

Abstract
The folding of proteins is directed by a variety of interactions, including hydrogen bonding, electrostatics, van der Waals' interactions, and the hydrophobic effect. We have argued previously that an n→π* interaction between carbonyl groups be added to this list. In an n→π* interaction, the lone pair (n) of one carbonyl oxygen overlaps with the π* antibonding orbital of another carbonyl group. The tendency of backbone carbonyl groups in proteins to engage in this interaction has consequences for the structures of folded proteins that we unveil herein. First, we employ density functional theory to demonstrate that the n→π* interaction causes the carbonyl carbon to deviate from planarity. Then, we detect this signature of the n→π* interaction in high-resolution structures of proteins. Finally, we demonstrate through natural population analysis that the n→π* interaction causes polarization of the electron density in carbonyl groups and detect that polarization in the electron density map of cholesterol oxidase, further validating the existence of n→π* interactions. We conclude that the n→π* interaction is operative in folded proteins.
Keywords: Bürgi–Dunitz trajectory, electron density, hyperconjugation, pyramidalization, stereoelectronic effect
Introduction
The three-dimensional structures of proteins enable their specific functions and arise largely from noncovalent interactions within and between polypeptide chains.1 These interactions include hydrogen bonding, electrostatics, van der Waals' interactions, and the hydrophobic effect.2 The current challenges in de novo structure prediction and protein design demonstrate that the understanding of these interactions is incomplete.3–5
We have argued previously that an n→π* interaction between two carbonyl groups can play a role in dictating protein conformation.6–11 In an n→π* interaction, the lone pair (n) of a carbonyl oxygen overlaps with the π* antibonding orbital of another carbonyl group [Fig. 1(a)]. This orbital overlap is possible when the putative donor forms a sub-van der Waals' contact with the acceptor (d < 3.22 Å) along the Bürgi–Dunitz trajectory for nucleophilic addition (θ ≈ 109°). The result of overlap between the n orbital of the donor and the π* orbital of the acceptor is the release of energy due to orbital mixing. The energy associated with this interaction varies with the geometry of the interacting groups, but we anticipate a typical n→π* interaction between amide bonds to contribute at least 0.27 kcal/mol.12 We have shown that numerous residues in folded proteins are oriented to take advantage of this energy release, suggesting that n→π* interactions could contribute significantly to the three-dimensional structure and conformational stability of proteins.13–15
Figure 1.
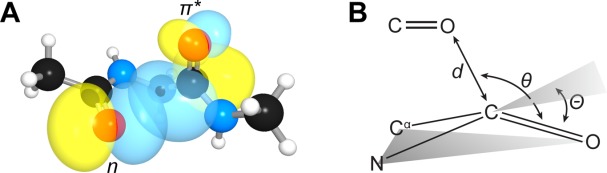
(a) Overlap of the n and π* orbitals in the backbone of bitter gourd trypsin inhibitor (PDB: 1vbw), residues 5–7. Image rendered with NBOView 1.1. (b) Geometric parameters that characterize an n→π* interaction.
An n→π* interaction results in population of the π* orbital of the acceptor carbonyl. A distinctive signature should arise in the structures of proteins. Specifically, as population of the π* orbital weakens the carbonyl π-bond, the acceptor should distort from a planar sp2 geometry toward a pyramidal sp3-like geometry [Fig. 1(b)]. Such deviations can be represented by the angle Θ at which the carbonyl group rises out of the plane of its substituents, as computed according to the method of Mazzarella and co-workers.16 Previously, we examined the pyramidalization of carbonyl groups in short, helical peptides from the Cambridge Structural Database.17 We observed that n→π* interactions engender pyramidalization of the carbonyl group toward its respective donor; helical residues not engaged in an n→π* interaction distort away from the putative donor so as to reduce Pauli repulsion. We have also detected the distortion instilled by an n→π* interaction in small molecules.10,12,18,19 Here, we ask the question: Is pyramidalization a signature in proteins as well?
Results and Discussion
We began our investigation with a foundational computational analysis. To evaluate the propensity of an amide carbonyl group to pyramidalize in the presence of an n→π* interaction, we employed density functional theory (DFT) at the B3LYP/6–311+G (2d,p) level of theory to optimize the structure of a model amide as a putative n→π* donor approaches along the Bürgi–Dunitz trajectory.20 To reduce the degrees of freedom in our analysis, we studied the structure of formamide as it was approached by formaldehyde along the Bürgi–Dunitz trajectory (d = 2.75–3.50 Å, θ = 110°); to simplify our analysis further, we restricted the geometry of the complex to ensure parallel orientation of the carbonyl groups. Then, we plotted the observed pyramidalization as a function of the n→π* interaction energy (En→π*) determined from second-order perturbation theory in the natural bond orbital analysis program NBO 5.9 [Fig. 2(a)].21 As expected, at high, stabilizing values of En→π*, which correspond to shorter donor–acceptor contacts, we observed greater pyramidalization. This correlation validates pyramidalization of backbone carbonyl carbon atoms as a signature of an n→π* interaction that could enable its detection in proteins.
Figure 2.
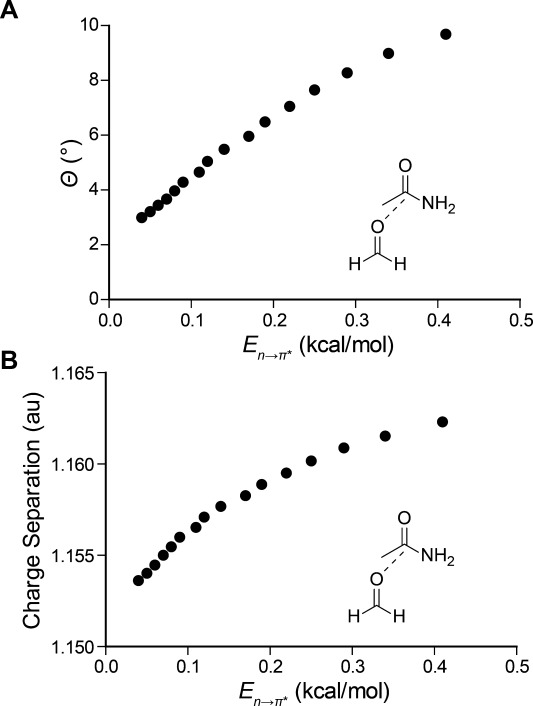
(a) Calculated pyramidalization and (b) carbonyl charge separation of formamide resulting from the approach of formaldehyde along the Bürgi–Dunitz trajectory. The value of En→π* was determined by second-order perturbation theory and charge separation is determined from natural population analysis (NPA), both as implemented by NBO 5.9.
Structural refinement can bias the atomic coordinates of peptide bonds toward planarity. Hence, we examined a nonredundant set (<25% pairwise sequence identity) of 192 protein crystal structures that were determined to sub-Å resolution to minimize the bias introduced by refinement.22,23 We identified residues that receive a backbone n→π* interaction using a geometric operational definition [Fig. 1(b)]. Specifically, when a carbonyl oxygen forms a sub-van der Waals' contact (d < 3.22 Å) with a carbonyl carbon along the Bürgi–Dunitz trajectory (95° < θ < 125°), we scored the carbonyl carbon as positive for receiving an n→π* interaction; those residues not meeting this criteria were scored as negative for receiving an n→π* interaction.
We sought to control for the effect of secondary structure in our analysis of carbonyl pyramidalization, recognizing that local conformation could contribute to pyramidalization.16,24 In particular, the prevalence of n→π* interactions appears to vary dramatically between different secondary structures, being observed with great frequency in α-helices, but rarely in β-sheets.13 To remove bias from local conformation, we examined the pyramidalization of the 3759 carbonyl groups from our high-resolution set of protein structures that were not assigned to any particular secondary structure by Kabsch and Sander criteria25 as implemented by PROMOTIF.26 Of these residues, 24% receive an n→π* interaction, a fraction that enables us to make an effective comparison between the geometries of residues that receive an n→π* interaction and those that do not.
We were able to observe a difference in the absolute pyramidalization of these two populations [Fig. 3(a)]. The absolute pyramidalization was found to be 0.32° higher on average in the presence of an n→π* interaction, indicating that the two populations are distorted differently (P < 0.00001). Thus, we conclude that n→π* interactions cause a distortion of the peptide bond in proteins. Moreover, for those carbonyl groups that accept an n→π* interaction, we observed that pyramidalization tends to occur toward the donor [Fig. 3(b)]. In the absence of an attractive interaction, one would expect the carbonyl group to distort away from the incoming oxygen so as to reduce unfavorable van der Waals' contacts and Pauli repulsion.11 We note that, even in these high-resolution structures, structural refinement likely enforces planarity upon the peptide bond; the true effect of the n→π* interaction on pyramidalization of protein carbonyl groups could be even greater than that observed herein.
Figure 3.
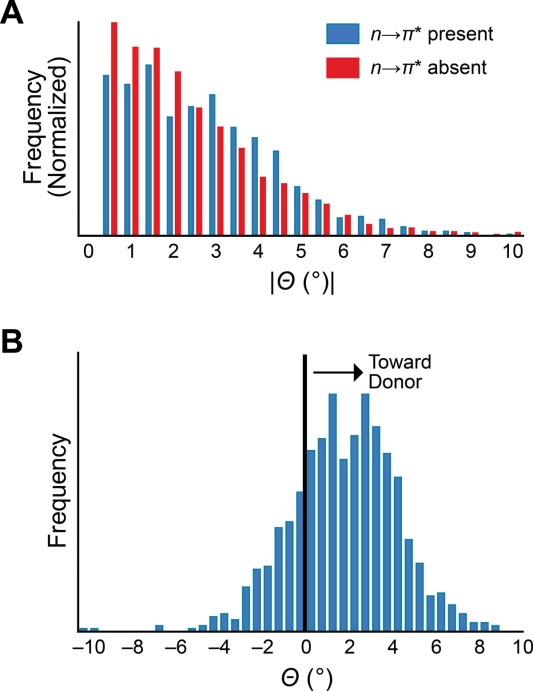
Pyramidalization of backbone carbonyl groups in proteins lacking secondary structure. (a) Absolute pyramidalization of residues receiving an n→π* interaction (blue, n = 896) versus those that do not (red, n = 2863). (b) Pyramidalization of residues receiving an n→π* interaction measured relative to the location of the n→π* donor, with Θ > 0 indicating pyramidalization toward the donor.
As the n→π* interaction involves donation of electron density to a carbonyl carbon, another signature of the n→π* interaction should be evident. Specifically, the approach of the nucleophilic n→π* donor oxygen should polarize the electron density of the acceptor amide carbonyl. To evaluate this hypothesis, we subjected formamide to natural population analysis (NPA) upon approach by the formaldehyde n→π* donor.27 NPA is a reliable method for assigning atomic charges and thus provides a measure of charge separation in the acceptor carbonyl group, which we plotted against the corresponding En→π* [Fig. 2(b)]. As expected, the degree of charge separation in the carbonyl group increases as the n→π* interaction grows stronger.
Finally, we searched for evidence of this signature in the electron density of a particular protein. Our analysis is based on a previous report of the electron density in carbonyl groups of cholesterol oxidase. In that work, Lario and Vrielink classified backbone carbonyl groups by the degree of charge separation observed between the carbonyl carbon and carbonyl oxygen in the electron density map [Fig. 4(a)].28 Here, if a clear separation in electron density is visible at 4.5σ, the carbonyl group is classified as a “gap”. In contrast, if no separation is evident at 5.5σ, the carbonyl group is classified as a “share”. Carbonyl groups for which separation of electron density was evident at 5.5σ but not 4.5σ were classified as “middle.” To control for the influence of secondary structure, we exclude residues assigned to helices or sheets by PROMOTIF. We then tabulated the relative abundance of each of these categories for residues in cholesterol oxidase that receive an n→π* interaction versus those that do not, according to our geometric criteria [Fig. 4(b)]. We observe that for carbonyl groups that receive an n→π* interaction, there is a lower proportion of “share” carbonyl groups and a higher proportion of “gap” carbonyl groups, as compared to those residues that do not receive an n→π* interaction. The greater proportion of “gap” carbonyl groups in the n→π* positive set is consistent with the notion that the n→π* interaction causes a polarization of the electron density of carbonyl groups.
Figure 4.
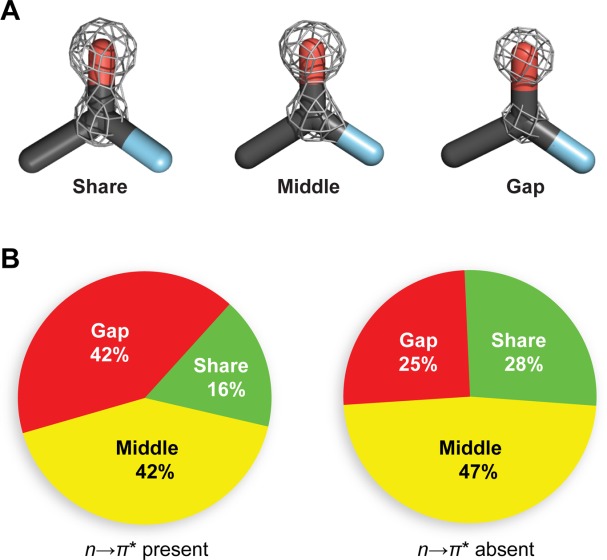
Experimental electron density around backbone carbonyl groups. (a) Prototypical examples of “gap”, “middle”, and “share” electronic distributions from residues Leu155, Val108, and Leu287 of cholesterol oxidase (PDB: 1n1p), respectively. (b) Relative proportion of “gap”, “middle”, and “share” electronic distributions for residues in cholesterol oxidase that receive an n→π* interaction (n = 50) and those that do not (n = 97).27
We conclude by noting two important implications of these observations. First, the polarization of electron density by the n→π* interaction could serve to increase the strength of hydrogen bonds that are critical for dictating secondary structure. Secondly, because polarization increases the nucleophilicity of a carbonyl oxygen, the polarization of carbonyl groups caused by n→π* donation should increase the strength of subsequent n→π* interactions, leading to cooperativity. These effects on carbonyl polarization could conspire to increase the thermal stability of the α-helix, which is stitched together by the action of both hydrogen bonding and n→π* interactions.13,28 The impact of n→π* interactions on the structure and the stability of proteins should thus extend beyond the direct effect of carbonyl attraction. Considering their sheer abundance, the impact of n→π* interactions on protein folding could be profound.
Methods
Computational chemistry
Geometry optimizations of the formaldehyde–formamide complex were conducted at the B3LYP/6-311+G(2d,p) level of theory as implemented in Gaussian 09.29 The distance between the formaldehyde oxygen and the formamide carbon was varied from 2.75 to 3.50 Å with the angle of approach of the formaldehyde oxygen to the formamide carbonyl constrained to 110° and the dihedral angle between the two carbonyl groups constrained to 180°. Optimized geometries were then subjected to NBO analysis at the B3LYP/6-311+G(2d,p) as implemented in NBO 5.9.30
Bioinformatics
A nonredundant set (<25% pairwise sequence identity) of 192 protein crystal structures (>40 residues, R < 20%) with resolution of 1.0 Å or better was culled from the PDB on 28 November 2012 using the PISCES server.22 Secondary structure assignments were made using Kabsch and Sander criteria25 as implemented in PROMOTIF.26 Residues modeled in multiple conformations of the peptide backbone were excluded from analysis.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 3.Fleishman SJ, Whitehead TA, Strauch E-M, Corn JE, Qin S, Zhou H-X, Mitchell JC, Demerdash ON, Takeda-Shitaka M, Terashi G, Moal IH, Li X, Bates PA, Zacharias M, Park H, Ko J-s, Lee H, Seok C, Bourquard T, Bernauer J, Poupon A, Azé J, Soner S, Kerem Ovalı Ş, Ozbek P, Ben Tal N, Haliloglu T, Hwang H, Vreven T, Pierce BG, Weng Z, Pérez-Cano L, Pons C, Fernández-Recio J, Jiang F, Yang F, Gong X, Cao L, Xu X, Liu B, Wang P, Li C, Wang C, Robert CH, Guharoy M, Liu S, Huang Y, Li L, Guo D, Chen Y, Xiao Y, London N, Itzhaki Z, Schueler-Furman O, Inbar Y, Potapov V, Cohen M, Schreiber G, Tsuchiya Y, Kanamori E, Standley DM, Nakamura H, Kinoshita K, Driggers CM, Hall RG, Morgan JL, Hsu VL, Zhan J, Yang Y, Zhou Y, Kastritis PL, Bonvin AMJJ, Zhang W, Camacho CJ, Kilambi KP, Sircar A, Gray JJ, Ohue M, Uchikoga N, Matsuzaki Y, Ishida T, Akiyama Y, Khashan R, Bush S, Fouches D, Tropsha A, Esquivel-Rodríguez J, Kihara D, Stranges PB, Jacak R, Kuhlman B, Huang S-Y, Zou X, Wodak SJ, Janin J, Baker D. Community-wide assessment of protein–interface modeling suggests improvements to design methodology. J Mol Biol. 2011;414:289–302. doi: 10.1016/j.jmb.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kryshtafovych A, Fidelis K, Moult J. CASP9 results compared to those of previous CASP experiments. Proteins 79 (Suppl. 2011;10):196–207. doi: 10.1002/prot.23182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stranges PB, Kuhlman B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013;22:74–82. doi: 10.1002/pro.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bretscher LE, Jenkins CL, Taylor KM, DeRider ML, Raines RT. Conformational stability of collagen relies on a stereoelectronic effect. J Am Chem Soc. 2001;123:777–778. doi: 10.1021/ja005542v. [DOI] [PubMed] [Google Scholar]
- 7.DeRider ML, Wilkens SJ, Waddell MJ, Bretscher LE, Weinhold F, Raines RT, Markley JL. Collagen stability: Insights from NMR spectroscopic and hybrid density functional computational investigations of the effect of electronegative substituents on prolyl ring conformations. J Am Chem Soc. 2002;124:2497–2505. doi: 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]
- 8.Hinderaker MP, Raines RT. An electronic effect on protein structure. Protein Sci. 2003;12:1188–1194. doi: 10.1110/ps.0241903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodges JA, Raines RT. Energetics of an n→π* interaction that impacts protein structure. Org Lett. 2006;8:4695–4697. doi: 10.1021/ol061569t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choudhary A, Gandla D, Krow GR, Raines RT. Nature of amide carbonyl–carbonyl interactions in proteins. J Am Chem Soc. 2009;131:7244–7246. doi: 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jakobsche CE, Choudhary A, Miller SJ, Raines RT. n→π* Interaction and nπ Pauli repulsion are antagonistic for protein stability. J Am Chem Soc. 2010;132:6651–6653. doi: 10.1021/ja100931y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newberry RW, VanVeller B, Guzei IA, Raines RT. n→π* Interactions of amides and thioamides: Implications for protein stability. J Am Chem Soc. 2013;135:7843–7846. doi: 10.1021/ja4033583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartlett GJ, Choudhary A, Raines RT, Woolfson DN. n→π* Interactions in proteins. Nat Chem Biol. 2010;6:615–620. doi: 10.1038/nchembio.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fufezan C. The role of Bürgi–Dunitz interactions in the structural stability of proteins. Proteins. 2010;78:2831–2838. doi: 10.1002/prot.22800. [DOI] [PubMed] [Google Scholar]
- 15.Bartlett GJ, Newberry RW, VanVeller B, Raines RT, Woolfson DN. Interplay of hydrogen bonds and n→π* interactions in proteins. J Am Chem Soc. 2013;135:18682–18688. doi: 10.1021/ja4106122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esposito L, Vitagliano L, Zagari A, Mazzarella L. Pyramidalization of backbone carbonyl carbon atoms in proteins. Protein Sci. 2001;9:2038–2042. [PMC free article] [PubMed] [Google Scholar]
- 17.Choudhary A, Raines RT. Signature of n→π* interactions in α-helices. Protein Sci. 2011;20:1077–1081. doi: 10.1002/pro.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choudhary A, Kamer KJ, Raines RT. An n→π* interaction in aspirin: Implications for structure and reactivity. J Org Chem. 2011;76:7933–7937. doi: 10.1021/jo201389d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choudhary A, Pua KH, Raines RT. Quantum mechanical origin of the conformational preferences of 4-thiaproline and its S-oxides. Amino Acids. 2011;41:181–186. doi: 10.1007/s00726-010-0504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bürgi HD, Dunitz JD, Shefter E. Chemical reaction paths. IV. Aspects of O···C=O interactions in crystals. Acta Crystallogr B. 1974;30:1517–1527. [Google Scholar]
- 21.Reed AE, Curtiss LA, Weinhold F. Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem Rev. 1988;88:899–926. [Google Scholar]
- 22.Wang G, Dunbrack RL. PISCES: A protein sequence culling server. Bioinformatics. 2003;19:1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 23.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Improta R, Vitagliano L, Esposito L. Peptide bond distortions from planarity: New insights from quantum mechanical calculations and peptide/protein crystal structures. PLoS One. 2011;6:1–10. doi: 10.1371/journal.pone.0024533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 26.Hutchinson EG, Thornton JM. PROMOTIF—A program to identify and analyze structural motifs in proteins. Protein Sci. 1996;5:212–220. doi: 10.1002/pro.5560050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed AE, Weinstock RB, Weinhold F. Natural population analysis. J Chem Phys. 1985;83:735–746. [Google Scholar]
- 28.Lario PI, Vrielink A. Atomic resolution density maps reveal secondary structure dependent differences in electronic distribution. J Am Chem Soc. 2003;125:12787–12794. doi: 10.1021/ja0289954. [DOI] [PubMed] [Google Scholar]
- 29.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.1. Wallingford: Gaussian, Inc; 2009. [Google Scholar]
- 30.Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F. NBO 5.9. Theoretical Chemistry Institute. Madison: University of Wisconsin–Madison; 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.