Biol. 2009;29:2828–40. doi: 10.1128/MCB.01830-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris EJ, Keramaris E, Rideout HJ, Slack RS, Dyson NJ, Stefanis L, Park DS. Cyclin-Dependent kinases and p53 pathways are activated independently and mediate Bax activation in neurons after DNA damage. J Neurosci. 2001;21:5017–5026. doi: 10.1523/JNEUROSCI.21-14-05017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung JH, Bunz F. Cdk2 is required for p53-independent G2/M checkpoint control. PLoS Genetics. 2010;6:e1000863. doi: 10.1371/journal.pgen.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson N, Shapiro GI. Cyclin-dependent kinases (Cdks) and the DNA damage response: rationale for cdk inhibitor-chemotherapy combinations as an anticancer strategy for solid tumors. Expert Opin Ther Targets. 2010;14:1199–212. doi: 10.1517/14728222.2010.525221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Golsteyn RM. Cdk1 and Cdk2 complexes (cyclin dependent kinases) in apoptosis: a role beyond the cell cycle. Cancer Lett. 2005;217:129–38. doi: 10.1016/j.canlet.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Ferguson M, Luciani MG, Finlan L, Rankin EM, Ibbotson S, Fersht A, Hupp TR. The development of a Cdk2-docking site peptide that inhibits p53 and sensitizes cells to death. Cell Cycle. 2004;3:80–89. [PubMed] [Google Scholar]
- 19.Wang Y, Prives C. Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin-dependent kinases. Nature. 1995;376:88–91. doi: 10.1038/376088a0. [DOI] [PubMed] [Google Scholar]
- 20.Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH. p14ARF links the tumor suppressors RB and p53. Nature. 1998;395:124–5. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- 21.Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–23. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 22.Elias BC, Bhattacharya S, Ray RM, Johnson LR. Polyamine-dependent activation of Rac1 is stimulated by focal adhesion-mediated Tiam1 activation. Cell Adh Migr. 2010;4:419–30. doi: 10.4161/cam.4.3.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattacharya S, Ray RM, Johnson LR. Integrin 3-mediated Src activation regulates apoptosis in IEC-6 cells via Akt and STAT3. Biochem J. 2006;397:437–447. doi: 10.1042/BJ20060256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng W, Viar MJ, Johnson LR. Polyamine depletion inhibits irradiation-induced apoptosis in intestinal epithelia. Am J Physiol Gastrointest Liver Physiol. 2005;289:G599–606. doi: 10.1152/ajpgi.00564.2004. [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharya S, Ray RM, Johnson LR. Basic helix-loop-helix protein E47-mediated p21Waf1/Cip1 gene expression regulates apoptosis of intestinal epithelial cells. Biochem J. 2007;407:243–254. doi: 10.1042/BJ20070293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhattacharya S, Ray RM, Johnson LR. Decreased apoptosis in polyamine depleted IEC-6 cells depends on Akt-mediated NF- B activation but not GSK3 activity. Apoptosis. 2005;10:759–776. doi: 10.1007/s10495-005-2943-3. [DOI] [PubMed] [Google Scholar]
- 27.Ray RM, Zimmerman BJ, McCormack SA, Patel TB, Johnson LR. Polyamine depletion arrests cell cycle and induces inhibitors p21(Waf1/Cip1), p27(Kip1), and p53 in IEC-6 cells. Am J Physiol. 1999;276:C684–91. doi: 10.1152/ajpcell.1999.276.3.C684. [DOI] [PubMed] [Google Scholar]
- 28.Quaroni A, Wands J, Trelstad R, Isselbacher KJ. Epithelial cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–265. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCormack SA, Viar MJ, Johnson LR. Polyamines are necessary for cell migration by a small intestinal crypt cell line. Am J Physiol. 1993;264:G367–74. doi: 10.1152/ajpgi.1993.264.2.G367. [DOI] [PubMed] [Google Scholar]
- 30.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 31.Ray RM, Bhattacharya S, Johnson LR. Mdm2 inhibition induces apoptosis in p53-defiecient human colon cancer cells by activating p73- and E2F1-mediated expression of PUMA and Siva-1. Apoptosis. 2011;16:35–44. doi: 10.1007/s10495-010-0538-0. [DOI] [PubMed] [Google Scholar]
- 32.Chassot AA, Lossaint G, Turchi L, Meneguzzi G, Fisher D, Ponzio G, Dulic V. Confluence-induced cell cycle exit involves pre-mitotic CDK inhibition by p27(Kip1) and cyclin D1 downregulation. Cell Cycle. 2008;7:2038–46. doi: 10.4161/cc.7.13.6233. [DOI] [PubMed] [Google Scholar]
- 33.Knudsen ES, Wang JY. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–8320. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- 34.Ray RM, Jin S, Bavaria MN, Johnson LR. Regulation of JNK activity in the apoptotic response of intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;300:G761–70. doi: 10.1152/ajpgi.00405.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim SG, Kim SN, Jong HS, Kim NK, Hong SH, Kim SJ, Bang YJ. Caspase-mediated Cdk2 activation is a critical step to execute transforming growth factor- 1-induced apoptosis in human gastric cancer cells. Oncogene. 2001;20:1254–1265. doi: 10.1038/sj.onc.1204203. [DOI] [PubMed] [Google Scholar]
- 36.Pucci B, Kasten M, Giordano A. Cell Cycle and apoptosis. Neoplasia. 2000;2:291–9. doi: 10.1038/sj.neo.7900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–91. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 38.Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35:25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- 39.Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–85. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 40.Maude SL, Enders GH. Cdk inhibition in human cells compromises chk1 function and activates a DNA damage response. Cancer Res. 2005;65:780–6. [PubMed] [Google Scholar]
- 41.Firsanov DV, Solovjeva LV, Svetlova MP. H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. Clin Epigenetics. 2011;2:283–97. doi: 10.1007/s13148-011-0044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu N, Libertini S, Zhang Y, Gillespie DA. Cdk phosphorylation of Chk1 regulates efficient Chk1 activation and multiple checkpoint proficiency. Biochem Biophys Res Commun. 2011;413:465–70. doi: 10.1016/j.bbrc.2011.08.119. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Y, Alvarez C, Doll R, Kurata H, Schebye XM, Parry D, Lees E. Intra-S-phase checkpoint activation by direct CDK2 inhibition. Mol Cell Biol. 2004;24:6268–6277. doi: 10.1128/MCB.24.14.6268-6277.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freeman AK, Monteiro AN. Phosphatases in the cellular response to DNA damage. Cell Commun Signal. 2010;8:27. doi: 10.1186/1478-811X-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. Embo Rep. 2003;4:671–7. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kotala V, Uldrijan S, Horky M, Trbusek M, Strnad M, Vojtesek B. Potent induction of wild-type p53-dependent transcription in tumor cells by a synthetic inhibitor of cyclin-dependent kinases. Cell Mol Life Sci. 2001;58:1333–9. doi: 10.1007/PL00000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carvajal LA, Manfredi JJ. Another fork in the road—life or death decisions by the tumor suppressor p53. EMBO Rep. 2013;14:414–21. doi: 10.1038/embor.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garner E, Raj K. Protective mechanisms of p53-p21-pRb proteins against DNA damage-induced cell death. Cell Cycle. 2008;7:277–82. doi: 10.4161/cc.7.3.5328. [DOI] [PubMed] [Google Scholar]
- 49.Wallick CJ, Gamper I, Thorne M, Feith DJ, Takasaki KY, Wilson SM, Seki JA, Pegg AE, Byus CV, Bachmann AS. Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine-inhibitor induced G1 cell cycle arrest in MYCN-amplified human neuroblastoma cells. Oncogene. 2005;24:5606–18. doi: 10.1038/sj.onc.1208808. [DOI] [PubMed] [Google Scholar]
- 50.Park DS, Farinelli SE, Greene LA. Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996;271:8161–9. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- 51.Appert-Collin A, Hugel B, Levy R, Niederhoffer N, Coupin G, Lombard Y, Andre P, Poindron P, Gies JP. Cyclin-dependent kinase inhibitors prevent apoptosis of postmitotic mouse motorneurons. Life Science. 2006;79:484–90. doi: 10.1016/j.lfs.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 52.Wu J, Kharebava G, Piao C, Stoica BA, Dinizo M, Sabirzhanov B, Hanscom M, Guanciale K, Faden AI. Inhibition of E2F1/CDK1 pathway attenuates neuronal apoptosis in vitro and confers neuroprotection after spinal cord injury in vivo. PLoS One. 2012;7:e42129. doi: 10.1371/journal.pone.0042129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002;23:417–25. doi: 10.1016/s0165-6147(02)02071-0. [DOI] [PubMed] [Google Scholar]
- 54.MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, Lane DP, Green SR. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65:5399–407. doi: 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]
- 55.Jin YH, Yoo KJ, Lee YH, Lee SK. Caspase 3-mediated cleavage of p21WAF1/CIP1 associated with the cyclin A-cyclin-dependent kinase 2 complex is a prerequisite for apoptosis in SK-HEP-1 cells. J Biol Chem. 2000;275:30256–63. doi: 10.1074/jbc.M001902200. [DOI] [PubMed] [Google Scholar]
- 56.Gervais JL, Seth P, Zhang H. Cleavage of CDK inhibitor p21(Cip1/Waf1) by caspases is an early event during DNA damage-induced apoptosis. J Biol Chem. 1998;273:19207–12. doi: 10.1074/jbc.273.30.19207. [DOI] [PubMed] [Google Scholar]
- 57.Shangary S, Wang S. Targeting the MDM2-p53 interaction for Cancer Therapy. Clin Cancer Res. 2008;14:5318–5324. doi: 10.1158/1078-0432.CCR-07-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arisan ED, Coker A, Palavan-Unsal N. Polyamine depletion enhances the roscovitine-induced apoptosis through the activation of mitochondria in HCT116 colon carcinoma cells. Amino Acids. 2012;42:655–65. doi: 10.1007/s00726-011-1040-x. [DOI] [PubMed] [Google Scholar]
- 59.Deng Y, Lin Y, Wu X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002;16:33–45. doi: 10.1101/gad.949602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kashkar H, Haefs C, Shin H, Hamilton-Dutoit SJ, Salvesen GS, Kronke M, Jurgensmeler JM. XIAP-mediated caspase inhibition in Hodgkin’s lymphoma-derived B cells. J Exp Med. 2003;198:341–7. doi: 10.1084/jem.20021279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gatsinzi T, Ivanova EV, Iverfeldt K. TRAIL resistance in human neurobasltoma SK-N-AS cells is dependent on protein kinase C and involves inhibition of caspase-3 proteolytic processing. J Neurooncol. 2012;109:503–12. doi: 10.1007/s11060-012-0932-2. [DOI] [PubMed] [Google Scholar]
- 62.Squires MS, Feltell RE, Wallis NG, Lewis EJ, Smith DM, Cross DM, Lyons JF, Thompson NT. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol Cancer Ther. 2009;8:324–32. doi: 10.1158/1535-7163.MCT-08-0890. [DOI] [PubMed] [Google Scholar]
- 63.Vesely J, Havlicek L, Strnad M, Blow JJ, Donella-Deana A, Pinna L, Letham DS, Kato J, Detivaud L, Leclerc S, et al. Inhibition of cyclin-dependent kinases by purine analogues. Eur J Biochem. 1994;224:771–86. doi: 10.1111/j.1432-1033.1994.00771.x. [DOI] [PubMed] [Google Scholar]
- 64.Waldman T, Zhang Y, Dillehay L, Yu J, Kinzler K, Vogelstein B, Williams J. Cell-cycle arrest versus cell death in cancer therapy. Nat Med. 1997;3:1034–6. doi: 10.1038/nm0997-1034. [DOI] [PubMed] [Google Scholar]