

Abstract
Multiple sclerosis is a chronic demyelinating disorder characterized by the infiltration of auto-reactive immune cells from the periphery into the central nervous system resulting in axonal injury and neuronal cell death. Experimental autoimmune encephalomyelitis represents the best characterized animal model as common clinical, histological, and immunological features are recapitulated. A label-free mass spectrometric proteomics approach was used to detect differences in protein abundance within specific fractions of disease-affected tissues including the soluble lysate derived from the spinal cord and membrane protein-enriched peripheral blood mononuclear cells. Tissues were harvested from actively induced experimental autoimmune encephalomyelitis mice and sham-induced (“vehicle” control) counterparts at the disease peak followed by subsequent analysis by nanoflow liquid chromatography tandem mass spectrometry. Relative protein quantitation was performed using both intensity- and fragmentation-based approaches. After statistical evaluation of the data, over 500 and 250 differentially abundant proteins were identified in the spinal cord and peripheral blood mononuclear cell data sets, respectively. More than half of these observations have not previously been linked to the disease. The biological significance of all candidate disease markers has been elucidated through rigorous literature searches, pathway analysis, and validation studies. Results from comprehensive targeted mass spectrometry analyses have confirmed the differential abundance of ∼200 candidate markers (≥twofold dysregulated expression) at a 70% success rate. This study is, to our knowledge, the first to examine the cell-surface proteome of peripheral blood mononuclear cells in experimental autoimmune encephalomyelitis. These data provide a unique mechanistic insight into the dynamics of peripheral immune cell infiltration into CNS-privileged sites at a molecular level and has identified several candidate markers, which represent promising targets for future multiple sclerosis therapies. The mass spectrometry proteomics data associated with this manuscript have been deposited to the ProteomeXchange Consortium with the data set identifier PXD000255.
Multiple sclerosis (MScl)1 is an inflammatory autoimmune condition, which targets the central nervous system (CNS) resulting in the onset of demyelinating events and irrevocable neurological deficits (1). Although the precise etiology and pathogenic features of the disease remain elusive, comprehensive epidemiological studies have revealed strong genetic and environmental determinants (2). MScl is widely considered as being a classical T-cell mediated autoimmune disease based on critical observations made on the quintessential animal model of CNS autoimmunity known as experimental autoimmune encephalomyelitis (EAE) (3).The disease can be actively induced in genetically susceptible animals (e.g. rodents, primates) by inoculation with an emulsion containing encephalitogenic myelin proteins (e.g. myelin oligodendrocyte protein, MOG) and an adjuvant. The ensuing disease mimics several clinical, histological, and immunological features of MScl including lower limb paralysis, breach of blood brain barrier (BBB) permeability, and inflammatory infiltration into the CNS (4, 5).
Advances in various “-omics”-based platforms such as proteomics and metabolomics has shed some light into the molecular events associated with EAE pathogenesis (6). Differential gene and protein expression profiles have been generated based on comparative analyses of healthy control and disease-affected tissues derived from clinical samples (7–18) and animal models (19–29). These biomarker discovery platforms include gel-based approaches such as two-dimensional gel electrophoresis (2D-GE) (10, 17, 30), 2D-difference image gel electrophoresis (2D-DIGE) (9, 14), as well as shotgun proteomics techniques (11, 13, 16, 31, 32) incorporating the use of label-free or stable isotope labeling LC-MS-based strategies for quantitative proteomic studies. In recent years there has been exponential growth in the use of these alternative gel-free shotgun proteomics strategies, which has been facilitated by advances in mass spectrometry instrumentation and computational capabilities. There are two fundamentally different approaches for performing label-free quantitation: (1) measuring the area under the chromatographic elution peak (AUC) based on each peptide precursor ion or the peptide signal intensity produced from the MS1 spectrum that correlates with peptide abundance in a complex mixture and (33, 34) (2) spectral counting (SC), which calculates the number of acquired fragment spectra (MS2) used to identify peptides from a given protein and thus is proportional to its abundance (35). The first strategy is generally considered to be more accurate, however, this assumes a high reproducibility is observed between chromatographic runs being compared and the sampling speed of the mass spectrometer is sufficient to record multiple data points across the chromatographic distribution of the analyte. The method of SC has traditionally been plagued with issues such as unreliable quantitation of low-abundance proteins and peptide bias given that it doesn't directly measure a physical property of the peptide (36, 37). However, efforts have been made to provide a better basis for quantification by adjusting counts with normalization factors that can take into consideration the length of proteins (38–40) or the number of observable tryptic peptides within a defined mass range (41, 42).
Here, we present an unbiased quantitative proteomics study involving both MS1-level and MS2 fragmentation-based label-free approaches to assess the unique repertoire of differentially abundant proteins contained within specific subcellular fractions of disease-affected tissues isolated from an MOG-EAE model of MScl. Several time-course studies on animal models of EAE support a caudal-to-rostral progression of disease driven by the vulnerability of the spinal cord to damage and the increased permeability of the BBB (43); Thus it is expected that quantifiable biochemical changes are taking place in this tissue. PBMCs are comprised of various lymphocyte populations including T and B cells, the major cellular components of the adaptive immune response in EAE and MScl and are known to infiltrate sites of CNS damage. Therefore, quantitative proteomic analysis of these cell-surface changes may provide insight into the immune-related pathways mediating disease progression, and facilitate the identification of early disease markers in the blood, a readily accessible tissue highly amenable to clinical sampling.
MATERIALS AND METHODS
EAE Induction and Tissue Collection
Female C57BL/6 mice (6–8 weeks old) were induced with EAE by immunization with 125 μg of MOG35–55 peptide (Canada Peptides, Canada) emulsified 1:1 (v/v) in CFA (Sigma) containing 4 mg/ml of Mycobacteria tuberculosis (H37RA) (Difco, Lawrence, KS). Mice received two intravenous (i.v.) injections of 300 ng Bordetella pertussis toxin (List Biological Laboratories Inc., Campbell, CA) the day of immunization. An identical pertussis toxin injection was repeated 3 days later. Control mice were age-matched and sham-injected, receiving all components of the injection, except for the MOG peptide. Eight mice were used per group and the animals were observed and weighed daily. Clinical assessment of EAE was performed with a modified scale derived from Giuliani and colleagues (44) (supplemental Fig. S1). In addition to the Giuliani scale, an assessment of the righting reflex of the mice was performed. The righting reflex capacity was graded from 0 to 2. Zero is given to normal righting reflex, 1 for a slow righting reflex, and 2 for a delay of more than 5 s in the righting reflex. Thus, this scale ranges from 0 (no-symptoms) to 16 (fully quadriplegic mouse with limp tail and significantly delayed righting reflex. Mice were sacrificed by carbon dioxide asphyxiation at peak disease onset (15 days postimmunization, dpi). Immediately following death, peripheral blood was collected from the mice before they were transcardially perfused with phosphate buffered saline (PBS). Spinal cord tissue was harvested from the mice, flash frozen, and stored at −80 °C. Peripheral blood was collected from all mice and transferred into capillary tubes coated with heparin. PBMCs were then isolated using a Ficoll gradient, with blood diluted 1:3 with sterile PBS and centrifuged at 1800 rpm for 25 mins at 4 °C. The buffy coat containing principally monocytes and lymphocytes (PBMCs) was isolated from the interface layer between the plasma and the Ficoll and transferred to a fresh tube. Cells were washed twice with PBS to remove traces of Ficoll before being pelleted by centrifugation. The pellet was stored at −80 °C. All animals were housed under specific pathogen-free conditions and all animal experimentation was performed in strict accordance with regulations set and approved by the institutional animal ethics committees.
Preparation of Soluble Protein Extracts from Spinal Cord Tissue
Frozen spinal cord tissue from EAE and sham mice were separately ground to a fine powder with liquid nitrogen in a precooled (on dry ice) ceramic mortar and pestle. Protein extracted from the tissue powder was then solubilized in ice-cold cell lysis buffer (50 mm Tris-HCl pH 7.5, 150 mm NaCl, 1X Complete EDTA-free protease inhibitor (Roche, Indianapolis, IN), 1.5 mm MgCl2, 1.5 mm CaCl2, 2 mm dithiothreitol (DTT, Thermo Scientific, Rockford, IL)). The homogenate was sonicated in a water bath for 10 mins prior to benchtop centrifugation at 20,817 × g, 4 °C for 15 mins to pellet cell debris and insoluble material. The supernatant containing the soluble proteins was collected and protein concentrations were determined by the modified Lowry method (DC Protein assay; Bio-Rad, Hercules, CA).
Preparation of Membrane-enriched Protein Fractions from PBMCs
Pelleted PBMCs (∼2.4 million cells per sample) from EAE and sham-induced mice were resuspended in 100 μl of hypotonic cell swelling buffer (10 mm Tris-HCl pH 7.4; 10 mm KCl, 1.5 mm MgCl2; 1.5 mm CaCl2; 2 mm DTT (Thermo Scientific), 1× Complete EDTA-free protease inhibitor (Roche). The resuspension was then subjected to sonication for 10 mins before benchtop centrifugation at 20,817 × g, 4 °C for 15 mins to pellet cell debris and insoluble material (i.e. membranes). The remaining supernatant (cytosolic protein fraction) was collected, and the membrane-containing pellet resuspended in hypotonic swelling buffer to a final volume of 50 μl containing 2% final concentration of lauryl maltose neopentyl glycol (MNG, Anatrace, Affymetrix (Maumee, OH, USA)). MNG detergent was used based on studies in our laboratory that have demonstrated that no additional detergent removal steps are required subsequent to FASP analysis, thus simplifying the overall protocol (Vuckovic et al., manuscript in preparation). The membrane-enriched lysate was sonicated for 10 mins then centrifuged at 20,817 × g at 4 °C for 10 mins before the remaining supernatant (membrane protein fraction) was collected and protein concentrations determined by the DC Protein Assay.
Protein Digestion by Filter-aided Sample Preparation (FASP)
Equal amounts of protein (80 μg) derived from six soluble spinal cord extracts and six membrane-enriched PBMCs (60 μg) were prepared for mass spectrometry analysis using the FASP protein digestion kit (Protein Discovery, Knoxville, TN) as per manufacturer's instructions (45, 46). Briefly, 12 protein samples (n = 3, each from EAE and sham group per tissue) were incubated with 5 mm DTT (Thermo Scientific) for 30 mins at room temperature (RT) followed by the addition of 200 μl 8 m urea/Tris-HCl, pH 8.5 (provided with the kit). The diluted samples were then added to the spin ultrafiltration devices [nominal molecular weight (Mw) cutoff of 30 kDa] and subjected to two rounds of benchtop centrifugation at 20,817 × g, 4 °C for 15 mins with an additional volume of 200 μl urea-Tris/HCl added before the second spin. Protein alkylation was performed with the addition of 1X iodoacetamide (IAA, provided with the kit) to the spin filters followed by incubation in the dark for 20 mins. The spin filters were then subjected to several wash and centrifugation steps, first with 100 μl Urea-Tris/HCl followed by 100 μl of 50 mm ammonium bicarbonate (NH4HCO3). Finally, sequence-grade modified trypsin (Roche) was added at an enzyme/substrate ratio of 1:50 to each spin filter. An additional 40 μl volume of 50 mm NH4HCO3 was added to each sample before overnight incubation at 37 °C with shaking. The following day, peptides were eluted by several rounds of washing with 50 mm NH4HCO3 and 0.5 m NaCl solutions followed by centrifugation.
Mass Spectrometry Analysis
Tryptic peptides were acidified with formic acid (FA) (2% final concentration) and analyzed by nanoflow liquid chromatography coupled to data-dependent tandem mass spectrometry (nLC-MS/MS) using the EASY-nLCTM system (Proxeon Biosystems, Odense, Denmark) connected to a hybrid LTQ-Orbitrap Velos instrument (Thermo Fisher Scientific, San Jose, CA) via a nanoelectrospray ion source. Digested samples (10 μl, ∼5 μg) were initially loaded onto a precolumn (75-μm i.d.), which consisted of a self-made frit and 2.5 cm of in-house packed Luna C18 (3-μm) material (Phenomenex, Torrence, CA) followed by separation on a 75-μm i.d. fused silica microcapillary column (Polymicro Technologies, Phoenix, AZ), which was pulled to a fine tip using a P-2000 laser puller (Sutter Instruments, Novato, CA) and packed in-house with 10 cm of Luna C18 (3-μm) material (Phenomenex). Tryptic peptides were eluted over a 105 min gradient at a flow rate of 300 nl/min using a water/acetonitrile (ACN) gradient (Solvent A, 95% water, 5% ACN and 0.1% FA; Solvent B, 95% ACN, 0.1% FA in water), consisting of 1 min 2% B, 2 mins change to 6% B, 72 mins gradient to 24% B, 16 mins to 90% B, followed by 5 mins hold at 90% B, 1 min gradient to 2% B, with final hold at 2% B for 8 mins. Full MS spectra were acquired in profile mode in the Orbitrap analyzer with a high resolution (r = 60,000) over a mass range of m/z 400–2000. Up to the 10 most intense ions in each full MS scan were sequentially isolated, fragmented, and analyzed in centroid mode within the linear ion trap part of the instrument, and monoisotopic precursor selection was enabled to preferentially select 2+ and 3+ charge states for fragmentation. Dynamic exclusion was enabled with a repeat count of 1, a repeat duration of 22.5 s, an exclusion list of 500 and exclusion duration of 20 s. All FASP samples were subjected to triplicate LC-MS/MS analysis.
Database Search and Protein Identification
Tandem mass spectra were extracted from .raw files and searched using the SEQUEST-PVM v.27 (rev.9) (47) database program against a mouse protein database downloaded as FASTA-formatted sequences from EBI-IPI (database v. 3.72), which contains 56,957 entries (with priority given to UniProt identifiers) as well as reverse decoy sequences to empirically assess the false identification rate. This search program was executed on a cluster computer to match the MS/MS spectra to the corresponding most highly correlated peptide sequences. Mass tolerances for precursor (MS) and product ions (MS/MS) were set to 3 and 0 m/z, respectively. Searches were performed with the enzyme selectivity set to trypsin with one missed cleavage allowed and protein modifications included fixed carbamidomethylation of cysteines (57 Da). Match likelihoods were assigned a statistical confidence score using the STATQUEST probabilistic model (48) and candidate peptide identifications were filtered using an estimated peptide confidence score of ≥ 95%. A 10 ppm high accuracy mass filter accounting for isotopic shifts in the spectra was applied post-SEQUEST analysis improving the fidelity of protein identifications.
Protein Quantification and Statistical Analysis
To estimate relative protein levels, spectral counts were transformed into normalized spectral abundance factors (NSAF) as previously described (49). Briefly, this involves dividing the spectral count (SC) of a protein by its length (Mw) normalizing this value to the sum of all SC/Mw for all proteins in each respective data set. To test for statistically significant abundance changes (p value ≤ 0.05), the Kolmogorov-Smirnov test (KS-test) was applied using R-studio v.0.94.011. Log2-fold changes of differentially abundant proteins were calculated by dividing the average EAE NSAF value by the corresponding sham value. In addition, label-free quantification was performed at the MS1-level using Progenesis LC-MS software v.4.1 (Nonlinear dynamics, Newcastle, U.K.). An extended description of the parameters used to perform Progenesis LC-MS analysis can be found in supplemental Methods.
Bioinformatics Analysis
The TMHMM 2.0 (50) software was used to predict the number of transmembrane helices (TMHs) for each putative membrane protein identified within the comparative PBMC study (http://www.cbs.dtu.dk/services/TMHMM/). Proteins with at least one TMH (exclusive of signal peptides) were considered integral membrane proteins (IMPs). Grand average of hydrophobicity (GRAVY) values of proteins were obtained using the GRAVY calculator tool (http://www.gravy-calculator.de/).
Pathway Enrichment Analyses
Two types of pathway enrichment analyses were performed on the spinal cord and PBMC spectral counting data sets to highlight common pathways, processes, or themes in the aberrant protein expression between disease and sham-induced mice. These tools include Gene Set Enrichment Analysis (GSEA) (http://www.broadinstitute.org/gsea/index.jsp) (51) and DAVID Bioinformatics Resources (http://david.abcc.ncifcrf.gov/) (52). An extended description of the parameters used to perform pathway analysis can be found in supplemental Methods. To better visualize the enrichment results, the output files from GSEA and DAVID were imported into Cytoscape network analysis environment via the Enrichment Map plugin (53).
Quantitative Western Blot Analysis
Soluble spinal cord lysates derived from EAE and sham mice (∼30 μg) were subjected to replicate SDS-PAGE analysis under denaturing conditions and electrophoretically transferred onto nitrocellulose membranes (GE Healthcare, Piscataway, NJ).Following transfer, the membranes were subsequently prepared using the Mouse Western blot kit (eBioscience, San Diego, CA) as per manufacturer's instructions. Briefly, Nitrocellulose membranes were blocked in an excess of TrueBlot blocking buffer (5% (w/v) skim milk powder, 0.05% TBS/Tween 20 (TBST)) for two hours at RT with gentle shaking. The following primary antibodies were also diluted in the blocking buffer and were either incubated for 1 h with gentle shaking (rabbit polyclonal anti-GAPDH, ab9485, AbCam Inc., 1:2500 dilution) or overnight at 4 °C with gentle shaking (mouse monoclonal anti-Ezrin, CPTC-Ezrin-1, DSHB, 1:100 dilution; mouse monoclonal anti-Moesin CPTC-MSN-1-s, DSHB, 1:100 dilution; mouse monoclonal anti-CLIC1, CPTC-CLIC1–1-s, DSHB, 1:100 dilution; mouse monoclonal anti-PSD93, Chapsyn-110, clone N18/30, NeuroMab 1:2000 dilution). After washing the membranes thoroughly in 0.05% TBST, the secondary anti-mouse IgG TrueBlot (eBioscience,1:1000 dilution) horseradish peroxidase-conjugated (HRP)-conjugated antibody was added to the respective membranes and incubated for 1 h at RT. Chemiluminescent detection of the membranes was performed using TrueBlot ECL reagents (eBioscience) and developed on film. The pixel intensities of the immunoreactive protein bands were calculated using Image Quant TL software v. 7.0 (GE Healthcare) and the relative densities were normalized against the total amount of GAPDH from the respective gels. Statistical analysis was performed using GraphPad Prism v. 5.0 (Graphpad Software Inc, San Diego, CA) with an unpaired two-tailed Student's t test and results were expressed as means ± S.E. of the mean (S.E.).
Label-free MRM-based Validation of Candidate Biomarkers
MRM transitions were designed for the ∼200 most differentially abundant proteins (absolute log2 ratio ≥1) from the spinal cord analysis and the top ∼100 from the PBMC data set using the open source software Skyline v.1.4 (54) (University of Washington, Seattle, WA). Three to four peptides per protein, and four transitions per peptide were selected primarily based on experimentally derived discovery data obtained on the Orbitrap Velos (Thermo Fisher Scientific) and 5600 TripleTOF (AB SCIEX, Concord, Canada) mass spectrometers. Some peptides were also derived in silico based on the online databases GPMDB (http://gpmdb.thegpm.org/index.html) and PeptideAtlas (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/GetProtein).Peptides (10 μl, ∼2 μg) derived from the original FASP analysis in the discovery phase, were analyzed using a nanoLC-2Dplus in combination with a cHiPLC™-nanoflex system (Eksigent) coupled to a 5500 QTRAP® mass spectrometer (AB SCIEX) operated at unit/unit resolution using the following experimental conditions: peptides were loaded onto a microfluidic trap column packed with ChromXP C18-CL 3 μm particles (120 Å nominal pore size; equilibrated in 0.1% FA/2% ACN) at 5 μl/min using an Eksigent NanoFlex cHiPLC system. An analytical (15 cm × 75 μm ChromXP C18-CL 3) microfluidic column was then switched in line and peptides were separated using linear gradient elution of 0–98% ACN over 60 mins (300 nl/min). Three biological replicates from each of the sham and EAE groups (both spinal cord and PBMC samples) were run in duplicate.
Three scheduled MRM methods were generated: one containing the transitions targeting proteins up-regulated in the diseased spinal cord (1186 transitions, 101 proteins, supplemental Table S11), another containing those down-regulated (1026 transitions, 93 proteins, Supplemental Table S12) and one method containing all the up-/down-regulated PBMC proteins (939 transitions, 74 proteins, supplemental Table S13). Each method was run with a target cycle time of 2.7 s, a retention-time window of 240 s, an interscan delay of 3 ms with information-dependent acquisition (IDA) criteria set to trigger full mass MS/MS spectra in full enhanced product ion (EPI) mode for the top two MRM transitions that exceeded 1000 counts. The IS voltage was set at 2300 V, curtain gas at 20 L/min, ion source gas at 20 L/min, high collision gas (CAD), and an interface heater temperature setting of 125 °C. A scan rate of 1000 Da/s was used for the EPI scan with a scanning m/z range of 100 to 1000 Da, a dynamic fill time, and the Q1 resolution set to unit. Optimal parameters for declustering potential (DP) and collision energy (CE) for an AB SCIEX instrument were taken from the recommended settings in Skyline. All data acquisition parameters used in these label-free MRM experiments can be found in (supplemental Tables S11–S13).
Peptide Quantitation by Label-free MRM
The acquired MRM data were imported into Skyline v.1.4 to visually assess extracted ion chromatogram, XIC) quality (intensity, noise level) and to facilitate the integration of all transitions. In addition, experimental MS/MS data were manually inspected using the PeakView software v.1.2.0.3 (AB SCIEX) to confirm the presence of targeted product ions thus confirming the correct peptide sequences were targeted supplemental Fig. S9. Finally, an exportable table (.csv file) containing the following sample information was generated: protein name, peptide sequence, replicate name, peptide and transition (precursor/product ion) information, peak area of transition. Microsoft Excel was then used to sum total areas of each of the four transitions per peptide quantified. To test for statistical differences in peak area between EAE and sham, an unpaired two-tailed Student's t test was performed for each peptide using GraphPad Prism v.5.0 (Graphpad Software Inc.) with results expressed as mean ± S.E. To account for multiple hypothesis testing (55), the Benjamini-Hochberg and Yeketuli false-discovery rate methods were used in R-studio v.0.94.011 to correct the t test p values for each peptide quantified by label-free MRM within individual experiments (experiment one: UP_SCORD; experiment 2: DOWN_SCORD; experiment 3: UP_DOWN_PBMCs). Further information as to how the label-free MRM experiments were performed and analyzed can be found in supplemental Methods.
RESULTS
Label-free Peptide Quantification Reveals Novel Disease Markers of EAE
The focus of this study was to identify candidate biomarkers within disease-affected tissues (spinal cord and PBMCs) of EAE-induced mice that distinguish between the sham-injected counterparts at the disease peak (15 dpi). A label-free quantitative proteomics approach involving two complementary forms of relative quantitation was selected: intensity-based (MS1) and fragmentation-based (MS2) (Fig. 1). Spectral counting analysis (MS2-based quantification) involved normalizing the peptide abundances via NSAF values that were then statistically compared between the EAE and sham-induced mice by the KS-test. The KS-test is ideally suited to proteomics studies because it makes no assumptions as to the underlying data distribution, in contrast to the t test, which assumes the data are normally distributed (56). We also compared the quantitation achieved by MS1 precursor intensities as an estimate of protein abundance. To achieve this, a commercially available software tool for analysis of label-free proteomics data at the MS1 level (Progenesis LC-MS) was used. Principal component analysis (PCA) was performed on the spinal cord and PBMC data acquired by the dual approaches to visually assess levels of variability and the overall groupings. As demonstrated in supplemental Fig. S2, there is a clear separation between the NSAF values from the diseased and control mice for both the spinal cord and PBMC tissues, with minimal technical and biological variability. The PCA plots of the peptide- and protein-level data from the spinal cord and PBMCs in Progenesis also illustrate good clustering (an indicator of low variance) in the biological replicates of each group (supplemental Fig. S3), in agreement with the spectral counting data.
Fig. 1.

Overview of the experimental design and label-free proteomics workflow used to identify candidate biomarkers within disease-affected tissues of the EAE and sham-induced mice. The soluble spinal cord extract and membrane-enriched fraction of the PBMCs were prepared for mass spectrometry analysis using the FASP method and the label-free data were acquired on a high resolution Orbitrap Velos mass spectrometer. Quantification of the data was performed by both intensity-based (MS1) and fragmentation-based (MS2) approaches. In the schematic illustration, there is a twofold difference in protein expression between theoretical samples A and B. Relative quantification takes place at the MS1 level via peak quantitation using the commercial Progenesis LC-MS software and differentially expressed proteins are quantified in the subsequent fragmentation step. Spectral counting quantitation and peptide identification occur simultaneously at the MS/MS (MS2) level where approximately double the number of spectra are identified for sample A.
A total of ∼1400 unique proteins (95% peptide confidence, 10 ppm mass filter) were identified from the comparative proteomics analysis of the spinal cord samples with a peptide FDR of 0.55%. After statistical evaluation of the NSAF data, 431 differentially abundant proteins were identified (KS-test, p value ≤ 0.05), with 197 proteins up-regulated and 234 down-regulated (Fig. 2A; supplemental Tables S1–S6). Of these 431 proteins, 233 exhibited a statistically significant ≥twofold change in expression, equivalent to a log2 ratio ≥1, with 120 proteins significantly up-regulated and 113 proteins down-regulated (Fig. 3A, Table I). In the PBMC comparison, just over 1000 unique protein identifications were made (95% peptide confidence, 10 ppm mass filter) with a peptide FDR of 0.61% and 207 statistically significant changes were discovered. Spectral counting analysis (KS-test, p value ≤ 0.05) revealed 122 proteins up-regulated and 85 down-regulated (Fig. 2B; supplemental Tables S7–S8). Overall 116 out of 207 proteins exhibited a statistically significant ≥twofold change in expression, with 55 proteins significantly up-regulated and 61 down-regulated (Fig. 3B, Table II). A total of 226 IMPs (containing 1 or more TMH) were identified by proteomic analysis of membrane-enriched PBMCs based on the TMHMM 2.0 software (50) with an average GRAVY score of −0.04 (supplemental Fig. S4). The GRAVY score is the sum of hydrophobicity values for each of all the amino acids in the protein, divided by the protein length (57).
Fig. 2.

Plot of the distribution of log2 ratio values representing differentially abundant proteins between EAE and sham-induced mice by relative protein quantitation via spectral counting. Representations of the proteins up-regulated (positive log2-fold changes) and down-regulated (negative log2-fold changes) in disease from the comparative proteomic analysis of (A) spinal cord and (B) membrane protein-enriched fraction of PBMCs.
Fig. 3.

Diagrammatic representation of the quantitative proteomics results obtained from label-free comparative analyses of EAE and sham-induced mice using MS1- and MS2-based approaches. A, Of the ∼1400 unique proteins identified in the spinal cord data set, 431 were deemed differentially abundant by spectral counting analysis, whereas 359 were found by Progenesis software. B, Of the ∼1000 unique proteins identified in the PBMC data set, 207 were deemed differentially abundant by spectral counting analysis, whereas 139 were found by Progenesis software.
Table I. A subset of proteins exhibiting differential expression in a comparative analysis of spinal cords isolated from EAE and sham-induced mice. Proteins were identified by label-free quantitation either by means of spectral counting or by comparisons of MS1-level spectra. Statistically significant proteins were determined by p ≤ 0.05. MRM-based validation was then performed on significantly changing proteins exhibiting log2-fold change of ≥1. NA refers to proteins that were not analysed by MRM. # UPEPS refers to the number of unique peptides used to make the protein identifications or were quantified by MRM. References are taken from previous studies on EAE and MScl.
| UniProt ID | Gene ID | Protein description | Spectral counting (MS2) |
Progenesis LC-MS (MS1) |
MRM |
References |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p value (KS-test) | %CV | log2-fold Δ | # UPEPS |
p value (ANOVA) | Max. fold Δ | # UPEPS for quant. | p value ≤ 0.05 (t-test), # UPEPS | ||||||
| EAE | Sham | EAE | MScl | ||||||||||
| ↑ EAE | |||||||||||||
| P07724 | Alb | Serum albumin | 4.11E-05 | 28.0 | 4.2 | 51 | 13 | 5.98E-04 | 17.0 | 14 | ✓, 3 | (24, 27, 29, 88, 89) | (30) |
| P29699 | Ahsg | Alpha-2-HS-glycoprotein | 2.47E-04 | 29.0 | 10.2 | 8 | 0 | 1.20E-03 | 10.1 | 3 | ✓, 3 | (24, 28) | (17, 90) |
| P12246 | Apcs | Serum amyloid P-component | 3.36E-04 | 4.19 | 1 | NA | (26) | (91) | |||||
| Q61176 | Arg1 | Arginase-1 | 2.47E-04 | 75.8 | 9.2 | 7 | 0 | 6.59E-03 | 5.44 | 3 | ✓, 3 | (19, 21, 92) | |
| P01887 | B2m | Beta-2-microglobulin | 3.96E-04 | 93.3 | 1 | NA | (25, 28) | (30) | |||||
| P01027 | C3 | Isoform Long of Complement C3 (Fragment) | 2.47E-04 | 52.4 | 9.2 | 26 | 0 | 1.54E-03 | 8.41 | 10 | ✓, 3 | (19, 21, 23, 24, 28, 93) | (11, 13, 94–98) |
| O35744 | Chi3l3 | Chitinase-3-like protein 3 | 1.63E-03 | 62.6 | 8.6 | 6 | 0 | 9.07E-03 | 8.13 | 6 | ✓, 3 | (19, 23) | |
| Q9Z1Q5 | Clic1 | Chloride intracellular channel protein 1 | 8.64E-03 | 82.3 | 8.1 | 4 | 0 | 8.02E-04 | 5.5 | 2 | ✓, 3 | (22, 26, 29) | |
| P26040 | Ezr | Ezrin | 2.47E-04 | 28.1 | 7.4 | 2 | 0 | 5.65E-03 | 3.09 | 1 | ✓, 2 | ||
| Q6WVG3 | Kctd12 | BTB/POZ domain-containing protein KCTD12 | 8.64E-03 | 56.9 | 6.7 | 1 | 0 | ✓, 3 | |||||
| P16110 | Lgals3 | Galectin-3 | 3.66E-02 | 86.2 | 7.4 | 1 | 0 | 1.34E-03 | 3.10 | 1 | ✓, 1 | (19, 21, 99) | (100) |
| P26041 | Msn | Moesin | 4.11E-05 | 16.4 | 1.1 | 19 | 12 | 5.36E-04 | 2.50 | 8 | ✓, 3 | (19, 24) | (101) |
| P21614 | Gc | Vitamin d-binding protein | 1.63E-03 | 61.1 | 8.5 | 5 | 0 | 6.32E-03 | 17.2 | 1 | ✓, 3 | (28) | (16, 17, 91) |
| Q99KQ4 | Nampt | Nicotinamide phosphoribosyltransferase | 2.47E-04 | 59.8 | 4.2 | 9 | 1 | 9.10E-04 | 3.17 | 4 | ✓, 2 | ||
| Q62422 | Ostf1 | Osteoclast-stimulating factor 1 | 3.66E-02 | 97.4 | 7.8 | 2 | 0 | ✓, 1 | |||||
| P29351-2 | Ptpn6 | Isoform 2 of Tyrosine-protein phosphatase nonreceptor type 6 | 3.66E-02 | 80.5 | 6.4 | 3 | 0 | 3.42E-03 | 3.69 | 3 | ✓, 2 | ||
| P42225 | Stat1 | Signal transducer and activator of transcription 1 | 2.47E-04 | 21.7 | 9.9 | 17 | 0 | 3.93E-06 | 4.97 | 7 | ✓, 3 | (19, 21, 23) | (12) |
| ↓ EAE | |||||||||||||
| Q8C8R3-2 | Ank2 | Isoform 2 of Ankyrin-2 | 8.64E-03 | 20.6 | −1.3 | 4 | 9 | 9.20E-04 | −2.26 | 3 | ✓, 3 | ||
| Q8R3P0 | Aspa | Aspartoacylase | 8.64E-03 | 22.4 | −1.1 | 4 | 6 | ✓, 3 | |||||
| Q06138 | Cab39 | Calcium-binding protein 39 | 8.64E-03 | 52.7 | −1.8 | 1 | 3 | 1.69E-03 | −4.05 | 2 | ✓, 3 | ||
| Q91XM9-1 | Dlg2 | Isoform 1 of Disks large homolog 2 | 2.47E-04 | 1.88 | −5.5 | 0 | 1 | ✓, 1 | |||||
| P11499 | Hsp90ab1 | Heat shock protein HSP 90-beta | 5.43E-05 | −3.22 | 3 | NA | |||||||
| O89112 | Lancl1 | LanC-like protein 1 | 1.63E-03 | 36.9 | −3.2 | 1 | 4 | 2.45E-03 | −1.58 | 1 | ✓, 3 | ||
| Q9JJK2 | Lancl2 | LanC-like protein 2 | 8.64E-03 | 29.8 | −2.2 | 1 | 2 | ✓, 1 | |||||
| P63085 | Mapk1 | Mitogen-activated protein kinase 1 | 1.10E-03 | −2.38 | 3 | NA | |||||||
| Q80W80 | Mapk10 | JNK3 beta2 protein kinase | 2.47E-04 | 35.4 | −6.7 | 0 | 1 | ✓, 1 | |||||
| P04370-4 | Mbp | Isoform 4 of Myelin basic protein | 2.47E-04 | 38.3 | −10.3 | 0 | 4 | ✓, 1 | |||||
| Q91XL9 | Osbpl1a | Oxysterol-binding protein-related protein 1 | 1.63E-03 | 33.6 | −4.7 | 1 | 4 | 6.70E-03 | −2.35 | 1 | ✓, 3 | ||
| P70296 | Pebp1 | Phosphatidylethanolamine-binding protein 1 | 1.27E-04 | −3.34 | 1 | NA | |||||||
| Q9QZ06 | Tollip | Toll-interacting protein | 8.64E-03 | 79.6 | −7.8 | 0 | 3 | 1.25E-02 | −1.87 | 1 | ✓, 2 | ||
Table II. A subset of proteins exhibiting differential expression in a comparative analysis of membrane-enriched PBMCs isolated from EAE and sham-induced mice. Proteins were identified by label-free quantitation either by means of spectral counting or by comparisons of MS1-level spectra. Statistically significant proteins were determined by p ≤ 0.05. MRM-based validation was then performed on significantly changing proteins exhibiting log2-fold change of ≥1. NA refers to proteins that were not analyzed by MRM. # UPEPS refers to the number of unique peptides used to make the protein identifications or were quantified by MRM. References are taken from previous studies on EAE and MScl. # refers to the proteins identified with increased abundance in the diseased spinal cord by MS1- and/or MS2-based quantitation.
| UniProt ID | Gene ID | Protein description | Spectral counting (MS2) |
Progenesis LC-MS (MS1) |
MRM |
References |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p value (KS-test) | %CV | Log2-fold Δ | # UPEPS |
p value (ANOVA) | Max fold Δ | # UPEPS for quant. | p value < 0.05 (t-test), # UPEPS | ||||||
| EAE | Sham | EAE | MScl | ||||||||||
| ↑ EAE | |||||||||||||
| O08538 | Angpt1 | Angiopoietin-1 | 3.66E-02 | 100 | 8.6 | 5 | 0 | ✓, 2 | (102) | ||||
| D3YVS6 | AU023871 | ITIM-receptor G6b-B | 8.64E-03 | 20.1 | 1.2 | 3 | 3 | ✓, 2 | |||||
| O08529 | Capn2 | Calpain-2 catalytic subunit | 2.47E-04 | 32.3 | 2.9 | 7 | 2 | ✓, 1 | (103) | (104) | |||
| Q9JL99 | Clec1b | C-type lectin domain family 1 member B | 3.36E-02 | 25.1 | 0.60 | 7 | 7 | 1.74E-02 | 1.64 | 2 | NA | ||
| P11276 | Fn1 | Fibronectin | 3.66E-02 | 71.9 | 2.7 | 4 | 3 | ✓, 1 | (105) | ||||
| P63038 | Hspd1 | Isoform 1 of 60 kDa heat shock protein, mitochondrial | 4.11E-05 | 20.8 | 1.5 | 24 | 16 | 2.84E-02 | 3.13 | 9 | ✓, 3 | ||
| P25911-2 | Lyn | Isoform LYN B of Tyrosine-protein kinase Lyn | 3.36E-02 | 29.8 | 0.50 | 16 | 14 | 2.11E-03 | 1.67 | 4 | NA | ||
| B1ARB3 | Pecam1 | Platelet endothelial cell adhesion molecule | 3.66E-02 | 63.7 | 2.8 | 4 | 2 | ✓, 1 | (106–108) | ||||
| Q62087 | Pon3 | Serum paraoxonase/lactonase 3 | 4.11E-05 | 17.2 | 1.2 | 6 | 3 | ✓, 1 | |||||
| P35441 | Thbs1 | Thrombospondin 1 | 4.11E-05 | 22.6 | 0.88 | 36 | 30 | 3.32E-02 | 1.47 | 17 | ✓, 1 | (102, 109) | (110) |
| Q3UZG1 | Trpc6 | Short transient receptor potential channel 6 | 8.64E-03 | 50.4 | 2.4 | 1 | 1 | ✓, 2 | |||||
| Q07235 | Serpine2 | Glia-derived nexin | 4.11E-05 | 27.6 | 1.7 | 8 | 6 | 1.77E-02 | 2.00 | 4 | ✓, 3 | (111) | |
| A3KGU9 | Spna2 | Spectrin alpha 2 | 7.40E-04 | 47.9 | 2.1 | 13 | 6 | ✓, 2 | |||||
| Q62261-2 | Sptbn1 | Isoform 2 of Spectrin beta chain, brain 1 | 7.40E-04 | 20.8 | 1.50 | 29 | 12 | 6.84E-03 | 1.64 | 18 | ✓, 3 | ||
| ↓ EAE | |||||||||||||
| P10107 | Anxa1 | Annexin A1 | 3.36E-02 | 31.0 | −1.0 | 7 | 10 | 3.66E-02 | −2.60 | 5 | ✓, 3 | ||
| P01027-1 | C3 # | Isoform Long of Complement C3 (Fragment) | 2.47E-04 | 47.7 | −3.4 | 3 | 9 | 2.58E-02 | −1.97 | 6 | ✓, 3 | ||
| Q3U1U4 | Itgam | Integrin alpha-M | 8.64E-03 | 60.7 | −2.3 | 1 | 7 | ✓, 3 | |||||
| P11672 | Lcn2 | Neutrophil gelatinase-associated lipocalin | 8.64E-03 | 55.7 | −2.3 | 2 | 5 | 3.06E-02 | −3.64 | 1 | ✓, 2 | (112) | |
| Q61233 | Lcp1# | Plastin-2 | 2.74E-04 | 45.3 | −3.8 | 3 | 14 | 9.17E-03 | −2.93 | 5 | ✓, 4 | ||
| P08071 | Ltf | Lactotransferrin | 8.64E-03 | 70.6 | −2.4 | 7 | 21 | 1.22E-02 | −3.14 | 3 | ✓, 4 | ||
| P26041 | Msn# | Moesin | 6.29E-03 | 25.9 | −1.1 | 3 | 8 | 1.29E-02 | −1.71 | 5 | ✓, 2 | ||
| Q62422 | Ostf1# | Osteoclast-stimulating factor 1 | 3.66E-02 | 78.7 | −7.9 | 0 | 2 | 2.45E-02 | −3.27 | 1 | ✓, 2 | ||
| Q61096 | Prtn3 | Myeloblastin | 8.64E-03 | 48.7 | −2.3 | 2 | 3 | 4.52E-02 | −3.90 | 2 | ✓, 2 | ||
| P27005 | S100a8 | Protein S100-A8 | 8.64E-03 | 39.4 | −1.8 | 3 | 3 | ✓, 2 | |||||
| P31725 | S100a9 | Protein S100-A9 | 1.63E-03 | 43.0 | −2.1 | 2 | 3 | 2.86E-02 | −4.19 | 1 | ✓, 2 | ||
| Q921I1 | Trf # | Serotransferrin | 2.47E-04 | 47.9 | −3.3 | 3 | 9 | 4.61E-4 | −2.94 | 5 | ✓, 3 | ||
| Q62351 | Tfrc | Transferrin receptor protein 1 | 2.47E-04 | 28.3 | −10.0 | 0 | 12 | 3.70E-03 | −9.00 | 5 | ✓, 3 | (113) | |
After statistical analysis of the Progenesis data, ∼360 differentially abundant proteins were identified in the spinal cord data set with one-way ANOVA p values ≤ 0.05, including 164 proteins significantly up-regulated and 195 down-regulated. A total of 183 proteins exhibited a ≥twofold change in expression, with 95 of these identified from all three technical replicates (Fig. 3A; supplemental Table S4). A total of 88 out of the 197 proteins originally identified as being up-regulated in the diseased spinal cord by spectral counting analysis were validated by MS1-based quantification. Moreover, an additional 69 novel proteins were identified as being up-regulated, such as serum amyloid P-component (Apcs), beta-2-microglobulin (B2m), and leukotriene A-4 hydrolase (Lta4h). A total of 92 out of the 234 proteins identified as being down-regulated in the diseased spinal cord by spectral counting analysis were validated by MS1-based quantification with an additional 103 novel proteins identified, including mitogen-activated protein kinase 1 (Mapk1), phosphatidylethanolamine-binding protein 1 (Pebp1), and heat shock protein HSP 90-beta (Hsp90ab1) (Table I).
After statistical analysis of the PBMC data from Progenesis, 139 differentially abundant proteins were identified with one-way ANOVA p values ≤ 0.05, including 43 proteins significantly up-regulated and 96 down-regulated. A total of 74 proteins exhibited a ≥twofold change in expression, with 43 of these identified from all three technical replicates (Fig. 3B; supplemental Table S9–S10). A total of 39 out of the 122 proteins identified as being up-regulated in diseased PBMCs by spectral counting analysis were validated by MS1-based quantification and an additional three novel proteins were identified. A total 56 out of the 85 proteins identified as being down-regulated in diseased PBMCs by spectral counting were validated by MS1-based quantification with an additional 23 novel down-regulated proteins identified. In comparison to previous quantitative proteomic studies on the spinal cord or CSF from rodents with EAE, there is an overlap of 44 proteins exhibiting a ≥twofold increase in expression from this study including many abundant blood proteins such as fetuin A, hemopexin, ceruloplasmin, and inflammatory markers beta-2-microglobulin and Ig gamma-2A chain C region (Fig. 4). However, there are more than 130 proteins that were exclusively identified in this study by MS1- and/or MS2-based quantitation, many which have yet to be linked to EAE or MScl. Moreover, there are 30 proteins that were originally found to be up-regulated in the diseased spinal cord by either MS1- and/or MS2-based quantitation and were also identified as being down-regulated within the diseased PBMCs, including complement C3, Ostf1, and serotransferrin (Table II).
Fig. 4.

Triple Venn diagrams illustrating the relationship between the numbers of candidate disease-specific markers identified within the diseased spinal cord/CSF from various quantitative proteomic analyses. Overall there are 44 proteins in common including 6 well-validated markers of EAE: complement C3, fetuin A, hemopexin and ceruloplasmin, beta 2 microglobulin, and Ig gamma-2A chain C region. However, a large proportion of proteins identified in this study by MS1- and MS2-based quantitation (exhibiting a ≥2-fold change in relative expression) that have yet to be linked to EAE or multiple sclerosis. Differences in the number of proteins observed between the three studies may be due the use of different rodent EAE models (MOG versus MBP-induced), varying capabilities of the mass spectrometry instrumentation and alternative methods of protein quantification.
Pathway enrichment analyses of the spectral counting data were performed by means of Gene Set Enrichment Analysis (GSEA) and the functional annotation tool from DAVID Bioinformatics Resources. They revealed the most statistically relevant and over-represented (enriched) biological annotations in the diseased spinal cord to be processes involving the immunoproteasome, complement and coagulation cascades and regulation of the actin cytoskeleton. The pathways that appear to be down-regulated relate to synaptic transmission involving neurotransmitter release and endocytosis (Fig. 5A, Table III). In the PBMC analysis, biological processes enriched in EAE mice include extracellular matrix remodeling and focal adhesion whereas those down-regulated include gene expression and DNA/RNA processing, antigen presentation and the mitochondrial respiratory chain (Fig. 5B, Table IV). These observations correlate well with the neuroinflammatory model of the disease (Fig. 6). The first stage of this model involves the activation of myelin-directed T helper cells by APCs in peripheral lymphoid organs, such as lymph nodes. The activated T cells then leave the lymphoid tissue and enter the systemic circulation via a permeable BBB, mediated by interactions between the cell adhesion molecules expressed by leukocytes such as integrins and those on the endothelium. T cells are subsequently re-activated in the CNS on MHC II molecules expressed on local APCs. This influx of activated T cells triggers a cascade of inflammatory responses (the activation of B cells and plasma cells to produce antibodies, the recruitment of other blood-derived immune cells, such as lymphocytes and monocytes, which then differentiate into macrophages and DCs. These cells signal the activation of microglia and the direct attack of the myelin sheath and the neuronal cells by T cells, through the production of inflammatory cytokines, which eventually leads to demyelination and neuronal cell death. Several proteins involved in mediating the entry of activated lymphocytes across the BBB were found to be elevated in the diseased spinal cord and the membrane-enriched fraction of PBMCs.
Fig. 5.

Enrichment map representations of the GSEA results obtained for the comparative proteomics analyses of spinal cord and membrane protein-enriched PBMC fraction isolated from EAE and sham-induced mice. Biological processes that are perturbed in (A) the spinal cord and (B) PBMCs are annotated. Node color and shading intensity represents the statistical significance of enrichment of a particular gene set, whereas larger node sizes indicate that more genes have contributed.
Table III. KEGG pathways significantly dysregulated in the comparative analysis of spinal cords isolated from EAE and sham-induced mice. Pathways were identified by DAVID Functional Annotation analysis and were ranked according to p value as determined by the EASE score. Counts and percentages (%) refer to the number and % of genes from the original input list that are categorized into given KEGG pathways. Fold enrichment refers to the magnitude of enrichment for each KEGG pathway as compared to the entire mouse genome which serves as the reference. The Benjamini scores are calculated to globally correct enrichment p values that were considered to be statistically significant if p ≤ 0.05.
| KEGG pathway | Count | % | p value | Fold enrichment | Benjamini | Genes |
|---|---|---|---|---|---|---|
| From genes ↑ EAE spinal cord | ||||||
| Proteasome | 9 | 0.32 | 5.56E-07 | 11.9 | 5.00E-05 | PSMB10, PSMC5, PSMA6, PSMB1, PSME1, PSME2, PSMD1, PSMC1, PSMA3, PSME2B-PS |
| Regulation of actin cytoskeleton | 13 | 0.46 | 1.42E-04 | 3.7 | 6.37E-03 | ACTB, ACTN4, ACTN1, MYH9, VCL, PFN1, ARPC1B, EZR, GSN, RAC1, CFL1, RHOA, MSN |
| Complement and coagulation cascades | 8 | 0.28 | 1.65E-04 | 6. 7 | 4.95E-03 | KNG1, FGG, FGA, SERPINA1B, FGB, C3, SERPINA1D, C1QC |
| Adherens junction | 7 | 0.25 | 1.21E-03 | 5.7 | 2.68E-02 | ACTB, PTPN6, ACTN4, RAC1, RHOA, ACTN1, VCL |
| Leukocyte transendothelial migration | 8 | 0.28 | 2.65E-03 | 4. 2 | 4.67E-02 | ACTB, EZR, ACTN4, RAC1, RHOA, ACTN1, MSN, VCL |
| From genes ↓ EAE spinal cord | ||||||
| Citrate cycle (TCA cycle) | 7 | 0.19 | 1.74E-05 | 12.1 | 1.90E-03 | SDHA, IDH3G, ACO1, ACLY, FH1, SUCLA2, MDH1 |
| Alanine, aspartate and glutamate metabolism | 6 | 0.17 | 2.31E-04 | 6.3 | 1.05E-02 | GLUL, ASPA, GOT1, GLUD1, ADSL, ABAT |
| Arginine and proline metabolism | 7 | 0.19 | 3.91E-04 | 7.1 | 1.06E-02 | GLUL, ALDH7A1, GOT1, CKMT2, GLUD1, CKMT1, ALDH2 |
| Pyruvate metabolism | 6 | 0.17 | 8.64E-04 | 7.8 | 1.87E-02 | ALDH7A1, PKM2, LOC100047228, ALDH2, GRHPR, ACAT2, MDH1 |
| Propanoate metabolism | 5 | 0.14 | 2.08E-03 | 8.9 | 3.72E-02 | ALDH7A1, ALDH2, ABAT, SUCLA2, ACAT2 |
| Glyoxylate and dicarboxylate metabolism | 4 | 0.11 | 2.88E-03 | 13.4 | 4.39E-02 | PGP, ACO1, GRHPR, MDH1 |
| Cysteine and methionine metabolism | 5 | 0.14 | 2.98E-03 | 8.1 | 3.99E-02 | GOT1, MAT2A, MTAP, AHCYL1, AHCYL2 |
| Endocytosis | 11 | 0.30 | 3.92E-03 | 2.9 | 4.65E-02 | DNM3, EPN3, AP2A2, AP2B1, AP2A1, DNAJC6, PDCD6IP, DNM1, SH3GL2, EHD3, AP2M1 |
Table IV. KEGG pathways significantly dysregulated in the comparative analysis of membrane-enriched PBMCs isolated from EAE and sham-induced mice. Pathways were identified by DAVID Functional Annotation analysis and were ranked according to p value as determined by the EASE score. Counts and percentages (%) refer to the number and % of genes from the original input list that are categorized into given KEGG pathways. Fold enrichment refers to the magnitude of enrichment for each KEGG pathway as compared to the entire mouse genome that serves as the reference. The Benjamini scores are calculated to globally correct enrichment p values, which were considered to be statistically significant if p ≤ 0.05.
| KEGG pathway | Count | % | p value | Fold enrichment | Benjamini | Genes |
|---|---|---|---|---|---|---|
| From genes ↑ EAE membrane-enriched PBMCs | ||||||
| ECM-receptor interaction | 10 | 0.48 | 9.19E-07 | 9.2 | 7.81E-05 | VWF, GP5, ITGA6, ITGA2, GP1BA, ITGB3, THBS1, ITGB1, ITGA2B, FN1 |
| Focal adhesion | 14 | 0.67 | 1.09E-06 | 5.4 | 4.63E-05 | ACTB, 2900073G15RIK, ITGA2, ITGB3, CAPN2, ITGB1, FLNA, PRKCB, VWF, ITGA6, LOC640441, GM6517, THBS1, PARVB, ITGA2B, FN1 |
| Butanoate metabolism | 7 | 0.33 | 6.35E-06 | 14.5 | 1.80E-04 | ACADS, OXCT1, PDHA1, HADH, ACAT1, HADHA, ALDH9A1 |
| Fatty acid metabolism | 7 | 0.33 | 2.05E-05 | 11.9 | 4.35E-04 | CPT2, ACADS, HADH, ACAA1A, ACAT1, HADHA, ALDH9A1 |
| Valine, leucine and isoleucine degradation | 7 | 0.33 | 2.33E-05 | 11.6 | 3.97E-04 | ACADS, OXCT1, HADH, ACAA1A, ACAT1, HADHA, ALDH9A1 |
| Citrate cycle (TCA cycle) | 6 | 0.29 | 4.07E-05 | 14.8 | 5.77E-04 | SDHA, IDH3B, PDHA1, FH1, OGDH, MDH2 |
| From genes ↓ EAE membrane-enriched PBMCs | ||||||
| Ribosome | 12 | 0.60 | 4.39E-11 | 16.9 | 1.58E-09 | RPL14, RPLP2, RPLP0, RPS27A, WDR89, UBA52, RPS28, RPL8, RPSA, RPS7, RPL30-PS2, UBC, UBB, RPL27A, RPL30, RPL24, RPL23 |
| Antigen processing and presentation | 9 | 0.45 | 3.91E-07 | 12.3 | 7.04E-06 | HSP90AB1, PDIA3, IFI30, CALR, TAPBP, PSME1, PSME2, PSME2B-PS, HSPA4, HSPA5 |
Fig. 6.
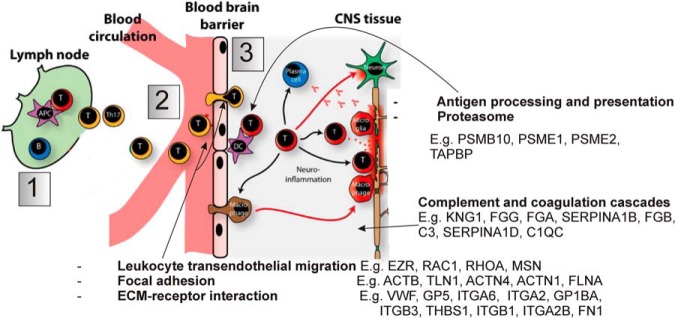
Schematic illustration of the neuroinflammatory model of demyelinating disease as is observed in EAE and MScl (1). Myelin-directed T helper cells are activated in the periphery by APCs. (2) The activated T cells enter the systemic circulation from the periphery via a permeable BBB, mediated by interactions between the cell adhesion molecules expressed by leukocytes such as integrins and those on the endothelium. (3) T cells are subsequently re-activated in the CNS on MHC II molecules expressed on local APCs. This influx of activated T cells triggers a cascade of inflammatory responses, ultimately leading to the recruitment of other blood-derived immune cells, such as lymphocytes and monocytes. These cells then differentiate into macrophages and DCs and produce inflammatory cytokines that act in concert with other stimuli to mediate demyelination and neuronal cell death. Some of the biological pathways found to be up-regulated in this study within the diseased spinal cord and the membrane-enriched fraction of PBMCs by gene enrichment analyses have been annotated. Fig. adapted from Ingwersen and colleagues (87).
Orthogonal Validation of Candidate Biomarkers by Western Blot Analysis
To further confirm the results obtained from the label-free proteomics analyses of the diseased and sham-induced spinal cord tissues, Western blot analysis was performed to measure the relative abundance of four differentially abundant proteins: moesin, ezrin, PSD93 or Dlg2 and Clic1. Bands of the expected molecular weights for these proteins were visualized in both EAE- and sham-induced controls (Fig. 7A). These data validate the observations made at the MS1- and MS2-level for ezrin and Clic1 during label-free quantitative proteomics. However, this observation did not reach statistical significance for moesin and Dlg2 (Fig. 7B). There is a clear shift in the amount of moesin present in the higher Mw band within the diseased mice compared with the sham. This difference suggests that the increased fold change detected by label-free mass spectrometry may be representative of the peptides derived from this higher Mw isoform.
Fig. 7.

Western blot analysis confirming the differential expression of select proteins in the spinal cord during EAE. A, Equal amounts of soluble spinal cord lysate (∼30 μg) derived from three biological replicates of each sham and EAE group were probed against Moesin (∼70 and 55 kDa), Ezrin (∼80 kDa), PSD93 or Dlg2 (110 kDa), Clic1 (∼27 kDa), and GAPDH (∼40 kDa). B, Relative densities of the bands were normalized to GAPDH from the same gel. Results are presented as mean ± S.E. for three biological replicates per group. A two-tailed t test was performed to identify significant differences between brain regions. * Significant difference with p ≤ 0.05; n.d. not detected.
Candidate Biomarker Validation by Targeted Mass Spectrometry (Label-free MRM)
A total of 194 out of 233 proteins exhibiting dysregulated protein expression (≥twofold change, p ≤ 0.05) in the spinal cord by spectral counting analysis were selected for targeted quantification by label-free MRM. Candidate proteins identified from Progenesis analysis were not included in MRM validation studies because they were identified subsequently as a means of establishing the reliability of MS2-based quantitation. A significant proportion of results (180/431 proteins in the spinal cord and 95/207 proteins in the PBMCs) overlapped between the spectral counting and Progenesis analyses. The differential abundance was confirmed for 136 of these proteins by the presence of one to four peptides with statistically significant differential expression (Student's t test, p ≤ 0.05) between EAE and sham groups (supplemental Figs. S5–S6). In contrast, comparable levels of six “housekeeping” proteins (i.e. proteins that were found to have negligible levels of differential expression by label-free proteomics analysis) were observed, confirming equal protein loading between samples. Examples of proteins with up-regulated expression confirmed in EAE spinal cord include Ostf1, moesin and ezrin (Fig. 8A) and those validated as being down-regulated include toll-interacting protein (Tollip), oxysterol-binding protein-related protein 1 (Osbpl1a) and lanC-like protein 1 (Lancl1) (Fig. 8B).
Fig. 8.


Targeted mass spectrometry analysis of a subset of differentially abundant proteins identified within the spinal cord tissue of EAE and sham-induced mice. Proteins validated to be (A) up-regulated and (B) down-regulated in EAE spinal cord or (C) “housekeeping” proteins (ENOB and PPIA) exhibiting similar levels of expression between sham and EAE mice Graphs are representative extracted ion chromatograms (XICs) for specific peptides derived from dysregulated proteins sampled from each group. Bar charts represent the average peak intensity (counts per second, cps) for peptides quantified in EAE (red) and sham (green) mice (three biological replicates per group, two technical replicates per peptide). A Student's t test was performed on the peak areas of each quantified peptide to test for statistical differences between EAE and sham with p values ≤ 0.05 considered statistically significant. n.s refers to peptides that were analyzed by MRM but were deemed not statistically significant.
From the PBMC analysis, 74 out of 116 proteins exhibiting dysregulated protein expression (≥twofold change) were similarly subjected to label-free MRM analysis. Similar levels of five “housekeeping” proteins were identified between samples, whereas differential abundance of 50 proteins was confirmed (supplemental Figs. S7–S8), including those up-regulated in disease such as short transient receptor potential channel 6 (Trpc6), glia-derived nexin (Serpine2), and angiopoietin-1 (Angpt1) (Fig. 9A), and others down-regulated in disease such as Ostf1, Transferrin receptor protein 1 (Tfrc) and Integrin alpha-M (Itgam) (Fig. 9B). Multiple hypothesis testing was performed on the t test p values calculated for each of the peptides quantified within individual MRM experiments. Only 17 out of 136 proteins previously determined to be differentially abundant in the spinal cord by an unpaired Student's t test were found not to be significantly different (p value ≥ 0.05) after applying the Benjamini-Hochberg and Yeketuli methods to control false discovery rates (supplemental Tables S14–S15). Examples include peptide/s derived from Calcium-binding protein 39 and Eukaryotic translation initiation factor 4H. Eight out of 50 proteins previously confirmed to be differentially abundant in the membrane protein-enriched fraction of PBMCs by unpaired Student's t-testing were also found not to be significantly different (p value ≥ 0.05) (supplemental Tables S16–S17). Examples include peptides derived from Serpine2, Trpc6, and coagulation factor XIII A chain.
Fig. 9.


Targeted mass spectrometry analysis of a subset of differentially abundant proteins identified within the membrane protein-enriched fraction of PBMCs isolated from EAE and sham-induced mice. Proteins validated to be up-regulated (A) and down-regulated (B) in EAE PBMCs or (C) “housekeeping” proteins (HSP70 and ALDH2) exhibiting similar levels of expression between sham and EAE mice Graphs are representative XICs for specific peptides derived from dysregulated proteins sampled from each group. Bar charts represent the average peak intensity (cps) for peptides quantified in EAE (red) and sham (green) mice (three biological replicates per group, two technical replicates per peptide). A Student's t test was performed on the peak areas of each quantified peptide to test for statistical differences between EAE and sham with p values ≤ 0.05 considered statistically significant. n.s refers to peptides that were analyzed by MRM but were deemed not statistically significant. * indicates that the t test p values for the peptide failed to reach statistical significance after multiple hypothesis testing.
DISCUSSION
Label-free Based Proteomic Identification and Subsequent Label-free MRM Validation of EAE Biomarkers
In this study we chose to combine methods of label-free quantification at the MS1- and MS2-levels to ensure the fidelity of the reported results, with protein abundance identified as being different by both techniques considered reliable. Comparative proteomics analyses of disease-affected tissues in EAE- and sham-induced mice revealed several hundred differentially abundant proteins by both spectral counting and intensity-based quantitation. Candidate markers of EAE were validated by Western blotting and targeted mass spectrometry (MRM) analyses. The increased abundance of ezrin and Clic1 proteins was confirmed by both Western blotting and MRM analyses, validating the label-free quantitative data. Western blotting is conceptually similar to MRM, however, it differs substantially in terms of the number of proteins that can be simultaneously detected and the overall reliability and quality of the results because of the reliance on antibodies of varying specificities (58). The specificity of an antibody dictates its usefulness in a Western blotting assay, whereas several parameters such as retention time, precursor and fragment ion m/z values are relied upon during MRM analysis, rendering it an inherently more sensitive technique. However, Western blots are still indeed useful for detecting specific protein isoforms or degradation products, as was found in this study with moesin and ezrin, observations that might be missed by the targeted MS approach. Scheduled MRM experiments offer an additional level of multiplexing capabilities with ≥1000 transitions that can be simultaneously analyzed, as only certain transitions related to a specific peptide are monitored during a time window around the peptide's elution time (59). Using this time-constrained feature, it allowed for significantly more candidates to be validated in this study without needing to repeatedly assay precious samples. Several rounds of optimization on the QQQ instrument were initially required to unequivocally determine the retention times of targeted peptides to perform the scheduling experiments. Label-free MRM assays were optimized for 194 of the 233 proteins exhibiting a ≥twofold change in expression within the spinal cord by spectral counting analysis, with 136 proteins confirmed to be statistically significant markers (Student's t test, p ≤ 0.05), equivalent to ∼70% success rate for MRM-based validation of candidates. Scheduled MRM assays were optimized for 74 of the 166 proteins exhibiting a ≥twofold change in expression in the PBMCs by spectral counting analysis, with 50 proteins confirmed to be statistically significant markers (Student's t test, p ≤ 0.05), also equivalent to ∼70% success rate for MRM-based validation of candidates. Scheduled MRM assays were performed in duplicate on three biological replicates per EAE and sham group to ensure the accuracy of validation. Moreover, multiple hypothesis testing of the p values obtained from the unpaired Student's t-tests confirmed that over ∼85% of the proteins are indeed differentially abundant in disease-affected tissues by targeted mass spectrometry.
Compared with previous quantitative proteomics studies on clinically derived MScl or EAE tissue, this is by far the most comprehensively validated data set, with hundreds of candidates subjected to label-free MRM analysis. Rosenling and colleagues recently identified 44 differentially abundant proteins within the CSF of a rodent EAE model of which only two were validated by MRM (28). In this study, three to four unique peptides served as protein surrogates in the MRM-based validation of candidate disease markers with the best four transitions per peptide monitored. The additional application of several constraints to the peptide-detection step including prior information on the chromatographic elution properties (i.e. retention time) dramatically reduced the likelihood of false positives and improved the quantitative accuracy, and ultimately the overall reliability of the results. There has been only one previously reported multiplexed MRM validation study that is on par with the scale of this current study in terms of the number of transitions targeted in a single scheduled MRM assay. Bisson and colleagues applied a scheduled MRM assay in combination with affinity purification to target 90 proteins (corresponding to 326 peptides or 1157 transitions) that were potentially GRB2-associated interactors (60). However, the MRM-based validation of candidate EAE markers described in this study is even more comprehensive given that three separate scheduled MRM assays were performed, each containing ∼1000 transitions.
Biological Significance of the Putative Disease Markers Detected in the Spinal Cord
Elevated levels of known protein members of the coagulation cascade such as fibrinogen (alpha, beta, and gamma isoforms) and kininogen-1, were identified by both spectral counting analysis and MS1-based quantification in the diseased spinal cord, observations that have previously been made by proteomic analyses of chronic active plaques isolated from post-mortem MScl brain tissue (12) and CSF from the MBP-EAE model (28). In concordance with other studies, we also identified increased levels of vitamin-d-binding protein (Vdbp), several protein members of the immunoproteasome, and multiple isoforms of immunoglobulin G (IgG) within the diseased spinal cord by both label-free quantification methods. Moreover, a subset of class I acute phase proteins that are known to increase in inflammatory conditions, including MScl (61), were found at elevated levels by both quantification methods in the diseased spinal cord. Their increased presence within this tissue may be caused by the breach in BBB permeability during disease onset and the subsequent extravasation into the CNS.
To our knowledge, this is the first study implicating the role of a number of proteins with EAE including osteoclast-stimulating factor 1 (Ostf1). A 7.8 log2-fold increase in Ostf1 expression was detected within the diseased spinal cord by spectral counting analysis, an observation that was subsequently validated by label-free MRM analysis. Ostf1 is produced by osteoclasts and functions to promote bone resorption by enhancing osteoclast formation and activity (62). One of the key osteoclast differentiation factors is the receptor activator of nuclear factor-κB (RANKL), which is not only expressed by bone-forming osteoblasts in response to osteoclastogenic factors (e.g. 1,25-dihydoxyvitamin D3), but also by activated T cells in response to pro-inflammatory cytokines (e.g. osteopontin) (63, 64). Osteopontin is produced by both immune cells (activated macrophages and T lymphocytes) (65, 66) as well as nonimmune cells including bone and brain cells (67). Ostf1 expression is stimulated by the inflammatory mediators tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) (68). Elevated levels of osteopontin have previously been detected in brain lesions (69), plasma (70, 71), and CSF (72) of MScl subjects. Another protein identified, Vdbp, is a plasma protein primarily responsible for the transport of vitamin D metabolites to target organs (73) and also plays a role in the activation of macrophages and osteoclasts (74). An 8.5 log2-fold increase in Vdbp expression was identified in diseased spinal cord by spectral counting analysis, whereas MS1-based quantitation revealed a 17.2-fold up-regulation. These observations have also been made in the CSF of an MBP-EAE model (28) and the CSF of secondary progressive (SP-) and relapsing remitting (RR-) MScl subjects, compared with controls (16, 17). These data suggest the dynamic crosstalk between the immune and skeletal systems plays an important role in EAE and MScl, and this process may be mediated through the osteoclast-activating abilities of Vdbp and Ostf1.
The Biological Significance of the Putative Disease Markers Detected in the Membrane-enriched PBMCs
Several proteins were identified with elevated expression in disease-affected PBMCs include short transient receptor potential channel 6 (Trpc6) and angiopoietin-1 (Angpt1). Proteins found to be down-regulated include transferrin receptor protein 1 (Tfrc) and integrin alpha-M (Itgam). Trpc6 is a Ca2+-permeable, nonselective cation channel of the transient receptor potential (TRP) superfamily that consists of six subgroups including TRPC (canonical),TRPV (vanilloid) channels, and TRPM (melastatin) channels (75). TRP channels act as cellular sensors, mediating calcium influx as well as the ion homeostasis of migrating cells (76). They are expressed in a wide range of tissues and cell types and are activated by multiple stimuli (77). Trpc6 and Trpv1 channels have been implicated in the lysophosphatidylcholine (LPC)-induced chemotaxis of human monocytes (78). Furthermore, administration of the Trpc channel inhibitor 2-aminoethoxydiphenyl borate (2-APB) resulted in the partial inhibition of LPC-activated currents. In our quantitative proteomics analysis of the membrane protein-enriched fraction of PBMCs, a 2.4 log2-fold increase in Trpc6 expression within diseased EAE mice by spectral counting analysis and validated by label-free MRM within diseased EAE mice. This event may be representative of activated monocytes trying to initiate and/or maintain their ability to migrate into the CNS. Therefore, the development of Trpc6 channel antagonists represents a novel therapeutic strategy for inhibiting disease progression in EAE and MScl. Recent studies have identified Trpm4 as being critically involved in calcium-mediated activation of channels, thus contributing to the destruction of axons and neurons in EAE and MScl. Moreover, Trpm4 knockout studies and pharmaceutical antagonism of Trpm4 in mice resulted in a reduction in EAE severity and disease progression, with neurons being protected against the toxic effects of Trpm4-mediated glutamate excitotoxicity (79).
Itgam, also known as CD18/CD11b or Mac-1, is a member of the β2-integrin family of adhesion molecules expressed on macrophages, microglia, and activated T cells. It plays a critical role in complement-mediated phagocytosis and cellular trafficking, and in MScl, it ultimately contributes to the development of demyelinating disease (80, 81). The differential regulation of β2-integrins on certain T cell subsets during the acute course of EAE has been shown to modulate the kinetics of EAE development by influencing T cell migration patterns into peripheral and CNS tissues (82). We identified a 2.3 log2-fold decrease in Itgam expression within disease-affected PBMCs by spectral counting and label-free MRM analyses within disease-affected PBMCs. This observation may be representative of T cell trafficking from the periphery into the spinal cord via a breached BBB in EAE thus resulting in decreased levels in the systemic circulation. A similar phenomenon has previously been reported in EAE mice at the onset of clinical symptoms whereby levels of pro-inflammatory monocytes (CD11b+CCR2+Ly6Chigh) circulating within the blood fall dramatically to levels observed in naïve mice, concomitant with their appearance within the spinal cord (83).
The analysis of proteomic data collected from spinal cord and PBMC tissue of EAE mice in tandem support with the established three-step model of EAE progression and the neuroinflammatory model of the disease: Initially, the peripheral activation and proliferation of myelin-specific TH1 or TH17 cells occurs and these cells migrate into the CNS across a permeable BBB, resulting in expansion of resident microglia and activation of astrocytes. Finally, blood-borne macrophages and neutrophils are recruited from the circulation, triggering a disease cascade resulting in CNS inflammation and neuronal cell death (84, 85). In summary, these proteomic data reveal the dynamics of immune cell infiltration from peripheral blood compartments into the CNS at a molecular level. This study identified specific proteins involved in inflammatory processes with decreased expression within PBMCs, and elevated levels detected within the spinal cord of EAE mice. These data demonstrate that the profile of immune cells is differentially regulated between the CNS and peripheral blood compartments during EAE (86).
CONCLUSIONS
A dual label-free comparative proteomics approach was employed to assess the repertoire of differentially abundant proteins within disease-affected tissues of actively induced EAE mice and their sham-induced counterparts. This is the first comparative proteomics analysis of this subcellular fraction derived from an animal model of MScl with which a novel mass spectrometry-compatible detergent, MNG, has been used to solubilize the membrane protein-enriched fraction of PBMCs. Additional validation by quantitative Western blotting and label-free MRM analysis were employed to confirm the differential abundance of ∼200 candidate disease-specific proteins, many of which have yet to be reported in the literature as being associated with EAE or MScl. A subset of these candidates also represent promising therapeutic targets including ion channels (Trpc6, Clic1) and enzymes crucial for the proper functioning of activated T cells (Cmpk2, Nampt). Future studies are now warranted to establish whether these biomarkers are relevant to the human condition. Here, the multiplexing capabilities of MRM analysis could be exploited in a quantitative proteomic screen of post-mortem lesional brain tissue and patient plasma/serum and CSF using a panel of the most sensitive and specific biomarkers originally identified within the mouse model. Routine testing of robust panels of markers will be immensely useful in the clinic to aid with disease diagnosis, for stratifying patients who differentially respond to drug treatment and for reliably predicting the clinical course of the disease once it is diagnosed. In summary, several candidate biomarkers of EAE have been identified in this study, which may have implications for early, presymptomatic diagnostics and may represent novel avenues for therapeutic intervention of MScl.
Supplementary Material
Acknowledgments
We wish to thank the Developmental Studies Hybridoma Bank (Iowa, USA) and the UC Davis/NIH NeuroMab Facility (California, USA) for providing access to the monoclonal antibodies used in this study. We thank Georgina Galicia and Jennifer Gommerman for access to the EAE and sham tissues and Paul Goulding for his assistance with Progenesis software data analysis. We also acknowledge the helpful PRIDE team at European Bioinformatics Institute for their assistance with data deposition.
Footnotes
Author contributions: L.F.D. and A.W.P. designed research; L.F.D. and N.P.C. performed research; N.P.C., J.B.O., V.F., and A.E. contributed new reagents or analytic tools; L.F.D., N.P.C., R.I., and A.W.P. analyzed data; L.F.D., A.E., and A.W.P. wrote the paper.
* This investigation was also supported in part by a pilot grant from the Multiple Sclerosis Scientific Research Foundation of Canada awarded to A. E. L.F.D. is supported by a Multiple Sclerosis Research Australia (MSRA) Postgraduate Research Scholarship. A.W.P. is a National Health and Medical Research Council of Australia (NH&MRC) Senior Research Fellow. A. E. is the Ontario Research Chair in Biomarkers.
 This article contains supplemental Methods, Figs. S1 to S9, and Tables S1 to S17.
This article contains supplemental Methods, Figs. S1 to S9, and Tables S1 to S17.
Disclosure statement: A.E. is the recipient of a research grant from Teva Pharmaceuticals. No other conflicts of interest are declared by the authors.
1 The abbreviations used are:
- MScl
- multiple sclerosis
- BBB
- blood-brain barrier
- CNS
- central nervous system
- CFA
- complete Freund's adjuvant
- CSF
- cerebrospinal fluid
- dpi
- days post immunisation
- EAE
- experimental autoimmune encephalomyelitis
- FASP
- filter-aided sample preparation
- IMP
- integral membrane protein
- MNG
- lauryl maltose neopentyl glycol
- MOG
- myelin oligodendrocyte glycoprotein
- MRM
- multiple reaction monitoring
- MS
- mass spectrometry
- PBMCs
- peripheral blood mononuclear cells
- SC
- spectral counting
- TMH
- transmembrane helix.
REFERENCES
- 1. Compston A., Coles A. (2002) Multiple sclerosis. Lancet 359, 1221–1231 [DOI] [PubMed] [Google Scholar]
- 2. Ascherio A., Munger K. L., Lunemann J. D. (2012) The initiation and prevention of multiple sclerosis. Nat. Rev. Neurol. 8, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ransohoff R. M. (2012) Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat. Neurosci. 15, 1074–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steinman L., Zamvil S. S. (2006) How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann. Neurol. 60, 12–21 [DOI] [PubMed] [Google Scholar]
- 5. Gold R., Linington C., Lassmann H. (2006) Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 129, 1953–1971 [DOI] [PubMed] [Google Scholar]
- 6. Dagley L. F., Emili A., Purcell A. W. (2013) Application of quantitative proteomics technologies to the biomarker discovery pipeline for multiple sclerosis. Proteomics Clin. Appl. 7, 91–108 [DOI] [PubMed] [Google Scholar]
- 7. Lock C., Hermans G., Pedotti R., Brendolan A., Schadt E., Garren H., Langer-Gould A., Strober S., Cannella B., Allard J., Klonowski P., Austin A., Lad N., Kaminski N., Galli S. J., Oksenberg J. R., Raine C. S., Heller R., Steinman L. (2002) Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 8, 500–508 [DOI] [PubMed] [Google Scholar]
- 8. Bomprezzi R., Ringner M., Kim S., Bittner M. L., Khan J., Chen Y., Elkahloun A., Yu A., Bielekova B., Meltzer P. S., Martin R., McFarland H. F., Trent J. M. (2003) Gene expression profile in multiple sclerosis patients and healthy controls: identifying pathways relevant to disease. Hum. Mol. Genet. 12, 2191–2199 [DOI] [PubMed] [Google Scholar]
- 9. Lehmensiek V., Sussmuth S. D., Tauscher G., Brettschneider J., Felk S., Gillardon F., Tumani H. (2007) Cerebrospinal fluid proteome profile in multiple sclerosis. Mult. Scler. 13, 840–849 [DOI] [PubMed] [Google Scholar]
- 10. Chiasserini D., Di Filippo M., Candeliere A., Susta F., Orvietani P. L., Calabresi P., Binaglia L., Sarchielli P. (2008) CSF proteome analysis in multiple sclerosis patients by two-dimensional electrophoresis. Eur. J. Neurol. 15, 998–1001 [DOI] [PubMed] [Google Scholar]
- 11. Stoop M. P., Dekker L. J., Titulaer M. K., Burgers P. C., Sillevis Smitt P. A., Luider T. M., Hintzen R. Q. (2008) Multiple sclerosis-related proteins identified in cerebrospinal fluid by advanced mass spectrometry. Proteomics 8, 1576–1585 [DOI] [PubMed] [Google Scholar]
- 12. Han M. H., Hwang S. I., Roy D. B., Lundgren D. H., Price J. V., Ousman S. S., Fernald G. H., Gerlitz B., Robinson W. H., Baranzini S. E., Grinnell B. W., Raine C. S., Sobel R. A., Han D. K., Steinman L. (2008) Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451, 1076–1081 [DOI] [PubMed] [Google Scholar]
- 13. Stoop M. P., Dekker L. J., Titulaer M. K., Lamers R. J., Burgers P. C., Sillevis Smitt P. A., van Gool A. J., Luider T. M., Hintzen R. Q. (2009) Quantitative matrix-assisted laser desorption ionization-fourier transform ion cyclotron resonance (MALDI-FT-ICR) peptide profiling and identification of multiple-sclerosis-related proteins. J. Proteome Res. 8, 1404–1414 [DOI] [PubMed] [Google Scholar]
- 14. Tumani H., Lehmensiek V., Rau D., Guttmann I., Tauscher G., Mogel H., Palm C., Hirt V., Suessmuth S. D., Sapunova-Meier I., Ludolph A. C., Brettschneider J. (2009) CSF proteome analysis in clinically isolated syndrome (CIS): candidate markers for conversion to definite multiple sclerosis. Neurosci. Lett. 452, 214–217 [DOI] [PubMed] [Google Scholar]
- 15. Stoop M. P., Singh V., Dekker L. J., Titulaer M. K., Stingl C., Burgers P. C., Sillevis Smitt P. A., Hintzen R. Q., Luider T. M. (2010) Proteomics comparison of cerebrospinal fluid of relapsing remitting and primary progressive multiple sclerosis. PLoS One 5, e12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Comabella M., Fernandez M., Martin R., Rivera-Vallve S., Borras E., Chiva C., Julia E., Rovira A., Canto E., Alvarez-Cermeno J. C., Villar L. M., Tintore M., Montalban X. (2010) Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain 133, 1082–1093 [DOI] [PubMed] [Google Scholar]
- 17. Ottervald J., Franzen B., Nilsson K., Andersson L. I., Khademi M., Eriksson B., Kjellstrom S., Marko-Varga G., Vegvari A., Harris R. A., Laurell T., Miliotis T., Matusevicius D., Salter H., Ferm M., Olsson T. (2010) Multiple sclerosis: Identification and clinical evaluation of novel CSF biomarkers. J. Proteomics. 73, 1117–1132 [DOI] [PubMed] [Google Scholar]
- 18. Ly L., Barnett M. H., Zheng Y. Z., Gulati T., Prineas J. W., Crossett B. (2011) Comprehensive tissue processing strategy for quantitative proteomics of formalin-fixed multiple sclerosis lesions. J. Proteome Res. 10, 4855–4868 [DOI] [PubMed] [Google Scholar]
- 19. Ibrahim S. M., Mix E., Bottcher T., Koczan D., Gold R., Rolfs A., Thiesen H. J. (2001) Gene expression profiling of the nervous system in murine experimental autoimmune encephalomyelitis. Brain 124, 1927–1938 [DOI] [PubMed] [Google Scholar]
- 20. Mix E., Ibrahim S., Pahnke J., Koczan D., Sina C., Bottcher T., Thiesen H. J., Rolfs A. (2004) Gene-expression profiling of the early stages of MOG-induced EAE proves EAE-resistance as an active process. J. Neuroimmunol. 151, 158–170 [DOI] [PubMed] [Google Scholar]
- 21. Alt C., Duvefelt K., Franzen B., Yang Y., Engelhardt B. (2005) Gene and protein expression profiling of the microvascular compartment in experimental autoimmune encephalomyelitis in C57Bl/6 and SJL mice. Brain Pathol. 15, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baranzini S. E., Bernard C. C., Oksenberg J. R. (2005) Modular transcriptional activity characterizes the initiation and progression of autoimmune encephalomyelitis. J. Immunol. 174, 7412–7422 [DOI] [PubMed] [Google Scholar]
- 23. Inglis H. R., Greer J. M., McCombe P. A. (2012) Gene expression in the spinal cord in female lewis rats with experimental autoimmune encephalomyelitis induced with myelin basic protein. PLoS One 7, e48555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu T., Donahue K. C., Hu J., Kurnellas M. P., Grant J. E., Li H., Elkabes S. (2007) Identification of differentially expressed proteins in experimental autoimmune encephalomyelitis (EAE) by proteomic analysis of the spinal cord. J. Proteome Res. 6, 2565–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jain M. R., Bian S., Liu T., Hu J., Elkabes S., Li H. (2009) Altered proteolytic events in experimental autoimmune encephalomyelitis discovered by iTRAQ shotgun proteomics analysis of spinal cord. Proteome Sci. 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jastorff A. M., Haegler K., Maccarrone G., Holsboer F., Weber F., Ziemssen T., Turck C. W. (2009) Regulation of proteins mediating neurodegeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Proteomics Clin. Appl. 3, 1273–1287 [DOI] [PubMed] [Google Scholar]
- 27. Linker R. A., Brechlin P., Jesse S., Steinacker P., Lee D. H., Asif A. R., Jahn O., Tumani H., Gold R., Otto M. (2009) Proteome profiling in murine models of multiple sclerosis: identification of stage specific markers and culprits for tissue damage. PLoS One 4, e7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rosenling T., Stoop M. P., Attali A., van Aken H., Suidgeest E., Christin C., Stingl C., Suits F., Horvatovich P., Hintzen R. Q., Tuinstra T., Bischoff R., Luider T. M. (2012) Profiling and identification of cerebrospinal fluid proteins in a rat EAE model of multiple sclerosis. J. Proteome Res. 11, 2048–2060 [DOI] [PubMed] [Google Scholar]
- 29. Vanheel A., Daniels R., Plaisance S., Baeten K., Hendriks J. J., Leprince P., Dumont D., Robben J., Brone B., Stinissen P., Noben J. P., Hellings N. (2012) Identification of protein networks involved in the disease course of experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. PLoS One 7, e35544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hammack B. N., Fung K. Y., Hunsucker S. W., Duncan M. W., Burgoon M. P., Owens G. P., Gilden D. H. (2004) Proteomic analysis of multiple sclerosis cerebrospinal fluid. Mult. Scler. 10, 245–260 [DOI] [PubMed] [Google Scholar]
- 31. Stoop M. P., Singh V., Dekker L. J., Titulaer M. K., Stingl C., Burgers P. C., Sillevis Smitt P. A., Hintzen R. Q., Luider T. M. (2010) Proteomics comparison of cerebrospinal fluid of relapsing remitting and primary progressive multiple sclerosis. PLoS One 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noben J. P., Dumont D., Kwasnikowska N., Verhaert P., Somers V., Hupperts R., Stinissen P., Robben J. (2006) Lumbar cerebrospinal fluid proteome in multiple sclerosis: characterization by ultrafiltration, liquid chromatography, and mass spectrometry. J. Proteome Res. 5, 1647–1657 [DOI] [PubMed] [Google Scholar]
- 33. Bondarenko P. V., Chelius D., Shaler T. A. (2002) Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed-phase liquid chromatography-tandem mass spectrometry. Anal. Chem. 74, 4741–4749 [DOI] [PubMed] [Google Scholar]
- 34. Chelius D., Bondarenko P. V. (2002) Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry. J. Proteome Res. 1, 317–323 [DOI] [PubMed] [Google Scholar]
- 35. Liu H., Sadygov R. G., Yates J. R., 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 36. Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 37. Colinge J., Chiappe D., Lagache S., Moniatte M., Bougueleret L. (2005) Differential proteomics via probabilistic peptide identification scores. Anal. Chem. 77, 596–606 [DOI] [PubMed] [Google Scholar]
- 38. Florens L., Carozza M. J., Swanson S. K., Fournier M., Coleman M. K., Workman J. L., Washburn M. P. (2006) Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 40, 303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 40. Zybailov B. L., Florens L., Washburn M. P. (2007) Quantitative shotgun proteomics using a protease with broad specificity and normalized spectral abundance factors. Mol. Biosyst. 3, 354–360 [DOI] [PubMed] [Google Scholar]
- 41. Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M. (2005) Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 42. Rappsilber J., Ryder U., Lamond A. I., Mann M. (2002) Large-scale proteomic analysis of the human spliceosome. Genome Res. 12, 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Butter C., Baker D., O'Neill J. K., Turk J. L. (1991) Mononuclear cell trafficking and plasma protein extravasation into the CNS during chronic relapsing experimental allergic encephalomyelitis in Biozzi AB/H mice. J. Neurol. Sci. 104, 9–12 [DOI] [PubMed] [Google Scholar]
- 44. Giuliani F., Metz L. M., Wilson T., Fan Y., Bar-Or A., Yong V. W. (2005) Additive effect of the combination of glatiramer acetate and minocycline in a model of MS. J. Neuroimmunol. 158, 213–221 [DOI] [PubMed] [Google Scholar]
- 45. Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 46. Manza L. L., Stamer S. L., Ham A. J., Codreanu S. G., Liebler D. C. (2005) Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 5, 1742–1745 [DOI] [PubMed] [Google Scholar]
- 47. Eng J. K., McCormack A. L., Yates Iii J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 48. Kislinger T., Rahman K., Radulovic D., Cox B., Rossant J., Emili A. (2003) PRISM, a generic large scale proteomic investigation strategy for mammals. Mol. Cell. Proteomics 2, 96–106 [DOI] [PubMed] [Google Scholar]
- 49. Mosley A. L., Florens L., Wen Z., Washburn M. P. (2009) A label free quantitative proteomic analysis of the Saccharomyces cerevisiae nucleus. J. Proteomics 72, 110–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sonnhammer E. L., von Heijne G., Krogh A. (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182 [PubMed] [Google Scholar]
- 51. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dennis G., Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 53. Merico D., Isserlin R., Stueker O., Emili A., Bader G. D. (2010) Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mani D. R., Abbatiello S. E., Carr S. A. (2012) Statistical characterization of multiple-reaction monitoring mass spectrometry (MRM-MS) assays for quantitative proteomics. BMC Bioinformatics 13, S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Levner I. (2005) Feature selection and nearest centroid classification for protein mass spectrometry. BMC Bioinformatics 6, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 58. Aebersold R., Burlingame A. L., Bradshaw R. A. (2013) Western blots versus selected reaction monitoring assays: time to turn the tables? Mol. Cell. Proteomics 12, 2381–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schreiber A., Pace N. (2010) Intelligent use of retention time during multiple reaction monitoring for faster and extended compound screening with higher sensitivity and better reproducibility. http://www.absciex.com/Documents/Downloads/Literature/mass-spectrometry-Multiple-Reaction-1282310.pdf [Google Scholar]
- 60. Bisson N., James D. A., Ivosev G., Tate S. A., Bonner R., Taylor L., Pawson T. (2011) Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat. Biotechnol. 29, 653–658 [DOI] [PubMed] [Google Scholar]
- 61. Gabay C., Kushner I. (1999) Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 340, 448–454 [DOI] [PubMed] [Google Scholar]
- 62. Reddy S., Devlin R., Menaa C., Nishimura R., Choi S. J., Dallas M., Yoneda T., Roodman G. D. (1998) Isolation and characterization of a cDNA clone encoding a novel peptide (OSF) that enhances osteoclast formation and bone resorption. J. Cell. Physiol. 177, 636–645 [DOI] [PubMed] [Google Scholar]
- 63. Theill L. E., Boyle W. J., Penninger J. M. (2002) RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu. Rev. Immunol. 20, 795–823 [DOI] [PubMed] [Google Scholar]
- 64. Kong Y. Y., Feige U., Sarosi I., Bolon B., Tafuri A., Morony S., Capparelli C., Li J., Elliott R., McCabe S., Wong T., Campagnuolo G., Moran E., Bogoch E. R., Van G., Nguyen L. T., Ohashi P. S., Lacey D. L., Fish E., Boyle W. J., Penninger J. M. (1999) Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402, 304–309 [DOI] [PubMed] [Google Scholar]
- 65. O'Regan A., Berman J. S. (2000) Osteopontin: a key cytokine in cell-mediated and granulomatous inflammation. Int. J. Exp. Pathol. 81, 373–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Patarca R., Freeman G. J., Singh R. P., Wei F. Y., Durfee T., Blattner F., Regnier D. C., Kozak C. A., Mock B. A., Morse H. C., 3rd, et al. (1989) Structural and functional studies of the early T lymphocyte activation 1 (Eta-1) gene. Definition of a novel T cell-dependent response associated with genetic resistance to bacterial infection. J. Exp. Med. 170, 145–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mazzali M., Kipari T., Ophascharoensuk V., Wesson J. A., Johnson R., Hughes J. (2002) Osteopontin–a molecule for all seasons. QJM 95, 3–13 [DOI] [PubMed] [Google Scholar]
- 68. Patarca R., Saavedra R. A., Cantor H. (1993) Molecular and cellular basis of genetic resistance to bacterial infection: the role of the early T-lymphocyte activation-1/osteopontin gene. Crit. Rev. Immunol. 13, 225–246 [PubMed] [Google Scholar]
- 69. Chabas D., Baranzini S. E., Mitchell D., Bernard C. C., Rittling S. R., Denhardt D. T., Sobel R. A., Lock C., Karpuj M., Pedotti R., Heller R., Oksenberg J. R., Steinman L. (2001) The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 294, 1731–1735 [DOI] [PubMed] [Google Scholar]
- 70. Vogt M. H., ten Kate J., Drent R. J., Polman C. H., Hupperts R. (2010) Increased osteopontin plasma levels in multiple sclerosis patients correlate with bone-specific markers. Mult. Scler. 16, 443–449 [DOI] [PubMed] [Google Scholar]
- 71. Vogt M. H., Lopatinskaya L., Smits M., Polman C. H., Nagelkerken L. (2003) Elevated osteopontin levels in active relapsing-remitting multiple sclerosis. Ann. Neurol. 53, 819–822 [DOI] [PubMed] [Google Scholar]
- 72. Braitch M., Nunan R., Niepel G., Edwards L. J., Constantinescu C. S. (2008) Increased osteopontin levels in the cerebrospinal fluid of patients with multiple sclerosis. Arch. Neurol 65, 633–635 [DOI] [PubMed] [Google Scholar]
- 73. Daiger S. P., Schanfield M. S., Cavalli-Sforza L. L. (1975) Group-specific component (Gc) proteins bind vitamin D and 25-hydroxyvitamin D. Proc. Natl. Acad. Sci. U.S.A. 72, 2076–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yamamoto N., Homma S., Millman I. (1991) Identification of the serum factor required for in vitro activation of macrophages. Role of vitamin D3-binding protein (group specific component, Gc) in lysophospholipid activation of mouse peritoneal macrophages. J. Immunol. 147, 273–280 [PubMed] [Google Scholar]
- 75. Ramsey I. S., Delling M., Clapham D. E. (2006) An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647 [DOI] [PubMed] [Google Scholar]
- 76. Schwab A., Nechyporuk-Zloy V., Fabian A., Stock C. (2007) Cells move when ions and water flow. Pflugers Arch. 453, 421–432 [DOI] [PubMed] [Google Scholar]
- 77. Nilius B. (2007) TRP channels in disease. Biochim. Biophys. Acta 1772, 805–812 [DOI] [PubMed] [Google Scholar]
- 78. Schilling T., Eder C. (2009) Nonselective cation channel activity is required for lysophosphatidylcholine-induced monocyte migration. J. Cell. Physiol. 221, 325–334 [DOI] [PubMed] [Google Scholar]
- 79. Schattling B., Steinbach K., Thies E., Kruse M., Menigoz A., Ufer F., Flockerzi V., Bruck W., Pongs O., Vennekens R., Kneussel M., Freichel M., Merkler D., Friese M. A. (2012) TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 18, 1805–1811 [DOI] [PubMed] [Google Scholar]
- 80. Bullard D. C., Hu X., Schoeb T. R., Axtell R. C., Raman C., Barnum S. R. (2005) Critical requirement of CD11b (Mac-1) on T cells and accessory cells for development of experimental autoimmune encephalomyelitis. J. Immunol. 175, 6327–6333 [DOI] [PubMed] [Google Scholar]
- 81. Hu X., Wohler J. E., Dugger K. J., Barnum S. R. (2010) beta2-integrins in demyelinating disease: not adhering to the paradigm. J. Leukoc. Biol. 87, 397–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Smith S. S., Barnum S. R. (2008) Differential expression of beta 2-integrins and cytokine production between gammadelta and alphabeta T cells in experimental autoimmune encephalomyelitis. J. Leukoc. Biol. 83, 71–79 [DOI] [PubMed] [Google Scholar]
- 83. Mishra M. K., Wang J., Silva C., Mack M., Yong V. W. (2012) Kinetics of proinflammatory monocytes in a model of multiple sclerosis and its perturbation by laquinimod. Am. J. Pathol. 181, 642–651 [DOI] [PubMed] [Google Scholar]
- 84. Arima Y., Harada M., Kamimura D., Park J. H., Kawano F., Yull F. E., Kawamoto T., Iwakura Y., Betz U. A., Marquez G., Blackwell T. S., Ohira Y., Hirano T., Murakami M. (2012) Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell 148, 447–457 [DOI] [PubMed] [Google Scholar]
- 85. Ajami B., Bennett J. L., Krieger C., McNagny K. M., Rossi F. M. (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 14, 1142–1149 [DOI] [PubMed] [Google Scholar]
- 86. Mikita J., Dubourdieu-Cassagno N., Deloire M. S., Vekris A., Biran M., Raffard G., Brochet B., Canron M. H., Franconi J. M., Boiziau C., Petry K. G. (2011) Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult. Scler. 17, 2–15 [DOI] [PubMed] [Google Scholar]
- 87. Ingwersen J., Aktas O., Kuery P., Kieseier B., Boyko A., Hartung H. P. (2012) Fingolimod in multiple sclerosis: mechanisms of action and clinical efficacy. Clin. Immunol. 142, 15–24 [DOI] [PubMed] [Google Scholar]
- 88. Morgan L., Shah B., Rivers L. E., Barden L., Groom A. J., Chung R., Higazi D., Desmond H., Smith T., Staddon J. M. (2007) Inflammation and dephosphorylation of the tight junction protein occludin in an experimental model of multiple sclerosis. Neuroscience 147, 664–673 [DOI] [PubMed] [Google Scholar]
- 89. Farias A. S., Martins-de-Souza D., Guimaraes L., Pradella F., Moraes A. S., Facchini G., Novello J. C., Santos L. M. (2012) Proteome analysis of spinal cord during the clinical course of monophasic experimental autoimmune encephalomyelitis. Proteomics 12, 2656–2662 [DOI] [PubMed] [Google Scholar]
- 90. Harris V. K., Donelan N., Yan Q. J., Clark K., Touray A., Rammal M., Sadiq S. A. (2013) Cerebrospinal fluid fetuin-A is a biomarker of active multiple sclerosis. Mult. Scler. [DOI] [PubMed] [Google Scholar]
- 91. Rithidech K. N., Honikel L., Milazzo M., Madigan D., Troxell R., Krupp L. B. (2009) Protein expression profiles in pediatric multiple sclerosis: potential biomarkers. Mult. Scler. 15, 455–464 [DOI] [PubMed] [Google Scholar]
- 92. Ahn M., Yang W., Kim H., Jin J. K., Moon C., Shin T. (2012) Immunohistochemical study of arginase-1 in the spinal cords of Lewis rats with experimental autoimmune encephalomyelitis. Brain Res. 1453, 77–86 [DOI] [PubMed] [Google Scholar]
- 93. Blanchet L., Smolinska A., Attali A., Stoop M. P., Ampt K. A., van Aken H., Suidgeest E., Tuinstra T., Wijmenga S. S., Luider T., Buydens L. M. (2011) Fusion of metabolomics and proteomics data for biomarkers discovery: case study on the experimental autoimmune encephalomyelitis. BMC Bioinformatics 12, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Barnett M. H., Parratt J. D. E., Cho E.-S., Prineas J. W. (2009) Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann. Neurol. 65, 32–46 [DOI] [PubMed] [Google Scholar]
- 95. Hedegaard C. J., Chen N., Sellebjerg F., Sorensen P. S., Leslie R. G., Bendtzen K., Nielsen C. H. (2009) Autoantibodies to myelin basic protein (MBP) in healthy individuals and in patients with multiple sclerosis: a role in regulating cytokine responses to MBP. Immunology 128, e451–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang B., Jiang Y., Yang Y., Peng F., Hu X. (2008) Correlation between serum thyroxine and complements in patients with multiple sclerosis and neuromyelitis optica. Neuro. Endocrinol. Lett. 29, 256–260 [PubMed] [Google Scholar]
- 97. Padilla-Docal B., Dorta-Contreras A. J., Fundora-Hernandez H., Noris-Garcia E., Bu-Coifiu-Fanego R., Gonzalez-Hernandez M., Rodriguez-Rey A. (2007) C3c intrathecal synthesis evaluation in patients with multiple sclerosis. Arq. Neuropsiquiatr. 65, 800–802 [DOI] [PubMed] [Google Scholar]
- 98. Jongen P. J., Lycklama a Nijeholt G., Lamers K. J., Doesburg W. H., Barkhof F., Lemmens W. A., Klasen I. S., Hommes O. R. (2007) Cerebrospinal fluid IgM index correlates with cranial MRI lesion load in patients with multiple sclerosis. Eur. Neurol. 58, 90–95 [DOI] [PubMed] [Google Scholar]
- 99. Reichert F., Rotshenker S. (1999) Galectin-3/MAC-2 in experimental allergic encephalomyelitis. Exp. Neurol. 160, 508–514 [DOI] [PubMed] [Google Scholar]
- 100. Stancic M., van Horssen J., Thijssen V. L., Gabius H. J., van der Valk P., Hoekstra D., Baron W. (2011) Increased expression of distinct galectins in multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 37, 654–671 [DOI] [PubMed] [Google Scholar]
- 101. Tajouri L., Mellick A. S., Ashton K. J., Tannenberg A. E., Nagra R. M., Tourtellotte W. W., Griffiths L. R. (2003) Quantitative and qualitative changes in gene expression patterns characterize the activity of plaques in multiple sclerosis. Brain Res. Mol. Brain Res. 119, 170–183 [DOI] [PubMed] [Google Scholar]
- 102. Roscoe W. A., Welsh M. E., Carter D. E., Karlik S. J. (2009) VEGF and angiogenesis in acute and chronic MOG((35–55)) peptide induced EAE. J. Neuroimmunol. 209, 6–15 [DOI] [PubMed] [Google Scholar]
- 103. Das A., Guyton M. K., Matzelle D. D., Ray S. K., Banik N. L. (2008) Time-dependent increases in protease activities for neuronal apoptosis in spinal cords of Lewis rats during development of acute experimental autoimmune encephalomyelitis. J. Neurosci. Res. 86, 2992–3001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Imam S. A., Guyton M. K., Haque A., Vandenbark A., Tyor W. R., Ray S. K., Banik N. L. (2007) Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. J. Neuroimmunol. 190, 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Milner R., Crocker S. J., Hung S., Wang X., Frausto R. F., del Zoppo G. J. (2007) Fibronectin- and vitronectin-induced microglial activation and matrix metalloproteinase-9 expression is mediated by integrins alpha5beta1 and alphavbeta5. J. Immunol. 178, 8158–8167 [DOI] [PubMed] [Google Scholar]
- 106. Losy J., Niezgoda A., Wender M. (1999) Increased serum levels of soluble PECAM-1 in multiple sclerosis patients with brain gadolinium-enhancing lesions. J. Neuroimmunol. 99, 169–172 [DOI] [PubMed] [Google Scholar]
- 107. Minagar A., Jy W., Jimenez J. J., Sheremata W. A., Mauro L. M., Mao W. W., Horstman L. L., Ahn Y. S. (2001) Elevated plasma endothelial microparticles in multiple sclerosis. Neurology 56, 1319–1324 [DOI] [PubMed] [Google Scholar]
- 108. Kuenz B., Lutterotti A., Khalil M., Ehling R., Gneiss C., Deisenhammer F., Reindl M., Berger T. (2005) Plasma levels of soluble adhesion molecules sPECAM-1, sP-selectin and sE-selectin are associated with relapsing-remitting disease course of multiple sclerosis. J. Neuroimmunol. 167, 143–149 [DOI] [PubMed] [Google Scholar]
- 109. Yang K., Vega J. L., Hadzipasic M., Schatzmann Peron J. P., Zhu B., Carrier Y., Masli S., Rizzo L. V., Weiner H. L. (2009) Deficiency of thrombospondin-1 reduces Th17 differentiation and attenuates experimental autoimmune encephalomyelitis. J. Autoimmun. 32, 94–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ferrandi C., Richard F., Tavano P., Hauben E., Barbie V., Gotteland J. P., Greco B., Fortunato M., Mariani M. F., Furlan R., Comi G., Martino G., Zaratin P. F. (2011) Characterization of immune cell subsets during the active phase of multiple sclerosis reveals disease and c-Jun N-terminal kinase pathway biomarkers. Mult. Scler. 17, 43–56 [DOI] [PubMed] [Google Scholar]
- 111. Chapman J. (2006) Thrombin in inflammatory brain diseases. Autoimmun. Rev. 5, 528–531 [DOI] [PubMed] [Google Scholar]
- 112. Berard J. L., Zarruk J. G., Arbour N., Prat A., Yong V. W., Jacques F. H., Akira S., David S. (2012) Lipocalin 2 is a novel immune mediator of experimental autoimmune encephalomyelitis pathogenesis and is modulated in multiple sclerosis. Glia 60, 1145–1159 [DOI] [PubMed] [Google Scholar]
- 113. Reder A. T., Genc K., Byskosh P. V., Porrini A. M. (1998) Monocyte activation in multiple sclerosis. Mult. Scler. 4, 162–168 [DOI] [PubMed] [Google Scholar]
- 114. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.