

Abstract
Peptidoglycan is the main component of the bacterial cell wall. It is a complex, three-dimensional mesh that surrounds the entire cell and is composed of strands of alternating glycan units crosslinked by short peptides. Its biosynthetic machinery has been, for the past five decades, a preferred target for the discovery of antibacterials. Synthesis of the peptidoglycan occurs sequentially within three cellular compartments (cytoplasm, membrane, and periplasm), and inhibitors of proteins that catalyze each stage have been identified, although not all are applicable for clinical use. A number of these antimicrobials, however, have been rendered inactive by resistance mechanisms. The employment of structural biology techniques has been instrumental in the understanding of such processes, as well as the development of strategies to overcome them. This review provides an overview of resistance mechanisms developed toward antibiotics that target bacterial cell wall precursors and its biosynthetic machinery. Strategies toward the development of novel inhibitors that could overcome resistance are also discussed.
Keywords: antibiotic resistance, bacterial cell wall, β-lactamases, Penicillin-Binding Proteins, fosfomycin, cycloserine, lipid II
Introduction
Peptidoglycan plays key roles in maintaining cell shape, providing an attachment site for surface-exposed virulence factors, and avoiding modifications in internal osmotic pressure.1 In rod-shaped cells, such as Escherichia coli, its biosynthesis can be described in two phases: elongation, when the lateral cell wall is formed, and division, leading to the generation of daughter cells.2 A common pool of precursors, synthesized in the cytoplasm mostly through the action of Mur enzymes, is required for both phases of peptidoglycan formation.3
The generation of the initial precursor, UDP-MurNAc, is catalyzed by the consecutive actions of MurA and MurB. MurA transfers enolpyruvate from phosphoenol pyruvate (PEP) to UDP-GlcNAc, thus forming UDP-GlcNAc-enolpyruvate (Fig. 1). As is the case for most enzymes involved in peptidoglycan biosynthesis, MurA is highly conserved among bacteria, is essential for cell survival, and has no human homolog. MurA is the target of fosfomycin, an antibiotic currently in clinical use for which bacterial resistance is well studied (see below). MurB subsequently reduces UDP-GlcNAc-enolpyruvate to UDP-MurNAc;3 although MurB has been extensively characterized, inhibitors for this enzyme are yet to be validated.4 Subsequently, the ATP-dependent Mur ligases MurC, MurD, MurE, and MurF, sequentially link five amino acid residues to UDP-MurNAc, forming the UDP-MurNAc-pentapeptide. These enzymes are structurally and functionally well known and have been the targets of great interest in the rational search for antibacterials, notably through the employment of high-throughput screens. The fact that they share similar active sites makes them attractive targets for multitargeted inhibitors, which could potentially reduce the likelihood of mutational resistance.5,6 However, and despite great efforts toward the identification of compounds with antibacterial activity, there are no antibiotics currently in use that target any of the Mur ligases.
Figure 1.
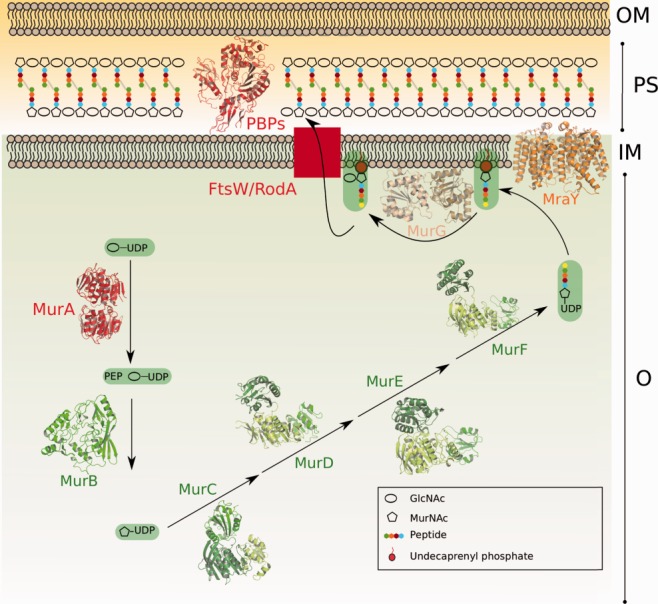
Schematic diagram of the cytoplasmic and membrane steps of the peptidoglycan biosynthetic pathway. The different domains of Mur enzymes are shown in shades of green. MurA and PBPs, which are the targets of antibiotics currently employed in hospital settings, are highlighted in red. OM, outer membrane; PS, periplasm; IM, inner membrane; C, cytoplasm.
MurF is the only Mur ligase which does not employ a single amino acid as substrate, but rather a dipeptide, d-Ala: d-Ala. Its synthesis requires racemization of l-Ala by the alanine racemase Alr, and subsequent condensation of two d-Ala molecules by Ddl, an alanine ligase.3 Both Alr and Ddl are targeted by the antibiotic d-cycloserine, used as a second-line therapeutic in the treatment of multidrug resistant tuberculosis.7
Membrane-linked steps include the transfer of the MurNAc-pentapeptide moiety to the undecaprenyl phosphate lipid carrier by MraY, an integral membrane protein whose crystal structure has recently been solved.8 This reaction generates Lipid I, to which MurG adds a GlcNAc moiety, resulting in Lipid II, the basic unit of the peptidoglycan polymer.9 Despite the fact that MraY is an essential enzyme and a known target of natural antibiotics, several of which present considerable antibacterial activity, no MraY inhibitors are presently used in the clinic.10
The translocation of Lipid II from cytoplasm to periplasm is mediated by flippases. This step requires the activity of proteins of the SEDS family (shape, elongation, division, sporulation), such as FtsW, RodA, and SpoVE,11 which have been shown to be essential in both Gram-negative and Gram-positive species.12,13 To date, no specific ligands have been developed to block their function. This is not the case for periplasmic proteins, such as Penicillin-Binding Proteins (PBPs), the macromolecular targets of β-lactam antibiotics. PBPs catalyze both the polymerization of Lipid II glycan chains and/or the crosslinking of stem peptides. It is the latter function, which recognizes the d-Ala:d-Ala moiety of the peptide, that is targeted by penicillin and its structural analogs.14 PBPs are accessible (since they are located outside of the cell membrane), metabolize molecules that do not exist in eukaryotes (amino acids with d-chirality), and have no mammalian homologs, and thus have been extensively studied through biochemical, structural, and microbiological techniques.15,16 Due to their extended employment in the clinic, different resistance mechanisms have arisen to counteract the targeting of PBPs by β-lactam antibiotics, underlining the importance for the search of novel molecules that do not carry the β-lactam ring.17
Despite the fact that the appearance of β-lactam resistant species has had dire consequences for the treatment of infections worldwide, the development of resistance has not been limited to this peptidoglycan biosynthesis step, and a number of other vastly employed antibiotics are now the targets of resistance mechanisms. This review aims at analyzing mechanisms developed by bacteria to inactivate or circumvent the action of drugs that target cell wall biosynthesis, with a main focus on the structural biology efforts that have been crucial toward the comprehension of such strategies. Novel molecules developed to counteract the action of resistant microbes are also highlighted.
Resistance to Antibiotics that Target Cytoplasmic Steps
Fosfomycin
Fosfomycin is a natural antibacterial produced by various Streptomyces and Pseudomonas species18,19 and is the only antibiotic currently in clinical use that targets a Mur enzyme; its broad-spectrum characteristics allow it to be employed against both Gram-positive and Gram-negative bacteria. This PEP mimetic (Fig. 2) irreversibly inhibits MurA by alkylating the highly conserved catalytic cysteine, in a step that is facilitated by the initial binding of UDP-GlcNAc to the “open” form of MurA.20 The resulting covalent adduct blocks catalysis, thus reducing the pool of peptidoglycan precursors. Crystal structures of multiple MurA–ligand complexes suggest that the mechanism of inhibition involves flexibility of a loop that lies in close proximity to the active site Cys residue, which can “trap” fosfomycin within the active site cleft.20–22
Figure 2.
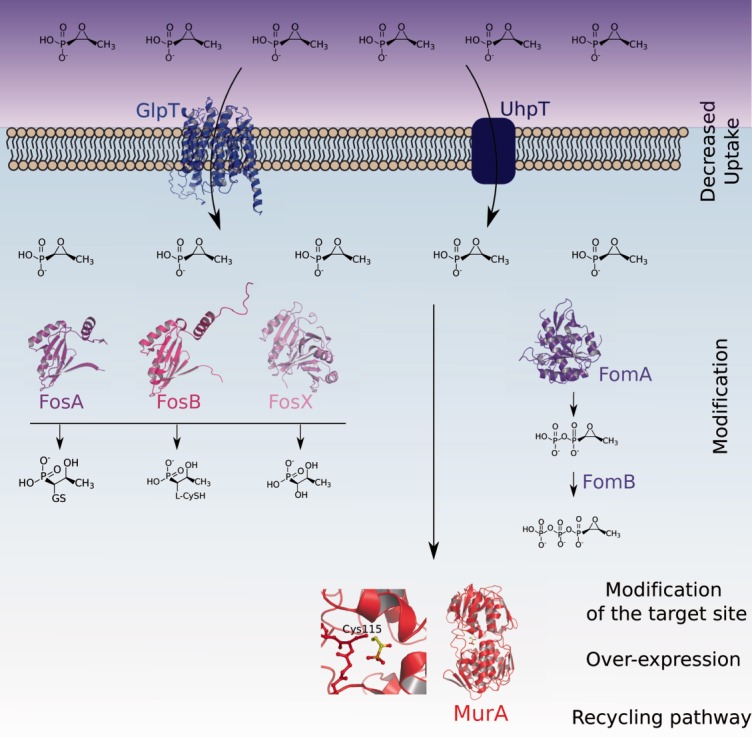
Mechanisms of fosfomycin resistance. Upon entry into the cell, fosfomycin can be phosphorylated by FomA/FomB, modified directly by FosA/FosB, or hydrolyzed by FosX. Other strategies include introduction of mutations within MurA, as well as its overexpression.
Interestingly, fosfomycin is a true textbook case involving a wide range of resistance mechanisms (Fig. 2), which include target modification, expression of antibiotic-degrading enzymes, reduced uptake, and rescue of the UDP-MurNAc biogenesis pathway. Resistance through modification of the catalytic site is naturally observed in fosfomycin-resistant species such as Mycobacterium tuberculosis, Chlamydia thrachomatix, and Borrelia burgdorferi,23–25 since their MurA homologs carry a Cys–Asp mutation that prevents alkylation by the antibiotic. Amino acid substitutions that are distant from the catalytic site have also been identified in E. coli clinical isolates and were shown to confer additional resistance to fosfomycin.26
FosA, FosB, and FosX, all inactivate fosfomycin through direct modification of its chemical structure. The thiol transferases FosA and FosB and the hydrolase FosX catalyze the opening of the epoxide ring of the antibiotic.27 FosA adds glutathione (GSH) directly to the oxirane ring of fosfomycin, generating an inactive form.28 Similarly, in Gram-positive species that do not produce GSH, such as Staphyloccocus aureus, FosB adds bacillithiol (BSH, a low-molecular mass thiol) or l-Cys to fosfomycin.29–31 The crystal structures of FosA and FosX, which display a large degree of structural similarity, have similar βαβββ motifs that harbor a metal-binding site within a cupped region. Notably, structures with bound product reveal that substrate binding is highly dependent on interaction with the Mn(II) center, and it is of interest that the metal itself has been proposed to play the role of acid catalyst in the fosfomycin hydrolysis reaction.27,28,31,32 In addition to FosA/FosB/FosX, a second group of enzymes was also shown to confer intrinsic resistance in fosfomycin-producing species. FomA, FomB, and FosC display sequence similarities to eukaryotic protein kinases and catalyze the phosphorylation of the antibiotic through an ATP and Mg++-dependent mechanism (Fig. 2).33–35 FomA is an open αβα sandwich, and in the structure with bound fosfomycin, the antibiotic is shown to interact with the active site through its phosphonate group. A flexible “lid” region was suggested to become structured upon antibiotic binding, thus promoting its optimal positioning for catalysis.34
Due to its hydrophobic nature, fosfomycin is imported through the inner bacterial membrane via two nutrient membrane transporters: the glycerol-3-phosphate transporter GlpT and the glucose-6-phosphate transporter UhpT (Fig. 2).36,37 Decreased expression or introduction of mutations in GlpT or UhpT can reduce fosfomycin uptake26 resulting in lower susceptibility.30,36 Most often, the defect in fosfomycin import is a consequence of complete deletion or a mutation resulting in a truncated form of the transporter,26 but it remains unclear whether single substitutions can lead to fosfomycin resistance as well. The structure of GlpT, which contains 12 transmembrane segments, was solved to 3.3 Å. It is closed at the periplasmic face and displays an open pore at the cytoplasmic one; presumably, GlpT could display a “rocker-switch” motion which would allow substrate to be translocated through the center of the channel.38 Despite the availability of this structural data, the precise mechanism of fosfomycin transfer through this transporter is still unclear.
Lastly, both transporters are known to be positively regulated by cyclic adenosine monophosphate (cAMP), and therefore lowering of intracellular cAMP concentrations through mutations in related genes can also induce resistance. This is the case for cyaA, which encodes adenyl cyclase, and ptsI, which is involved in the phosphoenolpyruvate:sugar phosphotransferase transport system.26,30,39,40
d-Cycloserine
d-cycloserine (seromycin), a cyclic structural analog of d-alanine, is a broad-spectrum antibiotic produced by some Streptomyces species.3,41 Despite the fact that adverse neurological side effects limit its use in regular chemotherapy regimens, it is routinely employed as a second-line drug for the treatment of multidrug resistant M. tuberculosis infections.42,43 d-cycloserine inhibits both Alr and Ddl.3,43
The major resistance mechanism involves the overexpression of AlrA.44,45 AlrA is a two-domain molecule consisting of an α/β barrel in its N-terminal region and a C-terminal β-strand rich domain. The cofactor pyridoxal-5′-phosphate is covalently associated to a lysine residue within the active site, located in the N-terminal domain. In the structure of the cycloserine-bound form, it becomes evident that the antibiotic breaks the bond between PLP and lysine and forms an alternative covalent bond with the cofactor, thus becoming directly linked to the active site;46 thus, overexpression of AlrA acts as a cycloserine “sink.” Additionally, CycA, an importer of the amino acids β-/l-/d-alanine, glycine, and d-serine, has also been linked to d-cycloserine uptake in E. coli and to the development of d-cycloserine resistance in mycobacterial BCG strains.41,47 However, the above-mentioned mechanisms are not sufficient to fully explain d-cycloserine resistance, and it is believed that additional strategies could be involved.47 In particular, mutations in a gene homologous to E. coli PBP4 were shown to confer resistance to d-cycloserine as well as to vancomycin in Mycobacterium smegmatis.48
The limited understanding of the resistance strategies toward d-cycloserine can be ascribed to the poor comprehension of the precise molecular mechanism of the drug itself. In particular, the prevalence of Alr or Ddl as the main target is still a matter of controversy, mostly due to the fact that both enzymes display complex regulatory mechanisms.45 Moreover, Baisa et al. recently reported that a mutation in dadA is linked to resistance in E. coli, suggesting that an additional mechanism of resistance could involve an antagonizing effect of d-cycloserine on d-amino acid dehydrogenase (DadA) activity.41
Developing agents against cytoplasmic targets: Mur enzymes as a case study
Mur enzymes are attractive antibacterial development targets due to the fact that they are highly conserved, most of them are essential, they are specific to bacteria, and are well characterized both structurally and enzymatically. However, despite the extensive effort that has been dedicated to the search for inhibitors of Mur enzymes (A–G) that display antibacterial activity and that could eventually be pursued for employment in the clinic, apart from fosfomycin no Mur inhibitors are employed either in hospital settings or are within the antibiotic development pipeline.
A number of natural and synthetic MurA inhibitors were discovered in the past few years through structure–activity relationship experimentation and high-throughput screening efforts.49 Interestingly, they present different modes of inhibition: covalent50,51 and non-covalent52 binding within the active site, or blocking of the transition from the open to the closed form, which is required for catalysis.53,54 Although efficient in vitro, most compounds appeared to have no or weak antibacterial activity, and/or were not specific to MurA. It is of note that a number of preliminary “hits” were only superficially characterized, but would deserve further investigation toward lead optimization, keeping in mind the high-domain flexibility of the enzyme in structure-based drug design approaches.49
The search for MurB inhibitors has yielded a number of hits;55–57 although none of the molecules could be shown to target MurB specifically in vivo, pyrazolidine analogs were reported to inhibit peptidoglycan biosynthesis.58 However, UDP-MurNAc is a strong feedback inhibitor of MurA, and MurB inhibition could prevent this feedback process;59 the consequent increase in the pool of UNAG-enolpyruvate produced by MurA could be competitive towards active site inhibitors of MurB.4 This prompted the suggestion that dual inhibitors of MurA and MurB could be more suitable; substituted thiazolyl ureas and pyrazolidinediones have been found to inhibit both enzymes and to display some antibacterial activity57,60 (but again there is no evidence that the inhibition of MurA/B is the only mechanism of action of these compounds). In addition, Kaur et al. recently identified three dual inhibitors of MurA and MurB from Acinetobacter baumannii using an in silico approach;61 their results remain to be validated by enzymatic assays.
Mur ligases (Mur enzymes C–F) have been the subject of a very significant effort toward the development of inhibitors, a process that has been aided by the availability of structural data for all enzymes from different species. MurD, for example, has been particularly well characterized by high-resolution crystal structures in complex with phosphinate-, rhodanine-, d-Glu-, and thiazolidine-based inhibitors, some of which display weak antibacterial activity.62–65
Mur ligases are three-domain molecules (Fig. 1); the small N-terminal domain recognizes the peptidoglycan, the central domain binds nucleotide, and the C-terminal domain binds to the incoming amino acid.5 This similarity is at the basis for the suggestion that a single compound could potentially inhibit all four ligases, thus preventing the development of drug resistance rapidly.4 In support of this idea, several compounds that inhibit more than one Mur ligase have been identified.63,66–68 To date, however, most of these compounds have shown little or no antibacterial activity. Notable exceptions are the MurF diarylquinolone inhibitors developed in the Bush lab, that generated an intracellular accumulation of UDP-MurNAc-tripeptide (and decrease of the pentapeptide) upon incubation with cells; however, the specific targeting of MurF within the cytoplasm was not shown.69
It is worthwhile mentioning that screens performed using industrial and commercial chemical libraries have had very limited success.4,42,70 This result has been partly attributed to the unsuitable nature of the chemical libraries employed71,72 but has also led to questions regarding the role played by the low permeability of the Gram-negative outer membrane in antibiotic intake, as well as the viability of Mur ligases as drug development targets.4 It is of note that the interdomain conformational flexibility of the ligases could be a drawback for inhibition assays in which the enzymes may not be in the same conformation as they adopt in vivo, as well as for docking approaches that may not be representative of the conformations that enzymes adopt in the cell. In addition, Mur enzymes could be members of a multiprotein complex whose arrangement limits the diffusion of inhibitors toward the active sites of the enzymes, thus providing a potential explanation as to why Mur ligases can be inhibited individually in vitro but not in vivo.15,73,74 The exact reason for weak bactericidal activity of Mur ligase inhibitors remains to be elucidated. Work on coupling Mur inhibitors to a transport carrier or efflux inhibitor is also worth considering.4,75,76
Resistance to Molecules that Target the Periplasm and Membrane-bound Steps
Glycopeptides
The glycopeptides vancomycin, teicoplanin, and telavancin are currently employed in hospital settings as last resort antibiotics for the treatment of multidrug resistant infections of Gram-positive cocci, the latter only having been approved in 2009.77,78 These molecules bind the d-Ala:d-Ala termini of peptidoglycan precursors, thus impeding proper transpeptidation and transglycosylation of peptidoglycan units.79,80
Resistance mechanisms associated with vancomycin, an antibiotic that has been employed for over 60 years, have been characterized in detail, and are mostly linked to the generation of alternate peptidoglycan precursors carrying d-Ala:d-Lac or d-Ala:d-Ser instead of d-Ala:d-Ala at their C-termini, for which vancomycin displays poor affinity.81 Seven van gene clusters (vanA, vanB, vanC, vanD, vanE, vanG, and vanL) are involved in resistance development. Genes within these clusters encode dehydrogenases that generate d-Lac from pyruvate (or serine racemases that generate d-Ser), ATP-dependent ligases that catalyze the formation of d-Ala:d-Lac(d-Ser), and d-d-peptidases that hydrolyze the d-Ala:d-Ala moiety which is necessary for the constitutive expression of the unmodified peptidoglycan.81,82
The structures of a number of Van enzymes have been solved in the presence of substrates and inhibitors. VanA from Enterococcus faecium reveals a fold that is similar to that of d-Ala:d-Ala ligase B (DdlB);83 notably, the recent structure of VanG, a d-Ala:d-Ser ligase, suggests that both VanA and VanG could have evolved from a common ancestral d-Ala:d-X ligase.84 Recently, vancomycin resistance has been tackled from a different point of view, that of the structural characterization of an enzyme that is essential for conjugative transfer of plasmids that confer drug resistance. The crystal structure of the nicking enzyme in S. aureus (NES) in complex with DNA indicates binding grooves that could be targeted for the development of novel inhibitors that could prevent plasmid propagation, the earliest stage of the resistance process.85
New glycopeptide mimics against Gram-positive pathogens
A number of glycopeptidic agents are presently in different steps of the antibiotic development pipeline. Oritavancin (The Medicines Company) is a lipoglycopeptide which showed promising results in Phase III clinical trials; it inhibits peptidoglycan biosynthesis both by interacting directly with the stem peptide and with its pentaglycine bridge. In addition, a central hydrophobic group allows for interaction and disruption of the cell membrane, as is the case for telavancin.86 This multiple mechanism of action confers activity against vancomycin-resistant organisms.87,88 Dalbavancin (developed by Durata), which is very efficient against multidrug resistant S. aureus (MRSA) as well as vancomycin-resistant strains,89 has completed Phase III clinical trials for skin infections. Both molecules still await FDA approval.90
β-Lactams
After its transfer to the periplasm, Lipid II is acted upon by PBPs, which catalyze the polymerization of the alternating MurNAc and GlcNAc chains (glycosyltransfer [GT]) and/or the crosslinking of the interchain stem peptides (transpeptidation [TP]). PBPs not only catalyze GT and TP reactions (activities addressed by class A and B high molecular mass enzymes), but low molecular mass PBPs are responsible for peptidic carboxypeptidation and endopeptidation, activities which regulate the level of crosslinking of stem peptides.15,16,91 PBPs thus play a key role in the formation of the cell wall, and inhibition of the transpeptidase/carboxypeptidase reactions through the action of β-lactam antibiotics has been the reason for β-lactams being amongst the most widely used antibiotics worldwide. β-Lactams display a broad-spectrum of antibacterial activity and share a common core, the highly reactive four-membered β-lactam ring (Fig. 3), and function by structurally mimicking the d-Ala:d-Ala moiety of the stem peptide to form an irreversible penicilloyl-β-lactam intermediate within the active site of PBPs.14 Blocking the transpeptidation reaction leads to weakening of the peptidoglycan and subsequent inhibition of cell growth or lysis. Since there seem to be at least one essential PBP in each bacterial species (in most cases, two), β-lactam use generally circumvents single mutation resistance mechanisms, requiring more complex strategies.4,92
Figure 3.
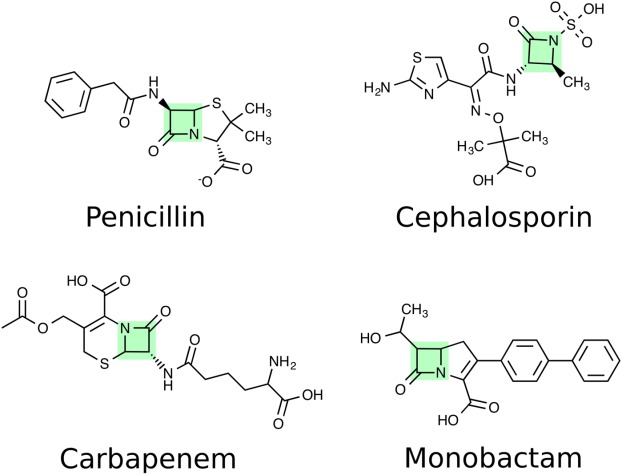
Chemical structures of the four clinically important β-lactam antibiotics. The common β-lactam ring is highlighted in green.
There are four clinically important groups of β-lactam antibiotics: penicillins, carbapenems, monobactams, and cephalosporins (Fig. 3); a broad range of derivatives has been developed for all four classes.93 The use of β-lactams has elicited the development of four major resistance mechanisms: (1) reduced membrane permeability or efflux increase through the action of multidrug efflux pumps; (2) expression of PBPs with reduced affinity to β-lactams or acquisition of “less sensitive variants”; (3) bypassing of the crosslinking step with l,d transpeptidases; (4) degradation of the antibiotic by β-lactamases. The mechanisms that involve enzymes that are related to peptidoglycan biosynthesis (thus 2, 3, and 4) will be described in more detail here.
Altering the target: modified PBPs
PBPs from pathogens such as Streptococcus pneumoniae have been well studied from drug-sensitive and-resistant strains through the employment of techniques ranging from genetics to structural biology. This study has brought to light two major β-lactam resistance mechanisms involving PBPs. For example, PBP2x and PBP2b, both class B enzymes, are major determinants of resistance and strains carrying tens of mutations throughout the pbp2x and pbp2b genes have been identified in a number of clinical strains. This effect is generated by homologous recombination events between closely related species in environments where antibiotic pressure is high.94–96
Class B PBPs are modular proteins that display a membrane-anchoring region, an N-terminal pedestal, and a C-terminal TP domain. The TP domain displays a β-sheet fold packed by helices on both sides; this fold is similar in all structures of PBPs solved to date and is a clear signature of the protein family [Fig. 4(a)]. In PBPs involved in β-lactam resistance, mutations can be present throughout the entire protein sequence; however, they are notably concentrated around the active site region. Such mutations have been shown to induce modifications in active site geometry, by modifying the β3/β4 region [red in Fig. 4(b,c)].97–99 Notably, a subtle modification in the position of β3 is also observed in the structure of PBP2a from S. aureus, an enzyme that is expressed in the case of an antibiotic challenge and that catalyzes stem peptide transpeptidation when the active site of the major class B PBP of the pathogen, PBP2, is inhibited by a β-lactam.100 Interestingly, a reduction in active site accessibility of PBP5fm is at the basis for β-lactam resistance in the naturally resistant pathogen E. faecium.101 It is of note that flexibility of the region in the vicinity of the active site has been recently suggested as playing a role the acylation reaction catalyzed by PBPs,102–104 and induced-fit conformational modifications have been associated to antibiotic recognition events.105
Figure 4.
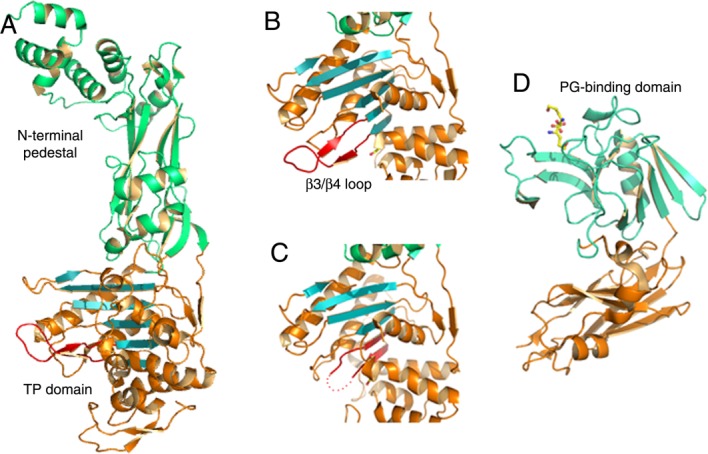
PBPs and l,d-transpeptidases recognize peptidoglycan and β-lactams through α/β folds. (A) PBP2b from S. pneumoniae folds into distinct domains, where the C-terminal, transpeptidase domain harbors the active site within an α/β fold, (B) Zoom of the β3/β4 region of PBP2b (indicated in red), which shows flexibility in a number of PBPs and peptidoglycan-recognizing enzymes. (C) Same region as in (B), but from PBP2b from a drug-resistant S. pneumoniae strain, indicating that the loop between β3/β4 could not be traced in the electron density map and is thus indicated with dots. (D) Structure of l,d transpeptidase from M. tuberculosis bound to a short region of a peptidoglycan substrate.
The development of non-β-lactam inhibitors of PBPs has been a strategy of choice, with the goal of circumventing (at least temporarily) the resistance process. A number of molecules have been developed, notably lactivicins,106,107 rhodanines,108 quinolones,109 and boronates.110,111 Of these, lactivicins and boronates have been shown to be able to not only inhibit specific PBPs but also eliminate drug-resistant bacteria.106,110 In addition, new fluorescence-based assays that can potentially be employed to test chemical libraries for PBP inhibitors are now available,112,113 paving the road to the exciting possibility of the identification of novel TP active site inhibitors.
In some organisms, low-β-lactam affinity PBPs can only proceed with transpeptidation if the substrate (the stem peptide) is branched. This is the case for methicillin resistance mediated by PBP2a in S. aureus, and penicillin resistance mediated by PBP2x in S. pneumoniae.114,115 The generation of crosslinks between adjacent stem peptides is catalyzed by nonribosomal peptidyl transferases that belong to the Fem protein family. Fem transferases transfer l-amino acids and Gly to peptidoglycan precursors directly from aminoacyl-tRNAs in a ribosome-independent manner, and have the ability to do so by employing either nucleotide precursors or lipid intermediates.116 Members of this family include FemABX from S. aureus, MurMN from S. pneumoniae, BppAiA2 from E. faecalis, and the well studied FemX from Weissella viridescens (FemXWv).
In S. aureus, transposon-mediated mutagenesis identified factors essential for methicillin resistance (genes femA and femB). The two genes were shown to be essential for the incorporation of glycine into pentaglycine cross-bridges, and introduction of mutations result in altered peptide bridges and an increase of susceptibility toward β-lactams.117 Shortened pentaglycine interpeptides result in a failure in crosslinking, and thus in loss of methicillin resistance mediated by PBP2a, prompting the interest in the development of Fem inhibitors. Interestingly, peptidyl-RNA conjugates were shown to inhibit FemXWv in vitro,118 and an inhibitor of S. aureus FemA potentiated the activity of imipenem against MRSA strains,119 indicating that it could be possible to develop inhibitors that could be employed in conjunction with β-lactams.
Bypassing a step in the pathway: l,d-transpeptidases (LDT)
Despite the fact that the transpeptidation reaction catalyzed by PBPs has been considered as being essential for peptidoglycan stability and bacterial survival, it can be bypassed by LDT, enzymes already identified in Enterococcus, Mycobacterium, and Clostridium spp. These enzymes employ a catalytic triad (His, Cys, Asp) in order to crosslink the third residues of neighboring stem peptides (3 → 3 bond), unlike PBPs, which catalyze a 4 → 3 bond. Notably, their differences also lie in the chirality of their substrates (l,d and d,d for l,d transpeptidases and PBPs, respectively).116,120,121 The structures of LDTs from different species reveal a two-domain molecule [Fig. 4(d)], with the catalytic domain displaying a mobile loop element in the vicinity of the substrate-binding site,122–124 which is reminiscent of PBPs, as mentioned above.
LDT production in E. faecium was shown to depend on activation of a d,d carboxypeptidase that generates tetrapeptides from the natural pentapeptides present in the peptidoglycan, thus providing the substrate for LDTs and asserting that they will catalyze all crosslinking reactions in these resistant species.125 Bypassing the PBP catalysis step by LDTs results in a very high level of resistance to ampicillin, and moderate levels toward ceftriaxone, in a mechanism that involves direct acylation of the active site Cys.126 Notably, in M. tuberculosis, LDT activity is the dominant strategy for peptidoglycan crosslinking during the chronic phases of infection, suggesting that a combination of β-lactams and LDT inhibitors could prove to be an excellent strategy for control of this pathogen.120
Destroying the antibiotic: β-lactamases
β-Lactamases hydrolyze the β-lactam ring of the antibiotic, thus inactivating it before it has the opportunity to block the PBP active site. They are the main antibiotic resistance mechanism in Gram-negative bacteria. To date, the Protein Data Bank includes more than 700 β-lactamase crystal structures, attesting to the importance of and interest in these enzymes. Upon expression, β-lactamases can have three main fates: secretion into the periplasm (in the case of Gram-negative organisms), association to the membrane, or secretion into the environment.127 The structural similarities observed between β-lactamases and PBPs have led to the hypothesis that the former enzymes arose as Streptomyces spp and other soil microorganisms secreted soluble forms of PBPs as a primary defense mechanism against exposure to increasing concentrations of β-lactams.128
β-Lactamases have been historically classified into four classes (A to D) based on sequence homologies.129 Classes A, C, and D display similar folds and harbor an active site serine required for the formation of an acyl–enzyme complex with the incoming β-lactam, followed by hydrolysis of the intermediate.130 Class B enzymes are metalloproteins that require one or two zinc ions to hydrolyze the β-lactam ring, and differ from the other classes in fold, sequence, and mechanistic details.131 An updated classification of these enzymes also takes substrate and inhibitor specificities into consideration,132 and ranks β-lactamases into group 1 (class C enzymes, including cephalosporinases); group 2 (classes A and D, including carbapenemases); and group 3, the metallo-β-lactamases.
Class A β-lactamases include penicillinases that are predominant in pathogens including staphylococci and enterococci. This class also includes the extended spectrum enzymes (ESBL). Classes A, C, and D β-lactamases share a common fold where the catalytic serine is embedded at the interface between two closely interacting domains, one of which is composed of a central β-strand surrounded by helices (and shows clear similarities to the TP domain of PBPs; green and orange, Fig. 5), and a second, mostly helical domain. TEM-1, and SHV-1 β-lactamases, clavulanate-resistant ESBLs, are frequently produced by clinical isolates, and have the ability to hydrolyze penicillins, monobactams, and a broad range of cephalosporins. The structures of several well-studied ESBLs, such as CTX-M, Toho-1, SHV-1, and KPC-2 have been solved in the presence of a number of ligands,133–136 providing key information for the potential development of inhibitors.
Figure 5.
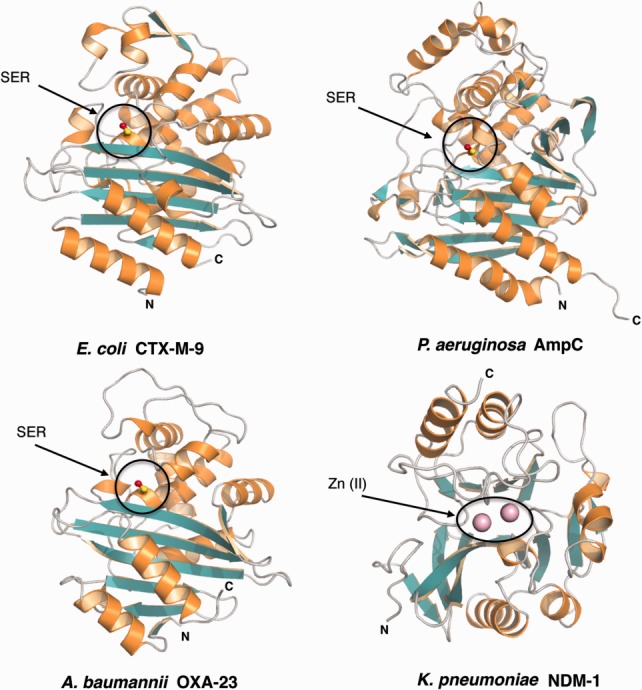
Structures of class A, C, D, and B β-lactamases. The conserved catalytic serine (SER) of classes A (CTX-M-9), C (AmpC), and D (OXA-23) β-lactamases is embedded at the interface between two closely interacting domains, shown in orange and green. NDM-1, a class B enzyme, differs from these serine-β-lactamases in fold and chemical mechanism. The enzyme shows a αβ/βα fold with an active site located at the edge of the β-sandwich. The active site is occupied by divalent zinc ions shown as pink spheres.
Class C (group 1) β-lactamases include enzymes that have the ability to hydrolyze a broad range of β-lactam antibiotics, including third generation cephalosporins. AmpC β-lactamases are representative of this class, and their crystal structures reveal a fold that is highly similar to that of class A enzymes, despite an active site pocket that is more open, resulting in their ability to better accommodate the hydrophobic moieties of cephalosporins.137 AmpC β-lactamases are resistant to all known classes of inhibitors, with the exception of boronic acid analogs. They can be chromosomally located or transmitted through mobile genetic elements; the chromosomally encoded β-lactamases are either constitutively expressed or require the presence of a specific regulatory system for induction.138 Strikingly, pathogens expressing inducible AmpC variants are of more acute clinical relevance due to the high-level expression of the enzyme upon exposure to β-lactams.139 Notably, this process requires a complex regulatory mechanism that involves enzymes of the cell wall recycling pathway that will be described below.140
Class D enzymes such as oxacillinases display not only the ability to hydrolyze carbapenems but also isoxazoyl β-lactams such as methicillin and oxacillin (hence the name, oxacillinases, or OXA β-lactamases).141 OXA-type β-lactamases are often encoded by genes located in integrons, but recently plasmid and transposons encoding OXA enzymes were reported in Gram-negative species.142 OXA β-lactamases show little sequence similarity to other classes of β-lactamases. A characteristic feature of OXA enzymes is the conserved carboxylated lysine in the active site, which most likely serves as a general base that activates the serine nucleophile.143,144 Furthermore, the high hydrophobic character and the larger size of the active site cleft of OXA-type enzymes frequently result in an extended spectrum of antimicrobial activity.143 Notably, class D β-lactamases of A. baumannii and Pseudomonas aeruginosa are at the source of well-documented failures in clinical treatment strategies.145
Metallo-β-lactamases (MBLs, class B, group 3) contain either one or two zinc ions within the active site in order to catalyze hydrolysis of the β-lactam ring. MBLs display the characteristic αβ/βα fold, with the active site located within a shallow groove at the interface between the two domains.146–149 These enzymes hydrolyze almost all known β-lactams with the exception of monobactams. Plasmid-encoded MBLs represent a major resistance mechanism of Gram-negative bacteria, including P. aeruginosa, A. baumannii, and enterobacteria.150 Recently, a novel class B enzyme, the New Delhi metallo-β-lactamase (NDM-1), was isolated from a Klebsiella pneumoniae strain that that was resistant to all β-lactams, even late-generation carbapenems including meropenem and imipenem.151 Since then, the number of cases of NDM-producing pathogens has increased drastically worldwide, and the term “superbug” was coined to indicate NDM-expressing bacteria.131,152 Strikingly, plasmids encoding NDM-1 often co-harbor genes encoding proteins involved in mechanisms of resistance to agents other than β-lactams, thus exacerbating the possibility of treatment failure.150
The crystal structure of NDM-1 reveals notable structural similarities to other MBLs153–155 (Fig. 5). The active site is located at the bottom of a shallow, hydrophobic pocket that is enlarged in respect to that of other MBLs, an observation which could potentially be at the basis of the broad substrate selectivity of this enzyme. The catalytic activity of NDM-1 also depends on the presence of two zinc ions that are located within different binding environments; Zn2+(I) is coordinated by three conserved histidine residues, while Zn2+(II) is ligated by an Asp–His–Cys triad. Catalytic activity requires mobility of two loops that are located in close vicinity to the active site; their flexibility has been linked to optimal substrate hydrolysis.156 Notably, the search for NDM inhibitors has led to the identification of natural compounds that bind to NDM-1 with high affinity, and could be eventually explored as leads.157
Mechanisms of inducible β-lactamases
Many bacteria induce β-lactamase expressing genes in the presence of high levels of antibiotics, and this phenomenon is now known to be tightly linked with the process of peptidoglycan precursor recycling. In Gram-negative organisms, two major mechanisms have been well characterized: the AmpG–AmpR–AmpC pathway, and the BlrA/BlrB two component regulatory system (Fig. 6).
Figure 6.
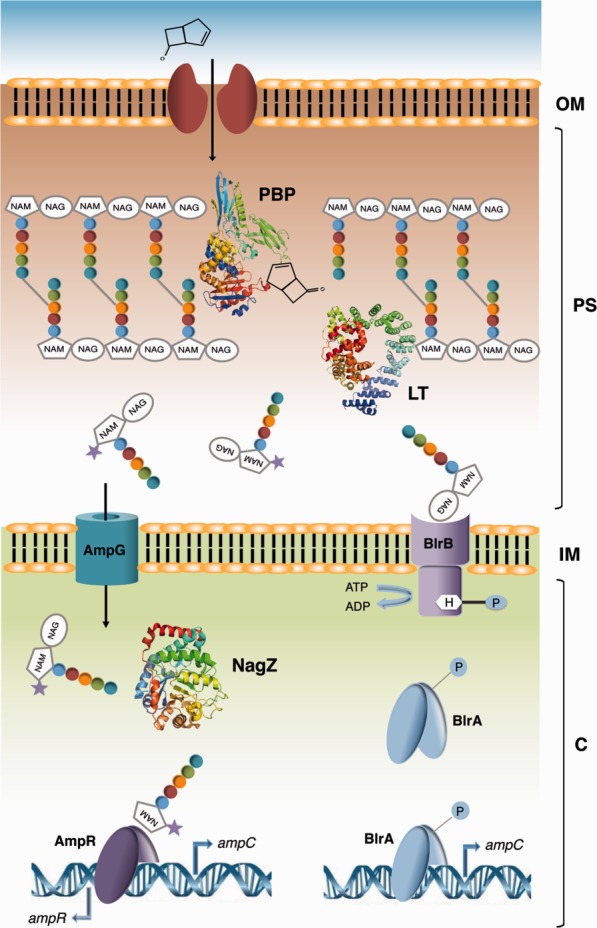
Schematic model of AmpC β-lactamase induction in Gram-negative organisms. The AmpG–AmpR–AmpC pathway as well as the BlrA/BlrB two component regulatory system are indicated. The presence of β-lactams results in excessive breakdown of the murein sacculus and thus in accumulation of muropeptides. This accumulation causes either the activation of AmpR (AmpG–AmpR–AmpC pathway shown on the left) or the phosphorylation of BlrA (BlrA/BlrB two component regulatory system shown on the right); in both situations, there is induction of the ampC gene. LT, lytic transglycosylase; PBP, Penicillin-Binding Protein.
Regulation of AmpC depends on the relative concentrations of cytoplasmic anhydromuropeptides. In the absence of β-lactam pressure, UDP-MurNAc-pentapeptide is bound to the transcriptional regulator AmpR, which inhibits expression of AmpC.138 Upon inhibition of PBPs by β-lactams, however, peptidoglycan biosynthesis is slowed down or blocked, while the activity of autolysins remains constant, resulting in accumulation of anhydromuropeptides in the periplasm. Muropeptides enter the cytoplasm through the AmpG permease, and their GlcNAc moiety is hydrolyzed by NagZ.158,159 The accumulation of anhydromuramyl peptides in the cytoplasm results in the displacement of UDP-MurNAc-pentapeptide from AmpR. AmpC is thus expressed and subsequently secreted to the periplasm, where it hydrolyzes the β-lactam ring of the antibiotic.138
A different regulatory mechanism was identified in bacterial species of the genus Aeromonas, which control the expression of AmpC using a two component regulatory system consisting of the sensor kinase BlrB and the response regulator BlrA (Fig. 6). Both proteins are closely related to the E. coli CreBC two-component regulatory system, which is involved in the regulation of key metabolic pathways in response to nutrient deprivation.160 The inhibition of the TP activity of PBPs by β-lactams results in the accumulation of disaccharide pentapeptides in the periplasm, inducing autophosphorylation of the kinase domain of BlrB. Transfer of the phosphate moiety to BlrA causes binding of the response regulator to the promotor region upstream of the genes that code for the Amp, Cep, and Imi β-lactamases, inducing expression.161
Despite the fact that both mechanism of induction display significant differences, both pathways seem to be induced by an intracellular increase of muropeptides and are thus linked to cell wall recycling. Consequently, enzymes involved in the β-lactamase induction pathway represent an attractive target for the development of effective inhibitors, which could facilitate therapeutic treatment in combination with classical β-lactams.
This is the case of the transmembrane protein AmpG. Inactivation of AmpG fully restored β-lactam susceptibility in resistant strains of P. aeruginosa, even minimizing the effect of an antibiotic efflux pump.162 Furthermore, employment of the AmpG inhibitor carbonyl cyanide m-chlorophenylhydrazone (CCCP) decreased the expression of AmpC in P. aeruginosa strains; combination of the inhibitor with β-lactams improved the MIC for specific pseudomonal strains.163 Interestingly, inhibition of the glucosaminidase NagZ also caused a 75% reduction in AmpC expression and an increase in β-lactam susceptibility in E. coli strains.158 The crystal structure of NagZ in complex with an inhibitor reveals that the molecule binds to the center of NagZ's TIM barrel. This high-resolution structure was essential for the development of novel glucose analogs with even higher selectivity for NagZ that also showed attenuation of AmpC expression in E. coli strains.164
Lastly, the transcriptional regulator AmpR could also be considered as a potential target for novel inhibitor development. Binding of the activator ligand to the active site of AmpR generates a conformational change leading to de-repression of the regulator. A potential approach for the development of inhibitors against AmpR would involve the identification of small molecules which bind with high affinity to its active site, causing the regulator to remain in its repressed state, resulting thus in steady inhibition of β-lactamase expression.140 The ubiquitous presence of proteins involved in regulation of peptidoglycan recycling in bacterial genomes suggests that the development of inhibitors of these enzymes could prove to be useful for combination therapy strategies.
Fighting β-lactamases
Although most β-lactam antibiotics are susceptible of being hydrolyzed by a subset of the 1300 β-lactamases that have been identified, new molecules that are able to at least partially circumvent this effect are being presently developed. BLA30072 (Basilea), a monocyclic β-lactam currently undergoing phase I clinical trials, is resistant to hydrolysis by MBLs, binds to distinct PBPs, and can kill P. aeruginosa and Acinetobacter that secrete these enzymes.165 In addition, combinations of different inhibitors are promising strategies. Ceftolozane is a cephalosporin that on its own is susceptible to extended spectrum β-lactamases, but when employed in addition to tazobactam is efficient against E. coli and P. aeruginosa.166 A number of other combinations are presently being explored, and it is likely that combination therapy will provide a solid approach for the development of anti-infectives, especially against Gram-negative pathogens.90
Conclusion
Bacteria have developed a number of resistance mechanisms to circumvent the targeting of its Achilles heel, the peptidoglycan biosynthetic machinery. However, the past few years have seen a substantial increase in the knowledge regarding not only the mechanism of action of cell wall-targeting antibiotics, but also in the development of new inhibitors that could target these resistance strategies. Structural biology will continue to play a critical role in the search for novel combination therapies that circumvent such resistance processes.
References
- 1.Höltje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.den Blaauwen T, de Pedro MA, Nguyen-Distèche M, Ayala JA. Morphogenesis of rod-shaped sacculi. FEMS Microbiol Rev. 2008;32:321–344. doi: 10.1111/j.1574-6976.2007.00090.x. [DOI] [PubMed] [Google Scholar]
- 3.Barreteau H, Kovac A, Boniface A, Sova M, Gobec S, Blanot D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:168–207. doi: 10.1111/j.1574-6976.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- 4.Silver LL. Viable screening targets related to the bacterial cell wall. Ann N Y Acad Sci. 2013;1277:29–53. doi: 10.1111/nyas.12006. [DOI] [PubMed] [Google Scholar]
- 5.Smith CA. Structure, function and dynmics in the mur family of bacterial cell wall ligases. J Mol Biol. 2006;362:640–655. doi: 10.1016/j.jmb.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 6.El Zoeiby A, Sanschagrin F, Levesque RC. Structure and function of the Mur enzymes: development of novel inhibitors. Mol Microbiol. 2003;47:1–12. doi: 10.1046/j.1365-2958.2003.03289.x. [DOI] [PubMed] [Google Scholar]
- 7.Caminero JA, Sotgiu G, Zumla A, Migliori GB. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect Dis. 2010;10:621–629. doi: 10.1016/S1473-3099(10)70139-0. [DOI] [PubMed] [Google Scholar]
- 8.Chung BC, Zhao J, Gillespie RA, Kwon DY, Guan Z, Hong J, Zhou P, Lee SY. Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science. 2013;34:1012–1016. doi: 10.1126/science.1236501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouhss A, Trunkfield AE, Bugg TDH, Mengin-Lecreulx D. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol Rev. 2008;32:208–233. doi: 10.1111/j.1574-6976.2007.00089.x. [DOI] [PubMed] [Google Scholar]
- 10.Winn MD, Goss RJM, Kimura K-I, Bugg TDH. Antimicrobial nucleoside antibiotics targeting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat Prod Rep. 2010;27:279–304. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- 11.Mohammadi T, van Dam V, Sijbrandi R, Vernet T, Zapun A, Bouhss A, Diepeveen-de Bruin M, Nguyen-Distèche M, de Kruijff B, Breukink E. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011;30:1425–1432. doi: 10.1038/emboj.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fay A, Meyer P, Dworkin J. Interactions between late-acting proteins required for peptidoglycan synthesis during sporulation. J Mol Biol. 2010;399:547–561. doi: 10.1016/j.jmb.2010.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang R, Ou HY, Zhang CT. DEG: a database of essential genes. Nucleic Acid Res. 2004;32:D271–D272. doi: 10.1093/nar/gkh024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tipper DJ, Strominger JL. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc Natl Acad Sci USA. 1965;54:1133–1141. doi: 10.1073/pnas.54.4.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lovering AL, Safadi SS, Strynadka NCJ. Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem. 2012;81:451–478. doi: 10.1146/annurev-biochem-061809-112742. [DOI] [PubMed] [Google Scholar]
- 16.Matteï P-J, Neves D, Dessen A. Bridging cell wall biosynthesis and bacterial morphogenesis. Curr Opin Struct Biol. 2010;20:749–766. doi: 10.1016/j.sbi.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 17.Zervosen A, Sauvage E, Frere J-M, Charlier P, Luxen A. Development of new drugs for an old target—the Penicillin Binding Proteins. Molecules. 2012;17:12478–12505. doi: 10.3390/molecules171112478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, Woodruff HB, Mata JM, Hernandez S, Mochales S. Phosphonomycin, a new antibiotic produced by strains of Streptomyces. Science. 1969;166:122–123. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- 19.Kim SY, Ju K-S, Metcalf WW, Evans BS, Kuzuyama T, van der Donk WA. Different biosynthetic pathways to fosfomycin in Pseudomonas syringae and Streptomyces species. Antimicrob Agents Chemother. 2012;56:4175–4183. doi: 10.1128/AAC.06478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bensen DC, Rodriguez S, Nix J, Cunningham ML, Tari LW. Structure of MurA (UDP-N-acetylglucosamine enolpyruvyl transferase) from Vibrio fischeri in complex with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Acta Crystallogr D Biol Crystallogr. 2012;F68:382–385. doi: 10.1107/S1744309112006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoon H-J, Lee SJ, Mikami B, Park H-J, Yoo J, Suh SW. Crystal structure of UDP-Nacetylglucosamine enolpyruvyl transferase from Haemophilus influenzae in complex with UDP-N-acetylglucosamine and fosfomycin. Proteins. 2008;71:1032–1037. doi: 10.1002/prot.21959. [DOI] [PubMed] [Google Scholar]
- 22.Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA, Duncan K. Structure of UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Structure. 1996;15:1465–1474. doi: 10.1016/s0969-2126(96)00153-0. [DOI] [PubMed] [Google Scholar]
- 23.De Smet KAL, Kempsell KE, Gallagher A, Duncan K, Young DB. Alteration of a single amino acid residue reverses fosfomycin resistance of recombinant MurA from Mycobacterium tuberculosis. Microbiology. 1999;145:3177–3184. doi: 10.1099/00221287-145-11-3177. [DOI] [PubMed] [Google Scholar]
- 24.McCoy A, Sandlin RC, Maurelli AT. In vitro and in vivo functional activity of chlamydia MurA, a UDP-N-Acetylglucosamine enolpyruvyl transferase involved in peptidoglycan synthesis and fosfomycin resistance. J Bacteriol. 2003;185:1218–1228. doi: 10.1128/JB.185.4.1218-1228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang S, Gilpin ME, Attia M, Ting Y-L, Berti P. Lyme disease enolpyruvyl-UDP-glcNAc synthase: fosfomycin-resistant MurA from Borrelia burgdorferi, a fosfomycin-sensitive mutant, and the catalytic role of the active site Asp. Biochemistry. 2011;50:2205–2212. doi: 10.1021/bi1017842. [DOI] [PubMed] [Google Scholar]
- 26.Takahata S, Ida T, Hiraishi T, Sakakibara S, Maebashi K, Terada S, Muratani T, Matsumoto T, Nakahama C, Tomono K. Molecular mechanisms of fosfomycin resistance in clinical isolates of Escherichia coli. Int J Antimicrob Agents. 2010;35:333–337. doi: 10.1016/j.ijantimicag.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 27.Fillgrove KL, Pakhomova S, Newcomer ME, Armstrong RN. Mechanistic diversity of fosfomycin resistance in pathogenic microorganisms. J Am Chem Soc. 2003;125:15730–15731. doi: 10.1021/ja039307z. [DOI] [PubMed] [Google Scholar]
- 28.Rife CL, Pharris RE, Newcomer ME, Armstrong RN. Crystal structure of a genomically encoded fosfomycin resistance protein (FosA) at 1.19 Å resolution by MAD phasing off the L-III edge of Tl+ J Am Chem Soc. 2002;124:11001–11003. doi: 10.1021/ja026879v. [DOI] [PubMed] [Google Scholar]
- 29.Roberts AA, Sharma SV, Strankman AW, Duran SR, Rawat M, Hamilton CJ. Mechanistic studies of FosB: a divalent-metal-dependent bacillithiol-S-transferase that mediates fosfomycin resistance in Staphylococcus aureus. Biochem J. 2013;451:69–79. doi: 10.1042/BJ20121541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karageorgopoulos DE, Wang R, Yu X-H, Falagas ME. Fosfomycin: evaluation of the published evidence on the emergence of antimicrobial resistance in Gram-negative pathogens. J Antimicrob Chemother. 2012;67:255–268. doi: 10.1093/jac/dkr466. [DOI] [PubMed] [Google Scholar]
- 31.Thompson MK, Keithly ME, Harp JM, Cook PD, Jagessar KL, Sulikowski GA, Armstrong RN. Structural and chemical aspects of resistance to the antibiotic, fosfomycin, conferred by FosB from Bacillus cereus. Biochemistry. 2013;52:7350–7362. doi: 10.1021/bi4009648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fillgrove KL, Pakhomova S, Schaab MR, Newcomer ME, Armstrong RN. Structure and mechanism of the genomically encoded fosfomycin resistance protein, FosX, from Listeria monocytogenes. Biochemistry. 2007;46:8110–8120. doi: 10.1021/bi700625p. [DOI] [PubMed] [Google Scholar]
- 33.García P, Arca P, Suárez JE. Product of fosC, a gene from Pseudomonas syringae, mediates fosfomycin resistance by using ATP as cosubstrate. Antimicrob Agents Chemother. 1995;39:1569–1573. doi: 10.1128/aac.39.7.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pakhomova S, Bartlett SG, Augustus A, Kuzuyama T, Newcomer ME. Crystal structure of fosfomycin resistance kinase FomA from Streptomyces wedmorensis. J Biol Chem. 2008;283:28518–28526. doi: 10.1074/jbc.M803709200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobayashi S, Kuzuyama T, Seto H. Characterization of the fomA and fomB gene products from Streptomyces wedmorensis, which confer fosfomycin resistance on Escherichia coli. Antimicrob Agents Chemother. 2000;44:647–650. doi: 10.1128/aac.44.3.647-650.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castaneda-Garcia A, Rodriguez-Rojas A, Guelfo JR, Blazquez J. The glycerol-3-phosphate permease GlpT Is the only fosfomycin transporter in Pseudomonas aeruginosa. J Bacteriol. 2009;191:6968–6974. doi: 10.1128/JB.00748-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santoro A, Cappello AR, Madeo M, Martello E, Iacopetta D, Dolce V. Interaction of fosfomycine with the glycerol-3-phosphate transporter of Escherichia coli. Biochim Biophys Acta. 2011;1810:1323–1329. doi: 10.1016/j.bbagen.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 38.Lemieux MJ, Huang Y, Wang DN. Crystal structure and mechanism of GlpT, the glycerol-3-phosphate transporter from E. coli. J Electron Microsc. 2005;54(Suppl 1):i43–i46. doi: 10.1093/jmicro/54.suppl_1.i43. [DOI] [PubMed] [Google Scholar]
- 39.Nilsson AI, Berg OG, Aspevall O, Kahlmeter G, Andersson DI. Biological costs and mechanisms of fosfomycin resistance in Escherichia coli. Antimicrob Agents Chemother. 2003;47:2850–2858. doi: 10.1128/AAC.47.9.2850-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakamoto Y, Furukawa S, Ogihara H, Yamasaki M. Fosmidomycin resistance in adenylate cyclase deficient (cya) mutants of Escherichia coli. Biosci Biotechnol Biochem. 2003;67:2030–2033. doi: 10.1271/bbb.67.2030. [DOI] [PubMed] [Google Scholar]
- 41.Baisa G, Stabo NJ, Welch RA. Characterization of Escherichia coli D-cycloserine transport and resistant mutants. J Bacteriol. 2013;195:1389–1399. doi: 10.1128/JB.01598-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kotnik M, Anderluh PS, Prezelj A. Development of novel inhibitors targeting intracellular steps of peptidoglycan biosynthesis. Curr Pharm Des. 2007;13:2283–2309. doi: 10.2174/138161207781368828. [DOI] [PubMed] [Google Scholar]
- 43.Prosser GA, Carvalho LPS. Kinetic mechanism and inhibition of Mycobacterium tuberculosis D-alanine:D-alanine ligase by the antibiotic D-cycloserine. FEBS J. 2013;280:1150–1166. doi: 10.1111/febs.12108. [DOI] [PubMed] [Google Scholar]
- 44.Cáceres NE, Harris NB, Wellehan JF, Feng Z, Kapur V, Barletta RG. Overexpression of the D-alanine racemase gene confers resistance do D-cycloserine in Mycobacterium smegmatis. J Bacteriol. 1997;179:5046–5055. doi: 10.1128/jb.179.16.5046-5055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng Z, Barletta RG. Roles of Mycobacterium smegmatis D-alanine:D-alanine ligase and D-alanine racemase in the mechanisms of action of and resistance to the peptidoglycan inhibitor D-cycloserine. Antimicrob Agents Chemother. 2003;47:283–291. doi: 10.1128/AAC.47.1.283-291.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noda M, Matoba Y, Kumagai T, Sugiyama M. Structural evidence that alanine racemase from a D-cycloserine-producing microorganism exhibits resistance to its own product. J Biol Chem. 2004;279:46153–46161. doi: 10.1074/jbc.M404605200. [DOI] [PubMed] [Google Scholar]
- 47.Chen JM, Uplekar S, Gordon SV, Cole ST. A point mutation in cycA partially contributes to the D-cycloserine resistance trait of Mycobacterium bovis BCG vaccine strains. PLoS One. 2012;7:e43467. doi: 10.1371/journal.pone.0043467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peteroy M, Severin A, Zhao F, Rosner D, Lopatin U, Scherman H, Belanger A, Harvey B, Hatfull GF, Brennan PJ, Connell ND. Characterization of a Mycobacterium smegmatis mutant that is simultaneously resistant to D-cycloserine and vancomycin. Antimicrob Agents Chemother. 2000;44:1701–1704. doi: 10.1128/aac.44.6.1701-1704.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gautam A, Rishi P, Tewari R. UDP-N-acetylglucosamine enoly pyruvyl transferase as a potential target for antibacterial chemotherapy: recent developments. Appl Microbiol Biotechnol. 2011;92:211–225. doi: 10.1007/s00253-011-3512-z. [DOI] [PubMed] [Google Scholar]
- 50.Han H, Yang Y, Olesen SH, Becker A, Betzi S, Schönbrunn E. The fungal product terreic acid is a covalent inhibitor of the bacterial cell wall biosynthetic enzyme UDP-N-acetylglucosamine 1-carboxyvinyltransferase (MurA) Biochemistry. 2010;49:4276–4282. doi: 10.1021/bi100365b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin BS, Han SG, Lee WK, Ryoo SW, Lee SJ, Suh SW, Yu YG. Inhibitory mechanism of novel inhibitors of UDP-N-acetylglucosamine enolpyruvyl transferase from Haemophilus influenzae. J Microbiol Biotechnol. 2009;19:1582–1589. [PubMed] [Google Scholar]
- 52.Baum EZ, Montenegro DA, Licata L, Turchi I, Webb GC, Foleno BD, Bush K. Identification and characterization of new inhibitors of the Escherichia coli MurA enzyme. Antimicrob Agents Chemother. 2001;45:3182–3188. doi: 10.1128/AAC.45.11.3182-3188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eschenbrug S, Priestman MA, Abdul-Latif FA, Delachaume C, Fassy F, Schönbrunn E. A novel inhibitor that suspends the induced fit mechanism of UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) J Biol Chem. 2005;280:14070–14075. doi: 10.1074/jbc.M414412200. [DOI] [PubMed] [Google Scholar]
- 54.Schonbrunn E, Eschenbrug S, Luger K, Kabsch W, Amrhein N. Structural basis for the interaction of the fluorescence probe 8-anilino-1-naphthalene sulfonate (ANS) with the antibiotic target MurA. Proc Natl Acad Sci USA. 2000;97:6345–6349. doi: 10.1073/pnas.120120397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bronson JJ, DenBleyker KL, Falk PJ, Mate RA, Ho H-T, Pucci MJ, Snyder LB. Discovery of the first antibacterial small molecule inhibitors of MurB. Bioorg Med Chem Lett. 2003;13:873–875. doi: 10.1016/s0960-894x(02)01076-4. [DOI] [PubMed] [Google Scholar]
- 56.Andres CJ, Bronson JJ, D'Andrea SV, Deshpande MS, Falke PJ, Grant-Young KA, Harte WE, Ho HT, Misco PF, Robertson JG, Stock D, Sun Y, Walsh AW. 4-Thiazolidinones: novel inhibitors of the bacterial enzyme MurB. Bioorg Med Chem Lett. 2000;10:715–717. doi: 10.1016/s0960-894x(00)00073-1. [DOI] [PubMed] [Google Scholar]
- 57.Kutterer KM, Davis JM, Singh G, Yang Y, Hu W, Severin A, Rasmussen BA, Krishnamurthy G, Failli A, Katz AH. 4-Alkyl and 4,4'-dialkyl 1,2-bis(4-chlorophenyl)pyrazolidine-3,5-dione derivatives as new inhibitors of bacterial cell wall biosynthesi. Bioorg Med Chem Lett. 2005;15:2527–2531. doi: 10.1016/j.bmcl.2005.03.058. [DOI] [PubMed] [Google Scholar]
- 58.Yang Y, Severin A, Chopra R, Krishnamurthy G, Singh G, Hu W, Keeney D, Svenson K, Petersen PJ, Labthavikul P, Shlaes DM, Rasmussen BA, Failli AA, Shumsky JS, Kutterer KM, Gilbert A, Mansour TS. 3,5-dioxopyrazolidines, novel inhibitors of UDP-N-acetylenolpyruvylglucosamine reductase (MurB) with activity against gram-positive bacteria. Antimicrob Agents Chemother. 2006;50:556–564. doi: 10.1128/AAC.50.2.556-564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mizyed S, Oddone A, Bryczynski B, Hughes DW, Berti PJ. UDP-N-acetylmuramic acid (UDP-MurNAc) is a potent inhibitor of MurA (enolpyruvyl-UDP-GlcNAc synthase) Biochemistry. 2005;44:4011–4017. doi: 10.1021/bi047704w. [DOI] [PubMed] [Google Scholar]
- 60.Francisco GD, Li Z, Albright JD, Eudy NH, Katz AH, Petersen PJ, Labthavikul P, Singh G, Yang Y, Rasmussen BA, Lin YI, Mansour TS. Phenyl thiazolyl urea and carbamate derivatives as new inhibitors of bacterial cell-wall biosynthesis. Bioorg Med Chem Lett. 2004;14:235–238. doi: 10.1016/j.bmcl.2003.09.082. [DOI] [PubMed] [Google Scholar]
- 61.Kaur N, Khokhar M, Jain V, Bharatam PV, Sandhir R, Tewari R. Identification of druggable targets for Acinetobacter baumannii via subtractive genomics and plausible inhibitors for MurA and MurB. Appl Biochem Biotechnol. 2013;171:417–436. doi: 10.1007/s12010-013-0372-2. [DOI] [PubMed] [Google Scholar]
- 62.Zidar N, Tomasic T, Sink R, Rupnik V, Kovac A, Turk S, Patin D, Blanot D, Contreras Martel C, Dessen A, Müller Premru M, Zega A, Gobec S, Peterlin Masic L, Kikelj D. Discovery of novel 5-benzylidenerhodanine and 5-benzylidenethiazolidine-2,3-dione inhibitors of MurD ligase. J Med Chem. 2010;53:6584–6594. doi: 10.1021/jm100285g. [DOI] [PubMed] [Google Scholar]
- 63.Tomašić T, Kovač A, Klebe G, Blanot D, Gobec S, Kikelj D, Mašič LP. Virtual screening for potential inhibitors of bacterial MurC and MurD ligases. J Mol Model. 2012;18:1063–1072. doi: 10.1007/s00894-011-1139-8. [DOI] [PubMed] [Google Scholar]
- 64.Tomašić T, Zidar N, Rupnik V, Kovac A, Blanot D, Gobec S, Kikelj D, Masic LP. Synthesis and biological evaluation of new glutamic acid-based inhibitors of MurD ligase. Bioorg Med Chem Lett. 2009;19:153–157. doi: 10.1016/j.bmcl.2008.10.129. [DOI] [PubMed] [Google Scholar]
- 65.Perdih A, Wolber G, Solmajer T. Molecular dynamics simulation and linear interaction energy study of D-Glu-based inhibitors of the MurD ligase. J Comput Aided Mol Des. 2013;27:723–738. doi: 10.1007/s10822-013-9673-3. [DOI] [PubMed] [Google Scholar]
- 66.Sova M, Kovac A, Turk S, Hrast M, Blanot D, Gobec S. Phosphorylated hydroxyethylamines as novel inhibitors of the bacterial cell wall biosynthesis enzymes MurC to MurF. Bioorg Chem. 2009;37:217–222. doi: 10.1016/j.bioorg.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 67.Mansour TS, Caufield CE, Rasmussen B, Chopra R, Krishnamurthy G, Morris KM, Svenson K, Bard J, Smeltzer C, Naughton S, Antane S, Yang Y, Severin A, Quagliato D, Petersen PJ, Singh G. Naphthyl tetronic acids as multi-target inhibitors of bacterial peptidoglycan biosynthesis. ChemMedChem. 2007;2:1414–1417. doi: 10.1002/cmdc.200700094. [DOI] [PubMed] [Google Scholar]
- 68.Perdih A, Kovac A, Wolber G, Blanot D, Gobec S, Solmajer T. Discovery of novel benzene 1,3-dicarboxylic acid inhibitors of bacterial MurD and MurE ligases by structure-based virtual screening approach. Bioorg Med Chem Lett. 2009;19:2668–2673. doi: 10.1016/j.bmcl.2009.03.141. [DOI] [PubMed] [Google Scholar]
- 69.Baum EZ, Crespo-Carbone SM, Foleno BD, Simon LD, Guillemont J, Macielag M, Bush K. MurF inhibitors with antibacterial activity: effect on muropeptide levels. Antimicrob Agents Chemother. 2009;53:3240–3247. doi: 10.1128/AAC.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zawadzke LE, Norcia M, Desbonnet CR, Wang H, Freeman-Cook K, Dougherty TJ. Identification of an inhibitor of the MurC enzyme, which catalyzes an essential step in the peptidoglycan precursor synthesis pathway. Assay Drug Dev Technol. 2008;6:95–103. doi: 10.1089/adt.2007.114. [DOI] [PubMed] [Google Scholar]
- 71.Chopra I. Discovery of antibacterial drugs in the 21st century. J Antimicrob Chemother. 2012;68:496–505. doi: 10.1093/jac/dks436. [DOI] [PubMed] [Google Scholar]
- 72.Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White CL, Kitich A, Gober JW. Positioning cell wall synthetic complexes by the bacterial morphogenetic proteins MreB and MreD. Mol Microbiol. 2010;76:616–633. doi: 10.1111/j.1365-2958.2010.07108.x. [DOI] [PubMed] [Google Scholar]
- 74.Favini-Stabile S, Contreras-Martel C, Thielens N, Dessen A. MreB and MurG as scaffolds for the cytoplasmic steps of peptidoglycan biosynthesis. Environ Microbiol. 2013;15:3218–3228. doi: 10.1111/1462-2920.12171. [DOI] [PubMed] [Google Scholar]
- 75.Lamers RP, Cavaliari JF, Burrow LL. The efflux inhibitor phenylalanine-arginine beta-naphthylamide (PAβN) permeabilizes the outer membrane of gram-negative bacteria. PLoS One. 2013;8:e60666. doi: 10.1371/journal.pone.0060666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Silver LL. Are natural products still the best source for antibacterial discovery? The bacterial entry factor. Expert Opin Drug Discov. 2008;3:487–500. doi: 10.1517/17460441.3.5.487. [DOI] [PubMed] [Google Scholar]
- 77.Stryjewski ME, Barriere SL, Rubinstein E, Genter FC, Lentnek AL, Magana-Aquino M, Luna CM, Niederman MS, Torres A, Corey GR. Telavancin versus vancomycin for bacteraemic hospital-acquired pneumonia. Int J Antimicrob Agents. 2013;42:367–369. doi: 10.1016/j.ijantimicag.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 78.Jafari-Saraf L, Wilson SE. Telavancin, a new lipoglycopeptide antimicrobial, in complicated skin and soft tissue infections. Infect Drug Resist. 2011;4:87–95. doi: 10.2147/IDR.S5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Vancomycin analogues active against vanA-resistant strains inhibit bacterial transglycosylase without binding substrate. Proc Natl Acad Sci USA. 2003;100:5658–5663. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pace JL, Yang G. Glycopeptides: update on an old successful antibiotic class. Biochem Pharmacol. 2006;71:968–980. doi: 10.1016/j.bcp.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 81.Depardieu F, Podglajen I, Leclercq R, Collatz E, Courvalin P. Modes and modulations of antibiotic resistance gene expression. Clin Microbiol Rev. 2007;20:79–114. doi: 10.1128/CMR.00015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 83.Roper DI, Huyton T, Vagin A, Dodson G. The molecular basis of vancomycin resistance in clinically relevant enterococci: crystal structure of D-alanyl-D-lactate ligase (VanA) Proc Natl Acad Sci USA. 2000;97:8921–8925. doi: 10.1073/pnas.150116497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meziane-Cherif D, Saul FA, Haouz A, Courvalin P. Structural and functional characterization of VanG d-Ala:d-Ser ligase associated with vancomycin resistance in Enterococcus faecalis. J Biol Chem. 2012;287:37583–37592. doi: 10.1074/jbc.M112.405522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Edwards JS, Betts L, Frazier ML, Pollet RM, Kwong SM, Walton WG, Ballentine WK, III, Huang JJ, Habibi S, Del Campo M, Meier JL, Dervan PB, Firth N, Redinbo MR. Molecular basis of antibiotic multiresistance transfer in Staphylococcus aureus. Proc Natl Acad Sci USA. 2013;110:2804–2809. doi: 10.1073/pnas.1219701110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhanel GG, Schweizer F, Karlowsky JA. Oritavancin: mechanism of action. Clin Infect Dis. 2012;54:S214–S219. doi: 10.1093/cid/cir920. [DOI] [PubMed] [Google Scholar]
- 87.Zhanel GG, Calic D, Schweizer F, Zelenitsky S, Adam H, Lagacé-Wiens PR, Rubinstein E, Gin AS, Hoban DJ, Karlowsky JA. New lipoglycopeptides: a comparative review of dalbavancin, oritavancin and telavancin. Drugs. 2010;70:859–886. doi: 10.2165/11534440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 88.Allen NE, Nicas TL. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol Rev. 2003;26:511–532. doi: 10.1111/j.1574-6976.2003.tb00628.x. [DOI] [PubMed] [Google Scholar]
- 89.Steiert M, Schmitz FJ. Dalbavancin (Biosearch Italia/Versicor) Curr Opin Investig Drugs. 2002;3:229–233. [PubMed] [Google Scholar]
- 90.Pucci MJ, Bush K. Investigational antimicrobial agent of 2013. Clin Microbiol Rev. 2013;26:792–821. doi: 10.1128/CMR.00033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 92.Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev. 2008;32:361–385. doi: 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 93.Moellering RC, Jr, Eliopoulos GM, Sentochnik DE. The carbapenems: new broad spectrum beta-lactam antibiotics. J Antimicrob Chemother. 1989;24 Suppl A:1–7. doi: 10.1093/jac/24.suppl_a.1. [DOI] [PubMed] [Google Scholar]
- 94.Hackenbeck R, Brückner R, Denapaite D, Maurer P. Molecular mechanisms of β-lactam resistance in Streptococcus pneumoniae. Future Microbiol. 2012;7:395–410. doi: 10.2217/fmb.12.2. [DOI] [PubMed] [Google Scholar]
- 95.Maurer P, Todorova K, Sauerbier J, Hakenbeck R. Mutations in Streptococcus pneumoniae penicillin-binding protein 2x: importance of the C-terminal penicillin-binding protein and serine/threonine kinase-associated domains for beta-lactam binding. Microb Drug Resist. 2012;18:314–321. doi: 10.1089/mdr.2012.0022. [DOI] [PubMed] [Google Scholar]
- 96.Sauerbier J, Maurer P, Rieger M, Hakenbeck R. Streptococcus pneumoniae R6 interspecies transformation: genetic analysis of penicillin resistance determinants and genome-wide recombination events. Mol Microbiol. 2012;86:692–706. doi: 10.1111/mmi.12009. [DOI] [PubMed] [Google Scholar]
- 97.Contreras-Martel C, Dahout-Gonzalez C, Dos Santos Martins A, Kotnik M, Dessen A. PBP active site flexibility as the key mechanism for beta-lactam resistance in pneumococci. J Mol Biol. 2009;387:899–909. doi: 10.1016/j.jmb.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 98.Contreras-Martel C, Job V, Di Guilmi AM, Vernet T, Dideberg O, Dessen A. Crystal structure of Penicillin-Binding Protein 1a (PBP1a) reveals a mutational hotspot implicated in β-lactam resistance in Streptococcus pneumoniae. J Mol Biol. 2006;355:684–696. doi: 10.1016/j.jmb.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 99.Job V, Carapito R, Vernet T, Dessen A, Zapun A. Common alterations in PBP1a from resistant Streptococcus pneumoniae decrease its reactivity towards beta-lactams: structural insights. J Biol Chem. 2008;283:4886–4894. doi: 10.1074/jbc.M706181200. [DOI] [PubMed] [Google Scholar]
- 100.Lim D, Strynadka NC. Structural basis for the beta lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat Struct Biol. 2002;9:870–876. doi: 10.1038/nsb858. [DOI] [PubMed] [Google Scholar]
- 101.Sauvage E, Kerff F, Fonze E, Herman R, Schoot B, Marquette JP, Taburet Y, Prevost D, Dumas J, Leonard G, Stefanic P, Coyette J, Charlier P. The 2.4 Å crystal structure of the penicillin-resistant penicillin-binding protein PBP5fm from Enterococcus faecium in complex with benzylpenicillin. Cell Mol Life Sci. 2002;59:1223–1232. doi: 10.1007/s00018-002-8500-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fedarovich A, Nicholas RA, Davies C. Unusual conformation of the SXN motif in the crystal strucutre of Penicillin-Binding Protein A from Mycobacterium tuberculosis. J Mol Biol. 2010;398:54–65. doi: 10.1016/j.jmb.2010.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fedarovich A, Nicholas RA, Davies C. The role of the β5-α11 loop in the active-site dynamics of acylated penicillin-binding protein A from Mycobacterium tuberculosis. J Mol Biol. 2012;418:316–330. doi: 10.1016/j.jmb.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Smith JD, Kumarasiri M, Zhang W, Hesek D, Lee M, Toth M, Vakulenko S, Fisher JF, Mobashery S, Chen Y. Structural analysis of the role of Pseudomonas aeruginosa penicillin-binding protein 5 in β-lactam resistance. Antimicrob Agents Chemother. 2013;57:3137–3146. doi: 10.1128/AAC.00505-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Han S, Zaniewski RP, Marr ES, Lacey BM, Tomaras AP, Evdokimov A, Miller JR, Shanmugasundaram V. Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2010;107:22002–22007. doi: 10.1073/pnas.1013092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Macheboeuf P, Fischer DS, Brown T, Jr, Zervosen A, Luxen A, Joris B, Dessen A, Schofield CJ. Structural and mechanistic basis of penicillin-binding protein inhibition by lactivicins. Nat Chem Biol. 2007;3:565–569. doi: 10.1038/nchembio.2007.21. [DOI] [PubMed] [Google Scholar]
- 107.Brown TJ, Charlier P, Herman R, Schofield CJ, Sauvage E. Structural basis for the interaction of lactivicins with serine beta-lactamases. J Med Chem. 2010;53:5890–5894. doi: 10.1021/jm100437u. [DOI] [PubMed] [Google Scholar]
- 108.Zervosen A, Lu WP, Chen Z, White RE, Demuth TP, Jr, Frère JM. Interactions between penicillin-binding proteins (PBPs) and two novel classes of PBP inhibitors, arylalkylidene rhodanines and arylalkylidene iminothiazolidin-4-ones. Antimicrob Agents Chemother. 2004;48:961–969. doi: 10.1128/AAC.48.3.961-969.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shilabin AG, Dzhekieva L, Misra P, Jayaram B, Pratt RF. 4-quinolones as noncovalent inhibitors of high molecular mass Penicillin-Binding Proteins. ACS Med Chem Lett. 2012;3:592–595. doi: 10.1021/ml3001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Contreras-Martel C, Amoroso A, Woon ECY, Zervosen A, Inglis S, Martins A, Verlaine O, Rydzik AM, Job V, Luxen A, Joris B, Schofield CJ, Dessen A. Structure-guided design of cell wall biosynthesis inhibitors that overcome β-lactam resistance in Staphylococcus aureus (MRSA) ACS Chem Biol. 2011;6:943–951. doi: 10.1021/cb2001846. [DOI] [PubMed] [Google Scholar]
- 111.Zervosen A, Herman R, Kerff F, Herman A, Bouillez A, Prati F, Pratt RF, Frère JM, Joris B, Luxen A, Charlier P, Sauvage E. Unexpected tricovalent binding mode of boronic acids within the active site of a penicillin-binding protein. J Am Chem Soc. 2011;133:10839–10848. doi: 10.1021/ja200696y. [DOI] [PubMed] [Google Scholar]
- 112.Fedarovich A, Djordjevic KA, Swanson SM, Peterson YK, Nicholas RA, Davies C. High-throughput screening for novel inhibitors of Neisseria gonorrhoeae penicillin-binding protein 2. PLoS One. 2012;7:e44918. doi: 10.1371/journal.pone.0044918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Inglis SR, Strieker M, Rydzik AM, Dessen A, Schofield CJ. A boronic-acid-based probe for fluorescence polarization assays with penicillin binding proteins and β-lactamases. Anal Biochem. 2012;420:41–47. doi: 10.1016/j.ab.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 114.Filipe SR, Tomasz A. Inhibition of the expression of penicillin resistance in Streptococcus pneumoniae by inactivation of cell wall muropeptide branching genes. Proc Natl Acad Sci USA. 2000;97:4891–4896. doi: 10.1073/pnas.080067697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Weber B, Ehlert K, Diehl A, Reichmann P, Labichinski H, Hakenbeck R. The fib locus in Streptococcus pneumoniae is required for peptidoglycan crosslinking and PBP-mediated beta-lactam resistance. FEMS Microbiol Lett. 2000;188:81–85. doi: 10.1111/j.1574-6968.2000.tb09172.x. [DOI] [PubMed] [Google Scholar]
- 116.Mainardi JL, Villet R, Bugg TD, Mayer C, Arthur M. Evolution of peptidoglycan biosynthesis under the selective pressure of antibiotics in Gram-positive bacteria. FEMS Microbiol Rev. 2008;32:386–408. doi: 10.1111/j.1574-6976.2007.00097.x. [DOI] [PubMed] [Google Scholar]
- 117.Berger-Bachi B, Tschierske M. Role of fem factors in methicillin resistance. Drug Resist Updat. 1998;1:325–335. doi: 10.1016/s1368-7646(98)80048-4. [DOI] [PubMed] [Google Scholar]
- 118.Fonvielle M, Chemama M, Villet R, Lecerf M, Bouhss A, Valéry JM, Ethève-Quelquejeu M, Arthur M. Aminoacyl-tRNA recognition by the FemXWv transferase for bacterial cell wall synthesis. Nucleic Acid Res. 2009;37:1589–1601. doi: 10.1093/nar/gkn1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Koyama N, Tokura Y, Munch D, Sahl HG, Schneider T, Ikeda H, Tomoda H. The nonantibiotic small molecule cyslabdan enhances the potency of β-lactams against MRSA by inhibiting pentaglycine interpeptide bridge synthesis. PLoS One. 2012;7:e48981. doi: 10.1371/journal.pone.0048981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gupta R, Lavollay M, Mainardi JL, Arthur M, Bishai WR, Lamichhane G. The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat Med. 2010;16:466–469. doi: 10.1038/nm.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mainardi JL, Fourgeaud M, Hugonnet JE, Dubost L, Brouard JP, Ouazzani J, Rice LB, Gutmann L, Arthur M. A novel peptidoglycan cross-linking enzyme for a beta-lactam-resistant transpeptidation pathway. J Biol Chem. 2005;280:38146–38152. doi: 10.1074/jbc.M507384200. [DOI] [PubMed] [Google Scholar]
- 122.Lecoq L, Dubée V, Triboulet S, Bougault C, Huggonet JE, Arthur M, Simorre JP. Structure of Enterococcus faecium L,D-transpeptidase acylated by ertapenem provides insight into the inactivation mechanism. ACS Chem Biol. 2013;8:1140–1146. doi: 10.1021/cb4001603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Biarrotte-Sorin S, Huggonet JE, Delfosse V, Mainardi JL, Gutmann L, Arthur M, Mayer C. Crystal structure of a novel β-lactam-insensitive peptidoglycan transpeptidase. J Mol Biol. 2006;359:533–538. doi: 10.1016/j.jmb.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 124.Erdemli SB, Gupta R, Bishai WR, Lamichhane G, Amzel LM, Bianchet MA. Targeting the cell wall of Mycobacterium tuberculosis: structure and mechanism of L,D-transpeptidase 2. Structure. 2012;20:2103–2115. doi: 10.1016/j.str.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mainardi JL, Morel V, Fourgeaud M, Cremniter J, Blanot D, Legrand R, Frehel C, Arthur M, Van Heijenoort J, Gutmann L. Balance between two transpeptidation mechanisms determines the expression of beta-lactam resistance in Enterococcus faecium. J Biol Chem. 2002;277:35801–35807. doi: 10.1074/jbc.M204319200. [DOI] [PubMed] [Google Scholar]
- 126.Triboulet S, Dubée V, Lecoq L, Bougault C, Mainardi JL, Rice LB, Ethève-Quelquejeu M, Gutmann L, Marie A, Dubost L, Hugonnet JE, Simorre JP, Arthur M. Kinetic features of L,D-transpeptidase inactivation critical for β-lactam antibacterial activity. PLoS One. 2013;8:e67831. doi: 10.1371/journal.pone.0067831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wilke MS, Lovering AL, Strynadka NC. Beta-lactam antibiotic resistance: a current structural perspective. Curr Opin Microbiol. 2005;8:525–533. doi: 10.1016/j.mib.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 128.Goffin C, Ghuysen JM. Biochemistry and comparative genomics of SXXK superfamily acyltransferases offer a clue to the mycobacterial paradox. Microb Mol Biol Rev. 2002;66:702–738. doi: 10.1128/MMBR.66.4.702-738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ambler RP. The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci. 1980;289:321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 130.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to beta-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 131.Cornaglia G, Giamarellou H, Rossolini GM. Metallo-β-lactamases: a last frontier for β-lactams? Lancet Infect Dis. 2011;11:381–393. doi: 10.1016/S1473-3099(11)70056-1. [DOI] [PubMed] [Google Scholar]
- 132.Bush K, Jacoby GA. Updated functional classification of β-lactamases. Antimicrob Agents Chemother. 2010;54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen Y, Shoichet B, Bonnet R. Structure, function, and inhibition along the reaction coordinate of CTX-M beta-lactamases. J Am Chem Soc. 2005;127:5423–5434. doi: 10.1021/ja042850a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shimamura T, Ibuka A, Fushinobu S, Wakagi T, Ishiguro M, Ishii Y, Matsuzawa H. Acyl-intermediate structures of the extended-spectrum class A beta-lactamase, Toho-1, in complex with cefotaxime, cephalothin, and benzylpenicillin. J Biol Chem. 2002;277:46601–46608. doi: 10.1074/jbc.M207884200. [DOI] [PubMed] [Google Scholar]
- 135.Nukaga M, Mayama K, Hujer AM, Bonomo RA, Know JR. Ultrahigh resolution structure of a class A beta-lactamase: on the mechanism and specificity of the extended-spectrum SHV-2 enzyme. J Mol Biol. 2003;328:289–301. doi: 10.1016/s0022-2836(03)00210-9. [DOI] [PubMed] [Google Scholar]
- 136.Petrella S, Ziental-Gelus N, Mayer C, Renard M, Jarlier V, Sougakoff W. Genetic and structural insights into the dissemination potential of the extremely broad-spectrum class A beta-lactamase KPC-2 identified in an Escherichia coli strain and an Enterobacter cloacae strain isolated from the same patient in France. Antimicrob Agents Chemother. 2008;52:3725–3736. doi: 10.1128/AAC.00163-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lobkovsky E, Moews PC, Liu H, Zhao H, Frere JM, Knox JR. Evolution of an enzyme activity: crystallographic structure at 2A resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class A penicillinase. Proc Natl Acad Sci USA. 1993;90:11257–11261. doi: 10.1073/pnas.90.23.11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jacoby GA. AmpC β-lactamases. Clin Microbiol Rev. 2009;22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Livermore DM. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis. 2002;34:634–640. doi: 10.1086/338782. [DOI] [PubMed] [Google Scholar]
- 140.Johnson JW, Fisher JF, Mobashery S. Bacterial cell wall recycling. Ann N Y Acad Sci. 2013;1277:54–75. doi: 10.1111/j.1749-6632.2012.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Babic M, Hujer AM, Bonomo RA. What's new in antibiotic resistance? Focus on beta-lactamases. Drug Resist Updat. 2006;9:142–156. doi: 10.1016/j.drup.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 142.Poirel L, Naas T, Nordmann P. Diversity, epidemiology, and genetics of class D β-lactamases. Antimicrob Agents Chemother. 2010;54:24–38. doi: 10.1128/AAC.01512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Paetzel M, Danel F, de Castro L, Mosimann SC, Page MGP, Strynadka NC. Crystal structure of the class D β-lactamase OXA-10. Nat Struct Mol Biol. 2000;7:918–925. doi: 10.1038/79688. [DOI] [PubMed] [Google Scholar]
- 144.Schneider KD, Bethel CR, Distler AM, Hujer AM, Bonomo RA, Leonard DA. Mutation of the active site carboxy-lysine (K70) of OXA-1 β-lactamase results in a deacylation-deficient enzyme. Biochemistry. 2009;48:6136–6145. doi: 10.1021/bi900448u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jacoby GA, Munoz-Price LS. The new β-lactamases. N Engl J Med. 2005;352:380–391. doi: 10.1056/NEJMra041359. [DOI] [PubMed] [Google Scholar]
- 146.Garau G, Bebrone C, Anne C, Galleni M, Frère JM, Dideberg O. A metallo-beta-lactamase enzyme in action: crystal structures of the monozinc carbapenemase CphA and its complex with biapenem. J Mol Biol. 2005;345:785–795. doi: 10.1016/j.jmb.2004.10.070. [DOI] [PubMed] [Google Scholar]
- 147.Garcia-Saez I, Docquier JD, Rossolini GM, Dideberg O. The three-dimensional structure of VIM-2, a Zn-ß-lactamase from Pseudomonas aeruginosa in its reduzed and oxidised form. J Mol Biol. 2008;375:604–611. doi: 10.1016/j.jmb.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 148.Carfi A, Parès S, Duee E, Galleni M, Duez C, Frère JM, Dideberg O. The 3-D structure of a zince metallo-beta-lactamase reveals a new type of protein fold. EMBO J. 1995;14:4914–4921. doi: 10.1002/j.1460-2075.1995.tb00174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Concha NO, Rasmussen BA, Bush K, Herzberg O. Crystal structure of the wide-spectrum binuclear zinc beta-lactamase from Bacteroides fragilis. Structure. 1996;4:823–836. doi: 10.1016/s0969-2126(96)00089-5. [DOI] [PubMed] [Google Scholar]
- 150.Palzkill T. Metallo-b-lactamase structure and function. Ann N Y Acad Sci. 2013;1277:91–104. doi: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kumarasamy KK, Toleman MA, Walsh TR, Bagaria J, Butt F, Balakrishnan R, Chaudhary U, Doumith M, Giske CG, Irfan S, Krishnan P, Kumar AV, Maharjan S, Mushtaq S, Noorie T, Paterson DL, Pearson A, Perry C, Pike R, Rao B, Ray U, Sarma JB, Sharma M, Sheridan E, Thirunarayan MA, Turton J, Upadhyay S, Warner M, Welfare W, Livermore DM, Woodford N. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis. 2010;10:597–602. doi: 10.1016/S1473-3099(10)70143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nordmann P, Poirel L, Walsh TR, Livermore DM. The emerging NDM carbapenemases. Trends Microbiol. 2011;19:588–595. doi: 10.1016/j.tim.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 153.Zhang H, Hao Q. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J. 2011;25:2574–2582. doi: 10.1096/fj.11-184036. [DOI] [PubMed] [Google Scholar]
- 154.Green VL, Verma A, Owens RJ, Phillips SE, Carr SB. Structure of New Delhi metallo-β-lactamase 1 (NDM-1) Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67:1160–1164. doi: 10.1107/S1744309111029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.King DT, Worrall LJ, Gruninger R, Strynadka NCJ. New Delhi Metallo-β-Lactamase: structural insights into β-Lactam recognition and inhibition. J Am Chem Soc. 2012;134:11362–11365. doi: 10.1021/ja303579d. [DOI] [PubMed] [Google Scholar]
- 156.Liang Z, Li L, Wang Y, Chen L, Kong X, Hong Y, Lan L, Zheng M, Guang-Yang C, Liu H, Shen X, Luo C, Li KK, Chen K, Jiang H. Molecular basis of NDM-1, a new antibiotic resistance determinant. PLoS One. 2011;6:e23606. doi: 10.1371/journal.pone.0023606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Thakur PK, Kumar J, Ray D, Anjum F, Hassan MI. Search of potential inhibitor against New Delhi metallo-beta-lactamase 1 from a series of antibacterial natural compounds. J Nat Sci Biol Med. 2013;4:51–56. doi: 10.4103/0976-9668.107260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Votsch W, Templin MF. Characterization of a β-N-acetylglucosaminidase of Escherichia coli and elucidation of its role in muropeptide recycling and β-lactamase induction. J Biol Chem. 2000;275:39032–39038. doi: 10.1074/jbc.M004797200. [DOI] [PubMed] [Google Scholar]
- 159.Cheng Q, Park JT. Substrate specificity of the AmpG permease required for recycling of cell wall anhydro-muropeptides. J Bacteriol. 2002;184:6434–6436. doi: 10.1128/JB.184.23.6434-6436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Avison MB, Horton RE, Walsh TR, Bennett PM. Escherichia coli CreBC is a global regulator of gene expression that responds to growth in minimal media. J Biol Chem. 2001;276:26955–26962. doi: 10.1074/jbc.M011186200. [DOI] [PubMed] [Google Scholar]
- 161.Tayler AE, Ayala JA, Niumsup P, Westphal K, Baker JA, Zhang L, Walsh TR, Wiedemann B, Bennett PM, Avison MB. Induction of β-lactamase production in Aeromonas hydrophila is responsive to β-lactam-mediated changes in peptidoglycan composition. Microbiology. 2010;156:2327–2335. doi: 10.1099/mic.0.035220-0. [DOI] [PubMed] [Google Scholar]
- 162.Zamorano L, Reeve TM, Juan C, Moya B, Cabot G, Vocadlo DJ, Mark BL, Oliver A. AmpG inactivation restores susceptibility of pan-β-lactam-resistant Pseudomonas aeruginosa clinical strains. Antimicrob Agents Chemother. 2011;55:1990–1996. doi: 10.1128/AAC.01688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang Y, Bao Q, Gagnon LA, Huletsky A, Oliver A, Jin S, Langaee T. ampG gene of Pseudomonas aeruginosa and its role in β-lactamase expression. Antimicrob Agents Chemother. 2010;54:4772–4779. doi: 10.1128/AAC.00009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Stubbs KA, Balcewich M, Mark BL, Vocadlo DJ. Small molecule inhibitors of a glycoside hydrolase attenuate inducible AmpC-mediated beta-lactam resistance. J Biol Chem. 2007;282:21382–21391. doi: 10.1074/jbc.M700084200. [DOI] [PubMed] [Google Scholar]
- 165.Mushtaq S, Warner M, Livermore D. Activity of the siderophore monobactam BAL30072 against multiresistant non-fermenters. J Antimicrob Chemother. 2010;65:266–270. doi: 10.1093/jac/dkp425. [DOI] [PubMed] [Google Scholar]
- 166.Livermore DM, Mushtaq S, Ge Y, Warner M. Chequerboard titration of cephalosporin CXA-101 (FR264205) and tazobactam versus beta-lactamase-producing Enterobacteriaceae. J Antimicrob Chemother. 2009;65:1972–1974. doi: 10.1093/jac/dkq248. [DOI] [PubMed] [Google Scholar]