

Abstract
Herpetic stromal keratitis is characterized by an inflammatory response that includes neutrophils, macrophages, NK cells and T cells. The factors that are responsible for this inflammation are pro-inflammatory cytokines and chemokines. Many of these factors have been defined for primary disease, but relatively few have been investigated during recurrent HSK. The present study was designed to determine the role that two of these factors, IL-6 and CXCL1 play during recurrent HSK. Results clearly indicate that unlike primary disease, IL-6 plays no role in recurrent HSK. However, the presence of CXCL1 is required for recurrent HSK as evidenced by the lack of corneal disease in mice treated with anti-CXCL1 antibody. This was confirmed using mice lacking the primary receptor for CXCL1, CXCR2. Corneal disease in this strain was significantly reduced compared to wild-type B6 controls. Unexpectedly, lack of disease occurs even though CXCL1 KO mice display increased viral shedding at the cornea. The primary mechanism that CXCL1 plays during disease is its ability to stimulate neutrophils to infiltrate the cornea following reactivation. This report provides further evidence that primary HSK and recurrent HSK possess overlapping yet distinct disease mechanisms.
INTRODUCTION
Herpetic stromal keratitis (HSK) is an infection of the cornea with herpes simplex virus 1 (HSV-1) and is the leading cause of infectious blindness in the Western world with one study determining a prevalence of HSV keratitis of 149/100,000 people (1). As with other herpes viruses, there are both primary and recurrent forms of the disease.
In humans, primary disease is rare, occurring mostly in children and the immunosuppressed. Primary disease is most often clinically asymptomatic, although in 1-6% of cases it presents as blepharo-conjunctivitis that heals without scarring (2). Primary disease begins typically by exposure through corneal or oral epithelium. The virus replicates in these cells and then travels via retrograde axonal transport in sensory neurons to the sensory ganglia (most often trigeminal) where it establishes latency. During latency, the viral genome is present, but few active virions are detected in these latently infected neurons (3). The dominant form of clinical disease is the result of reactivation of virus which is typically “triggered” by immunosuppressive events such as fever, menses, sunlight (UV), irradiation, stress, and trauma (4). Following reactivation, the virus travels via anterograde axonal transport back to the epithelial surface, and its replication and subsequent host immune response are responsible for observed symptoms that define most cases of corneal keratitis (5).
Recurrent disease in the cornea is an immunopathologic condition that is initiated by renewed presence of virus in the cornea which re-stimulates the immune response leading to inflammation of the cornea resulting in damage to the cornea. In humans, the inflammatory infiltrate in HSK is characterized by influx of a phenotypically diverse population of leukocytes consisting of lymphocytes, neutrophils, and mononuclear phagocytes (6,7). Animal studies have shown that the cell type found in greatest numbers in corneas displaying disease are neutrophils (8).
Typically, neutrophils follow chemokine and cytokine cues as to when and where to enter tissues in response to pathogens. In 2008, this lab demonstrated that pro-inflammatory cytokines CCL2 and CCL3 were unimportant in the pathogenesis of recurrent HSK. In fact, CCL3 deficient mice were shown to experience worse disease than wild type mice (9). In 2007, Lin et al. showed that neutrophils very quickly infiltrate the cornea in response to LPS administration, and that CXCL1/keratinocyte-derived chemokine (KC), produced by corneal stromal cells increased in parallel with neutrophilic infiltration (10). Previous studies also demonstrated that CXCL1 is upregulated in HSV-1 cornea infection and that it is crucial to this neutrophil infiltrate (10-13). One of the receptors for CXCL1, CXCR2 (14), has been shown to be important in controlling viral infection of the cornea (15). Banerjee, et al. reported that in the absence of CXCR2, there was minimal neutrophil influx during the first 7 days and that these mice exhibited increased IL-6 production that appeared to induce vascular endothelial factor production leading to worse HSK than was observed in wild-type mice (15). An additional chemokine, CXCL10 has more recently been reported to restrict viral replication in the cornea and to reduce the severity of primary HSK in a model using the RE strain of HSV-1 for infection (16).
In addition to CXCL1, HSV-1-infected human corneal epithelial cells also produce increased levels of the pro-inflammatory cytokine IL-6 following primary infection with HSV-1 (17). This mechanism responsible for increased proinflammatory cytokine production has been proposed to be through sequential activation of Toll-like receptors (11). In support of IL-6's role in primary HSK, Fenton, et al. demonstrated that IL-6 KO mice experience significantly decreased corneal opacity in primary HSK when compared to wild-type mice (12). Futhermore, they demonstrated that administration of exogenous IL-6 at time of infection restored disease to the same level as that experienced by wild type mice, confirming that IL-6 was crucial to developing primary HSK (12).
As previously described, the predominant form of HSK that affects humans is recurrent disease (1-7). Consequently, we decided to investigate whether CXCL1 and IL-6, which are so important during acute HSK, play similar roles during recurrent HSK. Our results demonstrate that CXCL1 is critical in developing recurrent HSK in mice. However, in contrast to what is seen in primary HSK (12,15-17), IL-6 has no role during recurrent HSK.
MATERIALS AND METHODS
Mice
Investigations with mice conformed to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. C57BL/6 (B6) were purchased from NCI and NIH inbred mice were obtained from Harlan OLAC (England). The B6.129S2(C)-Cxcr2tm1Mwm/J (B6-CXCR2 KO) mice and the B6.129S2-IL6tm1Kopf/J (B6-IL-6 KO) mice were obtained initially from Jackson Labs (Bar Harbor, ME) and bred in our colony.
Infection of Mice
6-12 week old mice were infected on the scarified cornea with 106 PFU HSV-1 McKrae strain as previously described (18). Each mouse received an intra-peritoneal (IP) injection of 0.5 ml pooled human serum (Sigma Chemicals, St. Louis MO; ED50 for virus neutralization = 1:1600) concurrent with infection. Administration of anti-HSV antibodies at the time of ocular infection has been shown to protect mice from death and corneal disease during primary infection, while allowing for the establishment of latency and subsequent reactivation of virus after corneal UV-B exposure. These antibodies are undetectable at the time of UV-B irradiation 5 weeks after primary infection. HSV positive eye swabs obtained three days after application of virus confirm primary infection.
UV-B irradiation and virus reactivation
Mice were reactivated from latency as previously described (18). Briefly, the eyes of all latently infected mice were examined for corneal opacity before irradiation, and only animals with clear corneas were used. At least 5 weeks after primary infection, the eyes of latently-infected and control mock-infected mice were exposed to 250 mJ/cm2 of UV-B light using a TM20 Chromato-Vu transilluminator (UVP, Inc., San Gabriel, CA), which emits UV-B at a peak wavelength of 302 nm. Irradiated mice were swabbed with sterile cotton applicators from day 0 to day 7, unless otherwise indicated. The swab material was cultured on VERO cells, as described above, in order to detect recurrent virus shedding from the cornea. Reactivation was defined as the finding of any HSV positive eye swab on any days post-UV-B exposure, with day 0 swabs serving as a control.
Clinical evaluation
On the designated days after viral infection or UV-B reactivation, a masked observer examined mouse eyes through a binocular-dissecting microscope in order to score clinical disease. Stromal opacification was rated on a scale of 0 to 4, where 0 indicates clear stroma, 1 indicates mild stromal opacification, 2 indicates moderate opacity with discernible iris features, 3 indicates dense opacity with loss of defined iris detail except pupil margins, and 4 indicates total opacity with no posterior view. Corneal neovascularization was evaluated as described (18,19) using a scale of 0-8, where each of four quadrants of the eye is evaluated for the density of vessels that have grown into them. Periocular disease was measured in a masked fashion on a semiquantitative scale as previously described (20). Note: Uninfected, UV-B irradiated control mice were used as a baseline for any effects due to UV-B irradiation.
Neutralization of CXCL1 and IL-6 by monoclonal antibody treatment
Mice receiving anti-KC (CXCL1) were injected IP with 40 μg of anti-KC (R&D Systems MAB 453, clone 48415). Mice receiving anti-IL-6 were injected IP with 100 μg anti-IL-6 (Southern Biotech, clone MP5-20F3). Infected control groups were injected IP with 100 μg (500 μg/mL, Sigma 14131) nonspecific control IgG. A single injection of these antibodies was performed on the same day that animals were reactivated with UV-B light.
Flow Cytometric Analysis
Cells were isolated from corneas as previously described (21). Briefly, corneas were excised at 18 and 23 dpi and incubated in PBS-EDTA at 37°C for 15 minutes at 37°C. Stromas were separated from overlying epithelium and digested in 84 U collagenase type 1 (Sigma-Aldrich, St. Louis, MO) per cornea for 2 hours at 37°C and then were triturated to form a single-cell suspension. Suspensions were filtered through a 40-μm cell strainer cap (BD Labware, Bedford, MA) and washed and then stained. Suspensions were stained with: PerCP-conjugated anti-CD45 (clone 30-F11) and Alexa Fluor700-Gr-1 (clone RB6-8C5) (from BioLegend, San Diego, CA); FITC conjugated anti-CD4 (clone RM4–5), PE-conjugated anti-CD8α (clone 53–6.7), PE-Cy7-conjugated anti-CD11c (clone HL3), (all BD PharMingen); eFluor450-conjugated CD11b (clone M1/70) (from eBiosciences, San Diego, CA). Cells were then analyzed on a flow cytometer (FACSAria with FACSDIVA data analysis software; BD Biosciences).
Statistics
All statistical analyses were performed with the aid of Sigma Stat for Windows, version 2.0 (Jandel, Corte Madera, CA). The log rank test was used to compare disease scores. Student's unpaired t-test was used to compare virus titer data. Fisher's exact X2 tests were used to compare limiting dilution assay data.
RESULTS
Previous studies evaluating the early production of chemokines and cytokines during primary HSK have indicated that several of them are critical to the development of disease (22). Some, but not all of these factors have been evaluated in recurrent HSK (23). The results of these later studies have been to illustrate that cytokines and chemokines that are important to the development of primary HSK may have very little to do with the development of recurrent HSK (23). As a consequence of our ongoing interest in evaluating these factors in recurrent HSK, we evaluated the role of CXCL1 (KC in mice) and IL-6 during recurrent HSK.
Inflammation is critical to the development of clinical disease in HSK. By blocking the factors that promote inflammatory cell migration, we hypothesized that this would lead to decrease the total inflammation in the eye a consequence would be the presumed decrease in clinical disease. We chose CXCL-1and IL-6 precisely because they have pro-inflammatory properties and as such provide logical targets for therapeutic intervention.
We and others have demonstrated that the presence of a significant infiltration of neutrophils into the cornea is associated with clinical disease in both primary (22,24,25) and recurrent (21) models of HSK. Consequently, our first molecular target was CXCL-1, a potent neutrophil chemokine (9-12). Our data demonstrates that antibody neutralization of CXCL-1 in vivo resulted in significantly lower clinical scores in both opacity and neovascularization during each week of clinical observation following reactivation as compared to treatment with a control antibody preparation (Figure 1A, 1B). At the same time we also targeted IL-6 with a neutralizing antibody towards this cytokine. Unlike what has been reported during primary HSK (11,17), neutralizing IL-6 did not lead to a decrease in recurrent HSK (Figures 1A, 1B). When these mice were assayed for shedding virus, as a measure of apparent reactivation rate, more mice treated with anti-CXCL1 had detectable virus than either anti-IL-6 or control antibody treated mice, however there were no differences in viral persistence between these different treatment groups (Table 1). This suggests that in the absence of CXCL1, more viruses are produced in the cornea following UV-B induced reactivation, but that virus is eventually cleared with the same kinetics.
Figure 1.
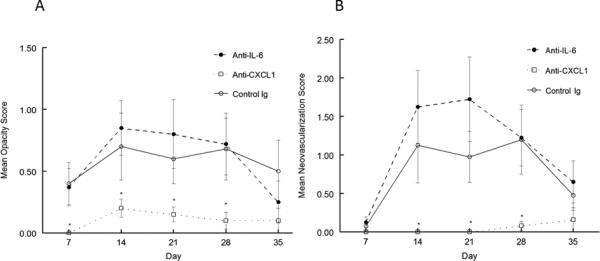
Recurrent HSK in anti-CXCL1 treated mice was significantly less than in anti-IL-6 treated and control antibody treated mice. Eyes of B6 mice were infected with 106 pfu of HSV-1, McKrae strain. Six weeks following infection mice were irradiated with UV-B to reactivate the latent infection. Mice were then treated with anti-CXCL1 (n=19), anti-IL-6 (n=20) or treated with control antibody (n=18). Corneal opacity (A) and corneal neovascularization (B) were measured and compared between these different treatments. Mice treated with anti-CXCL1 displayed significantly reduced corneal opacity and neovascularization than did either anti-IL-6 treated or control antibody treated mice from day 7 until day 35 (P<0.01-0.001). No significant differences were noted in between anti-IL-6 and control antibody treated mice.
Table 1.
| IgG | Anti-IL-6 | Anti-CXCL1 | |
|---|---|---|---|
| % Positive Swabs@ | 7% | 6% | 12% |
| Total Shedding Days# | 6 | 6 | 11 |
| Days Shedding/mouse† | 1.2 ± 0.11 | 1.0 ± .00 | 1.2 ± 0.10 |
| % Reactivation Rate‡ | 27.80% | 30% | 47.40% |
| Final Day Sheddingǂ | Day 5 | Day 5 | Day 5 |
| n | 18 | 20 | 19 |
The percent positive swabs is the percentage of virus-positive eye swabs (140 to 286 eye swabs per group) over the 10-day period following UV-B irradiation. (P<0.01 for anti-CXCL1 treated mice.)
Total shedding days number of days swab positive. (P<0.01 for CXCL1 treated mice.)
Days shedding/mouse is the number of days that a positive mouse shed virus. (No significance)
Percent reactivation rate is the percentage of mice that reactivated. (P<0.02 for CXCL1 treated mice.)
Final day shedding was the last day that a mouse was positive for a particular group.
Since the effect of targeting CXCL1 was so dramatic, we wished to confirm these results by performing similar studies in a different strain of mouse. We chose the inbred NIH strain, because this mouse strain has been used very successfully in recurrent models of HSK (23). As was seen in B6 mice, these mice also failed to generate significant levels of recurrent HSK disease when treated with a neutralizing antibody directed against CXCL1 (Figure 2A, 2B). It should be noted that there were no differences in viral shedding between anti-CXCL and control antibody treated mice as both demonstrated an apparent reactivation rate of about 70% (data not shown).
Figure 2.
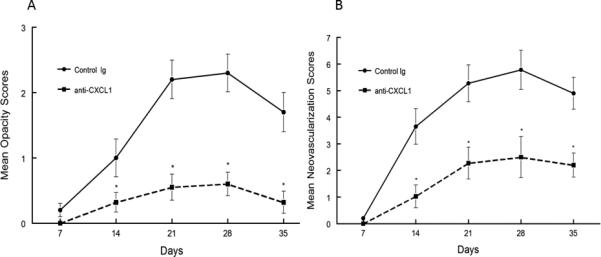
Recurrent HSK in anti-CXCL1 treated NIH mice was significantly less than in control antibody treated mice. Eyes of NIH mice were infected with 106 pfu of HSV-1, McKrae strain. Six weeks following infection mice were irradiated with UV-B to reactivate the latent infection. Mice were then treated with anti-CXCL1 (n=20) or treated with control antibody (n=20). Corneal opacity (A) and corneal neovascularization (B) were measured and compared between these different treatments. Mice treated with anti-CXCL1 displayed significantly reduced corneal opacity and neovascularization than did control antibody treated mice from day 7 until day 35 (P<0.01-0.001).
In order to provide further confirmation we decided to test gene targeted mice that do not express the primary receptor for CXCL1, namely CXCR2 (13,14) in our model of recurrent HSK. Results from these studies further emphasized that CXCL1 is needed for recurrent HSK, as B6-CXCR2 KO mice displayed significantly reduced recurrent HSK than did wild-type B6 mice (Figure 3A, 3B).
Figure 3.
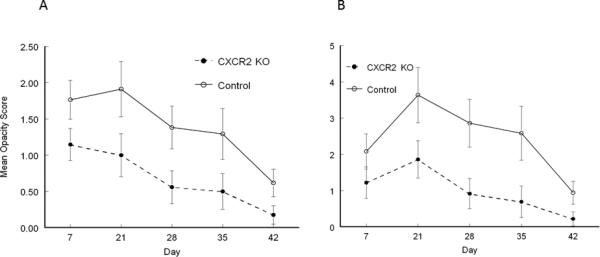
Recurrent HSK in B6-CXCR2 KO mice was significantly less than in wild-type B6 mice. Eyes of mice were infected with 106 pfu of HSV-1, McKrae strain. Six weeks following infection mice were irradiated with UV-B to reactivate the latent infection. B6-CXCR2 KO mice (n=19) were compared with wild-type B6 mice (n=18) for corneal opacity (A) and corneal neovascularization (B). CXCR2 mice demonstrated significantly reduced corneal opacity and neovascularization than did wild-type B6 mice from day 7 until day 35 (P<0.01-0.001).
Because our results for neutralizing IL-6 were so unexpected, we evaluated recurrent HSK in mice genetically incapable of producing this cytokine, B6-IL-6 KO mice. Results were similar to neutralization studies (Figures 1A, 1B), as recurrent HSK in B6-IL-6 KO mice was similar to that observed in wild-type B6 mice (Figure 4A, 4B). In fact, if anything, the trend though not statistically significant for virtually all time points, seemed to be towards increased recurrent HSK in these mice. Consequently, these data support the notion that IL-6 plays little, if any, critical role in the development of recurrent HSK.
Figure 4.
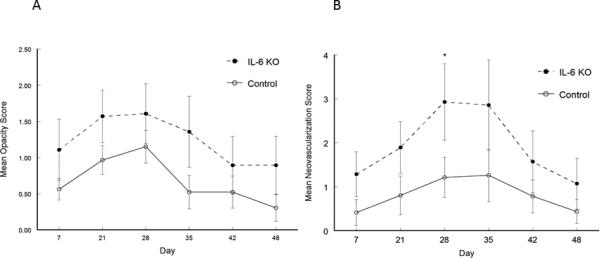
Recurrent HSK in B6-IL-6 KO mice was indistinguishable from that seen in wild-type B6 mice. Eyes of mice were infected with 106 pfu of HSV-1, McKrae strain. Six weeks following infection mice were irradiated with UV-B to reactivate the latent infection. B6-IL-6 KO mice (n=16) were compared with wild-type B6 mice (n=19) for corneal opacity (A) and corneal neovascularization (B). No significant differences were noted between these strains of mice for either corneal opacity or corneal neovascularization.
Since mice treated with anti-CXCL1 demonstrated its critical role during recurrent HSK, we decided to characterize the inflammatory infiltrate of B6 mice treated with anti-CXCL1 versus B6 mice treated with control IgG via flow cytometry. This analysis revealed two basic differences, the first being that the CD45+ infiltrate was less in anti-CXCL1 treated mice (4678±1046/cornea) than in B6 control antibody treated mice (36,723±8439/cornea). Indicating that overall inflammation by CD45+ cells is significantly affected by targeting CXCL1. The second difference was that analysis of the CD45+ infiltrate in mice treated with anti-CXCL1 displayed a significantly reduced percentage of Gr-1+ neutrophils (Figure 5, p = 0.019) and a non-statistically significant trend towards increased percentages of CD4+ T cells than did mice treated with control Ig (Figure 5). These data confirm the important role that CXCL1 plays in directing migration of neutrophils to corneas experiencing recrudescence of HSV-1.
Figure 5.
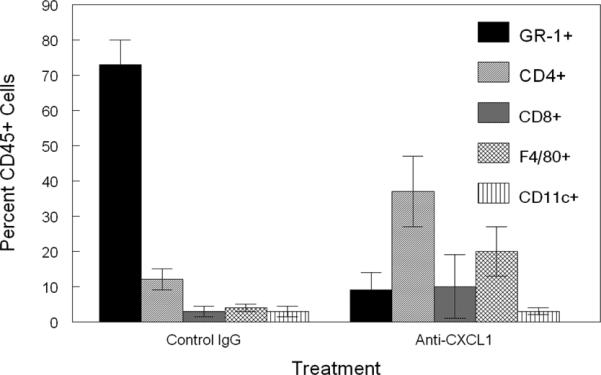
Inflammatory infiltrate from B6 mice treated with anti-CXCL1 demonstrates a significant reduction in Gr-1+ neutrophils compared to control antibody treated mice. Corneas were removed from latently infected B6 mice at days 17 and 23 following reactivation. These corneas were disaggregated into single-cell suspensions and stained with antibodies against: CD45, CD4, CD8α, Gr-1, CD11b, CD11c, and F4/80. Cells were then analyzed by flow cytometry. Data represents 8 to 10 corneas per group. Data indicates a significant decline in the percent Gr-1+ neutrophils between anti-CXCL1 treated and control IgG treated mice (P<0.01).
DISCUSSION
At present the treatment of choice for herpetic stromal keratitis (HSK) is a combination of anti-herpetic drugs to limit the replication of virus and steroids to prevent, or at least restrict, inflammation. In most cases this is effective in controlling the disease. However, such treatment comes with certain costs. One is that not all individuals respond the same to these treatments (1-6). In one study, up to 20% of patients retained some degree of corneal clouding (26) while in another study 30% had lingering corneal inflammation (27). Another is that it is possible for the virus to become refractory to the anti-viral treatment (5,28). And finally, steroid treatment itself has the potential of causing its own unwanted side effects (29-31). We have been studying recurrent HSK in order to better understand the factors that control disease such that we might identify more specific targets of therapeutic intervention. In this report we demonstrate that at least two strains of mice treated with anti-KC (CXCL1) displayed significant and strikingly lower incidence and severity of disease when compared to control antibody treated animals. Because of CXCL1/KC's established role as a chemoattractant for neutrophils, these results again confirm the importance of neutrophils in recurrent HSK pathology. They also support the idea that the majority of corneal disease pathology can be prevented with neutralization of CXCL1/KC. Given the fact that there are few effective treatments for ocular HSV-1 infections, this data suggests a possible new target for medical treatment of a disease which continues to be responsible for significant morbidity worldwide.
We further confirmed the importance of CXCL1 by using mice that do not express the CXCR2 chemokine receptor for which CXCL1 is known to bind. These mice also displayed significantly less disease than did wild-type B6 mice. Consequently, the case for CXCL1 being a critical factor leading to significant recurrent HSK disease is very compelling.
So while the case for targeting CXCL1 during recurrent HSK is strong, we did not observe similar results when targeting IL-6. This is in contrast to what has been reported during primary HSK (12,15,32,33) where IL-6 production appears to play a significant role during acute HSV-1 infection of the cornea and thus is involved in the disease pathology associated with primary HSK. Our study utilized both antibody neutralization and IL-6 gene targeted mice as a means of targeting IL-6. The results for both of these methods indicate that lack of functional IL-6 does not affect recurrent HSK disease incidence or progression in B6 mice. While it is possible IL-6 might play a more critical role in mice that display greater disease than do B6 mice, such as NIH mice, we have not observed that for other factors that we have tested in our recurrent model of HSK (19,21 and unpublished observations when assessing IFN-γ and IL-10). That said, these results concerning IL-6 were surprising, as this cytokine is found in abundance during both primary (12,15,32,33) and recurrent (34) HSK and thus it has been assumed that it is important in both forms of the disease. However, none of these studies actually neutralized IL-6 and thus did not evaluate its importance to the disease process in animals that can and do generate a normal IL-6 response. Another possible implication of these data concerns the potential role that Th17 cells might play in recurrent HSK. It is known that IL-6 is involved in the generation of Th17 cells (35) which have been implicated in primary HSK (36,37). Consequently, if Th17 or IL-17 itself is playing a significant role in recurrent HSK and IL-6 is not involved in recurrent HSK this implies that either Th17 cells are generated by a pathway that does not involve IL-6 or some other cell in the cornea is producing IL-17. Future studies need to be performed to address the role that IL-17 plays in recurrent HSK, particularly since Suryawanshi, et al. reported that IL-17A leads to increased production of CXCL1 during primary HSK which they demonstrated led to increased neutrophilic infiltration into the cornea (37).
It is interesting to note that while we did not observe an increase in the time that mice shed virus, we did note that the number of mice shedding virus was significantly greater in mice treated with neutralizing anti-CXCL1 antibody (47%) than in B6 mice treated with a control antibody (28%). One would have predicted that this might lead to increased corneal disease due to greater levels of virus in these CXCL1 neutralized mice as compared to control antibody-treated B6 mice. This was precisely the argument made for results reported for CXCL10 KO mice which displayed increased viral titers which they argued led to increased primary HSK (16). That said, that argument is not supported by our data for neutralizing CXCL1 nor did we see that in CXCR2 KO mice which also displayed increased numbers of mice shedding virus in spite of reduced recurrent HSK (data not shown). We would argue that the primary reason for our observations is that while targeting CXCL1 leads to increased virus shedding in the corneas, the fact that the numbers of neutrophils infiltrating the cornea is significantly reduced will lead to less disease. Particularly since we know that HSK requires a significant neutrophilic infiltrate of the cornea (21,24), less disease will be the consequence of fewer neutrophils. That without the recruitment of neutrophils to the cornea it does not matter if viral production is not controlled as efficiently because these corneas lack a critical cell required for HSK. In fact, reports have indicated that the dominant cells that remove virus from the cornea are T cells and NK cells and not neutrophils (38-40), thus viral clearance is not dependent on a robust recruitment of neutrophils.
Taken together, our results clearly indicate that CXCL1 is a major player in recurrent HSK. That therapeutic targeting of this chemokine significantly reduces recurrent disease. These results also indicate that IL-6, is not intimately involved in recurrent HSK and thus is not a useful target for therapeutic intervention.
Acknowledgements
We thank Stephanie Zobell and Chloe Potter for technical assistance in this work. We thank Dr. Robyn Klein, from the Department of Internal Medicine and Infectious Diseases at Washington University, for helpful discussions.
This work was supported by National Institutes of Health Grants EY16352 and EY21247 and an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology.
Footnotes
Disclosures
The authors have no financial conflicts of interest.
REFERENCES
- 1.Liesegang TJ, Melton LJ, Daly PJ, Ilstrup DM. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch. Ophthalmol. 1989;107:1155–1159. doi: 10.1001/archopht.1989.01070020221029. [DOI] [PubMed] [Google Scholar]
- 2.Umene K, Sakaoke H. Evolution of herpes simplex virus type 1 under herpesvirus evolutionary processes. Arch. Virol. 1999;144:637–656. doi: 10.1007/s007050050533. [DOI] [PubMed] [Google Scholar]
- 3.Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc. Natl. Acad, Sci. USA. 2002;99:978–983. doi: 10.1073/pnas.022301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuli S, Sonal S. Herpes Simplex Keratitis. In: Yanoff JS, Duker M, et al., editors. Ophthalmology. 3rd Edition Elsevier Inc.; 2009. © 2009. [Google Scholar]
- 5.Whitley RJ. Herpes simplex viruses. In: Fields BN, et al., editors. Fields virology. 3rd ed. Lippincott-Raven Publishers; Philadelphia: 1996. pp. 2297–2342. [Google Scholar]
- 6.Duan R, de Vries RD, Osterhaus AD, Remeijer L, Verjans GM. Acyclovir-Resistant Corneal HSV-1 Isolates from Patients with Herpetic Keratitis. J. Infect. Dis. 2008;198:659–663. doi: 10.1086/590668. [DOI] [PubMed] [Google Scholar]
- 7.Liesegang TJ. Ocular herpes simplex infection: pathogenesis and current therapy. Mayo Clin. Proc. 1998;63:1092–1105. doi: 10.1016/s0025-6196(12)65504-9. [DOI] [PubMed] [Google Scholar]
- 8.Thomas J, Gangappa S, Kanangat S, Rouse BT. On the Essential Involvement of Neutrophils in the Immunopathologic Disease Herpetic Stromal Keratitis. J. Immunol. 1997;158:1383–1391. [PubMed] [Google Scholar]
- 9.Stuart PM, Morris JE, Sidhu M, Keadle TL. CCL3 protects mice from corneal pathology during recurrent HSV-1 infection. Front. Biosci. 2008;13:4407–4415. doi: 10.2741/3013. [DOI] [PubMed] [Google Scholar]
- 10.Lin M, Carlson E, Diaconu E, Pearlman E. CXCL1/KC and CXCL5/LIX are selectively produced by corneal fibroblasts and mediate neutrophil infiltration to the corneal stroma in LPS keratitis. J. Leukoc. Biol. 2007;81:786–792. doi: 10.1189/jlb.0806502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Zhang J, Kumar A, Zheng M, Atherton SS, Yu FS. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology. 2006;117:167–176. doi: 10.1111/j.1365-2567.2005.02275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fenton R, Molesworth-Kenyon S, Oakes JE, Lausch RN. Linkage of IL-6 with Neutrophil Chemoattractant Expression in Virus-Induced Ocular Inflammation. Invest. Ophthalmol. Vis. Sci. 2002;43:737–743. [PubMed] [Google Scholar]
- 13.Duan R, Remeijer L, van Dun JM, Osterhaus AD, Verjans GM. Granulocyte macrophage colony-stimulating factor expression in human herpetic stromal keratitis: Implications for the role of neutrophils in HSK. Invest. Ophthalmol. Vis. Sci. 2007;48:277–284. doi: 10.1167/iovs.06-0053. [DOI] [PubMed] [Google Scholar]
- 14.Tsai HH, Frost E, To V, Robinson S, Ffrench-Constant C, Geertman R, Ransohoff RM, Miller RH. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell. 2002;110:373–383. doi: 10.1016/s0092-8674(02)00838-3. [DOI] [PubMed] [Google Scholar]
- 15.Banerjee K, Biswas PS, Kim B, Lee S, Rouse BT. CXCR2−/− mice show enhanced susceptibility to herpetic stromal keratitis: a role for IL-6-induced neovascularization. J. Immunol. 2004;172:1237–1245. doi: 10.4049/jimmunol.172.2.1237. [DOI] [PubMed] [Google Scholar]
- 16.Shen FH, Wang SW, Yeh TM, Tung YY, Hsu SM, Chen SH. Absence of CXCL10 aggravates herpes stromal keratitis with reduced primary neutrophil influx in mice. J. Virol. 2013;87:8502–8510. doi: 10.1128/JVI.01198-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staats HF, Lausch RN. Cytokine expression in vivo during murine herpetic stromal keratitis: effect of protective antibody therapy. J. Immunol. 1993;151:277–283. [PubMed] [Google Scholar]
- 18.Keadle TL, Morrison LA, Morris JE, Pepose JS, Stuart PM. Therapeutic immunization with a virion host shutoff-defective, replication in competent herpes simplex virus type 1 strain limits recurrent herpetic ocular infection. J. Virol. 2002;76:3615–3625. doi: 10.1128/JVI.76.8.3615-3625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuart PM, Summers BC, Morris JE, Morrison LA, Leib DA. CD8(+) T cells control corneal disease following ocular infection with herpes simplex virus type 1. J. Gen. Virol. 2004;85:2055–2063. doi: 10.1099/vir.0.80049-0. [DOI] [PubMed] [Google Scholar]
- 20.Smith TJ, Ackland-Berglund CE, Leib DA. Herpes simplex virus virion host shutoff (vhs) activity alters periocular disease in mice. J. Virol. 2000;74:3598–3604. doi: 10.1128/jvi.74.8.3598-3604.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris JE, Zobell S, Yin XT, Zakeri H, Summers BC, Leib DA, Stuart PM. Mice with Mutations in Fas and Fas Ligand Demonstrate Increased Herpetic Stromal Keratitis following Corneal Infection with HSV-1. J. Immunol. 2012;188:793–799. doi: 10.4049/jimmunol.1102251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rowe AM, St Leger AJ, Jeon S, Dhaliwal DK, Knickelbein JE, Hendricks RL. Herpes Keratitis. Prog. Retin. Eye Res. 2013;32:88–101. doi: 10.1016/j.preteyeres.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stuart PM, Keadle TL. Recurrent herpetic stromal keratitis in mice, a model for studying human HSK. Clin. Dev. Immunol. 2012;2012:728480. doi: 10.1155/2012/728480. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Divito SJ, Hendricks RL. Activated inflammatory infiltrate in HSV-1-infected corneas without herpes stromal keratitis. Invest. Ophthalmol. Vis. Sci. 2008;49:1488–1495. doi: 10.1167/iovs.07-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suryawanshi A, Veiga-Parga T, Reddy PB, Rajasagi NK, Rouse BT. IL-17A differentially regulates corneal vascular endothelial growth factor (VEGF)-A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J. Immunol. 2012;188:3434–3446. doi: 10.4049/jimmunol.1102602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Croxtall JD. Ganciclovir ophthalmic gel 0.15%: in acute herpetic keratitis (dendritic ulcers). Drugs. 2011;71:603–610. doi: 10.2165/11207240-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 27.Wilhelmus KR, Mitchell BM, Jones DB. Photographic monitoring of herpes simplex virus keratitis during anti-inflammatory treatment. Arch. Ophthalmol. 2011;129:252–253. doi: 10.1001/archophthalmol.2010.366. [DOI] [PubMed] [Google Scholar]
- 28.Duan R, de Vries RD, van Dun JM, van Loenen FB, Osterhaus AD, Remeijer L, Verjans GM. Acyclovir susceptibility and genetic characteristics of sequential herpes simplex virus type 1 corneal isolates from patients with recurrent herpetic keratitis. J. Infect. Dis. 2009;200:1402–1414. doi: 10.1086/606028. [DOI] [PubMed] [Google Scholar]
- 29.Munjal VP, Dhir SP, Jain IS, Gangwar DN, D'souza M. Topical corticosteroids and cataract. Indian J. Ophthalmol. 1984;32:478–480. [PubMed] [Google Scholar]
- 30.Dada T, Nair S, Dhawan M. Steroid-induced glaucoma. J. Cur. Glaucoma Prac. 2009;3:33–38. [Google Scholar]
- 31.Renfro L, Snow JS. Ocular effects of topical and systemic steroids. Dermatol. Clin. 1992;10:505–512. [PubMed] [Google Scholar]
- 32.Thomas J, Kanangat S, Rouse BT. Herpes simplex virus replication-induced expression of chemokines and proinflammatory cytokines in the eye: implications in herpetic stromal keratitis. J. Interferon Cytokine Res. 1998;18:681–690. doi: 10.1089/jir.1998.18.681. [DOI] [PubMed] [Google Scholar]
- 33.Terasaka Y, Miyazaki D, Yakura K, Haruki T, Inoue Y. Induction of IL-6 in transcriptional networks in corneal epithelial cells after herpes simplex virus type 1 infection. Invest. Ophthalmol. Vis. Sci. 2009;51:2441–2449. doi: 10.1167/iovs.09-4624. [DOI] [PubMed] [Google Scholar]
- 34.Stumpf TH, Shimeld C, Easty DL, Hill TJ. Cytokine production in a murine model of recurrent herpetic stromal keratitis. Invest. Ophthalmol. Vis. Sci. 2001;42:372–378. [PubMed] [Google Scholar]
- 35.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 36.Suryawanshi A, Veiga-Parga T, Rajasagi NK, Reddy PB, Sehrawat S, Sharma S, Rouse BT. Role of IL-17 and Th17 cells in herpes simplex virus-induced corneal immunopathology. J. Immunol. 2011;187:1919–1930. doi: 10.4049/jimmunol.1100736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suryawanshi A, Veiga-Parga T, Reddy PB, Rajasagi NK, Rouse BT. IL-17A differentially regulates corneal vascular endothelial growth factor (VEGF)-A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J. Immunol. 2012;188:3434–3446. doi: 10.4049/jimmunol.1102602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molesworth-Kenyon SJ, Popham N, Milam A, Oakes JE, Lausch RN. Resident Corneal Cells Communicate with Neutrophils Leading to the Production of IP-10 during the Primary Inflammatory Response to HSV-1 Infection. Int. J. Inflam. 2012:810359. doi: 10.1155/2012/810359. 2012. 10 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conrady CD, Zheng M, Stone DU, Carr DJ. CD8+ T cells suppress viral replication in the cornea but contribute to VEGF-C-induced lymphatic vessel genesis. J. Immunol. 2012;189:425–432. doi: 10.4049/jimmunol.1200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frank GM, Buela KA, Maker DM, Harvey SA, Hendricks RL. Early responding dendritic cells direct the local NK response to control herpes simplex virus 1 infection within the cornea. J. Immunol. 2012;188:1350–1359. doi: 10.4049/jimmunol.1101968. [DOI] [PMC free article] [PubMed] [Google Scholar]