

Abstract
Sepsis is a major cause of mortality, and dysregulation of the immune response plays a central role in this syndrome. Hydrogen sulfide (H2S), a recently discovered gaso-transmitter, is endogenously generated by many cell types, regulating a number of physiologic processes and pathophysiologic conditions. Here we report that H2S increased survival after experimental sepsis induced by cecal ligation and puncture (CLP) in mice. Exogenous H2S decreased the systemic inflammatory response, reduced apoptosis in the spleen, and accelerated bacterial eradication. We found that CHOP, a mediator of the endoplasmic reticulum (ER) stress response, was elevated in several organs after CLP and its expression was inhibited by H2S treatment. Using CHOP knockout (KO) mice, we demonstrated for the first time that genetic deletion of Chop increased survival after lipopolysaccharide (LPS) injection or CLP. CHOP KO mice displayed diminished splenic caspase-3 activation and apoptosis, decreased cytokine production and augmented bacterial clearance. Furthermore, septic CHOP KO mice treated with H2S showed no additive survival benefit compared to septic CHOP KO mice. Finally, we showed that H2S inhibited CHOP expression in macrophages by a mechanism involving Nrf2 activation. In conclusion, our findings show a protective effect of H2S treatment afforded, at least partially, by inhibition of CHOP expression. The data reveal a major negative role for the transcription factor CHOP in overall survival during sepsis and suggest a new target for clinical intervention as well potential strategies for treatment.
Introduction
Sepsis is a systemic inflammatory response to suspected or identified infection. Severe sepsis and septic shock remain the tenth most common cause of death in intensive care units with over 210,000 annual deaths in the United States (1, 2). This high mortality rate demonstrates the lack of effective therapies. Therefore, a deeper understanding of the pathophysiology of this complex syndrome is necessary to develop new and more effective treatments.
The response to infection depends on the initial host-microbial interaction. Activation of the innate immune system directs eradication of the infection while limiting the inflammatory response. At the molecular level, several signal transduction pathways are induced as well as transcription factors such as nuclear factor (NF)-κB, which is a master regulator of several inflammatory mediators (3).
However, a new role is emerging for endoplasmic reticulum (ER) stress signaling and the unfolded protein response (UPR) in the regulation of inflammation and innate immunity (4, 5). Specific ER stress mediators have been shown to increase the inflammatory response of dendritic cells and macrophages (6–8). Additionally, signaling from the UPR intersects with major inflammatory pathways, such as JNK and NF-κB (9). The ER exerts a central role in synthesis, maturation and trafficking of proteins. Under stressful cellular conditions, accompanied by protein misfolding, alterations in ER Ca2+, or oxidative stress, the ER elicits a complex adaptive response by activation of the UPR to reestablish cellular homeostasis. Ultimately, if the ER stress is not alleviated and cellular function is compromised, apoptosis is initiated.
C/EBP homologous protein 10 (CHOP) is a transcription factor and mediator of the UPR that plays a major role in inducing apoptosis (4, 9, 10). However, evidence indicates a new role for CHOP in the inflammatory response (11–13). CHOP expression is triggered in lungs of mice injected with lipopolysaccharide (LPS) and after hemorrhagic trauma (14, 15). This transcription factor is crucial for induction of caspase-11 (16) that is involved in maturation of pro-IL-1β through caspase-1 (17). Accordingly, genetic loss of Chop results in decreased LPS-induced secretion of IL-1β (14). Thus, it is plausible that CHOP may play a major role in the pathogenesis of sepsis, although direct evidence is lacking.
Hydrogen sulfide (H2S) is a biologically active gas serving as a messenger molecule (18). H2S is endogenously generated in many cell types, mainly from cysteine, by two enzymes: cystathionine-β-synthase (CBS) and cystathionine γ-lyase (CSE). It has a pivotal role in modulating cardiovascular function, under physiologic and pathologic conditions. Mice lacking the CSE gene develop hypertension (19), while cardiac specific over-expression of CSE protects against myocardial ischemia-reperfusion injury (20). The role exerted by H2S in inflammation remains controversial because it has been reported to have both pro-inflammatory and anti-inflammatory effects (18). In animal models of sepsis, both the pharmacological inhibition of H2S synthesis, as well as its administration have been shown to improve survival of septic mice (21, 22).
The present investigation examined the therapeutic benefit of administration of H2S during polymicrobial sepsis induced by cecal ligation and puncture (CLP) in mice, and the mechanistic basis for this effect. Exogenous H2S inhibited CHOP expression and genetic deletion of Chop identified this transcription factor as a key molecule in the pathogenesis of sepsis.
Materials and methods
Cecal ligation and puncture (CLP) and lipopolysaccharide (LPS) injection
CLP protocol
Male wild-type mice (6–8 weeks old) C57BL6/J (B6) and CHOP KO on B6 background were obtained from Jackson Laboratory (Bar Harbor, ME). Mice were anesthetized with isoflurane via a vaporizer at 1.5–2.5 minimum alveolar concentrations (MAC). Under sterile conditions, a 1.5-cm incision was made in the lower abdominal region and the cecum was exposed. The distal portion of the cecum was completely ligated 1 cm from the end with a 3-0 silk suture and punctured once with an 18-gauge needle then replaced in the peritoneal cavity. Subsequently, the peritoneal wall and skin were closed with double sutures. A 1-ml subcutaneous injection of sterile saline (0.9%) was administrated to mice after surgery. Sham mice underwent abdominal incision and cecal exposure without ligation and puncture. After the procedure, mice had access to water and food ad libitum. Survival was monitored for eight days, while for cytokines, chemokines, CFU and organ harvesting, animals were sacrificed after 5 or 18 hrs depending on the time point analyzed.
LPS injection
Mice were injected intraperitoneally with a dose of 25 mg/kg of LPS (from Escherichia coli 055:B5) and survival monitored for eight days. All procedures were carried out in accordance with the Johns Hopkins University Animal Care and Use Committee approved protocols that conformed to the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health.
Hydrogen sulfide (H2S) was generated by dissolving NaHS into saline just before use (21). One hundred micro liter of the solution was subcutaneously injected in mice one hour before CLP (100 μmol/kg). In the H2S post-treatment survival, mice received H2S subcutaneously 2 hrs after induction of sepsis, and H2S was administrated again after 24 hrs for the following 2 days.
Bacterial counts
Peritoneal lavage was performed with 5 ml of sterile PBS. Serial dilution of peritoneal fluid and blood were plated on brain and heart infusion agar plates (BD, NJ USA) at 37°C. Bacterial colonies were counted after 24 h.
Cytokine measurements
Animals were sacrificed at the time specified in the figures and blood was drawn by cardiac puncture. Plasma was diluted and assessed for cytokine levels by ELISA (eBioscience Inc, San Diego, CA).
Myeloperoxidase (MPO) activity
Leukocyte accumulation in the lung was measured as previously described with some modification (23). Briefly, lung tissue was homogenized in a solution containing 0.5% hexa-decyl-trimethyl-ammonium bromide and centrifuged for 30 min at 20,000 × g at 4°C. An aliquot of the supernatant was then incubated with a solution of tetra-methyl-benzidine (1.6 mM) and 0.1 mM H2O2. The rate of change in absorbance was measured at 405 nm. MPO activity was defined as the quantity of enzyme degrading 1 μmol hydrogen peroxide/min at 25°C and expressed in units per g weight (U g−1 of tissue).
Aspartate aminotransferase (AST)
AST levels were measured as an indicator of liver damage from mouse serum using the Vet Ace Clinical Chemistry analyzer system.
Isolation of peritoneal macrophages and RAW 264.7 culture
Cells were isolated from mice as previously described (24), plated on cover slips and non adherent cells were washed off after 2 hrs. The day after, cells were stimulated with LPS (10 μg/ml) alone or in combination with H2S (200 μM) for 18 hrs. RAW 264.7 mouse macrophages were cultured in DMEM medium with penicillin, streptomycin and 10% FBS. All cells were maintained at 37°C in the presence of 5% CO2 in a humidified incubator.
RNA Extraction and qPCR analysis
For measurement of mRNA levels, total RNA was extracted from cells using Trizol Reagent (Life Technologies Corporation). RNA was reverse transcribed into cDNA using oligo (dT) 20 primers and SuperScript™ III First-Strand Synthesis System (Life Technologies Corporation). The resulting cDNA was amplified using the SYBR Green Real-Time PCR and detected on an ABI 7500 system (Life Technologies Corporation) with the following amplification conditions: 95 °C for 10 min and then 40 cycles of 95 °C for 15 s and 60 °C for 60 s. Gene expression was calculated relative to the housekeeping gene β-actin using the ΔΔCt algorithm (primers used: CHOP FOR: GAGCCAGAATAACAGCCGGA, CHOP REV: CTGTCAGCCAAGCTAGGGAC)
Western blotting assay
Harvested organs were flash frozen in liquid nitrogen and then stored at −80°C. Tissue was homogenized in RIPA buffer with protease and phosphatase inhibitors and protein content determined. Immunoblotting was performed as previously described (25). Briefly, proteins were separated on a gradient SDS-PAGE, transferred to a PVDF membrane and blocked with 5% milk buffer. Membranes were incubated with primary antibody against: CHOP, Nrf2, ATF4 or AFT6 (Santa Cruz Biotechnology Inc. CA), activated caspase 3 (Cell Signaling Technology, Inc. MA), or tubulin (Abcam, MA) overnight at 4°C. After washing, membranes were incubated with specific secondary horseradish peroxidase-conjugated antibodies for 1 hr, incubated with HRP substrate and signal was detected with Kodak Image Station 4000B.
Histology and immunohistochemistry
Fixed specimens were embedded in paraffin and stained with hematoxylin–eosin for light microscopic evaluation. Activation of Caspase-3 was detected by immunoperoxidase staining, using an antibody that only recognizes the activated form. CHOP expression was identified by using a rabbit anti-mouse antibody against CHOP (Santa Cruz Biotechnology Inc. CA). Sections were counterstained with hematoxylin. Slides were viewed with the inverted microscope Axio Observer.A1 (Zeiss) and images were acquired with Olympus DP72 color camera using CellSense software.
Peritoneal macrophages were fixed in cold methanol and immunostaining carried out as previously described (26). A rabbit anti-mouse antibody (against CHOP) was used as primary antibody at 4°C overnight. After washing, fluorescent labeled secondary antibody was added for 1 hr at room temperature. Cells were washed again, mounted, and fluorescence was observed with a confocal (Zeiss LSM 510 Meta) microscope.
Data analysis
Data were expressed as mean ± SD and statistical significance was determined by unpaired t-test with Welch’s correction, P < 0.05 was considered significant. Animal survival was analyzed by Kaplan-Meier survival analysis and log rank test.
Results
Hydrogen sulfide increases survival after induction of sepsis
Previous in vivo studies assessing the impact of exogenous H2S during sepsis have reported conflicting results, revealing both a protective and a deleterious effect of H2S (21, 22). We induced sepsis by performing cecal ligation and puncture (CLP), a widely used animal model of polymicrobial sepsis that has the advantage of closely resembling sepsis in humans (27). One group of mice was treated subcutaneously with sodium hydrosulphide (NaHS, 100 μmol/kg) to generate H2S. One hour later, severe sepsis was induced by single puncture with an 18-gauge needle in the H2S- treated and untreated groups. We carried out a mortality study and monitored survival for eight days. Remarkably, we found that >80% of the septic mice treated with H2S survived (17 of 21 mice) compared to <25% of untreated septic mice (5 of 22 mice, P< 0.0001) (Fig. 1 A). Thus, adopting the same dosage and delivery of H2S used in the study by Spiller and coworkers, we confirmed a significant increase in survival of septic mice treated with H2S (21).
Figure 1. H2S increases survival after sepsis.

A) Survival study of mice monitored for 8 days after CLP. A group of mice received sodium hydrosulphide (NaHS, 100 mmol/kg) as an H2S donor subcutaneously (CLP+H2S) or saline and after 1 h CLP was carried out (CLP n=21, CLP+H2S n=22. * P=0.0001). Data are pooled from three different experiments). B) Sepsis was induced in mice by CLP, and 2h later, H2S was administrated as described above (both groups n=6. * P=0.01).
To assess the potential therapeutic use of H2S, we evaluated the efficacy of H2S treatment after induction of sepsis. For this purpose, sepsis was induced by CLP and after 2 hrs H2S was administered subcutaneously. We found that H2S is equally effective in reducing mortality when given after induction of sepsis (Fig. 1 B).
Hydrogen sulfide reduces inflammation
Given the relevance of cytokines in regulating the inflammatory response (28, 29), we next evaluated the levels of the pro-inflammatory TNFα and the anti-inflammatory IL-10 in the plasma of septic mice by ELISA. Blood levels of TNFα were increased 5 hrs after CLP as expected. H2S treatment attenuated the increase by about 50% (Fig. 2 A). TNFα levels continued to rise 18 hrs after CLP to ten times the 5 hour level, and H2S blunted this increase. Also, plasma levels of IL-10 were elevated 5h after CLP, with a further modest rise at 18h. H2S attenuated the increase in IL-10 levels as well. Then, we determined the concentration of these mediators in the peritoneum, the site of infection. A peritoneal lavage was carried out and TNFα and IL-10 levels were determined at 5 and 18 hrs post CLP. Similar patterns of change in the levels of these cytokines were found in the peritoneal cavity (Fig. 2 B) as in the plasma. Thus, H2S treatment significantly lowered both TNFα and IL-10 levels in the blood and at the infected site.
Figure 2. H2S decreases inflammation.

A) Plasma TNFα and IL-10 levels were measured at 5 h and 18 h after CLP by an ELISA (Sham n=3–5, CLP and CLP+H2S n=6–11) Data are from individual mice from two to three experiments. B) Levels of cytokines were measured in the peritoneal lavage (Sham n=3–4, CLP and CLP+H2S n=6–8). C) Left panel: MPO activity from lung homogenized measured at 18 h after CLP. Right panel: AST measured from plasma at 18 h after CLP (Sham n=2, CLP and CLP+H2S n=4–8). Data shown are from individual mice from two different experiments (* P≤0.05).
Then, we assessed MPO activity as a measure of neutrophil infiltration in the lung 18 hrs after CLP induction (Fig. 2 C, left panel). Septic mice showed a significant increase in MPO activity in the lung compared to sham animals. Treatment with H2S lowered this activity to basal levels. In addition, we assessed liver function by determining AST levels (Fig. 2 C, right panel). We found that CLP increased the level of AST compared to sham, however, H2S treatment did not prevent this increase.
Apoptosis is decreased in septic H2S-treated mice
Next, we evaluated morphologic changes in the lung, liver and spleen using H&E staining at 18h after CLP. The histological sections of lung from septic mice showed signs of injury including thickening of the alveolar septa and interstitial edema (Supplemental Fig. 1, upper panels). In the liver sections, swelling of hepatocytes and infiltrating neutrophils were observed (Supplemental Fig. 1 middle panels). Spleen from septic mice displayed numerous macrophages containing phagocytized karyorrhectic debris likely from apoptotic cells in the white pulp (Supplemental Fig. 1, bottom panel). To verify cell death by apoptosis, we carried out immunostaining of spleen sections using an antibody to detect activated caspase-3 (Fig. 3 A). We also examined caspase-3 activation in tissue extracts by immunoblotting (Fig. 3 B). Caspase-3 activation was induced in the white pulp of spleen from septic mice. H2S decreased its cleavage and prevented most of the above described histological changes in organs due to CLP. Caspase-3 activation was also examined in lung and liver of septic mice by immunostaining of tissue sections and by Western blotting analysis. However, no sign of caspase-3 activation in lung or liver of septic mice were detected, at least 18 hrs after CLP (data not shown).
Figure 3. Apoptosis is reduced by H2S treatment.
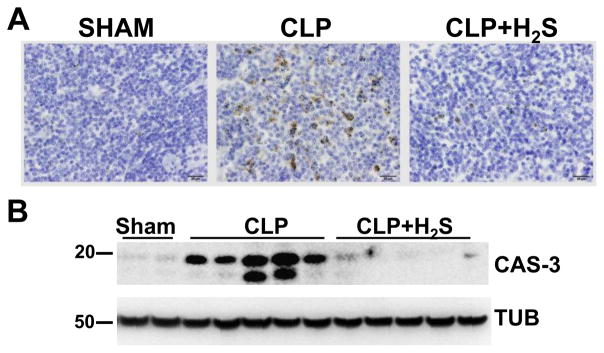
A) Activated Caspase-3 immunoperoxidase staining was performed on spleen sections 18h after CLP (40 X objective, bar 20 mm). B) Lysates from spleens 18 h after CLP were subjected to Western blotting assay to detect activated Caspase-3 (CAS-3). Tubulin (TUB) was used as a loading control. Samples represent individual mouse spleen from 2 sham, 5 CLP and 5 CLP+H2S.
Hydrogen sulfide increases bacterial clearance
Bacterial clearance is an important component in the resolution of infection. Therefore, we next evaluated the number of live bacteria in the blood and in the peritoneal cavity, and how H2S treatment would modulate bacterial load (Fig. 4). H2S did not significantly alter the number of live bacteria 5 hrs after CLP, either in blood or peritoneum. At 18 hrs, the CLP group displayed an increase in live bacteria while H2S treatment resulted in an almost complete elimination of the bacterial burden in both blood and peritoneal cavity. Thus, H2S treatment was associated with a significantly enhanced rate of bacterial clearance.
Figure 4. H2S increases bacterial clearance.

Whole blood (upper panels) and peritoneal lavage (lower panels) were assessed for viable bacteria by colony-forming unit (CFU) assay at 5h and 18h after CLP. Samples from single mice were streaked on brain and heart infusion agar plates for 24 h and then colonies were counted (n=4–6). Data are a representative from two independent experiments with similar results. (* P≤0.05).
CHOP expression is reduced by H2S treatment in vivo
Recent findings have highlighted links between the inflammatory response and the ER stress signaling pathways (4, 30). CHOP is emerging as a new key molecule involved in the inflammatory response (11–13). Additionally, it has been reported that H2S can reduce ER stress in cardiac tissue (31) and in a neuroblastoma cell line (32). However, there are no studies testing the impact of H2S on CHOP modulation during inflammation or sepsis. To elucidate the mechanism of H2S protection, we first examined whether CHOP is expressed in vivo after CLP and whether H2S would modulate its expression. CLP-induced sepsis led to a significantly increased expression of CHOP in spleen, lung and liver of septic mice 18 hrs after CLP, compared to sham operated mice (Fig. 5 A). H2S treatment in vivo was associated with decreased expression of CHOP in all of these organs.
Figure 5. H2S decreases CHOP expression in vivo.

A) Spleens, lungs and livers harvested 18h after CLP were homogenized and analyzed by Western blot for expression of CHOP. Tubulin (Tub) was used as a loading control. Samples analyzed are from single mice (Sham n=2–3, CLP and CLP+H2S n=5). Data are a representative of two independent experiments with similar results. B) Representative immunohystochemistry of CHOP expression in sham and CLP mice.
Then, to investigate which cell type in the organs expressed CHOP, we performed immunostaining of tissue sections (Fig. 5 B). The lungs of septic mice display CHOP expression in a disseminated pattern, probably both in inflammatory cells and lung parenchymal cells. Indeed, a previous study shows that alveolar type I and type II cells express CHOP after LPS injection (14). In the liver, CHOP staining did not appear in hepatocytes or endothelial cells. Cells expressing CHOP are most likely tissue macrophages such as Kupffer cells and infiltrating inflammatory cells. In the spleen, CHOP staining was evident in the sinusoidal areas outside the white pulp where macrophages mostly reside. Higher magnification shows that the CHOP-positive cells were large round cells with abundant cytoplasm, consistent with the morphology of macrophages. Thus, under septic conditions, CHOP was expressed mainly by inflammatory and reticulo-endothelial cells in these organs.
H2S treatment in vivo results in down-regulation of other ER stress mediators
We also investigated if other members of the ER stress response were affected by H2S treatment in vivo. Activation of the canonical unfolded protein response (UPR) is mediated by three specific ER membrane proteins including PERK (PKR-like eukaryotic initiation factor 2a kinase), and ATF6 (activating transcription factor-6). Activation of the PERK pathway is implicated in attenuation of global translation initiation while increasing translation of specific transcription factors, such as ATF4, which is a main inducer of CHOP transcription. ATF6 signaling has a major role in chaperone induction and may also induce CHOP expression (9). Induction of these pathways is complex and modulates numerous proteins intersecting with a variety of inflammatory and stress signaling. In the spleen we found that H2S decreased CLP induced ATF6 activation as well as ATF4 (Fig. 6). Thus, other members of the UPR were, directly or indirectly, modulated by H2S in vivo.
Figure 6. H2S decreases other ER stress response proteins expression in vivo.
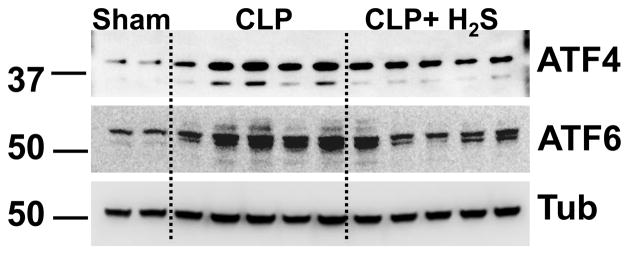
Spleens harvested 18h after CLP were homogenized and analyzed by Western blot for expression of ATF4 and ATF6. Tubulin (Tub) was used as a loading control. Samples analyzed are from single mice.
Genetic deletion of Chop increased survival after endotoxemia or CLP-induced sepsis
To determine whether CHOP is an important mediator of the inflammatory response, we used CHOP KO mice. A previous study showed that CHOP plays a role in the pathogenesis of lung inflammation after LPS injection (14); however, its role in the overall outcome of mice treated with LPS has not been established thus far. Thus, we first sought to test whether CHOP KO mice would be more resistant to LPS induced mortality (Fig. 7). To address this, we injected WT mice and CHOP KO mice with LPS intraperitoneally (25 mg/kg) and observed them for eight days. Survival of CHOP KO mice (87.5%) was significantly higher than WT mice (12.5%, p=0.001).
Figure 7. Genetic deletion of Chop protects against LPS induced mortality.

The survival rate of WT and CHOP KO mice (n=8) was observed for 8 days after intraperitoneal injection of 25 mg/kg of LPS (n=8, * P=0.001).
Next we investigated whether CHOP KO mice would be more resistant to CLP induced sepsis. We found that septic CHOP KO mice showed significantly improved survival compared to septic WT mice (P=0.01, Fig. 8). Furthermore, we tested whether there would be an additive effect produced by H2S treatment in combination with Chop genetic deletion. Treatment of septic CHOP KO mice with H2S did not show significant additional benefit compared to septic CHOP KO mice (P=0.9). These results clearly demonstrate that CHOP is a major negative factor in LPS and CLP-induced mortality, and provides evidence that H2S increased survival is mediated through CHOP inhibition.
Figure 8. Genetic deletion of Chop protects against CLP induced mortality.

The survival rate of WT and CHOP KO mice (8 mice per group) was observed for 8 days after induction of sepsis by CLP (n=8–10 *, P=0.01). A group of CHOP KO mice was also treated with H2S 2h prior to CLP (CHOP KO vs CHOP KO+H2S P=0.9).
Next, we examined the general histologic profile of liver, spleen and lung, 18 hrs post CLP in WT and CHOP KO mice (Supplemental Fig. 2). Septic CHOP KO mice displayed a lesser degree of tissue damage compared to septic WT mice, corroborating that inhibiting CHOP expression increases survival. We confirmed apoptosis by immunostaining of sections of spleen using an antibody to detect activated caspase-3 (Fig. 9). While septic WT mice displayed activation of Caspase-3, CHOP KO mice were almost completely negative. Then, we explored whether CHOP is involved in the release of TNFα and IL-10 in septic mice. The levels of these cytokines were significantly reduced in septic KO mice compared to septic WT mice, both in plasma and peritoneal cavity (Fig. 10 A–B). Lastly, we determined bacterial burden in CHOP KO. After CLP, bacterial clearance was markedly improved in septic CHOP KO mice compared to WT animals, in the blood as well as the peritoneum (Fig. 10 C).
Figure 9. Septic CHOP KO mice display reduced apoptosis.
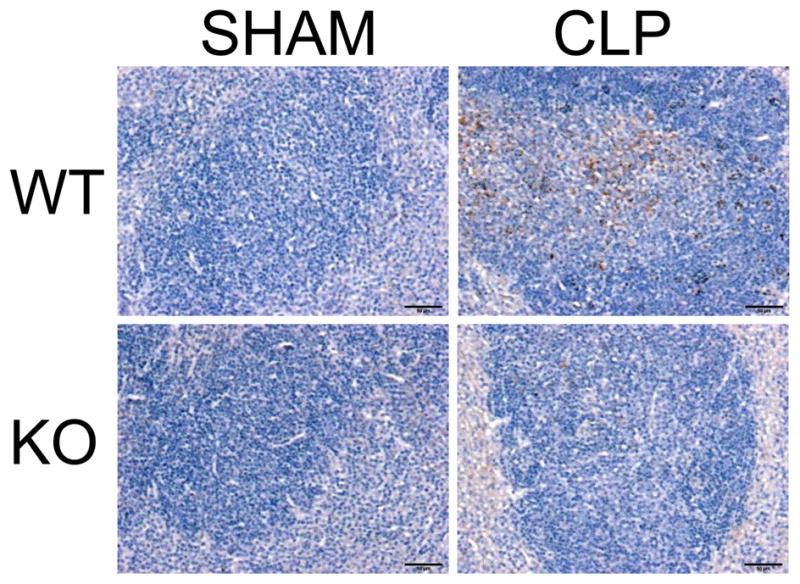
Representative activated Caspase-3 immunoperoxidase staining performed on spleen sections 18 h after CLP.
Figure 10. Decreased TNFα and IL-10 and increased bacterial clearance in CHOP KO mice.

A) TNFα and IL-10 levels were measured by ELISA in the plasma of sham operated and septic (CLP) mice from WT and CHOP KO groups 18h after CLP (n=4–6). B) Levels of cytokines in the peritoneum of septic WT and CHOP KO mice (n=4–6). C) Colony-forming unit (CFU) assay was performed on blood and peritoneum lavage at 18h after CLP. Samples from single mice were streaked on brain and heart infusion agar plates for 24 h and then colonies were counted (n=4–5, * P≤0.05).
Thus, genetic deletion of Chop increases survival after CLP by reducing the systemic inflammatory response, decreasing apoptosis, and improving bacterial clearance.
H2S inhibits CHOP expression in macrophages by modulation of Nrf2
To explore the mechanism by which H2S reduces CHOP expression, we used macrophages, since these cells play a central role in the innate immune response. In addition, our data suggest that this cell type shows increased CHOP expression during sepsis (Fig. 5B).
First, we assessed CHOP expression and the effects of H2S in the macrophage cell line RAW264.7 and in peritoneal mouse macrophages. RAW cells were stimulated with LPS and with constituents of gram positive bacterial peptidoglycan (PGN). Incubation of cells for 18h with these inducers led to CHOP expression. H2S treatment blunted this effect (Fig. 11 A), along with a reduction of CHOP mRNA transcript (Fig. 11 B). Immunostaining of peritoneal macrophages demonstrated that LPS treatment resulted in increased CHOP expression, in agreement with a previous study (16). H2S treatment reduced CHOP expression in primary macrophages (Fig. 11 C). Next, we investigated whether H2S inhibits the ER stress induced by tunicamycin (TN) treatment, a widely-used inducer of the ER stress response. H2S was able to reduce CHOP expression by TN (Fig. 11D). Thus, H2S decreased CHOP expression in macrophages stimulated with bacterial products and with a direct ER stress inducer.
Figure 11. H2S treatment decreases CHOP expression in macrophages in a Nrf2 dependent manner.

A) Representative Western blot of CHOP. RAW cells were incubated for 18h with LPS (L, 10 mg/ml) or PGN (P, 10 mg/ml) with or without H2S (200 mM). Densitometric analysis is shown in the graph below and it was calculated by ratio of CHOP over tubulin (Tub) and normalized to LPS (n=3, p≤0.01, * vs LPS alone; ** vs PGN alone). B) CHOP mRNA is reduced after incubation of cells with LPS and/or H2S for 6h (n=2). C) Representative confocal image of peritoneal macrophages incubated with LPS and/or H2S for 18h and then stained for expression of CHOP. (n=2) D) RAW cells were treated with tunicamicyn (TN) for 3h with or without H2S (n=3). E) Cells were incubated for the time indicates with and Nrf2 was then evaluated (n=2). F) RAW cells were transfected with scrambled oligo (siCon) or siRNA for Nrf2 (siNrf2) for 24h and then incubated with H2S for 18h (n≥3). G) After transfection with siCon or siNrf2, RAW cells were incubated with LPS with (L+H) or without H2S for 12h. Densitometric analysis is shown in the graph (n=3, p≤0.01, #vs LPS, *vs Bas and **vs LPS+H2S of siCon group).
Subsequently, we investigated the mechanism by which H2S decreases CHOP expression. A recent study shows that the protective effect of H2S in the heart is mediated by increased Nrf2 levels (33). Nrf2 is a transcription factor implicated in improving cell survival following ER stress and it has been shown to inhibit CHOP expression (34, 35). We first examined whether H2S induces Nrf2 in macrophages. We incubated RAW cells with H2S for up to 24 h and assessed Nrf2 expression. Nrf2 is induced after 1 hour of H2S treatment, increased with time from 1 h to 8 h, and started to decrease at 24 (Fig. 11 E).
To establish whether H2S decreases CHOP expression through Nrf2 activation, we used silencing RNA (siRNA) directed against Nrf2 (siNrf2). RAW cells were transfected with a scrambled oligo (siCon) or siNrf2 for 24 h and then incubated with medium (Bas) or H2S for 18 hrs. Nrf2 was effectively down-regulated by the siNrf2 compared to siCon (Fig. 11 F). Then, we stimulated the cells with LPS in the presence or absence of H2S. In the siCon group, LPS-induced CHOP expression was decreased by H2S treatment. In the siNrf2 group, the basal level of CHOP was increased and treatment with H2S did not decrease LPS-induced CHOP expression (Fig. 11 G). Our data indicate that Nrf2 modulates CHOP expression under basal conditions and it is a major regulator of the H2S-mediated decrease in CHOP expression.
Discussion
In this study, we report that administration of H2S markedly improves survival of experimental polymicrobial sepsis, given prior to the onset and more importantly even 2 hrs after CLP. The improved outcome is associated with decreased cytokine production, reduced apoptosis, and increased bacterial clearance. CLP induced CHOP expression, but this effect was attenuated by H2S treatment. We found that CHOP is a major regulator of the inflammatory response since CHOP KO mice are significantly resistant to LPS- and CLP-induced mortality. We also demonstrate that the protection afforded by genetic deletion of Chop is not affected by H2S treatment, suggesting that CHOP may be a key mediator of H2S-induced protection. Finally, we demonstrated that H2S inhibition of CHOP expression is mediated by Nrf2 activation in macrophages.
Two previous investigations on the effect of exogenous H2S on experimental sepsis led to conflicting results. Zhang and colleagues showed that H2S injection into septic mice results in increased mortality (22), whereas Spiller and coworkers reported a beneficial effect (21). The divergent outcome of these studies may be explained by the different route of H2S administration and the dosage used. While a beneficial effect was observed when H2S is administered subcutaneously using a dose of 100 μmol/kg (about 5.6 mg/kg), an intravenous bolus (10 mg/kg) resulted in increased mortality. As the study from Spiller and colleagues shows, an intravenous bolus of H2S provokes a significant decrease in mean arterial pressure while subcutaneous administration avoids this effect. This latter is likely due to its slow release into the circulation (21). Therefore, during sepsis an already compromised vascular tone could be even further dysregulated by a direct intravenous bolus of H2S, which induces vasorelaxation in different vascular beds (18). Thus, although other factors may also have a role, our study confirms the primary importance of the route of administration in dictating whether H2S is beneficial or deleterious in the setting of septic shock.
Our study shows that H2S treatment resulted in decreased production of the pro-inflammatory TNFα as well as decreased leukocyte infiltration in the lungs and liver, in line with other studies (21, 36–38). Since IL-10 is known to have anti-inflammatory properties (39), we were surprised to find that H2S treatment decreased the release of this cytokine. Previously, H2S has been reported to increase IL-10 levels in vitro and in a model of acute lung injury (37, 40), although H2S decreased IL-10 levels during hepatic ischemic-reperfusion injury (41). It is plausible that the impact of H2S on IL-10 levels varies depending on the cell type or animal model used. However, and more importantly, it has been shown that neutrophils from septic patients have reduced bactericidal activity when treated with IL-10, leading to impaired host defense with overgrowth or persistence of pathogens (39, 42–44). Moreover, neutralization of IL-10 in septic mice improves survival (45), and enhances phagocytosis by macrophages (46). Thus, the attenuated release of this cytokine could be important, since the overproduction of this cytokine, together with other factors, could be responsible for the immune-suppressive state characteristic of sepsis (47). Therefore, H2S treatment may help to maintain an effective but not overactive host immune response leading to increased survival.
Another important aspect of our study is that treatment with H2S was associated with a virtually complete inhibition of apoptosis induced by CLP. H2S is known to decrease apoptosis in different cell types such as neurons (48), cardiomyocytes (20) and cancer cells (49). However, this effect in a model of experimental sepsis has not been reported before. Recent studies have highlighted that apoptosis during sepsis plays an important role. Indeed, septic spleens from patient autopsies and from animal studies show widespread lymphocyte apoptosis during sepsis (50–55). Conversely, inhibition of lymphocyte apoptosis in experimental models leads to increased survival (53, 54, 56–58). Among the mechanisms of the anti-apoptotic effect of H2S, the preservation of mitochondrial function through the opening of the KATP channels has been shown to play an important role (18). Nevertheless, during sepsis both the death receptor pathway and mitochondrial-mediated apoptosis have been described, as well as cross talk between these two pathways (51, 54, 55, 57, 59). Taken together our results suggest that the beneficial effect of H2S is likely due to modulation of different pathways that are often interconnected, targeting apoptosis as well as inflammatory mediators and resulting in increased survival.
Septic mice exhibited increased expression of CHOP and treatment with H2S was associated with its down-regulation. Although CHOP is a major inducer of apoptosis in response to ER stress (4, 9, 10), recent evidence identifies a new role for CHOP as a mediator of the inflammatory response (14–17). Our data demonstrates that this transcription factor plays a pivotal role in the pathogenesis of sepsis. Indeed, CHOP KO mice displayed virtually no apoptosis, augmented bacterial clearance, and increased survival after CLP or LPS injection. All of the beneficial features observed in mice with genetic deletion of Chop were also seen in septic WT mice treated with H2S. Furthermore, when we administered H2S to septic CHOP KO mice there was no additional improvement in survival compared to septic untreated CHOP KO mice. This evidence supports the concept that suppression of CHOP may be a major factor in the beneficial effect afforded by H2S. It is important to note that we observed no co-localization of Caspase-3 activation with CHOP expression in the spleen of septic mice. While Caspase-3 activation was mainly in the white pulp of the spleen, CHOP expression was present in the outer marginal zones of this organ. Additionally, we also identified augmented CHOP expression in lung and liver, however, no detectable activation of Caspase-3 was observed. These data together suggest that CHOP may not play a direct role in the apoptotic events we observed in the spleen of septic mice, indicating that its role could be primarily involved with the inflammatory response per se. As shown by Endo and colleagues, CHOP has a critical role in the induction of Caspase-11, which is needed for production of IL-1β. In this regard, our data add new insights supporting the concept that CHOP contributes to the inflammatory response, in agreement with other studies (11–14, 16, 17). Importantly, we show for the first time that genetic deletion of Chop leads to increased survival after CLP or LPS injection.
Whether H2S directly affects CHOP expression is currently unknown. One possible target of H2S was demonstrated in a study by Lefer and colleagues who found that in myocardial ischemic injury, H2S treatment provides protection in the heart through Nrf2 signaling (33), a master regulator of cytoprotective genes. Furthermore, mouse embryonic fibroblasts deficient in Nrf2 display augmented CHOP expression after glucose deprivation (60). Thus, we hypothesized that H2S could inhibit LPS induced CHOP by increasing Nrf2 signaling. Our results show that H2S increases Nrf2 levels in macrophages, and that silencing of Nrf2 abolishes the inhibitory effect of H2S on LPS induced CHOP up-regulation. A molecular mechanism by which H2S modulates Nrf2 has been recently described. Under basal conditions, Nrf2 is maintained in the cytoplasm by binding to its negative regulator Keap1. H2S S-sulfhydrates Keap1 at specific cysteine residues, resulting in dissociation of Keap1 from Nrf2 and enhanced Nrf2 nuclear translocation and transcriptional activity (61).
How may CHOP regulate the inflammatory response? CHOP is an atypical transcription factor since it can inhibit or enhance transcription of a target gene depending on the interaction with other transcription factors (62, 63). A very recent study demonstrates that CHOP can modulate NF-κB activation by decreasing IκB degradation and p65 translocation. This mechanism leads to a decrease in NF-κB target genes involved in apoptosis and inflammation (11). Thus, modulation of CHOP by H2S can interfere with different pathways involved in the immune response during sepsis.
In summary, here we report that H2S treatment increased survival in a model of experimental sepsis by reducing the inflammatory response and the immunosuppressive state induced by apoptosis of immune cells, leading to a more effective and balanced innate and acquired immune response. Our data also highlights a major role of CHOP, which may act as an amplifier of the inflammatory response in the pathogenesis of sepsis, and the ability of H2S treatment to counter CHOP signaling. Finally, H2S inhibits CHOP expression at least partially through a mechanism involving Nrf2 activation. Sepsis is a complex and dynamic syndrome. These results increase our knowledge of the pathogenetic mechanisms of sepsis and simultaneously shed light on new targets and suggest innovative strategies for the treatment of this syndrome.
Supplementary Material
Acknowledgments
We would like to tank Dr. F. Spiller (Federal University of Florianopolis, SC, Brazil) and Dr. F Cunha (School of Medicine of Ribeirao Preto, University of Sao Paulo, SP, Brazil) for their helpful advice on CLP procedure and H2S treatment.
This work was supported by a grant from American Heart Association (0830395N) to M.F. and by a grant from the National Institutes of Health (5R01HL039752) to C.S. and Robert Garret Fund.
Abbreviation used in this paper
- CLP
cecal ligation and puncture
- CHOP
C/EBP homologous protein 10
- ER
endoplasmic reticulum
- H2S
hydrogen sulfide
Footnotes
The authors have no conflicting financial interests.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Zingarelli B. Nuclear factor-kappaB. Crit Care Med. 2005;33:S414–416. doi: 10.1097/01.ccm.0000186079.88909.94. [DOI] [PubMed] [Google Scholar]
- 4.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009;60:2633–2643. doi: 10.1002/art.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwakoshi NN, Lee AH, Glimcher LH. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev. 2003;194:29–38. doi: 10.1034/j.1600-065x.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- 8.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 11.Allagnat F, Fukaya M, Nogueira TC, Delaroche D, Welsh N, Marselli L, Marchetti P, Haefliger JA, Eizirik DL, Cardozo AK. C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in beta-cells. Cell Death Differ. 2012 doi: 10.1038/cdd.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K, Hokimoto S, Yamamuro M, Iwawaki T, Gotoh T, Ogawa H, Oike Y. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1124–1132. doi: 10.1161/ATVBAHA.111.224519. [DOI] [PubMed] [Google Scholar]
- 13.Suyama K, Ohmuraya M, Hirota M, Ozaki N, Ida S, Endo M, Araki K, Gotoh T, Baba H, Yamamura K. C/EBP homologous protein is crucial for the acceleration of experimental pancreatitis. Biochem Biophys Res Commun. 2008;367:176–182. doi: 10.1016/j.bbrc.2007.12.132. [DOI] [PubMed] [Google Scholar]
- 14.Endo M, Oyadomari S, Suga M, Mori M, Gotoh T. The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem. 2005;138:501–507. doi: 10.1093/jb/mvi143. [DOI] [PubMed] [Google Scholar]
- 15.Jian B, Hsieh CH, Chen J, Choudhry M, Bland K, Chaudry I, Raju R. Activation of endoplasmic reticulum stress response following trauma-hemorrhage. Biochim Biophys Acta. 2008;1782:621–626. doi: 10.1016/j.bbadis.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Endo M, Mori M, Akira S, Gotoh T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J Immunol. 2006;176:6245–6253. doi: 10.4049/jimmunol.176.10.6245. [DOI] [PubMed] [Google Scholar]
- 17.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 18.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 19.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spiller F, Orrico MI, Nascimento DC, Czaikoski PG, Souto FO, Alves-Filho JC, Freitas A, Carlos D, Montenegro MF, Neto AF, Ferreira SH, Rossi MA, Hothersall JS, Assreuy J, Cunha FQ. Hydrogen sulfide improves neutrophil migration and survival in sepsis via K+ATP channel activation. Am J Respir Crit Care Med. 2010;182:360–368. doi: 10.1164/rccm.200907-1145OC. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Zhi L, Moore PK, Bhatia M. Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1193–1201. doi: 10.1152/ajplung.00489.2005. [DOI] [PubMed] [Google Scholar]
- 23.Squadrito F, Altavilla D, Squadrito G, Ferlito M, Campo GM, Arlotta M, Grimaldi S, Quartarone C, Saitta A, Caputi AP. Tacrolimus suppresses tumour necrosis factor-alpha and protects against splanchnic artery occlusion shock. Br J Pharmacol. 1999;127:498–504. doi: 10.1038/sj.bjp.0702528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fulton WB, Reeves RH, Takeya M, De Maio A. A quantitative trait loci analysis to map genes involved in lipopolysaccharide-induced inflammatory response: identification of macrophage scavenger receptor 1 as a candidate gene. J Immunol. 2006;176:3767–3773. doi: 10.4049/jimmunol.176.6.3767. [DOI] [PubMed] [Google Scholar]
- 25.Ferlito M, Irani K, Faraday N, Lowenstein CJ. Nitric oxide inhibits exocytosis of cytolytic granules from lymphokine-activated killer cells. Proc Natl Acad Sci U S A. 2006;103:11689–11694. doi: 10.1073/pnas.0600275103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferlito M, Fulton WB, Zauher MA, Marban E, Steenbergen C, Lowenstein CJ. VAMP-1, VAMP-2, and syntaxin-4 regulate ANP release from cardiac myocytes. J Mol Cell Cardiol. 2010;49:791–800. doi: 10.1016/j.yjmcc.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bone RC. Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS) Ann Intern Med. 1996;125:680–687. doi: 10.7326/0003-4819-125-8-199610150-00009. [DOI] [PubMed] [Google Scholar]
- 29.Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol. 2006;177:1967–1974. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- 30.Martinon F, Glimcher LH. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr Opin Immunol. 2010;23:35–40. doi: 10.1016/j.coi.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei H, Zhang R, Jin H, Liu D, Tang X, Tang C, Du J. Hydrogen sulfide attenuates hyperhomocysteinemia-induced cardiomyocytic endoplasmic reticulum stress in rats. Antioxid Redox Signal. 2009;12:1079–1091. doi: 10.1089/ars.2009.2898. [DOI] [PubMed] [Google Scholar]
- 32.Xie L, Tiong CX, Bian JS. Hydrogen sulfide protects SH-SY5Y cells against 6-hydroxydopamine-induced endoplasmic reticulum stress. Am J Physiol Cell Physiol. 2012;303:C81–91. doi: 10.1152/ajpcell.00281.2011. [DOI] [PubMed] [Google Scholar]
- 33.Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105:365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol. 2006;38:317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 35.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Salto-Tellez M, Tan CH, Whiteman M, Moore PK. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic Biol Med. 2009;47:103–113. doi: 10.1016/j.freeradbiomed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 37.Whiteman M, Li L, Rose P, Tan CH, Parkinson DB, Moore PK. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid Redox Signal. 2009;12:1147–1154. doi: 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho HH, Antoniv TT, Ji JD, Ivashkiv LB. Lipopolysaccharide-induced expression of matrix metalloproteinases in human monocytes is suppressed by IFN-gamma via superinduction of ATF-3 and suppression of AP-1. J Immunol. 2008;181:5089–5097. doi: 10.4049/jimmunol.181.7.5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2010;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- 40.Li T, Zhao B, Wang C, Wang H, Liu Z, Li W, Jin H, Tang C, Du J. Regulatory effects of hydrogen sulfide on IL-6, IL-8 and IL-10 levels in the plasma and pulmonary tissue of rats with acute lung injury. Exp Biol Med (Maywood) 2008;233:1081–1087. doi: 10.3181/0712-RM-354. [DOI] [PubMed] [Google Scholar]
- 41.Kang K, Zhao M, Jiang H, Tan G, Pan S, Sun X. Role of hydrogen sulfide in hepatic ischemia-reperfusion-induced injury in rats. Liver Transpl. 2009;15:1306–1314. doi: 10.1002/lt.21810. [DOI] [PubMed] [Google Scholar]
- 42.Hiraki S, Ono S, Tsujimoto H, Kinoshita M, Takahata R, Miyazaki H, Saitoh D, Hase K. Neutralization of interleukin-10 or transforming growth factor-beta decreases the percentages of CD4+ CD25+ Foxp3+ regulatory T cells in septic mice, thereby leading to an improved survival. Surgery. 2012;151:313–322. doi: 10.1016/j.surg.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 43.Laichalk LL, Danforth JM, Standiford TJ. Interleukin-10 inhibits neutrophil phagocytic and bactericidal activity. FEMS Immunol Med Microbiol. 1996;15:181–187. doi: 10.1111/j.1574-695X.1996.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 44.Lyons A, Kelly JL, Rodrick ML, Mannick JA, Lederer JA. Major injury induces increased production of interleukin-10 by cells of the immune system with a negative impact on resistance to infection. Ann Surg. 1997;226:450–458. doi: 10.1097/00000658-199710000-00006. discussion 458–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hiraki S, Ono S, Kinoshita M, Tsujimoto H, Takahata R, Miyazaki H, Saitoh D, Seki S, Hase K. Neutralization of IL-10 restores the downregulation of IL-18 receptor on natural killer cells and interferon-gamma production in septic mice, thus leading to an improved survival. Shock. 2011;37:177–182. doi: 10.1097/SHK.0b013e31823f18ad. [DOI] [PubMed] [Google Scholar]
- 46.Steinhauser ML, Hogaboam CM, Kunkel SL, Lukacs NW, Strieter RM, Standiford TJ. IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J Immunol. 1999;162:392–399. [PubMed] [Google Scholar]
- 47.Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: A complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med. 2002;30:S58–S63. [PubMed] [Google Scholar]
- 48.Tang XQ, Yang CT, Chen J, Yin WL, Tian SW, Hu B, Feng JQ, Li YJ. Effect of hydrogen sulphide on beta-amyloid-induced damage in PC12 cells. Clin Exp Pharmacol Physiol. 2008;35:180–186. doi: 10.1111/j.1440-1681.2007.04799.x. [DOI] [PubMed] [Google Scholar]
- 49.Rose P, Moore PK, Ming SH, Nam OC, Armstrong JS, Whiteman M. Hydrogen sulfide protects colon cancer cells from chemopreventative agent beta-phenylethyl isothiocyanate induced apoptosis. World J Gastroenterol. 2005;11:3990–3997. doi: 10.3748/wjg.v11.i26.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung CS, Song GY, Lomas J, Simms HH, Chaudry IH, Ayala A. Inhibition of Fas/Fas ligand signaling improves septic survival: differential effects on macrophage apoptotic and functional capacity. J Leukoc Biol. 2003;74:344–351. doi: 10.1189/jlb.0102006. [DOI] [PubMed] [Google Scholar]
- 51.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813–822. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 52.Hiramatsu M, Hotchkiss RS, Karl IE, Buchman TG. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and gut by an endotoxin and TNF-independent pathway. Shock. 1997;7:247–253. doi: 10.1097/00024382-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A. 1999;96:14541–14546. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ayala A, Perl M, Venet F, Lomas-Neira J, Swan R, Chung CS. Apoptosis in sepsis: mechanisms, clinical impact and potential therapeutic targets. Curr Pharm Des. 2008;14:1853–1859. doi: 10.2174/138161208784980617. [DOI] [PubMed] [Google Scholar]
- 55.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 56.Peck-Palmer OM, Unsinger J, Chang KC, McDonough JS, Perlman H, McDunn JE, Hotchkiss RS. Modulation of the Bcl-2 family blocks sepsis-induced depletion of dendritic cells and macrophages. Shock. 2009;31:359–366. doi: 10.1097/SHK.0b013e31818ba2a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwulst SJ, Muenzer JT, Peck-Palmer OM, Chang KC, Davis CG, McDonough JS, Osborne DF, Walton AH, Unsinger J, McDunn JE, Hotchkiss RS. Bim siRNA decreases lymphocyte apoptosis and improves survival in sepsis. Shock. 2008;30:127–134. doi: 10.1097/shk.0b013e318162cf17. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, Wan X, Deng X, Cai Z. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care. 2010;14:R220. doi: 10.1186/cc9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hotchkiss RS, Osmon SB, Chang KC, Wagner TH, Coopersmith CM, Karl IE. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J Immunol. 2005;174:5110–5118. doi: 10.4049/jimmunol.174.8.5110. [DOI] [PubMed] [Google Scholar]
- 60.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279:20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 61.Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, Khaper N, Wu L, Wang R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal. 2013;18:1906–1919. doi: 10.1089/ars.2012.4645. [DOI] [PubMed] [Google Scholar]
- 62.Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- 63.Ubeda M, Wang XZ, Zinszner H, Wu I, Habener JF, Ron D. Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol Cell Biol. 1996;16:1479–1489. doi: 10.1128/mcb.16.4.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.