

Abstract
Background
Lipoprotein(a) [Lp(a)] is an LDL-like particle largely independent of known risk factors and predictive of cardiovascular disease (CVD). Statins may offset the risk associated with elevated Lp(a), but it is unknown if Lp(a) is a determinant of residual risk in the setting of low LDL-cholesterol after potent statin therapy.
Methods and Results
Baseline and on-treatment Lp(a) concentrations were assessed in 9,612 multiethnic JUPITER trial participants before and after random allocation to rosuvastatin 20 mg/day or placebo, with outcomes reported for whites (N=7,746). Lp(a) concentrations (nmol/L) were highest in blacks (median [25th–75th percentile] 60 [34–100]), then Asians (38 [18–60]), hispanics (24 [11–46]), and whites (23 [10–50]); p<0.001. While the median change in Lp(a) with rosuvastatin and placebo was zero, rosuvastatin nonetheless resulted in a small but statistically significant positive shift in the overall Lp(a) distribution (p<0.0001). Baseline Lp(a) concentrations were associated with incident CVD: adjusted hazard ratio (HR) per 1-SD increment in Ln[Lp(a)] 1.18 (95%CI 1.03 – 1.34; p=0.02). Similarly, on-statin Lp(a) concentrations were associated with residual risk of CVD: adjusted HR 1.27 (95%CI 1.01 – 1.59; p=0.04), which was independent of LDL-cholesterol and other factors. Rosuvastatin significantly reduced incident CVD among participants with baseline Lp(a)≥median (HR 0.62, 0.43–0.90) and Lp(a)<median (HR 0.46, 0.30–0.72), with no evidence of interaction. Similar results were obtained when analyses included non-whites.
Conclusion
Among white JUPITER participants treated with potent statin therapy, Lp(a) was a significant determinant of residual risk. The magnitude of relative risk reduction with rosuvastatin was similar among participants with high or low Lp(a).
Keywords: Lipoproteins, Statins, Risk Factors
Medical therapies, including statins, have demonstrated efficacy in the prevention of cardiovascular events across a wide spectrum of baseline risk.1 However, substantial residual risk has fostered interest in identifying the underlying risk factors in hopes of identifying novel targets of therapy. Lipoprotein(a) [Lp(a)] is a low density lipoprotein (LDL)-like particle with apolipoprotein B covalently linked to apoliprotein(a) by a single disulfide bond.2
Since its initial description by Berg in 1963 as a variant of LDL, the Lp(a) molecule has generated interest regarding its potential proatherogenic or prothrombotic role in human disease.3 Circulating concentrations of Lp(a) differ widely across individuals and ethnic subgroups, mediated in large part by genetic variation at the LPA gene locus.2 Individuals contain highly polymorphic copy numbers of the Kringle IV-type 2 domain, with lower numbers relating to smaller apoliprotein(a) size and increased plasma Lp(a) concentrations.4 Robust associations between Lp(a) and cardiovascular disease (CVD) outcomes have been noted in previous studies conducted in general populations, with Lp(a) concentrations providing small, statistically significant improvement in risk prediction when added to conventional risk factors.5, 6 Recent Mendelian randomization studies have linked genetic variations at the LPA locus to both circulating plasma concentrations and the risk of CVD, supporting a possible causal role of Lp(a) in CVD pathogenesis.7, 8
Previous studies have suggested that statin therapy may attenuate the risk associated with Lp(a), although current data addressing this common clinical question remains very limited.9 After the completion of the Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) trial but before obtaining the Lp(a) measurements, we prespecified the hypothesis that the residual risk of CVD may be related in part to increased Lp(a) concentrations. Therefore, we determined the association of baseline and on-treatment Lp(a) concentrations with incident CVD events in the context of potent rosuvastatin therapy and very low achieved LDL-cholesterol concentrations in JUPITER.
Methods
Study Population
JUPITER (Clinical Trial.gov number NCT00239681) was a primary prevention randomized, double-blind, placebo-controlled trial investigating whether rosuvastatin 20 mg per day would decrease incident CVD in 17,802 asymptomatic individuals with LDL-cholesterol (LDL-C) < 130 mg/dL and a high-sensitivity C-reactive protein (hsCRP) ≥ 2.0 mg/L.10 Exclusion criteria for the JUPITER trial were diabetes, previous or current use of lipid-lowering therapy, or triglycerides greater than 500 mg/dL. The trial protocol stipulated both a baseline and 12-month visit for blood draws and immediate trial assays. Study participants were requested but not required to provide samples for additional phenotyping: 11,953 participants provided these additional samples at both baseline and one year, and of these, 9,612 had sufficient sample remaining for Lp(a) assessment. Owing to ethnic variation in Lp(a) concentrations and the smaller proportion of non-white participants in JUPITER, the primary outcomes analysis is reported among white participants (n = 7,746) with subsequent sensitivity analyses that included all 9,612 white and non-white participants. A small number of samples failed assay quality control criteria (< 0.2%), leading to an effective size of 7,730 and 7,739 individuals for the baseline and on-statin white cohort, respectively.
Laboratory Measurements
Lipid, apolipoprotein, and hsCRP values were assayed in a core laboratory as previously described.10–12 Consistent with previous JUPITER biomarker analyses, on-treatment Lp(a) concentrations were defined as values obtained after one year of randomized treatment.11–14 Lp(a) concentrations were measured in a blinded manner at Quest Diagnostics Nichols Institute (San Juan Capistrano, CA) using a commercially available assay (Randox Laboratories; Crumlin, Co. Antrim, United Kingdom) that is not affected by Kringle IV type-2 repeats. Given substantial interindividual variability in the number of Kringle IV type-2 repeats and thus Lp(a) molecular weight, values were measured and reported in nmol/L to reflect the concentration of Lp(a) particles. This methodology of Lp(a) assessment is in accordance with a recent National Heart, Lung, and Blood Institute workshop recommendation.15 An assessment of five standard samples across a broad range of Lp(a) concentrations indicated that conversion to mg/dL can be approximated by dividing nmol/L values by 2.15 (r2 = 0.998 for linearity). Mean coefficient of variation for the assay was 3.5%, 4.0% and 2.6 % at Lp(a) concentrations of 38, 60 and 138 nmol/L, respectively.
Outcomes
Our primary outcome was the pre-specified JUPITER trial primary endpoint, a composite CVD endpoint that included incident myocardial infarction, stroke, hospitalization for unstable angina, arterial revascularization, or cardiovascular death. In the current analysis, we also examined a combined endpoint of CVD and all-cause mortality consistent with prior analyses of lipids and residual risk in JUPITER.12 Endpoint criteria have been described previously; all were adjudicated by an independent committee blinded to treatment assignment.10
Statistical Analysis
Statistical analyses were performed with SAS version 9.1 (SAS Institute Inc., Cary, North Carolina). Medians, 25th, and 75th percentiles were calculated for continuous variables. The significance of variation in Lp(a) values across categorical clinical characteristics was assessed using the nonparametric Wilcoxon rank sum or Kruskal-Wallis one-way analysis of variance tests. Spearman coefficients were used to express the magnitude of correlation between baseline and on-treatment biomarkers with corresponding Lp(a) concentrations.
Tests of outcomes were performed by calculating incidence rates per 100 person-years, with exposure time calculated as the time from randomization to occurrence of the primary endpoint or the date of death, last study visit, withdrawal, or loss to follow-up. Consistent with previous JUPITER biomarker analyses, on-treatment Lp(a) concentrations were defined as values obtained at one year of treatment.1–14 As in prior reports, we decided a priori to include all postrandomization events in the on-treatment analysis of associations with incident events given minimal impact of statin therapy on Lp(a) and that any such change would have occurred within the first few weeks of randomization. Cox proportional hazard regression models were used to calculate hazard ratios (HRs) and 95% confidence intervals (CI) for first CVD. Hazard ratios are reported both per standard deviation (SD) increment in the natural logarithm (ln) of Lp(a) expressed as a continuous variable, and according to Lp(a) quartiles. P values for trend were obtained by including quartile number as a variable in the regression model. Regression models were adjusted for age, gender, and treatment group, with subsequent additional adjustment for smoking status, family history of premature coronary disease, body mass index, systolic blood pressure, fasting glucose, HDL-cholesterol, LDL-cholesterol, and log transformed values for triglycerides and hsCRP. Results were also unaffected in a sensitivity analysis that removed family history of premature coronary disease from the adjusted model. Similar analyses were subsequently conducted using an expanded endpoint of the primary endpoint plus all-cause mortality. Additional sensitivity analyses were performed including non-white participants. We assessed for non-linearity in the association of Lp(a) and outcomes by repeating the analyses after adding a quadratic term to the models. The quadratic terms were not statistically significant.
The risk reduction for the primary endpoint with rosuvastatin therapy was calculated in participant subgroups dichotomized by the median baseline Lp(a) concentration to assess for heterogeneity of effect. Statistical tests for interaction between Lp(a) concentration and treatment allocation in relation to outcomes were obtained using likelihood ratio tests. Cutpoint analysis implemented a threshold of 50 mg/dL (approximately 108 nmol/L using a correction factor of 2.15) as well as the 90th percentile of Lp(a) in accordance with the recommendations of a recent expert panel and a previous cohort analysis respectively.16, 17 All P-values were two-tailed with a value < 0.05 considered to indicate statistical significance.
Results
Lp(a) concentrations were greatest in black participants (n = 853; median 60 nmol/L), then Asians(n=138; median 38 nmol/L), then hispanics (n = 784; median 24 nmol/L) and whites (n = 7730 ; median 23 nmol/L), as displayed in Supplemental Table 1. Subsequent analyses were thus restricted to white participants unless otherwise noted.
Baseline characteristics of the white JUPITER Lp(a) cohort were similar to those in which Lp(a) was not available and the overall study population, except for a slightly decreased prevalence of metabolic syndrome in patients included in the present analysis (Table 1). As shown in Table 2, women had higher Lp(a) concentrations than men (26 vs 22 nmol/L, p<0.0001). Participants with metabolic syndrome had lower Lp(a) compared with those without metabolic syndrome (20 vs 25 nmol/L, p<0.0001). As anticipated, Lp(a) was weakly correlated with other risk factors at baseline and on-statin treatment (Supplemental Table 2). Spearman correlation coefficients between baseline Lp(a) and LDL-cholesterol, apolipoprotein B, and hsCRP were 0.13, 0.08, and 0.04, respectively.
Table 1.
Clinical characteristics of white participants in the JUPITER Lp(a) cohort and overall study population
| Lp(a) Cohort (N= 7,746)* | Lp(a) Unavailable (N = 4,937) | Overall Cohort (N = 17,802) | |
|---|---|---|---|
| Median (25th – 75th %) or N (%) | Median (25th – 75th %) or N (%) | Median (25th – 75th %) or N (%) | |
| Age, yr | 66 (60 – 71) | 66 (60 – 71) | 66 (60 – 71) |
| Female Sex | 2574 (33%) | 1623 (33%) | 6801 (38%) |
| Rosuvastatin group | 3882 (50%) | 2476 (50%) | 8901 (50%) |
| BMI, kg/m2 | 28 (25 – 32) | 29 (26 – 32) | 28 (25 – 32) |
| SBP, mm Hg | 135 (125– 146) | 134 (125 – 145) | 134 (124 – 145) |
| DBP, mm Hg | 80 (74 – 86) | 80 (75 – 87) | 80 (75 – 87) |
| Current smoker | 1111 (14%) | 733 (15%) | 2820 (16%) |
| FH of premature CHD | 1054 (14%) | 656 (13%) | 2045 (11.5%) |
| Metabolic syndrome | 2892 (38%) | 2151 (44%) | 7375 (42%) |
| Aspirin use | 1463 (19%) | 929 (19%) | 2958 (16.6%) |
| hsCRP, mg/L | 4.0 (2.7 – 6.4) | 4.1 (2.8 – 6.6) | 4.3 (2.9 – 7.1) |
| Lipoprotein(a), nmol/L | 23 (10 – 50) | ----- | ----- |
| LDL-cholesterol, mg/dL | 110 (96 – 120) | 110 (97 – 120) | 108 (94 – 119) |
| HDL-cholesterol, mg/dL | 50 (41 – 61) | 48 (40 – 59) | 49 (40 – 60) |
| Triglycerides, mg/dL | 114 (82 – 160) | 120 (87 – 174) | 118 (85 – 169) |
| Total cholesterol, mg/dL | 187 (172 – 201) | 187 (171 – 201) | 185 (169 – 200) |
| Glucose, mg/dL | 95 (89 – 101) | 96 (89 – 104) | 94 (88 – 102) |
| Glycated hemoglobin, % | 5.6 (5.4 – 5.8) | 5.7 (5.4 – 5.9) | 5.7 (5.5 – 5.9) |
| GFR, ml/min/1.73 m2 of body surface area | 73 (65 – 82) | 72 (64 – 81) | 74 (65 – 84) |
Baseline Lp(a) measurements available on 7730 white participants. Family history of premature coronary disease defined as diagnosis of the disease in a male first-degree relative before the age of 55 years or in a female first-degree relative before the age of 65 years.
Table 2.
Baseline lipoprotein(a) concentration according to clinical subgroups among white JUPITER particpants
| N | Median (25th – 75th %) |
P-value | ||
|---|---|---|---|---|
| Sex | Men | 5172 | 22 (10 – 47) | < 0.0001 |
| Women | 2574 | 26 (12 – 53) | ||
| Treatment group | Placebo | 3864 | 23 (10 – 48) | 0.96 |
| Rosuvastatin | 3882 | 24 (10 – 51) | ||
| Current smoker | No | 6635 | 23 (10 – 49) | 0.61 |
| Yes | 1111 | 25 (10 – 52) | ||
| FH of premature CHD | No | 6666 | 23 (10 – 49) | 0.09 |
| Yes | 1054 | 25 (11 – 53) | ||
| Metabolic syndrome | No | 4788 | 25 (11 – 53) | < 0.0001 |
| Yes | 2892 | 20 (10 – 44) | ||
| Aspirin use | No | 6283 | 23 (10 – 49) | 0.16 |
| Yes | 1463 | 23 (11 – 54) |
Family history of premature coronary disease defined as diagnosis of the disease in a male first-degree relative before the age of 55 years or in a female first-degree relative before the age of 65 years.
Among the placebo group, Lp(a) concentrations at baseline and twelve months were stable and highly self-correlated (Spearman r = 0.95; intraclass correlation coefficient 0.93 [95% CI 0.89–0.97]). Similar results were noted in the rosuvastatin arm, with Spearman r = 0.95 and intraclass correlation coefficient 0.92 (95% CI 0.87 – 0.97). While the median change in Lp(a) with rosuvastatin and placebo was zero, rosuvastatin nonetheless resulted in a small but statistically significant positive shift in the overall Lp(a) distribution; 25th, 75th percentile change in Lp(a): (−1, 5) for rosuvastatin, and (−3, 2) for placebo, p<0.0001. No relationship was noted between change in LDL-cholesterol and change in Lp(a) with statin therapy (Spearman r = 0.02; p=0.14).
Incident cardiovascular events according to baseline Lp(a) concentrations
During a median follow-up of 2.0 years, the primary and expanded CVD endpoints occurred in 210 and 283 white JUPITER participants respectively. Baseline Lp(a) was associated with increased risk of CVD (Table 3), with fully adjusted HR per 1-SD increment in ln Lp(a) (representing an approximately 2.5-fold increment in Lp(a)) of 1.18 (95%CI 1.03 – 1.35) and 1.21 (95%CI 1.08 – 1.36) for the primary and expanded endpoint respectively. Incidence rates and HRs also indicated a statistically significant increased risk in the quartile of patients with the highest Lp(a) concentrations (> 50 nmol/L) as compared to those in the referent quartile with the lowest Lp(a) values, with adjusted HR of 1.64 (95% CI 1.12 – 2.41) for the primary endpoint and 1.61 (95%CI 1.16 – 2.25) for the expanded endpoint. The association of baseline Lp(a) with CVD did not differ according to randomized treatment group, with no significant interaction in an unadjusted model including treatment group and Lp(a) as a continuous variable (p-interaction = 0.80; Supplemental Tables 3 and 4) or quartile number (p-interaction =0.80). Furthermore, the association of Lp(a) with CVD was similar across clinically relevant clinical subgroups as displayed in Supplemental Table 5 (p-interaction > 0.05 for all). The observed relationship was somewhat stronger in participants with baseline hsCRP below the cohort median of 4.0 mg/L, with HR 1.32 (95%CI 1.10 – 1.59), as compared with those equal to or above the median, HR 1.05 (95%CI 0.88 – 1.26), although this interaction did not achieve statistical significance in formal interaction testing (p-interaction = 0.09).
Table 3.
Association between baseline lipoprotein(a) and incident CVD among white JUPITER participants
| Quartile One | Quartile Two | Quartile Three | Quartile Four | P-trend | HR/SD increment |
P-value | |
|---|---|---|---|---|---|---|---|
| Range (nmol/L) | ≤ 10 | 11 – 23 | 24 – 49 | ≥ 50 | |||
| Primary Endpoint | |||||||
| # of events / N | 44 / 1991 | 50 / 1884 | 45 / 1957 | 71 / 1898 | 210 / 7730 | ||
| Incidence rate, per 100 person years | 0.99 | 1.17 | 1.02 | 1.62 | 0.02 | 1.20 | |
| Model One | 1.00 | 1.18 (0.79 – 1.78) P = 0.44 |
1.04 (0.68 – 1.58) P = 0.87 |
1.70 (1.16 – 2.47) P = 0.006 |
0.01 | 1.19 (1.05 – 1.36) |
0.008 |
| Model Two | 1.00 | 1.19 (0.79 – 1.79) P = 0.40 |
1.02 (0.67 – 1.56) P = 0.93 |
1.64 (1.12 – 2.41) P = 0.01 |
0.02 | 1.18 (1.03 – 1.35) |
0.02 |
| Primary Endpoint Plus Total Mortality | |||||||
| # of events / N | 59 / 1991 | 63 / 1884 | 67 / 1957 | 94 / 1898 | 283 / 7730 | ||
| Incidence rate, per 100 person years | 1.32 | 1.47 | 1.53 | 2.14 | 0.004 | 1.62 | |
| Model One | 1.00 | 1.11 (0.78 – 1.59) P = 0.56 |
1.15 (0.81 – 1.63) P = 0.44 |
1.66 (1.20 – 2.29) P = 0.002 |
0.002 | 1.22 (1.09 – 1.37) |
0.0005 |
| Model Two | 1.00 | 1.12 (0.78 – 1.60) P = 0.54 |
1.14 (0.80 – 1.63) P = 0.47 |
1.61 (1.16 – 2.25) P = 0.005 |
0.005 | 1.21 (1.08 – 1.36) |
0.001 |
Hazard ratios are expressed per 1-SD increment in ln Lp(a), with 1-SD representing an approximately 2.5-fold increment in Lp(a). N = 7,730 reflective of number of white participants with baseline Lp(a) value available.
Model One: Adjusted for age, gender, and treatment group.
Model Two: Adjusted for age, gender, treatment group, smoking, family history of premature coronary disease, body mass index, systolic blood pressure, fasting glucose, HDL-cholesterol, LDL-cholesterol, ln triglycerides, ln hsCRP
Residual risk according to on-statin Lp(a) concentrations
Among patients allocated to rosuvastatin, greater on-treatment Lp(a) concentrations were similarly associated with residual risk of CVD, with adjusted HR of 1.27 for each SD change in Lp(a) (95%CI 1.01 – 1.59) for the primary endpoint and 1.29 (95% CI 1.07–1.56) for the expanded endpoint (Table 4). Quartile analysis showed directionally consistent results. Additional models that examined on-treatment Lp(a) adjusted for on-statin (instead of baseline) concentrations of HDL-cholesterol, LDL-cholesterol, ln triglycerides, and ln hsCRP yielded similar results (Supplemental Table 6).
Table 4.
Association between on-statin lipoprotein(a) and residual risk among white JUPITER participants randomly allocated to rosuvastatin
| Quartile One | Quartile Two | Quartile Three | Quartile Four | P-trend | HR/SD | P-value | |
|---|---|---|---|---|---|---|---|
| Range (nmol/L) | ≤ 10 | 11 – 23 | 24 – 53 | ≥ 54 | |||
| Primary Endpoint | |||||||
| # of events / N | 19 / 1068 | 10 / 872 | 22 / 984 | 24 / 953 | 75 / 3877 | ||
| Incidence rate, per 100 person years | 0.79 | 0.52 | 0.98 | 1.10 | 0.13 | 0.86 | |
| Model One | 1.00 | 0.65 (0.30 – 1.39) P = 0.26 |
1.16 (0.63 – 2.15) P = 0.63 |
1.47 (0.81 – 2.69) P = 0.21 |
0.10 | 1.29 (1.03 – 1.60) |
0.02 |
| Model Two | 1.00 | 0.64 (0.30 – 1.39) P = 0.26 |
1.17 (0.63 – 2.18) P = 0.62 |
1.37 (0.73 – 2.57) P = 0.32 |
0.17 | 1.27 (1.01 – 1.59) |
0.04 |
| Primary Endpoint Plus Total Mortality | |||||||
| # of events / N | 23 / 1068 | 17 / 872 | 31 / 984 | 35 / 953 | 106 / 3877 | ||
| Incidence rate, per 100 person years | 0.96 | 0.88 | 1.38 | 1.61 | 0.02 | 1.21 | |
| Model One | 1.00 | 0.90 (0.48 – 1.69) P = 0.75 |
1.33 (0.77 – 2.28) P = 0.31 |
1.75 (1.03 – 2.97) P = 0.04 |
0.02 | 1.30 (1.08 – 1.56) |
0.006 |
| Model Two | 1.00 | 0.91 (0.48 – 1.70) P = 0.65 |
1.35 (0.78 – 2.32) P = 0.37 |
1.71 (0.99 – 2.95) P = 0.06 |
0.03 | 1.29 (1.07 – 1.56) |
0.01 |
Hazard ratios are expressed per 1-SD increment in ln Lp(a), with 1-SD representing an approximately 2.5-fold increment in Lp(a).
Model One: Adjusted for age and gender.
Model Two: Adjusted for age, gender, smoking, family history of premature coronary disease, body mass index, systolic blood pressure, glucose, and on-treatment levels of HDL-cholesterol, LDL-cholesterol, ln triglycerides, and ln hsCRP
Threshold analysis
The previously recommended threshold of 50 mg/dL (approximately 108 nmol/L) was exceeded by 11% of white participants at baseline. Compared with participants whose baseline Lp(a) was <50 mg/dL, those with Lp(a) ≥50 mg/dL had increased risk of CVD, with fully adjusted HRs of 1.57 (95% CI 1.08 – 2.27; p=0.02) and 1.69 (95%CI 1.24 – 2.31; p=0.001) for the primary and expanded endpoints, respectively. Similarly, participants whose on-statin Lp(a) exceeded this threshold (13% of the cohort) exhibited a trend towards increased risk for the primary (HR 1.67; 95% CI 0.93 – 3.02; p=0.09) and expanded endpoint (HR 1.54; 95%CI 0.93 – 2.55; p=0.09). A similar analysis dichotomized white participants based on the 90th percentile value (116 nmol/L at baseline, 134 nmol/L in the on-statin group). Compared with white participants whose Lp(a) was <90th percentile, individuals with baseline Lp(a) ≥ 90th percentile had a trend towards increase risk of the primary endpoint (adjusted HR 1.48; 95%CI 1.00 – 2.20; p=0.05) which was statistical significant for the expanded endpoint (adjusted HR 1.64; 95%CI 1.18 – 2.27; p=0.003). On-statin analyses indicated similar results (primary endpoint adjusted HR 1.96; 95%CI 1.04 – 3.67; p=0.04; expanded endpoint adjusted HR 1.75; 1.02 – 3.00; p=0.04).
Lp(a) associations in multiethnic cohort and by ethnic subgroups
A sensitivity analysis was conducted in the multiethnic cohort including all participants with Lp(a) concentrations available. The baseline cohort included 9,591 multiethnic participants, in whom the primary and expanded endpoints occurred in 234 and 327 participants respectively. The adjusted HRs per 1-SD (roughly 2.5-fold) increase were 1.19 (95%CI 1.04 – 1.35; P = 0.01) for the primary endpoint and 1.19 (95%CI 1.06 – 1.32; P = 0.002) for the expanded endpoint. A similar analysis was conducted using the on-statin subgroup involving 4,797 participants with 81 primary and 118 expanded endpoints observed. The adjusted HRs per 1-SD increase were 1.29 (95%CI 1.03 – 1.61; P = 0.02) and 1.25 (95%CI 1.04 – 1.50; P = 0.02) for the primary and expanded endpoints.
Few primary events occurred in blacks (n=13) or hispanics (n=6), limiting power to explore relationships with incident events. The adjusted HRs per 1-SD increment were 1.43 (95% CI 0.69 – 2.98; P = 0.34) in black participants and 1.23 (95%CI 0.55 – 2.75) in hispanic participants in this cohort. There was no evidence of interaction by ethnicity when model involved all ethnic groups (p-interaction = 0.52) or in an additional analysis that dichotomized participants as white vs. nonwhite (p-interaction = 0.37).
Efficacy of rosuvastatin according to baseline Lp(a)
Rosuvastatin had similar efficacy in reducing the incidence of the primary and expanded endpoints in participant subgroups with above or below median baseline Lp(a) concentrations (Figure), p-interaction = 0.33 and 0.10 for the primary and expanded endpoint respectively.
Figure 1.
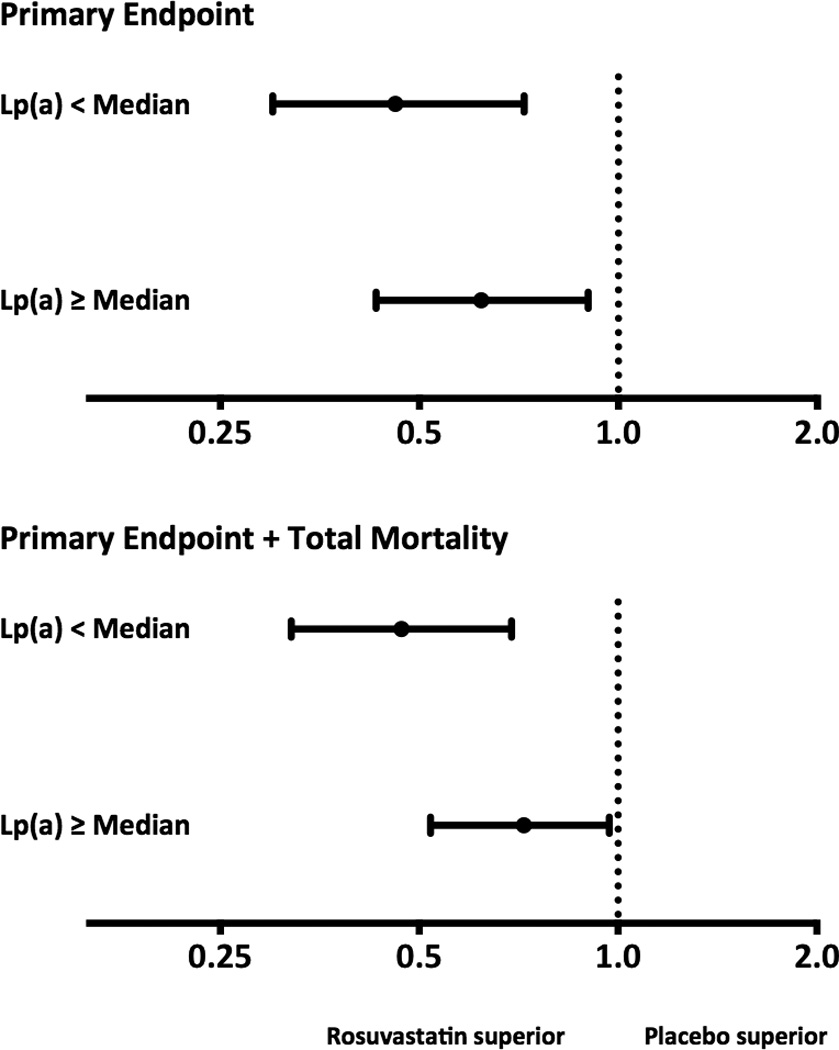
Efficacy of rosuvastatin according to baseline lipoprotein(a) concentration. Hazard ratios and 95% confidence intervals according to intention-to-treat analysis for the primary endpoint (top) and the expanded endpoint (bottom) by baseline lipoprotein(a) concentrations. For the primary endpoint, hazard ratio with rosuvastatin therapy was 0.47 (95%CI 0.30 – 0.72) for participants with baseline Lp(a) concentration below the median and 0.62 (95%CI 0.43 – 0.90) in those above the median (p-interaction = 0.33). Similarly, for the expanded endpoint, hazard ratios were 0.46 (95%CI 0.32 – 0.69) and 0.72 (95%CI 0.52 – 0.97) for those below and above the median respectively (p-interaction = 0.10).
Discussion
This evaluation from the JUPITER trial among participants with initially low LDL-cholesterol and elevated hsCRP demonstrates that baseline Lp(a) concentrations were associated with increased CVD risk. In addition, among white participants randomly allocated to potent statin therapy who achieved very low LDL-cholesterol (median on-treatment LDL-cholesterol 54 mg/dl), baseline and on-statin Lp(a) concentrations were associated with residual risk of CVD. This was independent of other risk factors, including LDL-cholesterol. Rosuvastatin had similar efficacy in reducing CVD regardless of baseline Lp(a). Threshold analyses using previously proposed clinical cutpoints demonstrated potential utility in identifying asymptomatic individuals at increased CVD risk. This increased risk in the context of robust LDL-cholesterol and CRP-lowering with statin therapy reinforces growing interest in targeting Lp(a) for residual risk assessment and potential modulation for therapeutic gain.
Our data complement recent studies demonstrating that genetic polymorphisms conferring higher Lp(a) concentrations were associated with increased risk for atherosclerotic events, thus supporting the notion that lifelong elevation in Lp(a) may be causally associated with CVD.7, 8 However, the relative contribution of multiple potential mechanisms remains unclear.2 Apolipoprotein(a) may lead to a prothrombotic state based on interference with plasminogen activation. Additional data has demonstrated an effect of Lp(a) on endothelial cell permeability, adhesion molecule expression, and regulation of vascular proliferation.2 Lp(a) also serves as a carrier of oxidized phospholipids which may propagate atherosclerosis via inflammatory pathways. Indeed, Lp(a) concentrations are increased in humans with a broad range of inflammatory conditions.18–20
The results from this study are broadly consistent with a recent individual participant meta-analysis that noted adjusted risk ratios per 1-SD increment of ln Lp(a) for coronary disease of 1.13 (95% CI 1.09 – 1.18).5 We confirmed the substantially increased Lp(a) concentrations in blacks and modest elevations in women noted in previous reports.5 Lp(a) concentrations were highest in blacks, followed by Asians, then hispanics and whites. Although our study included fewer nonwhite than white participants, a recent analysis from the Atherosclerosis Risk in Communities study found similar associations with CVD among whites and blacks.21 We also note lower concentration of Lp(a) in participants with the metabolic syndrome, a finding that is consistent with previous data indicating an inverse relationship between baseline Lp(a) and incident diabetes in two large population-based cohorts.22
Prior data regarding the relationship between Lp(a) and CVD outcomes in the setting of statin therapy is limited and inconsistent. An analysis of the Familial Atherosclerosis Treatment Study involving 146 males with both hypercholesterolemia and a family history of premature coronary disease suggested that baseline Lp(a) concentrations were associated with coronary disease severity but that the impact of high Lp(a) on clinical events was attenuated if LDL-cholesterol reduction >10% was achieved pharmacologically, a concept that was not supported by the current JUPITER results.9 In the Scandinavian Simvastatin Survival Study of secondary prevention among individuals with hypercholesterolemia, baseline Lp(a) concentrations were moderately higher in patients who ultimately suffered a major coronary event or all-cause death, but on-treatment Lp(a) concentrations were not measured.23 The Air Force/Texas Coronary Atherosclerosis Prevention Study (21 coronary events) reported an HR of 1.15 (95% CI 0.72 – 1.84) per 3.5-fold increase (roughly one SD) in baseline Lp(a) concentrations, a point estimate similar in magnitude to the current analysis.5 By contrast, null associations were noted in a small case control study (108 cases) from the West of Scotland Coronary Prevention Study.24 These disparate findings may reflect varying methodologies in Lp(a) measurement and mixed adherence to current recommendations to use size-independent metrics of Lp(a).
Therapeutic targeting of Lp(a) concentrations to achieve cardiovascular risk reduction is not currently practiced clinically. Niacin is known to decrease Lp(a) by up to 40%, in addition to other effects on lipids.25 The recently completed AIM-HIGH and HPS2-THRIVE studies, which failed to demonstrate clinical benefit with the addition of niacin or niacin/laropiprant to LDL-reduction therapy, may afford an opportunity for additional analyses of these interventions in subgroups of participants with elevated Lp(a).26–27
Beyond niacin, multiple novel agents currently in various stages of development have been noted to decrease Lp(a) concentrations. For example, the cholesteryl ester transfer protein inhibitor anacetrapib decreased Lp(a) by 36% and is currently being evaluated in the Phase III Randomized EValuation of the Effects of Anacetrapib Through Lipid-modification trial (NCT01252953).28 Inhibition of proprotein convertase subtilisin/kinexin 9 (PCSK9) has also demonstrated moderate ability to decrease Lp(a) in addition to LDL-cholesterol reduction.29 Mipomersen, an antisense oligonucleotide targeting apolipoprotein B also decreased Lp(a) by 17% and was approved recently by the Food and Drug Administration for the treatment of familial hypercholesterolemia.30 Interventions that specifically target Lp(a) are not available at present, although an antisense oligonucleotide directed against Kringle IV repeats demonstrated ability to reduce Lp(a) in transgenic murine models.31 Enthusiasm has increased for an intervention trial that selectively enrolls patients with elevated Lp(a) concentrations, although no specific trial plans have been announced.2
Current study limitations include the two-year median length of follow-up in the JUPITER trial related to the study’s early termination due to clinical benefit. Generalizability may be limited beyond the population studied, specifically asymptomatic and nondiabetic participants meeting LDL-cholesterol and hsCRP eligibility criteria. Strengths of the study include the large number of participants with randomized baseline and on-treatment Lp(a) concentrations assayed with a validated immunoassay independent of kringle IV type-2 repeats, detailed baseline cardiovascular risk assessment, prospective endpoint adjudication, and the use of potent statin therapy with very low achieved LDL-cholesterol concentrations.
Conclusions
In this cohort of asymptomatic white JUPITER participants with low LDL-cholesterol and elevated hsCRP, Lp(a) was a significant determinant of residual risk. Furthermore, the efficacy of rosuvastatin in reducing CVD was similar among participants with high or low Lp(a) concentrations. Future studies are needed to directly assess the impact of specifically lowering Lp(a) concentrations for potentially reducing residual risk.
Supplementary Material
Acknowledgments
The authors thank James Morton, PharmD (Clinical Programs Manager) and William Patten (Clinical Development Department), Quest Diagnostics Nichols Institute San Juan Capistrano, for their diligent assistance.
Funding Sources: JUPITER was financially supported by AstraZeneca, who collected trial data and monitored sites but had no role in the design or conduct of the current study, including data analysis or interpretation, drafting or editing this report, or in preparation, review or the decision to submit the manuscript for publication. Quest Diagnostics Nichols Institute San Juan Capistrano absorbed the cost of performing the Lp(a) measurements and performed them in a blinded manner. Research reported in this publication was supported in part by the National Heart, Lung, And Blood Institute (NHLBI) of the National Institutes of Health under Award Number R01HL117861 to Dr Mora. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest Disclosures: Dr. Mora has received research support from AstraZeneca, Atherotec Diagnostics, and the NHLBI, and served as a consultant to Pfizer, Quest Diagnostics, and Genzyme, and received speaker honoraria from AstraZeneca, Abbott, and the National Lipid Association for educational (non-promotional) activities. Dr. Everett has received investigatorinitiated grant support from Roche Diagnostics. Drs Caulfield, Hantash, and Wohlgemuth are employees of Quest Diagnostics. Dr. Ridker has received research grant support from AstraZeneca, Novartis, Amgen, and NHLBI and has served as a consultant to Genzyme, Jannsen, Aegerion, ISIS, Vascular Biogenics, Boeringer, Pfizer, and Merck. Dr. Ridker is listed as a coinventor on patents held by the Brigham and Women's Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease that have been licensed to AstraZeneca and Seimens.
Footnotes
This manuscript will be presented in part as an oral research abstract on November 17, 2013 at the American Heart Association Annual Scientific Sessions in Dallas, Texas.
References
- 1.Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C Cholesterol Treatment Trialists' (CTT) Collaborators. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsimikas S, Hall JL. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol. 2012;60:716–721. doi: 10.1016/j.jacc.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 3.Berg K. A new serum type system in man—the LP system. ActaPatholMicrobiol Scand. 1963;59:369–382. doi: 10.1111/j.1699-0463.1963.tb01808.x. [DOI] [PubMed] [Google Scholar]
- 4.Koschinsky ML, Beisiegel U, Henne-Bruns D, Eaton DL, Lawn RM. Apolipoprotein(a) size heterogeneity is related to variable number of repeat sequences in its mRNA. Biochemistry. 1990;29:640–644. doi: 10.1021/bi00455a007. [DOI] [PubMed] [Google Scholar]
- 5.Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, White IR, Marcovina SM, Collins R, Thompson SG, Danesh J Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The Emerging Risk Factors Collaboration*. Lipid-Related Markers and Cardiovascular Disease Prediction. JAMA. 2012;307:2499–2506. doi: 10.1001/jama.2012.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveira A, Malarstig A, Green FR, Lathrop M, Gigante B, Leander K, de Faire U, Seedorf U, Hamsten A, Collins R, Watkins H, Farrall M PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 8.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 9.Maher VM, Brown BG, Marcovina SM, Hillger LA, Zhao XQ, Albers JJ. Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein(a) JAMA. 1995;274:1771–1774. [PubMed] [Google Scholar]
- 10.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ,Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 11.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, KasteleinJJ, Koenig W, Libby P, Lorenzatti AJ, Macfadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ JUPITER Trial Study Group. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–1182. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- 12.Mora S, Glynn RJ, Boekholdt SM, Nordestgaard BG, Kastelein JJ, Ridker PM. On-treatment non-high-density lipoprotein cholesterol, apolipoprotein B, triglycerides, and lipid ratios in relation to residual vascular risk after treatment with potent statin therapy: JUPITER (justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin) J Am Coll Cardiol. 2012;59:1521–1528. doi: 10.1016/j.jacc.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ridker PM, Genest J, Boekholdt SM, Libby P, Gotto AM, Nordestgaard BG, Mora S, MacFadyen JG, Glynn RJ, Kastelein JJ JUPITER Trial Study Group. HDL cholesterol and residual risk of first cardiovascular events after treatment with potent statin therapy: an analysis from the JUPITER trial. Lancet. 2010;376:333–339. doi: 10.1016/S0140-6736(10)60713-1. [DOI] [PubMed] [Google Scholar]
- 14.Ridker PM, MacFadyen JG, Wolfert RL, Koenig W. Relationship of lipoprotein-associated phospholipase A2 mass and activity with incident vascular events among primary prevention patients allocated to placebo or to statin therapy: an analysis from the JUPITER trial. Clin Chem. 2012;58:877–886. doi: 10.1373/clinchem.2011.180281. [DOI] [PubMed] [Google Scholar]
- 15.Marcovina SM, Koschinsky ML, Albers JJ, Skarlatos S. Report of the National Heart, Lung, and Blood Institute Workshop on Lipoprotein(a) and Cardiovascular Disease: recent advances and future directions. Clin Chem. 2003;49:1785–1796. doi: 10.1373/clinchem.2003.023689. [DOI] [PubMed] [Google Scholar]
- 16.Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgözoglu L, Tybjærg-Hansen A European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA. 2006;296:1363–1370. doi: 10.1001/jama.296.11.1363. [DOI] [PubMed] [Google Scholar]
- 18.Maeda S, Abe A, Seishima M, Makino K, Noma A, Kawade M. Transient changes of serum lipoprotein(a) as an acute phase protein. Atherosclerosis. 1989;78:145–150. doi: 10.1016/0021-9150(89)90218-9. [DOI] [PubMed] [Google Scholar]
- 19.Pietrzak A, Kadzielewski J, Janowski K, Roliński J, Krasowska D, Chodorowska G, Paszkowski T, Kapeć E, Jastrzebska I, Tabarkiewicz J, Lotti T. Lipoprotein (a) in patients with psoriasis: associations with lipid profiles and disease severity. Int J Dermatol. 2009;48:379–387. doi: 10.1111/j.1365-4632.2009.03994.x. [DOI] [PubMed] [Google Scholar]
- 20.Sari RA, Polat MF, Taysi S, Bakan E, Capoğlu I. Serum lipoprotein(a) level and its clinical significance in patients with systemic lupus erythematosus. Clin Rheumatol. 2002;21:520–524. doi: 10.1007/s100670200127. [DOI] [PubMed] [Google Scholar]
- 21.Virani SS, Brautbar A, Davis BC, Nambi V, Hoogeveen RC, Sharrett AR, Coresh J, Mosley TH, Morrisett JD, Catellier DJ, Folsom AR, Boerwinkle E, Ballantyne CM. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125:241–249. doi: 10.1161/CIRCULATIONAHA.111.045120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mora S, Kamstrup PR, Rifai N, Nordestgaard BG, Buring JE, Ridker PM. Lipoprotein(a) and risk of type 2 diabetes. Clin Chem. 2010;56:1252–1260. doi: 10.1373/clinchem.2010.146779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berg K, Dahlén G, Christophersen B, Cook T, Kjekshus J, Pedersen T. Lp(a) lipoprotein level predicts survival and major coronary events in the Scandinavian Simvastatin Survival Study. Clin Genet. 1997;52:254–261. doi: 10.1111/j.1399-0004.1997.tb04342.x. [DOI] [PubMed] [Google Scholar]
- 24.Gaw A, Brown EA, Docherty G, Ford I. Is lipoprotein(a)-cholesterol a better predictor of vascular disease events than total lipoprotein(a) mass? A nested case control study from the West of Scotland Coronary Prevention Study. Atherosclerosis. 2000;148:95–100. doi: 10.1016/s0021-9150(99)00259-2. [DOI] [PubMed] [Google Scholar]
- 25.Carlson LA, Hamsten A, Asplund A. Pronounced lowering of serum levels of lipoprotein Lp(a) in hyperlipidaemic subjects treated with nicotinic acid. J Intern Med. 1989;226:271–276. doi: 10.1111/j.1365-2796.1989.tb01393.x. [DOI] [PubMed] [Google Scholar]
- 26.Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 27. [Accessed January 11, 2013];Merck Press Release. Available at: http://www.mercknewsroom.com/pressrelease/prescription-medicine-news/merck-announces-hps2-thrive-study-tredaptive-extendedrelea.
- 28.Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, Zafarino J, Mitchel Y, Barter P. Determining the Efficacy and Tolerability Investigators. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 29.Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, Lisbon E, Gutierrez M, Webb C, Wu R, Du Y, Kranz T, Gasparino E, Swergold GD. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–1118. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- 30.Akdim F, Visser ME, Tribble DL, Baker BF, Stroes ES, Yu R, Flaim JD, Su J, Stein EA, Kastelein JJ. Effect of mipomersen, an apolipoprotein B synthesis inhibitor, on low-density lipoprotein cholesterol in patients with familial hypercholesterolemia. Am J Cardiol. 2010;105:1413–1419. doi: 10.1016/j.amjcard.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Merki E, Graham M, Taleb A, Leibundgut G, Yang X, Miller ER, Fu W, MullickAE,Lee R, Willeit P, Crooke RM, Witztum JL, Tsimikas S. Antisense oligonucleotide lowers plasma levels of apolipoprotein (a) and lipoprotein (a) in transgenic mice. J Am Coll Cardiol. 2011;57:1611–1621. doi: 10.1016/j.jacc.2010.10.052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.