

Highlights
-
•
The PKD1 gene, mutated in ADPKD is developmentally regulated in the kidney.
-
•
Renal organ cultures of two distinct Pkd1 mutants display normal UB branching.
-
•
Glomeruli fail to properly develop in Pkd1 mutant renal organ cultures.
-
•
Defective endothelial cell migration likely accounts for glomerulogenesis defects.
-
•
PI3kinase inhibitors phenocopy the Pkd1 phenotype, VEGF minimally improves it.
Keywords: Polycystic kidney disease, Kidney development, Endothelial cell migration, Glomerulogenesis
Abstract
The PKD1 gene is essential for a number of biological functions, and its loss-of-function causes autosomal dominant polycystic kidney disease (ADPKD). The gene is developmentally regulated and believed to play an essential role in renal development. Previous studies have shown that manipulating murine renal organ cultures with dominant-negative forms of the Pkd1 gene impaired ureteric bud (UB) branching. In the current study, we analyzed different stages of renal development in two distinct mouse models carrying either a null mutation or inactivation of the last two exons of Pkd1. Surprisingly, metanephric explants from Pkd1-deleted kidneys harvested at day E11.5 did not show defects of UB branching and elongation, estimated by cytokeratin staining on fixed tissues or by Hoxb7-GFP time-lapse imaging. However, renal explants from Pkd1-mutants isolated at day E14.5 showed impaired nephrogenesis. Notably, we observed cell migratory defects in the developing endothelial compartment. Previous studies had implicated the Pkd1 gene in controlling cell migration and collagen deposition through PI3 kinases. In line with these studies, our results show that wild-type explants treated with PI3-kinase inhibitors recapitulate the endothelial defects observed in Pkd1 mutants, whereas treatment with VEGF only partially rescued the defects. Our data are consistent with a role for the Pkd1 gene in the endothelium that may be required for proper nephrogenesis.
1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common monogenic disorders with an incidence of 1/500–1/1000. The disease is characterized by the formation of bilateral fluid-filled cysts, which originate from every segment of the nephron [1], [2]. ADPKD is, however, a systemic disorder because cyst formation is observed in other organs, such as the liver and the pancreas. In addition, numerous vascular abnormalities were reported including intracranial aneurysms, aortic root dilatation, aneurysm and mitral valve prolapses [2]. The disease is caused by a mutation in the PKD1 gene in 85% of the cases and in the PKD2 gene in the remaining cases [1]. The PKD1 gene product, Polycystin-1 (PC-1), is a large plasma membrane protein (approximately 520 kDa) that forms a functional complex with the PKD2 gene product Polycystin-2 (PC-2), a non-selective cation channel [2]. The polycystin complex functions in the primary cilium, where it is believed to act as a mechanosensor that controls the Ca2+ influx provoked by mechanical stimuli [3].
Numerous mouse models carrying null and/or hypomorphic alleles of the Pkd1 gene have been generated, which share very similar phenotypes [4], [5], [6], [7], [8], [9], [10]. The mice harboring homozygous mutations inactivating the Pkd1 gene die during the fetal stage and invariably develop renal cysts by E15.5. These homozygous mutant embryos also show cardiovascular and heart defects, hydrops fetalis, and bone defects [4], [7], [8]. Mice heterozygous for the mutations survive after birth and display late-onset polycystic kidney and liver disease with very few renal cysts [4], [5], [6], [7], [8].
The Pkd1 gene is developmentally regulated and is postulated to be an important player in renal development. Development of the kidney initiates when an outpouching of the Wolffian duct, called the ureteric bud (UB), invades the metanephric mesenchyme (MM) at around day E11.5 in the mouse. After the UB invades the MM, it undergoes branching followed by elongation of the collecting duct [11]. Each UB tip will induce condensation of the metanephric mesenchyme, which eventually epithelializes to form the renal tubule. Once the initial stages of mesenchymal condensation have occurred, precursors of the endothelial compartment migrate towards the nascent tubule and eventually form the glomerular tuft.
Two previous studies had implied a role for the Pkd1 gene and its product PC-1 in UB branching and early morphogenesis during renal development using two different strategies to impair PC-1 function. Treatment of kidney rudiments with a peptide derived from a highly conserved sequence in the PKD repeats of PC-1 (WDGFDG) or the overexpression of the C-terminal domain of PC-1 in the UB prevented correct branching morphogenesis in a model of ex vivo embryonic murine kidney explants [12], [13].
In the current study, we have performed similar renal organ culture experiments to investigate the role of the Pkd1 gene in renal development. Rather than manipulate wild-type organ cultures, we chose to use mouse models in which the Pkd1 gene has been genetically inactivated. Renal explants derived from Pkd1 mutant mice do not develop cysts ex vivo unless analogues of cAMP are added to the culture medium [14]. We show that renal rudiments harvested at day E11.5, Pkd1−/− kidneys do not show defects in UB branching and elongation. However, in Pkd1−/− E14.5 explants, we observed impaired glomerulogenesis, possibly due to defective endothelial cell migration.
2. Materials and methods
2.1. Mice crosses
All animal care and experimental protocols were conducted in accordance with the guidelines provided by the Italian Ministry of Health, upon approval of a specific protocol (IACUC-548) by the institutional care and use ethical committee (I.A.C.U.C.) at the San Raffaele Scientific Institute.
The Pkd1 null mouse model and the Pkd1ΔC mouse model were previously described [9], [10]. For the culture of kidneys at 11.5 days Pkd1 Bl6/57 were crossed whereas culture of kidneys at 14.5 days mice were 50% FVB–50% Bl6/57. The mice Pkd1ΔC/+ in 100% Bl6/57 were crossed to harvest the kidneys at day E14.5. The Hoxb7-GFP mice were kindly provided by Dr. Frank Costantini [15]. Noon the day when the vaginal plug was observed was considered to be embryonic day E0.5.
2.2. Embryonic kidney explant cultures
Kidney rudiments have been isolated from mouse embryos at day E11.5 or at day E14.5 in CO2 independant medium (Gibco Cat #18045-088) kept on ice and cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and Penicillin/streptomycin on polyester membrane filters of 0.4 µm pore size, (Transwell®, Costar). After 3 days, the tissues were fixed in −20 °C methanol 10 min at room temperature and processed for whole mount immunofluorescence. For PI3kinase inhibition or VEGF treatment, cultures were incubated with 10 μM LY294002 (Promega, Madison, WI) or 25 ng/ml VEGF (R&D).
For time-lapse video microscopy, explants were recorded using a Nikon Elipse TE2000 microscope equipped with an Evolution VF digital camera. The control unit allows mixing of humidified air with CO2 at defined ratios (typically 95% air/5% CO2). A water bath regulated by a software maintains the temperature at 37 °C.
2.3. Immunofluorescence
For Immunofluorescence, sections or rudiments were rehydrated, permeabilised in PBS/0.1% Triton X-100 blocked with 5% goat serum (Sigma)/3% BSA/PBS, incubated with primary antibodies 1 h at 37 °C or overnight at 4 °C. Kidneys are washed in PBS, incubated with 1/1000 Alexa Fluor-488 or Alexa Fluor-594 conjugated secondary antibody (Molecular Probe) for 1 h RT, washed in PBS, mounted in glycerol/anti-fade/PBS. Digital images were obtained with a Zeiss microscope. Primary antibodies used were anti pan-cytokeratin from Sigma, (St. Louis, MO; C2562) Laminin from Sigma (St. Louis, MO; C2562) L9393), E-cadherin (BD Transduction Laboratories 610181), WT1 from Santa Cruz Biotechnology (Santa Cruz, CA; sc192), NCAM (Chemicon international MAB310).
2.4. Statistical analysis of results
Student’s t test was used to analyze differences between groups.
3. Results
3.1. No early defects in UB branching of Pkd1−/− kidneys
We investigated whether early nephrogenesis was altered in a previously described Pkd1-null mouse model [9]. Ex-vivo organ cultures of metanephric explants from control and mutant embryos were initiated at day E11.5, cultured for 3 days and stained for cytokeratin, a marker of the UB. A quantification analysis did not show defects in UB branching and elongation in Pkd1−/− kidneys compared to the Pkd1+/+ and the Pkd1+/− kidneys (Fig. 1A). Likewise, the number of ureteric bud tips was not statistically different (Fig. 1F). Pkd1+/+, Pkd1+/−, and Pkd1−/− explants were stained for cytokeratin and laminin, which revealed comma-shaped and S-shaped bodies. Pkd1−/− explants did not display major defects in early nephrogenesis at this stage compared to control explants (Fig. 1B and C). To more carefully study potentially mild defects and to measure kinetic parameters of branching morphogenesis, we performed real-time analysis on the Pkd1-null mice intercrossed with Hoxb7-GFP transgenic mice, which expressed the green fluorescent protein (GFP) reporter specifically in the UB epithelium [15]. Kidneys harvested at day E11.5 were placed in culture, and the branching of the UB was followed for 3 days using time-lapse video microscopy [15], [16]. Based on conventional standards, the initial bud that evaginated from the Wolffian duct was considered a first-generation event, and the following characteristic “T-shape” was considered as time 0 [16]. Seven images of each culture of Pkd1+/+ and Pkd1−/− metanephric explants at 10-h intervals (from 0 to 60 h) were analyzed (Fig. 1D and E, respectively). These images showed no differences in kinetics in the branching morphogenesis between Pkd1+/+ and Pkd1−/− organ cultures isolated from littermates (movies 1 and 2, respectively; Pkd1+/+ n = 4, Pkd1+/− n = 8, and Pkd1−/− n = 13 in four independent litters).
Fig. 1.

No early UB branching defects in Pkd1−/− compared to Pkd1+/+ and Pkd1+/− explants. (A–E) Metanephric explants from embryos of 11.5 days were cultured for 3 days. (A) Staining for cytokeratin (green), marker of the UB epithelium in Pkd1+/+, Pkd1+/− and Pkd1−/−renal explants shows similar UB elongation and branching. (B) Laminin staining (red), which marks the basement membranes of epithelializing structures, of the kidney rudiments isolated at day E11.5 and cultured for 3 days, does not show defects of early nephrogenesis. (C) Merge of cytokeratin and laminin. (D and E) Time-lapse video microscopy for up to 60 h of Pkd1+/+(D) or Pkd1−/−. (E) Hoxb7-GFP transgenic kidneys isolated at E11.5 show normal kinetics of UB branching and elongation of Pkd1−/− explants. (F) Quantification of UB tips number of renal explants isolated at E11.5 and cultured for 3 days. Means ± SD are given; n.s.: no significant; n indicates the number of kidneys. Counts were performed on four independent litters. (Bar = 500 μm). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Defective nephrogenesis in Pkd1−/− explants
Next, we stained explants for WT1, which is expressed in the condensing mesenchyme and in precursors of podocytes. This staining revealed a reduced number of glomeruli in Pkd1−/− explants (Fig. 2C, F and H) compared to those in the Pkd1+/+ (Fig. 2A, D and G) and Pkd1+/− explants (Fig. 2B and E). There was a significant decrease in the number of glomeruli (a 52% reduction) in the Pkd1−/− explants compared to that of the Pkd1+/+ explants, respectively 91 +/- 17 and 189 +/- 31 (Fig. 2I). However, the number of condensed mesenchyme was increased in the Pkd1−/− kidneys (Fig. 2F) as compared to the controls (Fig. 2D and E), suggesting that the delay in nephron maturation is not caused by defects in the mesenchymal condensation. Similar results were obtained when using the Pkd1ΔC/ΔC mouse model [10] (not shown), excluding the possibility that the mouse model itself, and not the defective Pkd1 gene, results in the observed defects. Because Pkd1ΔC/ΔC mice survived for longer times in a pure genetic background [10], this second set of experiments was conducted in a pure Bl6/C57 background to exclude the possibility that the genetic background influenced the outcome.
Fig. 2.

Impaired nephrogenesis in Pkd1−/− cultured kidneys. (A–F) Kidneys withdrawn at E14.5 and cultured for 72 h were stained for WT1 (red), marker of condensed mesenchyme (arrow) and glomeruli (arrowhead). Two different magnifications of Pkd1+/+ (A and D), Pkd1+/− (B and E) and Pkd1−/− (C and F) revealed decreased glomeruli (arrowheads) and increased condensed mesenchyme (arrows) in Pkd1−/− as compared to Pkd1+/− and Pkd1+/+ explants. (G and H) Co-staining of cytokeratin (green) and WT1 (red) showing more abundant glomeruli in Pkd1+/+ kidneys (G) compared to the Pkd1−/− explants (H), whereas there is more condensed mesenchyme in the mutant compared to the wt. (I) The number of the glomeruli in Pkd1−/− kidneys is significantly impaired compared to the Pkd1+/+ and Pkd1+/− kidneys. Means ± SD are given; n.s.: no significant P > 0.05; ∗∗∗P < 0.001. n indicated corresponds to the number of kidneys. Experiments were carried out on three independent litters. (A–C) Bar = 500 μm, (F–H) Bar = 200 μm). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Defective endothelial cell migration in Pkd1−/− explants
Correct nephrogenesis requires precise crosstalk between the UB branching and the condensing mesenchyme. However, neither UB branching nor the structures derived from the condensing mesenchyme (comma- and S-shape bodies) were grossly affected in the Pkd1 mutant kidneys (Fig. 1, Supplementary Fig. 1 and Fig. 3A). A third essential component for glomerulogenesis is the endothelium, which migrates toward the nascent glomerulus to contribute to its maturation (Fig. 3A) [17]. We have previously reported that PC-1 mediates cell migration in epithelial cells and fibroblasts [18]. Furthermore, a recent study has shown that PC-1 plays an essential role in the endothelial compartment [19]. Therefore, we hypothesized that the defective endothelial cell migration in the developing kidney accounts for the defective glomerulogenesis. Staining with anti-CD31 identified an interconnection of endothelial cells with WT-1-positive cells in the renal rudiments isolated at E14.5 and cultured for 3 days (Fig. 3B and movie 3). In addition, an abundant number of CD31-positive cells were migrating on the filter that surrounded the renal cultures. Notably, CD31-positive migrating cells were very abundant in Pkd1+/+ explants (Fig. 3C and C′), whereas they were impaired in Pkd1−/− explants (Fig. 3D and D′; n = 9; seven independent litters). Quantification revealed that cells migrating out of the renal explants were strongly reduced in the Pkd1−/− explants compared to controls (Fig. 3E) and that CD31-positive cells were more affected than the other non-endothelial cells (CD31-negative, DAPI-positive, Fig. 3E). These results indicate a defect in the migration of renal embryonic endothelial cells that may cause defective glomerulogenesis (Fig. 3A).
Fig. 3.

Impaired endothelial migration out of the Pkd1−/− cultured kidneys. (A) Schematic representation of early nephrogenesis. The UB undergoes a series of branching events (1). Each UB tip will induce mesenchymal condensation, comma (2) and s-shaped bodies (3) formation. The S-shaped body secretes VEGF attracting endothelial cells (E.C.) which eventually form the glomerular tuft (4). The last event might be affected in Pkd1 mutant explants. (B) Kidneys isolated at E14.5 and cultured for 72 h were stained for CD31 (green), marker of endothelial cells, and WT-1 (red), marker of maturing podocytes. (C and D) Kidneys isolated at E14.5 and cultured for 72 h were stained for CD31, marker of endothelial cells. (C) CD31-positive cells migrated out of the renal explants in Pkd1+/+ kidneys (arrows) whereas the DAPI staining shows numerous cells migrating out of the cultures (arrowheads). (D) CD31-positive cells did not migrate out of the renal Pkd1−/− explants or formed only short protrusions (arrow). Non-endothelial cells were also impaired in their migration, but to a lower extent (arrowhead). (C′ and D′) Zoom of the CD31 positive protrusions of the Pkd1+/+ kidneys (C′) and Pkd1−/− (D′). (E) The quantity of endothelial cells out of the explants was monitored measuring the area positive for CD31. The area of the Pkd1−/− kidneys was significantly reduced compared to the area of the Pkd1+/+ kidneys. The migration rates of non-endothelial cells (DAPI-positive, CD31-negative) was established by measuring the distance (in μm) migrated from the renal rudiment. Means ± SD are given; ∗∗∗P < 0.001. n indicated corresponds to the number of kidneys. Bar = 100 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.4. PI3-kinase inhibitors phenocopy the defective endothelial cell migration and glomerulogenesis observed in Pkd1−/− explants
We previously reported that PC-1 induces cell migration of epithelial cells and fibroblasts in a phosphatidylinositol-3-kinase (PI3-kinase)-dependent manner [18]. Therefore, we hypothesized that the absence of the Pkd1 gene in endothelial cells might cause the defective PI3-kinase-dependent migration of the CD31-positive population of cells. Treatment of the renal organ cultures with 10 μM LY294002, a PI3-kinase inhibitor (Fig. 4B), completely abolished the movement of endothelial cells out of the kidney and caused defective nephrogenesis (Fig. 4E and F). These observations were similar to those made in Pkd1−/− organ cultures (Fig. 3D and E). Notably, treating Pkd1−/− renal organ cultures with VEGF promoted elongation and migration of the few CD31-positive cells out of the organ cultures (Fig. 4K). However, VEGF treatment did not completely rescue the migration phenotype, which suggested that the CD31-positive cells response to VEGF is blunted in Pkd1 mutant explants.
Fig. 4.

Treatment of renal explants with PI3-kinase inhibitors copies defective Pkd1−/− endothelial cell migration. (A–G) Pkd1+/+ renal explants isolated at E14.5 were cultured for 72 h in the presence (B, D and F) or absence (A, C and E) of the PI3kinase inhibitor LY294002 (LY) at 10 μM. Staining for CD31 shows reduced CD31-positive cells migrating out of the LY treated explants (B and D) as compared to the control (A and C). The CD31-positive area of the LY treated explants was significantly reduced compared to the area of the control kidneys (G). Staining for WT1 shows reduced glomeruli (arrowheads) and increased condensed mesenchyme (arrows) in LY treated explant (F) compared to the control (E). (C and D) Zoom of the CD31 positive protrusions of the Pkd1+/+ kidneys (C) and LY (D) treated renal explants. (G) The endothelial cells migrating out of the explants were quantified measuring the area positive for CD31 in LY-treated explants compared to the control. (H–K) Renal explants of Pkd1+/+ (H and I) and Pkd1−/− (J and K) kidneys cultured for 72 h with VEGF 25 ng/ml (I and K) or without VEGF (H and J) and stained for CD31. The phenotype of impaired migration in Pkd1−/− renal explants was not rescued by the VEGF treatment, although the protrusions are longer. Means ± SD are given; ∗∗∗P < 0.001.n corresponds to the number of kidneys. Bar = 100 μm.
4. Discussion
Previous studies have proposed that PC-1 may play a major role in early kidney development and UB branching morphogenesis. Microinjection into the developing UB of a lentiviral construct containing a putative dominant-negative form of PC-1, which encodes a myristoylated form of its intracellular C-terminus fused to GFP (pMyr-GFP-C-terminal), significantly decreased ureteric bud elongation and branching morphogenesis in metanephric explants cultured at day E11.5 [13].
In a second study, the treatment of kidney rudiments in the presence of a peptide, which was derived from a highly conserved sequence in the PKD repeats of PC-1 (WDGFDG), prevented proper branching morphogenesis in a model of ex vivo embryonic murine kidney explants [12]. The authors concluded that ureteric bud branching required correct PC-1 function. There was no direct evidence for a role for the Pkd1 gene in UB branching using genetically modified mice. In the current study, we have used two different mouse models that carry the inactivation of the Pkd1 gene. Our results show that PC-1 is not involved in the initial steps of kidney development, which include UB branching and early nephrogenesis. Furthermore, crossing Pkd1 mutants with a transgenic line harboring a Hoxb7-GFP transgene allowed us to perform time-lapse analysis of UB branching in Pkd1 mutant kidneys. Our results demonstrate that there are no differences in the kinetics or morphology of UB branching.
Our findings cannot exclude a role for PC-1 in UB branching morphogenesis in vivo, which are much harder to quantify. However, our results strongly suggest that previous work that also used renal organ cultures may have introduced artifactual effects due to the manipulations performed and/or interference with genes other than Pkd1 itself.
We have found that functional Pkd1 is essential in the later stages of renal development. The numbers of condensed mesenchymal vesicles and the comma- and S-shaped bodies were not decreased in two different Pkd1 mutant mouse models, suggesting that mesenchymal condensation occurred normally, as previously postulated [4]. However, the subsequent maturation of the S-shaped bodies into glomeruli is specifically impaired in Pkd1 mutant kidneys. Our data suggest that this effect may be caused by the defective migration of endothelial cells to form the mature glomerulus. Endothelial cell migration is necessary for proper nephrogenesis to occur. During glomerulogenesis, the podocyte precursors attract the endothelial cells into the proximal cleft by secreting the chemoattractant VEGF [17], [20]. In line with this, inactivation of VEGF-A in the glomeruli induces defects in endothelial cell migration, differentiation and survival, which ultimately inhibits the development of a filtration barrier [21]. Our current study shows that Pkd1-mutant endothelial cells (CD31-positive) derived from the metanephric organ cultures are impaired in their capability to migrate. Treatment with VEGF enhances elongation and migration of the few endothelial cells that successfully migrate out of the rudiment. However, VEGF does not significantly rescue the migration phenotype. The expression levels of VEGF-A in these mutant organ cultures did not appear to be altered as compared to wild-type organ cultures (not shown). Taken together, these results suggest that the defective production or secretion of VEGF by the proximal cleft is unlikely to be the primary cause of failure in migration. However, the defects in VEGF-A sensitivity by Pkd1 mutant endothelial cells may be a plausible explanation to account, at least in part, for the effect on cell migration.
The endothelial phenotype that we describe in the current study is in line with the dramatic vascular phenotype observed in all of the Pkd1 mutant models [7], [6], [22], [9]. In addition, PC-1 localizes to primary cilia and mediates the sensitivity of endothelial cells to fluid shear stress by regulating calcium signaling and nitric oxide production [23]. Furthermore, a recent study has provided definitive evidence for a primary role of the Pkd1 gene in the endothelial compartment by inactivating this gene using a Tie2-Cre line [19]. Our current work shows that defects in the endothelial compartment of the developing kidney might result in the defective glomerulogenesis in Pkd1 mutant mice.
Garcia-Gonzalez et al. did not report the development of defective glomerulogenesis; however, it should be noted that a careful analysis of the glomeruli in these mice was not performed, and it was not determined if Tie2 is expressed by the endothelium in the developing kidneys, whereas Tie1 has been shown to be very abundant. Future studies should focus on inactivating the Pkd1 gene using a line carrying a Cre-recombinase expressed by the angioblasts of the developing kidney.
Our previous work has shown that PC-1 regulates cell migration both in epithelial cells and in fibroblasts [18] through a mechanism that depends on PI3-kinase-mediated regulation of the actin cytoskeleton. Our current work shows that PC-1 is essential for cell migration in endothelial cells in a process that also likely depends on PI3-kinase. Notably, a recent study has shown that morpholinos against the two Pkd1 orthologues in Zebrafish (Pkd1a and b) results in the defective production and deposition of collagen through a mechanism that requires PI3-kinase [24]. In their study, Mangos et al. observed that, similar to our current work, inhibitors of PI3-kinase activity phenocopy the effect of a lack of functional Pkd1, which reinforces the idea that PI3-kinase might be an essential mediator of PC-1 activity.
In conclusion, our study has uncovered a potential function for PC-1 in the endothelial compartment in the developing kidney, which is likely important for proper glomerulogenesis.
Statement of competing financial interests
The authors declare to have no competing financial interests.
Acknowledgments
The authors are grateful to other members of the Boletta lab for helpful discussions and to Dr. F. Costantini for providing the Hoxb7-GFP mice and help with setting up the system. This work was supported by the PKD Foundation (PKD145b2r) and by Telethon-Italy (GGP12183) to AB.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bbrc.2014.01.068.
Appendix A. Supplementary data
Supplementary Figure 1.
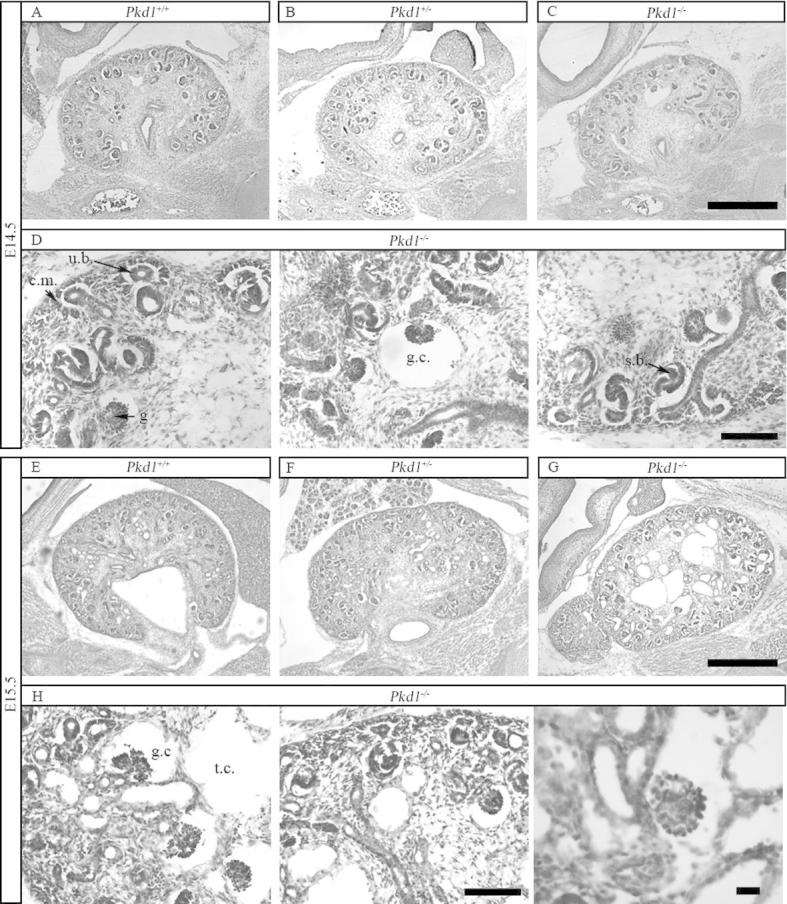
Hematoxylin and Eosin analysis of kidneys. (A–C) Hematoxylin-Eosin staining of Pkd1+/+, Pkd1+/− and Pkd1−/− kidneys at day E14.5 shows no major developmental defects of Pkd1−/− kidneys. Bar = 500 μm. (D) Condensed mesenchyme (cm) surrounding the ureteric bud (ub) as well as more advanced structures such as coma-shaped and s-shaped bodies (ssb) can be identified in Pkd1−/− kidneys. Some rare glomerular cysts are already present (gc). Bar = 500 μm. (E–G) Hematoxylin-Eosin staining of kidneys Pkd1+/+, Pkd1+/− and Pkd1−/− at day E15.5 shows formation of both tubular and glomerular cysts in Pkd1−/− kidneys. Bar = 500 μm. (H) Epithelializing structures and both glomerular (gc) and tubular cysts (tc) are visible in Pkd1−/− kidneys. Bar = 100 μm on the left, 20 μm on the right.
Pkd1+/+: Hoxb7-GFP renal explants were isolated at E11.5 and cultured for 3 days. Images were taken every 30 min and the movie contains 100 frames.
Pkd1−/−: Hoxb7-GFP renal explants were isolated at E11.5 and cultured for 3 days. Images were taken every 30 min and the movie contains 100 frames.
Pkd1+/+ renal explants isolated at E14.5 and cultured for 3 days were stained for WT1 (red) and CD31 (green). Images were capture using a Perkin Elmer UltraVIEW Spinning Disk Confocal Microscope. The movie is composed by 34 frames corresponding to the z stacks captured at 1 m for a total of over a 33 μm depth.
References
- 1.Gabow P.A. Autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 1993;22:511–512. doi: 10.1016/s0272-6386(12)80921-8. [DOI] [PubMed] [Google Scholar]
- 2.Torres V.E., Harris P.C., Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 3.Nauli S.M., Alenghat F.J., Luo Y., Williams E., Vassilev P., Li X., Elia A.E., Lu W., Brown E.M., Quinn S.J., Ingber D.E., Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 4.Lu W., Peissel B., Babakhanlou H., Pavlova A., Geng L., Fan X., Larson C., Brent G., Zhou J. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat. Genet. 1997;17:179–181. doi: 10.1038/ng1097-179. [DOI] [PubMed] [Google Scholar]
- 5.Lu W., Shen X., Pavlova A., Lakkis M., Ward C.J., Pritchard L., Harris P.C., Genest D.R., Perez-Atayde A.R., Zhou J. Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum. Mol. Genet. 2001;10:2385–2396. doi: 10.1093/hmg/10.21.2385. [DOI] [PubMed] [Google Scholar]
- 6.Kim K., Drummond I., Ibraghimov-Beskrovnaya O., Klinger K., Arnaout M.A. Polycystin 1 is required for the structural integrity of blood vessels. Proc. Natl. Acad. Sci. USA. 2000;97:1731–1736. doi: 10.1073/pnas.040550097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulter C., Mulroy S., Webb S., Fleming S., Brindle K., Sandford R. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc. Natl. Acad. Sci. USA. 2001;98:12174–12179. doi: 10.1073/pnas.211191098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muto S., Aiba A., Saito Y., Nakao K., Nakamura K., Tomita K., Kitamura T., Kurabayashi M., Nagai R., Higashihara E., Harris P.C., Katsuki M., Horie S. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum. Mol. Genet. 2002;11:1731–1742. doi: 10.1093/hmg/11.15.1731. [DOI] [PubMed] [Google Scholar]
- 9.Piontek K.B., Huso D.L., Grinberg A., Liu L., Bedja D., Zhao H., Gabrielson K., Qian F., Mei C., Westphal H., Germino G.G. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J. Am. Soc. Nephrol. 2004;15:3035–3043. doi: 10.1097/01.ASN.0000144204.01352.86. [DOI] [PubMed] [Google Scholar]
- 10.Wodarczyk C., Rowe I., Chiaravalli M., Pema M., Qian F., Boletta A. A novel mouse model reveals that polycystin-1 deficiency in ependyma and choroid plexus results in dysfunctional cilia and hydrocephalus. PLoS ONE. 2009 doi: 10.1371/journal.pone.0007137. (http://dx.plos.org/10.1371/journal.pone.0007137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costantini F., Kopan R. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev. Cell. 2010;18:698–712. doi: 10.1016/j.devcel.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Adelsberg J. Peptides from the PKD repeats of polycystin, the PKD1 gene product, modulate pattern formation in the developing kidney. Dev. Genet. 1999;24:299–308. doi: 10.1002/(SICI)1520-6408(1999)24:3/4<299::AID-DVG13>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 13.Polgar K., Burrow C.R., Hyink D.P., Fernandez H., Thornton K., Li X., Gusella G.L., Wilson P.D. Disruption of polycystin-1 function interferes with branching morphogenesis of the ureteric bud in developing mouse kidneys. Dev. Biol. 2005;286:16–30. doi: 10.1016/j.ydbio.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 14.Magenheimer B.S., St John P.L., Isom K.S., Abrahamson D.R., De Lisle R.C., Wallace D.P., Maser R.L., Grantham J.J., Calvet J.P. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+), K(+), 2Cl(−) Co-transporter-dependent cystic dilation. J. Am. Soc. Nephrol. 2006;17:3424–3437. doi: 10.1681/ASN.2006030295. [DOI] [PubMed] [Google Scholar]
- 15.Srinivas S., Goldberg M.R., Watanabe T., D’Agati V., Al-Awqati Q., Costantini F. Expression of green fluorescent protein in the ureteric bud of transgenic mice: a new tool for the analysis of ureteric bud morphogenesis. Dev. Genet. 1999;24:241–251. doi: 10.1002/(SICI)1520-6408(1999)24:3/4<241::AID-DVG7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe T., Costantini F. Real-time analysis of ureteric bud branching morphogenesis in vitro. Dev. Biol. 2004;271:98–108. doi: 10.1016/j.ydbio.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 17.Schedl A. Renal abnormalities and their developmental origin. Nat. Rev. Genet. 2007;8:791–802. doi: 10.1038/nrg2205. [DOI] [PubMed] [Google Scholar]
- 18.Boca M., D’Amato L., Distefano G., Polishchuk R.S., Germino G.G., Boletta A. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3beta-dependent cell cell mechanical adhesion. Mol. Biol. Cell. 2007;18:4050–4061. doi: 10.1091/mbc.E07-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Gonzalez M.A., Outeda P., Zhou Q., Zhou F., Menezes L.F., Qian F., Huso D.L., Germino G.G., Piontek K.B., Watnick T. Pkd1 and Pkd2 are required for normal placental development. PLoS ONE. 2010;5:e12821. doi: 10.1371/journal.pone.0012821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tufro A. VEGF spatially directs angiogenesis during metanephric development in vitro. Dev. Biol. 2000;227:558–566. doi: 10.1006/dbio.2000.9845. [DOI] [PubMed] [Google Scholar]
- 21.Eremina V., Wong M.A., Cui S., Schwartz L., Quaggin S.E. Glomerular-specific gene excision in vivo. J. Am. Soc. Nephrol. 2002;13:788–793. doi: 10.1681/ASN.V133788. [DOI] [PubMed] [Google Scholar]
- 22.Guay-Woodford L.M. Murine models of polycystic kidney disease: molecular and therapeutic insights. Am. J. Physiol. Renal Physiol. 2003;285:F1034–F1049. doi: 10.1152/ajprenal.00195.2003. [DOI] [PubMed] [Google Scholar]
- 23.Nauli S.M., Kawanabe Y., Kaminski J.J., Pearce W.J., Ingber D.E., Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mangos S., Lam P.Y., Zhao A., Liu Y., Mudumana S., Vasilyev A., Liu A., Drummond I.A. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis. Model Mech. 2010;3:354–365. doi: 10.1242/dmm.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pkd1+/+: Hoxb7-GFP renal explants were isolated at E11.5 and cultured for 3 days. Images were taken every 30 min and the movie contains 100 frames.
Pkd1−/−: Hoxb7-GFP renal explants were isolated at E11.5 and cultured for 3 days. Images were taken every 30 min and the movie contains 100 frames.
Pkd1+/+ renal explants isolated at E14.5 and cultured for 3 days were stained for WT1 (red) and CD31 (green). Images were capture using a Perkin Elmer UltraVIEW Spinning Disk Confocal Microscope. The movie is composed by 34 frames corresponding to the z stacks captured at 1 m for a total of over a 33 μm depth.