

Abstract
Controlling angiotensin AT1 receptor function has been shown to be protective for many pathophysiological disorders. Although estrogen metabolite, 2-methoxyestradiol (2ME2) can down-regulate angiotensin AT1 receptor expression independently of nuclear receptors, no specific cellular targets have been identified. This study was focused on identification and validation of a cellular target responsible for 2ME2-mediated angiotensin AT1 receptor down-regulation in a continuously passaged rat liver epithelial cell line. Cell membranes were isolated and used to determine 2ME2 specific binding. Cell membranes exposed to [3H]2ME2 showed specific saturable binding, which was found to be pertussis toxin (PTx) sensitive. Under similar conditions, G-protein coupled receptor 30 (GPR30) agonist (G1) and antagonist (G15) inhibited 2ME2 specific binding. In these cells GPR30 was found localized to endoplasmic reticulum (ER) membranes. In intact cells, G1 down-regulated angiotensin AT1 receptor expression and this effect was reversed by G15. Furthermore, 2ME2 mediated activation of epidermal growth factor receptor (EGFR) followed by ERK1/2 phosphorylation, an essential signaling step in angiotensin AT1 receptor down-regulation, was abrogated by G15, suggesting that this signal is GPR30 dependent. Additionally, EGF was found to independently down-regulate angiotensin AT1 receptor in an ERK1/2-dependent manner. In summary, our results demonstrate for the first time that 2ME2 down-regulation of angiotensin AT1 receptor is dependent on ER membrane-associated GRP30. Moreover, this effect is facilitated by GPR30 dependent transactivation of EGFR and ERK1/2 phosphorylation. This study provides further understanding of the physiological significance of 2ME2 and its role in modulating angiotensin AT1 receptor expression.
Keywords: Angiotensin II, angiotensin AT1 receptor, 2-Methoxyestradiol (2ME2), G-Protein coupled receptor 30 (GPR30), MAP-Kinase, Epidermal growth factor receptor (EGFR)
1. INTRODUCTION
Angiotensin II, the principal hormone in the renin-angiotensin endocrine axis, signals through at least two primary G-protein coupled receptors, of which angiotensin AT1 receptor is the principal isoform associated with cellular damage and multiple cardiovascular disease states. Therefore inhibition or more specifically down-regulation of angiotensin AT1 receptor is shown to be protective in nearly all cardiovascular disorders (Parrish et al. 2011).
Menopausal transition results in the loss of roughly 60% endogenous estrogen production in women, leading to significant physiological changes and vasomotor symptoms (Lisabeth and Bushnell, 2012). Importantly, the prevalence of hypertension in post-menopausal females is actually higher than males of similar age, though the general prevalence of hypertension in the population as a whole is greater among males (Pimenta, 2012). In order to alleviate post-menopausal vasomotor symptoms, as well as other conditions resulting from the loss of endogenous estrogen production, one may initiate hormone replacement therapy (HRT) involving exogenous supplementation of estrogen (Yang et al., 2011) or a combination of estrogen and progestin/progesterone (Gadducci et al, 2009). Interestingly, select retrospective analyses suggested that women receiving HRT derive some cardioprotective benefits as compared to women not receiving HRT (Wren, 2009), though clinical studies such as HERS and WHI failed to demonstrate conclusively that HRT was sufficiently cardioprotective after finding increased cancer and thromboembolic risks. Estrogen principally exerts its physiological effects through binding and activation of nuclear estrogen receptors (ER), of which there are two general isoforms, ER3 and ER3 (Al-Bader et al., 2011). Activation of these receptors results in transcriptional regulation of target genes with ER response elements. However, it has been shown that estrogens are also capable of exerting effects independent of nuclear estrogen receptors, allowing estrogenic signaling to be divided into genomic and non-genomic signaling pathways (Kumar et al. 2011). For example, investigation in the non-genomic signaling pathway has shown that estrogen may activate extracellular signal regulated kinases 1 and 2 (ERK1/2) through a low affinity G-protein coupled receptor (GPR30) to mediate its effects (Filardo et al. 2000). However GPR30 is considered an orphan receptor as the specific endogenous ligand remains unidentified.
2-methoxyestradiol (2ME2) is a final end product of estrogen metabolism. Existing evidence of 2ME2’s effect on cardiovascular physiology has been erratic, with studies indicating that 2ME2 possesses antimitogenic effects in vascular tissue (Dubey and Jackson, 2009), reduces HMG Co-A reductase expression (Barchiesi et al., 2010; Masi et al., 2012), and serum 2ME2 level has a positive correlation with HDL levels (Masi et al., 2012). However, a binding site or specific mechanism has not been attributed to its effects. Previously, we have shown that 2ME2 specifically down-regulates angiotensin AT1 receptor gene transcription and protein expression in rat liver epithelial cells and aortic smooth muscle cells (Koganti et al., 2012). Moreover, this effect was not affected by estrogen receptor blocker (Fulvestrant), suggesting that 2ME2’s down-regulatory effect was unrelated to classical estrogen signaling. The primary focus of the current study was to elucidate a 2ME2 specific binding site and down-stream signal transduction using a cellular model with high endogenous expression of angiotensin AT1 receptor (rat liver epithelial cells). We proposed that 2ME2 activates GPR30, initiating a complex signal transduction cascade leading to down-regulation of angiotensin AT1 receptor.
2. MATERIALS AND METHODS
2.1. Materials
Richter’s improved minimal essential medium (RIMEM) was obtained from Cellgro-Mediatech Inc. (Manassas, VA). [3H]2ME2 was from American Radiolabeled Chemicals, Inc. (St.Louis, Mo). Goat anti-rabbit Alexa-Flour 488® secondary IgG, 4′6′diamidino-2-phenylindole-dihydrochloride (DAPI), and Prolong Gold Anti-Fade® were from Invitrogen (Carlsbad, CA). DRAQ 5 was obtained from Thermo Scientific (Rockford, IL). Disposable culture tubes for binding assays were from Kimble Glass, Inc. (Vineland, NJ). Nitrocellulose membrane filters (0.45μm) were from Millipore (Billerica, MA). Fetal bovine serum (FBS) was from Equitech-Bio, Inc. (Kerrville, TX). GPR30 agonist G1 and antagonist G15 were from Tocris Biosciences (Bristol, UK). MAP Kinase inhibitor PD98059 and 2-methoxyestradiol were from Calbiochem (La Jolla, CA). Anti-ERK1/2 antibodies were from Cell Signaling (Danvers, MA). Anti-GPR30 and anti-β-actin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-calnexin antibody was from Abcam (Cambridge, MA). Electrophoresis reagents were from Bio-Rad (Hercules, CA) and all other chemicals and molecular biology grade reagents were purchased from Sigma (St. Louis, MO) or Fisher Scientific (Fairlawn, NJ).
2.2. Methods
2.2.1. Cell culture
Continuously passaged rat liver epithelial cells (WB) were kindly provided by Dr. H. Shelton Earp, University of North Carolina at Chapel Hill (Chapel Hill, NC). Cells were maintained in Richter’s improved minimal essential media containing 10% fetal bovine serum (FBS) and 50 μg/ml gentamicin (complete medium) at 37°C in 5% CO2 under 100% humidity. For the studies, cells were grown to 85–90% confluence (cells cultured from passages 21–28). All treatments were initiated in fresh complete medium and cells were incubated for indicated times. The control plates were replenished with complete medium.
2.2.2. Total membrane and ER specific membrane isolation
Total membrane was isolated using previously described method (Tuma and Hubbard, 1999). Briefly, cells were grown until 90% confluent and washed with Hanks buffer (HBSS). Cells were then collected by scraping in 50mM Tris-HCl, pH 7.4. and homogenized using a Wheaton® glass dounce tissue grinder with a “B” pestle for 10 strokes. The homogenate was centrifuged at 1000xg for 7 min. The resultant supernatant was centrifuged at 45,000xg for 20 min; the membrane pellet obtained was resuspended in 50mM Tris-HCl, pH 7.4 buffer and centrifugation and pellet resuspension was repeated. Membranes were immediately used for receptor binding studies. For isolation of ER membranes (Rieder and Emr, 1999) cells were grown until 90% confluent, after which cells were trypsinized and collected by centrifugation at 2500xg. The cell pellet was suspended in 4 ml MTE buffer (270 mM mannitol, 10mM Tris-HCl, 0.1mM EDTA pH 7.4), which also contained a Complete Mini Protease inhibitor cocktail tablet (Roche) and 1 mM PMSF. The suspension was homogenized using a Wheaton glass dounce tissue grinder with a “B” pestle for 10 strokes. The resultant homogenate was centrifuged at 700xg for 10 minutes at 4°C. The supernatant was subjected to high speed centrifugation at 15,000xg for 10 minutes at 4°C. The supernatant thus obtained was further purified by sucrose gradient centrifugation; specifically, the supernatant was layered on 3 progressive sucrose solutions (1.3 M, 1.5 M, and 2M sucrose in 10mM Tris-HCl, pH 7) and subjected to centrifugation at 100,000xg for 45 min at 4°C. The purified ER obtained as a white circular band was removed and resuspended in 1X PBS and was immediately used for radioligand binding assay.
2.2.3. [3H]2-methoxyestradiol Binding Assay
[3H]2ME2 binding studies were performed in triplicate using total or ER-enriched membranes. Briefly, membrane samples were placed in disposable borosilicate culture tubes, and incubated with 0.8 nM [3H]2ME2 for 1 h at 22°C in binding buffer (50 mM Tris-HCl pH 7.4, 120 mM NaCl, 4 mM KCl, 1mM CaCl2, 10 μg/ml bacitracin, 0.25% BSA, and 2 mg/ml dextrose). Non-specific binding was determined in the presence of 10 μM unlabeled 2ME2. Specific [3H]2ME2 binding was defined as that portion of the total binding displaced by unlabeled 2ME2. To determine GPR30 and G-protein coupling specificity [3H]2ME2 binding was performed in the presence of G15 (10 μM, specific GPR30 antagonist) and CTx (10μg/ml), or PTx (100ng/ml), respectively. Total reaction volume was 500μl. Upon completion of 1 h incubation at 22°C with [3H]2ME2, the reaction volume was passed through presoaked membrane filter using a filtration unit (Millipore). The membranes were washed three times with 2 ml of binding buffer without BSA. The filters were then transferred to counting vials, in which radioactivity was determined using a Beckman scintillation spectrometer in 10 ml Scintiverse® scintillation fluid.
2.2.4. Radioligand Angiotensin II binding studies in intact cells
[3H]AngII binding studies were performed in triplicate on WB cells as described previously (Thekkumkara et al., 1998). Briefly, cells were grown to 70–80% confluence in 6-well plates and treated with different agents for 24 hours. Cells were washed with phosphate buffered saline (PBS) and incubated at 22°C for1 hour with 0.05 nM [3H]AngII in binding buffer (50mM Tris-HCl pH 7.4, 120 mM NaCl, 4mM KCl, 1mM CaCl2, 10 μg/ml bacitracin, 0.25% BSA, and 2 mg/ml dextrose). In order to confirm specific binding, selected group of wells were incubated with [3H]AngII in the presence of unlabeled AngII (1μM). Cells were washed 3 times with chilled PBS to remove non-specifically bound [3H]AngII and lysed in 0.1 % Triton-X-100 for 1 h. The lysates were collected and transferred to counting vials, and radioactivity was determined using a Beckman auto-gamma scintillation spectrometer. Specific binding was defined as that portion of the total binding displaced by 1 μM unlabeled AngII. Protein concentrations were determined by using Bio-Rad protein assay system (Bradford, 1976).
2.2.5. Confocal Microscopy
For this analysis we used the method of dual-antibody immunofluorescent staining described previously (Kumar et al., 1999). Cells were grown on sterile coverslips until 75–80% confluent, after which the cells were washed three times with room temperature 1X PBS and fixed with 3% paraformaldehyde in PBS for 30 minutes at 22°C. Cells were permeabilized with 0.2% Triton X-100 dissolved in 1X PBS for 5 minutes and washed 3 times with 22°C 1X PBS. Cells were blocked in 5% goat serum (1X PBS) for 1 h and washed 3 times with 1X PBS before incubating with primary antibodies (anti-GPR30 and anti-Calnexin; each 1:100) for 45 minutes. Cells were then washed an additional 3 times with 1X PBS (10 min for each wash) on a shaker incubator at 22°C. Cells were incubated with secondary antibody: FITC labeled anti-rabbit IgG (for GPR30), Alexa-fluor® 594 labeled anti-mouse IgG (for calnexin) both at a dilution of 1:300 for 1 h at 22°C in the dark, followed by 3 additional washes with 1X PBS (10 min each). Nuclei were stained with DRAQ5 (5 μM) for 5 min and cells underwent a final wash for 30 min in 1X PBS. Coverslips were sealed onto slides with prolong Gold Antifade mounting medium (Invitrogen, Carlsbad, CA) and visualized and photographed with a Leica® DM IRE2 confocal microscope utilizing a TCS SL three-channel laser and a 60X objective.
2.2.6. Immunoflourescence Microscopy
Using the method described previously (Koganti et al., 2012), WB cells were grown in chamber slides (Lab-Tek, Naperville, IL) to 75–80% confluence and exposed to agents separately or in combination as described in the figure legends. Following 1 h incubation at 37°C, cells were washed with ice-cold PBS, fixed with 3% paraformaldehyde in PBS for 30 minutes at 22°C. Cells were washed 3 times with PBS, blocked in 5% goat serum for 1 h at 22°C, and incubated with 1:100 rabbit polyclonal anti-ERK1/2 or anti-phospho-ERK1/2 IgG overnight at 4°C. Cells were then washed 5 times with PBS and incubated with 1:500 Alexa-Fluor® 488 labeled secondary anti-rabbit IgG for 1 h at 22°C protected from light, and followed by washing. Nuclei were stained with 1 nM DAPI for 5 min and washed with PBS. Slides were sealed with Prolong Gold Antifade™ mounting medium, and were then visualized and photographed with a fluorescence microscope (Olympus IX70) using a 60X objective.
2.2.7. Data Analysis
Results are presented as mean ± S.E.M. and the value of P<0.05 was considered statistically significant. Analyses performed include parametric t-test, first testing for Gaussian distribution, and one-way analysis of variance (ANOVA) with post-hoc Bonferroni analysis where appropriate. Values are normalized to mg of protein determined by Bio-Rad R DC protein assay system based on the Bradford method (Bradford, 1976). Data were analyzed using the GraphPad Prism software.
3. RESULTS
3.1. 2-methoxyestradiol shows specific binding to GPR30 in WB cells
Total cell membrane isolates were prepared from semi-confluent WB cells in culture and exposed to [3H]2ME2 in the presence or absence of unlabeled 2ME2 to determine specific binding. Addition of 10 μM unlabeled 2ME2 displaced 73.6±3.8% (P<0.001, n=3) of total [3H]2ME2 binding, suggesting that 2ME2 binding is specific in membranes of cultured WB cells [Fig. 1]. To further determine the identity of the 2ME2 specific binding site, membrane 2ME2 specific binding was performed in the presence or absence of the G-protein coupled receptor 30 (GPR30) agonist G1 and antagonist G15 (Li et al., 2012). Addition of 10 μM unlabeled G1 displaced 85.1±2.0% (P<0.001, n=3) of total [3H]2ME2 binding [Fig 2A], while addition of 10 μM unlabeled G15 displaced 82.9±1.3% (P<0.001 n=3) of total [3H]2ME2 binding [Fig 2B]. These results indicate that 2ME2 specific binding is displaced by blockade or mutual competition of GPR30, and therefore suggests that the specific binding site of 2ME2 is GPR30. Saturation binding isotherms of specific [3H]2ME2 binding (10 μM 2ME2 displaceable) indicate a [3H]2ME2 specific binding Kd of approximately 10.1 nM and a Bmax of 14.0 nmol/mg protein (R2=0.914, n=3), demonstrating that 2ME2 binds with relative high affinity to its specific binding site [Fig. 3A]. As GPR30 is a G-protein coupled orphan receptor, in the next study we determined G-protein specificity. Cell membranes were exposed to one of two toxins, cholera toxin (CTx) and pertussis toxin (PTx) shown to be specific for selective inhibition of Gαs and Gαi isoforms respectively (Finlay et al., 2010). 10 μg/ml CTx or 100 ng/ml PTx were incubated for 15 minutes prior to [3H]2ME2 membrane binding. [3H]2ME2 specific binding was not significantly reduced in CTx treated membrane preparations (mean difference in control and CTx treated membranes 20.8±20.1%, P=0.36, n=3); however, PTx treatment resulted in a 69.7±14.3% (P=<0.01, n=3) reduction in [3H]2ME2 total binding, indicating that binding with GPR30 in WB cells is primarily PTx-sensitive and is predominantly associated with Gαi-protein [Fig. 3B].
Fig. 1. [3H]2ME2 displays specific binding in WB cell membrane preparations.
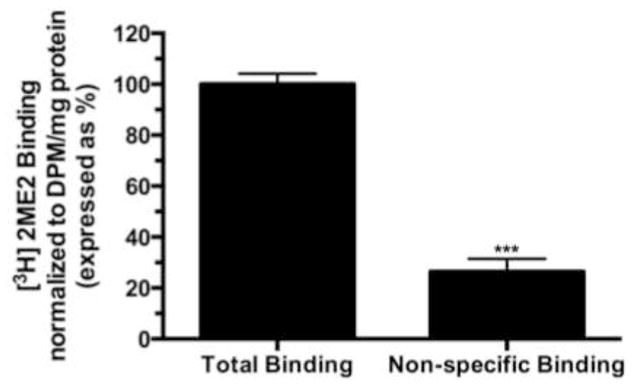
Total binding corresponds to total binding detected after addition of 20 nCi of [3H]2ME2. Non-specific binding corresponds to detectable binding after addition of 10 μM 2ME2. Data are expressed as mean ± S.E.M, ***P<0.001, n=3.
Fig. 2. GPR30 selective agents displace specific [3H]2ME2 binding.
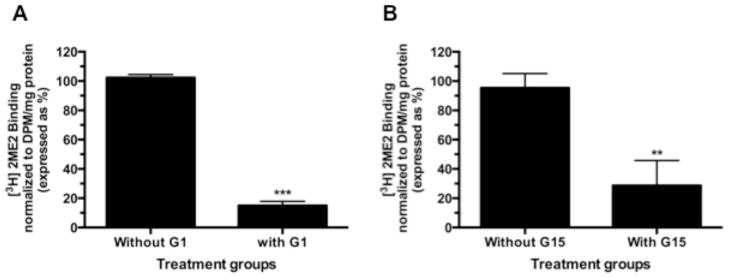
(A) 10 μM unlabeled G1 (GPR30 agonist) significantly displaces 20 nCi [3H]2ME2 specific binding. (B) 10 μM unlabeled G15 (GPR30 antagonist) significantly displaces 20 nCi [3H]2ME2 specific binding. Data are expressed as mean ± S.E.M, **P<0.01. ***P<0.001, n=3.
Fig. 3. 2ME2 binds to a Gαi associated GPCR with high affinity.
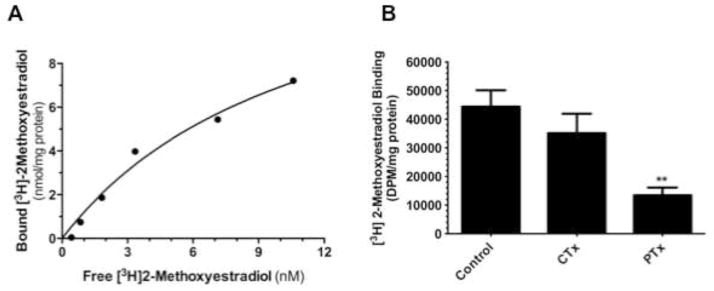
(A) Saturation binding isotherm of specific [3H]2ME2 binding (10 μM unlabeled 2ME2 displaceable) Kd-10.1 nM, Bmax-14.0 nmol/mg protein (R2=0.914, n=3). (B) [3H]2ME2 specific binding in the presence of CTx and PTx. Data are expressed as mean ± S.E.M, **P<0.01.
3.2. Localization of GPR30 at the Endoplasmic Reticulum
We conducted analyses to verify cellular localization of GPR30 in order to establish the site of interaction of 2ME2. We performed dual-antibody directed confocal immunofluorescence microscopy with immunoglobulins targeting GPR30 and the endoplasmic reticulum (ER). The results indicate that there is significant co-localization (orange) of the GPR30 specific signal (green) with endoplasmic reticulum specific marker calnexin directed immunostaining (red) [Fig. 4A]. Furthermore, membrane fractionation by sucrose gradient centrifugation to enrich ER membranes followed by [3H]2ME2 membrane binding analysis revealed that 10 μM unlabeled 2ME2 displaced 40.4±4.4% (P<0.001, n=4) of total [3H]2ME2 binding in ER-enriched membrane preperations [Fig. 4B], indicating that a significant proportion of total binding to the endoplasmic reticulum was specific, supporting our results from confocal microscopy demonstrating GPR30 localization to the ER.
Fig. 4. GPR30 in WB cells is primarily expressed in endoplasmic reticulum (ER) membranes.


(A) Confocal microscopy analysis of immunostained GPR30 (green) and calnexin (red) show significant colocalization of signal (orange) in overlay image. (B) Sucrose gradient isolated ER membranes shows significant [3H]2ME2 specific binding. Data are expressed as mean ± S.E.M, ***P<0.001.
3.3. 2-methoxyestradiol dependent down-regulation of angiotensin AT1 receptor is mediated by GPR30 activation
Based on our results that 2ME2 binding of GPR30 leads to G-protein signal activation, we next conducted an angiotensin AT1 receptor binding analysis to determine whether GPR30-selective agonist activation would result in angiotensin AT1 receptor down-regulation. Cells were exposed to G1 for 24 hours. All concentrations of G1 utilized for this study resulted in significant and dose-dependent down-regulation of AngII binding (reduction at 1 μM: 17.5±3.9% [P=<0.01, n=3]; reduction at 1.5 μM: 35.0±5.1% [P<0.001, n=3]; reduction at 2 μM: 61.8±3.3% [P<0.001, n=3]) [Fig. 5]. We next determined how GPR30 activation mediates angiotensin AT1 receptor down-regulation. 2ME2 mediated down-regulation is shown to be ERK1/2 phosphorylation dependent (Verenich and Gerk, 2010); therefore, we determined GPR30’s role in ERK1/2 phosphorylation and subsequent nuclear translocation by immunofluorescent microscopy after treatment with 2ME2 with or without GPR30-specific antagonist G15 [Fig. 6A & 6B]. The results indicate that 1 μM 2ME2-mediated ERK1/2 translocation was entirely abrogated in the presence of 10 μM G15. These results were performed in parallel with a MEK inhibitor control to demonstrate that MEK and thus ERK1/2 inhibition displayed a similar pattern as that achieved by treatment with G15. We performed an additional series of [3H]AngII binding studies in order to demonstrate that G1 or 2ME2 antagonism by G15 or MEK inhibition using PD98059 would result in restoration of angiotensin AT1 receptor expression, and thus demonstrate that the down-regulatory effect of either agent is through GPR30 activation and ERK1/2 phosphorylation [Fig. 7A & 7B]. The results indicate that 1 μM G1 and 2ME2 both significantly reduce [3H]AngII binding (G1 induced mean reduction of 56.6±4.9%, P<0.001, n=3; 2ME2 induced mean reduction of 72.0±4.4%, P<0.001, n=3). Moreover, upon treatment with G15, binding was significantly restored in G1 and 2ME2 treated groups (G1+G15 treatment group mean AngII binding increased from G1 alone treatment group 94.5±5.7%, P<0.001, n=3; 2ME2+G15 treatment group mean AngII binding increased from 2ME2 alone treatment group 41.2±3.2%, P<0.001, n=3). Moreover, G15 treatment paralleled the effect of treatment with the MEK inhibitor PD98059 in a way that there was no significant difference between PD98059 and G15 treated groups (P=0.41 and P=0.74 for G1 and 2ME2 comparisons respectively, n=3), demonstrating that 2ME2 and G1 result in GPR30 dependent activation of ERK1/2.
Fig. 5. GPR30 agonist G1 independently down-regulates angiotensin AT1 receptor expression in intact WB cells.

[3H]AngII binding assay demonstrates concentration dependent down-regulation of angiotensin AT1 receptor expression in the presence of G1. Data are expressed as mean ± S.E.M, ***P<0.001.
Fig. 6. GPR30 antagonist G15 blocks 2ME2 mediated ERK1/2 phosphorylation and translocation to WB nuclei.


(6A) Anti-ERK1/2 IgG labeled with FITC labeled secondary anti-rabbit IgG (green) shows increased translocation to the nucleus (blue – DAPI stained) in 1 μM 2ME2 compared to control (panels A & B). GPR30 antagonist G15 effectively blocks ERK1/2 translocation in 2ME2 treated cells showing no significant difference from G15 treated cells alone (panels C & D). MEK inhibitor PD98059 was used as a control for ERK1/2 blockade (panels E & F). (6B) Similar treatment conditions as figure 6A, but with phospho-ERK1/2 specific antibody confirming phospho-ERK1/2 nuclear translocation.
Fig. 7. G1 and 2ME2 mediated angiotensin AT1 receptor down-regulation is reversed by treatment with G15.
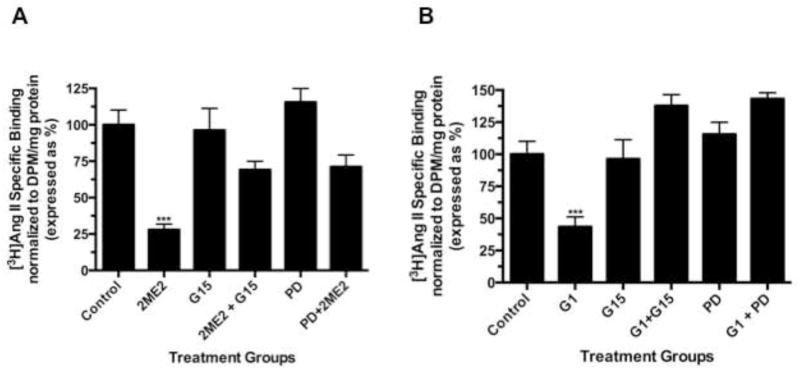
(A) G1 significantly reduced [3H]AngII binding, and angiotensin AT1 receptor expression was restored to control levels or greater with treatment with G15 or MEK inhibitor PD98059. (B) 2ME2 significantly reduced [3H]AngII binding, and angiotensin AT1 receptor expression was restored significantly with treatment with G15 or MEK inhibitor PD98059. Data are expressed as mean ± S.E.M, ***P<0.001.
3.4. GPR30 mediated ERK1/2 phosphorylation and angiotensin AT1 receptor down-regulation involves EGFR transactivation
While data in this study indicates that GPR30 activation by 2ME2 or G1 results in ERK1/2 phosphorylation and nuclear translocation, we had yet to make a link between GPR30 stimulation and MEK/ERK signaling. Previous studies have demonstrated coordinated transactivation via GPR30 of EGFR (Filardo, 2002; Filardo et al., 2008). Therefore, we determined whether EGFR stimulation with EGF results in angiotensin AT1 receptor down-regulation, presumably through ERK1/2 activation. The results demonstrate that there is a dose dependent down-regulatory effect in [3H]AngII binding by addition of EGF (mean reduction from control in 10 ng/ml treatment group 4.25±6.5%, P=0.546; in 100 ng/ml treatment group 28.1±2.5%, P<0.001; in 1000 ng/ml treatment group 62.6±2.6%, P<0.001, n=3) [Fig. 8]. To confirm the effect on ERK1/2 phosphorylation and translocation, immunfluorescent microscopy analysis revealed that 1000 ng/ml EGF treatment resulted in significant ERK1/2 activation [Fig. 9A & 9B]. Moreover, this effect was effectively inhibited using 10 nM EGFR inhibitor AG1478, as well as 20 μM MEK inhibitor PD98059. These data demonstrated that EGF can independently activate ERK1/2 and result in angiotensin AT1 receptor down-regulation; however, in order to associate EGFR signaling with GPR30 activation, we performed an additional [3H]AngII binding study to demonstrate that GPR30 and EGFR are downstream of 2ME2 induced signal transduction [Fig. 10]. The results indicate that while 1 μM 2ME2 and EGF treatment resulted in similar reductions of [3H]AngII binding (mean reduction of binding compared to untreated control of 49.3±8.6%, P<0.01 and 51.9±7.1% for 2ME2 and EGF treated groups respectively), 35 μM G15 significantly restored AngII binding only in 2ME2 treated cells (difference between means of untreated control and 2ME2+G15 8.87±7.1%, P=0.27 [restoration of 40.5±5.5%, P<0.001 compared to 2ME2 treated group]; difference between means of untreated control and EGF+G15 43.6±7.3%, P=0.004 [no significant restoration 8.34±4.9%, P=0.139 compared to EGF treated group]). However, we observed significant restoration of AngII binding for both 2ME2 and EGF treatment groups by using 10 nM EGFR inhibitor AG1478 (difference between means of untreated control and 2ME2+AG1478 1.15±8.3%, P=0.9 [restoration of 48.2±6.3%, P<0.001 compared to 2ME2 treated group]; difference between means of untreated control and EGF+AG1478 4.46±13.9%, P=0.77 [restoration of 47.5±9.9%, p=0.003 compared to EGF treated group]). These data suggest that 2ME2 and not EGF signaling may be effectively inhibited by GPR30 antagonist G15, and either signal is effectively inhibited by EGFR inhibition by AG1478. Therefore, 2ME2 induced activation of GPR30 leads to EGFR transactivation, thus leading to activation of the MEK-ERK1/2 signaling pathway and subsequent angiotensin AT1 receptor down-regulation.
Fig. 8. Epidermal growth factor (EGF) independently down-regulates angiotensin AT1 receptor expression in intact WB cells.

[3H]AngII binding assay demonstrates concentration dependent down-regulation of angiotensin AT1 receptor expression in the presence of EGF. Data are expressed as mean ± S.E.M, ***P<0.001.
Fig. 9. EGFR inhibitor AG1478 blocks 2ME2 and EGF mediated ERK1/2 phosphorylation and translocation to WB nuclei.


(9A) Anti-ERK1/2 IgG labeled with FITC labeled secondary anti-rabbit IgG (green) shows increased translocation to the nucleus (blue – DAPI stained) in 1 μM 2ME2 (panel B) and 1 μM EGF (panel C) compared to control (panel A). EGFR inhibitor AG1478 effectively blocks ERK1/2 translocation in EGF (panel E) and 2ME2 (panel F) treated cells showing no significant difference from AG1478 treated cells alone (panel D). (9B) Similar treatment conditions as figure 9A, but with phospho-ERK1/2 specific antibody confirming phospho-ERK1/2 nuclear translocation.
Fig. 10. EGFR inhibition reverses EGF and 2ME2 mediated angiotensin AT1 receptor down-regulation while G15 is not effective in reversing EGF-mediated effects.

1 μM 2ME2 and EGF significantly down-regulate angiotensin AT1 receptor expression. 35 μM G15 restores 2ME2 treated cells. EGF+G15 showed no significant difference in angiotensin AT1 receptor expression from EGF alone treated cells. 10 nM AG1478 signficantly restored all down-regulation by 2ME2 and EGF. Data are expressed as mean ± S.E.M, **P<0.01. ***P<0.001, n=3.
4. DISCUSSION
In recent years, significant effort has been directed towards understanding the beneficial effects of estrogen; however, empirical clinical evidence from direct estrogen supplementation is enigmatic, providing support that estrogen possesses both increased benefit as well as increased risk to patients (Gadducci et al., 2009). Therefore, investigators are currently focused on understanding the role of estrogen metabolites as possible candidates for beneficial estrogenic signaling (Mueck and Seeger, 2010; Tofovic, 2010). In a recently published study, we demonstrated that 2ME2 is capable of down-regulating angiotensin AT1 receptor expression, a potential risk factor for many pathophysiological disorders (Koganti et al., 2012). The study showed that 2ME2 was not acting through classical nuclear estrogen receptors, as treatment with the ER antagonist ICI182780 did not restore 2ME2 induced angiotensin AT1 receptor down-regulation. The purpose of the current study was to identify the novel cellular target and target-mediated signaling mechanism(s) by which 2ME2 down-regulates angiotensin AT1 receptor. The results of this study showed specific binding of radio-labeled 2ME2 to GPR30, a Gαi associated G-protein coupled receptor, which was found predominantly localized to endoplasmic reticulum. Moreover, selective activation of GPR30 by agonist G1 resulted in down-regulation of angiotensin AT1 receptor, and the observed effect was reversed by GPR30 antagonist G15.
Based upon existing literature, the role of GPR30 in cardiovascular physiology is not well understood. Research largely has focused upon GPR30 expression and the effect of agonism and antagonism in cancer cells (Filardo and Thomas, 2012), whereas our study examined its effects not associated to anti-mitogenic potentiation. 17-β-Estradiol, considered by many to be the endogenous ligand for this receptor, has been shown to induce vasodilatory effects (Meyer et al., 2011); however, a recent study found that 17-β-Estradiol induced vasodilation in rat aorta but was not prevented by the application of the GPR30 antagonist G15 (Seok et al., 2012). Interestingly, this study demonstrated that G1, applied in the same manner as 17-β-Estradiol, induced vasodilation, and this effect was reversible with the addition of G15, suggesting that GPR30 activation can result in relaxation of smooth muscle, and this effect is not mediated by 17-β-Estradiol. Proposed mechanisms of 17-β-Estradiol-mediated vasodilation largely involve the increased production of nitric oxide (Nilsson et al., 2011; Qiao et al., 2008), though GPR30 associated effects are often contradictory, as seen above, and no mechanism of 2ME2 mediated cardioprotective effect has yet been proposed. Moreover, in contrast to previous reports suggesting estradiol’s cardioprotective effects, implementation has been limited due to the increased incidence of cancer reported due to estrogen use in hormone replacement therapy (Gapstur et al, 1999; Magnusson et al., 1999). Studies utilizing GPR30 knockout models have shown that deletion of GPR30 increases blood pressure (Mårtensson et al., 2009); however, association between GPR30 and angiotensin AT1 receptor has yet to be studied. Observations from less direct studies have shown that G1 administration lowers blood pressure in a model of estrogen depleted mRen2.Lewis rats with hyper-activated renin-angiotensin signaling (Lindsey et al., 2009). Therefore, it is entirely possible that G1 interferes with angiotensin AT1 receptor expression, as demonstrated in this study, and may be a principal effect of GPR30 activation.
Identification of 2ME2 as a ligand for GPR30 is central to our overall hypothesis, as no study has clearly identified this metabolite, which bears 2000-fold less activation potential for estrogen receptors (Sibonga et al., 2003), as a ligand for GPR30. Studies aimed at establishing the binding affinities of steroid compounds for specific targets are difficult, as the tracers are highly lipophilic and often result in non-specific binding in lipid-rich membrane preparations with low-level target receptor expression (Thomas et al., 2010). Inevitably, reports of relative affinities of steroids, 17-β-Estradiol in particular, have been highly variable, ranging from low nanomolar (Kuiper et al., 1996; Pang et al., 2008; Liu et al., 2009) to no affinity for GPR30 (Pedram et al., 2006; Kang et al., 2010). Therefore, the findings of this study must be considered significant by the novel demonstration of specific binding of 2ME2, an agent with no known affinity for this receptor. In our previous study, we demonstrated that ICI182780 treatment failed to reverse 2ME2’s down-regulatory effect on angiotensin AT1 receptor, and also showed that ICI182780 treatment alone down-regulated the angiotensin AT1 receptor independently of 2ME2 (Koganti et al., 2012). When 2ME2 and ICI182780 were combined, the effects were neither additive nor synergistic. ICI182780 (Fulvestrant) is an effective ERα/ERβ antagonist when used in vitro and in vivo, but it also functions as an agonist for GPR30, providing indirect evidence of participation of GPR30 (Langer et al., 2010). However, data presented in this study provides direct evidence of GPR30-mediated angiotensin AT1 receptor down-regulation. GPR30 is not differentially expressed among males and females, and treatment with 2ME2 may be considered equally efficacious in both genders, though additional studies must be performed (Delbeck et al., 2011; Meyer et al., 2011). Thus, 2ME2’s effect as a GPR30 agonist may be clinically significant, particularly in light of recent observations that GPR30 agonism has been shown to be protective in cardiovascular tissue (Chakrabarti and Davidge, 2012). 2ME2 has proven effective as a chemotherapeutic adjunct (Kumar et al., 2001), making it a promising candidate for additional translational studies. 2ME2 is non-feminizing, and can be considered beneficial irrespective of gender (Dantas and Sandberg, 2006). 2ME2 is non-toxic to normal cells, and according to clinical data produces no cytotoxicity in cancer patients (Pribluda et al., 2000).
Furthermore, our studies shows that GPR30 activation leads to transactivation of EGFR, phosphorylating and translocating ERK1/2 and down-regulating angiotensin AT1 receptor. Epidermal growth factor (EGF) receptor tyrosine kinases participate in proliferation, migration, differentiation, and survival (Holbro and Hynes, 2004). EGFR over-expression is correlated with a poor prognosis in select cancers (Bhola and Grandis, 2008). However, a number of studies have shown the involvement of EGFR activation and its effect on enhanced vascular relaxation, though the attributable mechanisms differ much in the same way as the postulated mechanisms of vasorelaxation for estrogen (Harris et al., 1990; Matsumoto et al., 2001; McEwen et al., 2009; Zhou et al., 2009). Significant evidence exists for GPCR involvement in transactivation of EGFR (Gschwind et al., 2003; Paolillo and Schinelli, 2008). A specific crosstalk between GPR30 and EGFR activation has been reported by numerous studies, many of which were directed to understanding GPR30 signaling upon the mitogenic contribution of GPR30 ligands (Albanito et al., 2007; Pupo et al., 2012). The possible activation of EGFR by GPR30 involves activation of MMPs or ADAMs to cleave EGF precursor ligands to activate EGFR tyrosine kinase phosphorylation (Filardo and Thomas, 2005; Ohtsu et al., 2006); however, the exact role of these signaling intermediates in this particular model requires further investigation. Upon stimulation, EGFR is a potent activator of ERK1/2 (Yamashita and Shimada, 2012). Our data shows that ERK1/2 is activated upon stimulation by 2ME2 and G1, and is effectively inhibited by antagonism of GPR30, EGFR, and MEK. Sequential stimulation or blockade of each intermediate transducer indicates the progressive signal from GPR30 to EGFR to ERK1/2 and eventual angiotensin AT1 receptor down-regulation [Fig. 11]. Based on our previous study, 2ME2 induced down-regulation of angiotensin AT1 receptor is through transcriptional repression; however, the mechanism following phosphorylation and translocation of ERK1/2 to the nucleus in angiotensin AT1 receptor transcriptional repression requires further investigation.
Fig. 11.

Schematic representation of the proposed mechanism of 2ME2 initiated signal transduction intermediates in angiotensin AT1 receptor down-regulation.
In conclusion, this study was conducted to identify and validate a distinct site of action for the initiation of 2ME2’s down-regulatory effect on angiotensin AT1 receptor. Further determination of the mechanism of GPR30 initiated and ERK1/2 mediated effects should prove valuable, both in elucidating 2ME2’s general mechanism in angiotensin AT1 receptor regulation, and demonstrating a novel regulatory pathway that may prove exploitable in the future by additional compounds. As we have found that GPR30 agonists 2ME2, G1, and ICI182780 are all down-regulators of angiotensin AT1 receptor, there is a potential role for pharmacological application of all three agents that may be elucidated in translational studies.
Acknowledgments
This research was funded by NIH Grant #DK072140. The authors have nothing to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Bader MD, Malatiali SA, Redzic ZB. Expression of estrogen receptor α and β in rat astrocytes in primary culture: effects of hypoxia and glucose deprivation. Physiol Res. 2011;60(6):951–60. doi: 10.33549/physiolres.932167. [DOI] [PubMed] [Google Scholar]
- Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Andò S, Maggiolini M. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007;67(4):1859–66. doi: 10.1158/0008-5472.CAN-06-2909. [DOI] [PubMed] [Google Scholar]
- Barchiesi F, Lucchinetti E, Zaugg M, Ogunshola OO, Wright M, Meyer M, Rosselli M, Schaufelberger S, Gillespie DG, Jackson EK, Dubey RK. Candidate genes and mechanisms for 2-methoxyestradiol-mediated vasoprotection. Hypertension. 2010;56(5):964–72. doi: 10.1161/HYPERTENSIONAHA.110.152298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhola NE, Grandis JR. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front Biosci. 2008;13:1857–65. doi: 10.2741/2805. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Davidge ST. G-protein coupled receptor 30 (GPR30): a novel regulator of endothelial inflammation. PLoS One. 2012;7(12):e52357. doi: 10.1371/journal.pone.0052357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas AP, Sandberg K. Does 2-methoxyestradiol represent the new and improved hormone replacement therapy for atherosclerosis? Circ Res. 2006;99(3):234–7. doi: 10.1161/01.RES.0000236802.00855.cd. [DOI] [PubMed] [Google Scholar]
- Delbeck M, Golz S, Vonk R, Janssen W, Hucho T, Isensee J, Schäfer S, Otto C. Impaired left-ventricular cardiac function in male GPR30-deficient mice. Mol Med Rep. 2011;4(1):37–40. doi: 10.3892/mmr.2010.402. [DOI] [PubMed] [Google Scholar]
- Dubey RK, Jackson EK. Potential vascular actions of 2-methoxyestradiol. Trends Endocrinol Metab. 2009;20(8):374–9. doi: 10.1016/j.tem.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14(10):1649–60. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Sabo E. Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids. 2008;73(9–10):870–3. doi: 10.1016/j.steroids.2007.12.025. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab. 2005;16(8):362–7. doi: 10.1016/j.tem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Thomas P. Minireview: G Protein-Coupled Estrogen Receptor-1, GPER-1: Its Mechanism of Action and Role in Female Reproductive Cancer, Renal and Vascular Physiology. Endocrinology. 2012 Jul;153(7):2953–62. doi: 10.1210/en.2012-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo EJ. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol. 2002;80(2):231–8. doi: 10.1016/s0960-0760(01)00190-x. [DOI] [PubMed] [Google Scholar]
- Finlay TM, Jayanth P, Amith SR, Gilmour A, Guzzo C, Gee K, Beyaert R, Szewczuk MR. Thymoquinone from nutraceutical black cumin oil activates Neu4 sialidase in live macrophage, dendritic, and normal and type I sialidosis human fibroblast cells via GPCR Galphai proteins and matrix metalloproteinase-9. Glycoconj J. 2010;27(3):329–48. doi: 10.1007/s10719-010-9281-6. [DOI] [PubMed] [Google Scholar]
- Gadducci A, Biglia N, Cosio S, Sismondi P, Genazzani AR. Progestagen component in combined hormone replacement therapy in postmenopausal women and breast cancer risk: a debated clinical issue. Gynecol Endocrinol. 2009;25(12):807–15. doi: 10.3109/09513590903056878. [DOI] [PubMed] [Google Scholar]
- Gapstur SM, Morrow M, Sellers TA. Hormone replacement therapy and risk of breast cancer with a favorable histology: results of the Iowa Women’s Health Study. JAMA. 1999;281(22):2091–7. doi: 10.1001/jama.281.22.2091. [DOI] [PubMed] [Google Scholar]
- Gschwind A, Hart S, Fischer OM, Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 2003;22(10):2411–21. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RC, Munger KA, Badr KF, Takahashi K. Mediation of renal vascular effects of epidermal growth factor by arachidonate metabolites. FASEB J. 1990;4(6):1654–60. doi: 10.1096/fasebj.4.6.2138579. [DOI] [PubMed] [Google Scholar]
- Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- Kang L, Zhang X, Xie Y, Tu Y, Wang D, Liu Z, Wang ZY. Involvement of estrogen receptor variant ER-alpha36, not GPR30, in nongenomic estrogen signaling. Mol Endocrinol. 2010;24(4):709–21. doi: 10.1210/me.2009-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koganti S, Snyder R, Thekkumkara T. Pharmacologic effects of 2-methoxyestradiol on angiotensin type 1 receptor down-regulation in rat liver epithelial and aortic smooth muscle cells. Gend Med. 2012;9(2):76–93. doi: 10.1016/j.genm.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93(12):5925–30. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar AP, Garcia GE, Slaga TJ. 2-methoxyestradiol blocks cell-cycle progression at G(2)/M phase and inhibits growth of human prostate cancer cells. Mol Carcinog. 2001;31(3):111–24. doi: 10.1002/mc.1046. [DOI] [PubMed] [Google Scholar]
- Kumar R, Balhuizen A, Amisten S, Lundquist I, Salehi A. Insulinotropic and antidiabetic effects of 17β-estradiol and the GPR30 agonist G-1 on human pancreatic islets. Endocrinology. 2011;152(7):2568–79. doi: 10.1210/en.2010-1361. [DOI] [PubMed] [Google Scholar]
- Kumar RK, Chapple CC, Hunter N. Improved double immunofluorescence for confocal laser scanning microscopy. J Histochem Cytochem. 1999;47(9):1213–8. doi: 10.1177/002215549904700913. [DOI] [PubMed] [Google Scholar]
- Langer G, Bader B, Meoli L, Isensee J, Delbeck M, Noppinger PR, Otto C. A critical review of fundamental controversies in the field of GPR30 research. Steroids. 2010;75(8–9):603–10. doi: 10.1016/j.steroids.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Li ZL, Liu JC, Liu SB, Li XQ, Yi DH, Zhao MG. Improvement of vascular function by acute and chronic treatment with the GPR30 agonist G1 in experimental diabetes mellitus. PLoS One. 2012;7(6):e38787. doi: 10.1371/journal.pone.0038787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology. 2009;150(8):3753–8. doi: 10.1210/en.2008-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisabeth L, Bushnell C. Stroke risk in women: the role of menopause and hormone therapy. Lancet Neurol. 2012;11(1):82–91. doi: 10.1016/S1474-4422(11)70269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhu P, Sham KW, Yuen JM, Xie C, Zhang Y, Liu Y, Li S, Huang X, Cheng CH, Lin H. Identification of a membrane estrogen receptor in zebrafish with homology to mammalian GPER and its high expression in early germ cells of the testis. Biol Reprod. 2009;80(6):1253–61. doi: 10.1095/biolreprod.108.070250. [DOI] [PubMed] [Google Scholar]
- Magnusson C, Baron JA, Correia N, Bergström R, Adami HO, Persson I. Breast-cancer risk following long-term oestrogen- and oestrogen-progestin-replacement therapy. Int J Cancer. 1999;81(3):339–44. doi: 10.1002/(sici)1097-0215(19990505)81:3<339::aid-ijc5>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Mårtensson UE, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grände PO, Owman C, Rosen CJ, Adamo ML, Lundquist I, Rorsman P, Nilsson BO, Ohlsson C, Olde B, Leeb-Lundberg LM. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology. 2009;150(2):687–98. doi: 10.1210/en.2008-0623. [DOI] [PubMed] [Google Scholar]
- Masi CM, Hawkley LC, Cacioppo JT. Serum 2-methoxyestradiol, an estrogen metabolite, is positively associated with serum HDL-C in a population-based sample. Lipids. 2012;47(1):35–8. doi: 10.1007/s11745-011-3600-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Kanamoto K, Kawakubo K, Aomi H, Matsumoto T, Ibayashi S, Fujishima M. Gastroprotective and vasodilatory effects of epidermal growth factor: the role of sensory afferent neurons. Am J Physiol Gastrointest Liver Physiol. 2001;280(5):G897–903. doi: 10.1152/ajpgi.2001.280.5.G897. [DOI] [PubMed] [Google Scholar]
- McEwen ST, Balus SF, Durand MJ, Lombard JH. Angiotensin II maintains cerebral vascular relaxation via EGF receptor transactivation and ERK1/2. Am J Physiol Heart Circ Physiol. 2009;297(4):H1296–303. doi: 10.1152/ajpheart.01325.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Prossnitz ER, Barton M. The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function. Vascul Pharmacol. 2011;55(1–3):17–25. doi: 10.1016/j.vph.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueck AO, Seeger H. 2-Methoxyestradiol--biology and mechanism of action. Steroids. 2010;75(10):625–31. doi: 10.1016/j.steroids.2010.02.016. [DOI] [PubMed] [Google Scholar]
- Nilsson BO, Olde B, Leeb-Lundberg LM. G protein-coupled oestrogen receptor 1 (GPER1)/GPR30: a new player in cardiovascular and metabolic oestrogenic signalling. Br J Pharmacol. 2011;163(6):1131–9. doi: 10.1111/j.1476-5381.2011.01235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291(1):C1–10. doi: 10.1152/ajpcell.00620.2005. [DOI] [PubMed] [Google Scholar]
- Paolillo M, Schinelli S. Therapeutic targeting of g-protein coupled receptor-mediated epidermal growth factor receptor transactivation in human glioma brain tumors. Mini Rev Med Chem. 2008;8(13):1418–28. doi: 10.2174/138955708786369500. [DOI] [PubMed] [Google Scholar]
- Parrish MR, Wallace K, Tam Tam KB, Herse F, Weimer A, Wenzel K, Wallukat G, Ray LF, Arany M, Cockrell K, Martin JN, Dechend R, LaMarca B. Hypertension in response to AT1-AA: role of reactive oxygen species in pregnancy-induced hypertension. Am J Hypertens. 2011;24(7):835–40. doi: 10.1038/ajh.2011.62. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. 2006;20(9):1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- Pimenta E. Hypertension in women. Hypertens Res. 2012;35(2):148–52. doi: 10.1038/hr.2011.190. [DOI] [PubMed] [Google Scholar]
- Pribluda VS, Gubish ER, Jr, Lavallee TM, Treston A, Swartz GM, Green SJ. 2-Methoxyestradiol: an endogenous antiangiogenic and antiproliferative drug candidate. Cancer Metastasis Rev. 2000;19(1–2):173–9. doi: 10.1023/a:1026543018478. [DOI] [PubMed] [Google Scholar]
- Pupo M, Pisano A, Lappano R, Santolla MF, De Francesco EM, Abonante S, Rosano C, Maggiolini M. Bisphenol A Induces Gene Expression Changes and Proliferative Effects through GPER in Breast Cancer Cells and Cancer-Associated Fibroblasts. Environ Health Perspect. 2012;120(8):1177–82. doi: 10.1289/ehp.1104526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao X, McConnell KR, Khalil RA. Sex steroids and vascular responses in hypertension and aging. Gend Med. 2008;5 (Suppl A):S46–64. doi: 10.1016/j.genm.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Rieder SE, Emr SD. In: Current Protocols in Cell Biology. Bonifacino JS, DAsso M, Harford JB, Lippincott-Schwartz J, Yamada KM, editors. John Wiley; New York: 1999. p. 3.2.36. [Google Scholar]
- Seok YM, Jang EJ, Reiser O, Hager M, Kim IK. 17β-Estradiol induces vasorelaxation in a G-protein-coupled receptor 30-independent manner. Naunyn Schmiedebergs Arch Pharmacol. 2012 Jun 12; doi: 10.1007/s00210-012-0770-y. Epub ahead of print. [DOI] [PubMed]
- Sibonga JD, Lotinun S, Evans GL, Pribluda VS, Green SJ, Turner RT. Dose-response effects of 2-methoxyestradiol on estrogen target tissues in the ovariectomized rat. Endocrinology. 2003;144(3):785–92. doi: 10.1210/en.2002-220632. [DOI] [PubMed] [Google Scholar]
- Thekkumkara TJ, Cookson R, Linas SL. Angiotensin (AT1A) receptor-mediated increases in transcellular sodium transport in proximal tubule cells. Am J Physiol. 1998;274(5 Pt 2):F897–905. doi: 10.1152/ajprenal.1998.274.5.F897. [DOI] [PubMed] [Google Scholar]
- Thomas P, Alyea R, Pang Y, Peyton C, Dong J, Berg AH. Conserved estrogen binding and signaling functions of the G protein-coupled estrogen receptor 1 (GPER) in mammals and fish. Steroids. 2010;75(8–9):595–602. doi: 10.1016/j.steroids.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofovic SP. Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol. 2010;56(6):696–708. doi: 10.1097/FJC.0b013e3181f9ea8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma PL, Hubbard AL. In: Current Protocols in Cell Biology. Bonifacino JS, DAsso M, Harford JB, Lippincott-Schwartz J, Yamada KM, editors. John Wiley; New York: 1999. pp. 3.2.1–3.2.5. [Google Scholar]
- Wren BG. The benefits of oestrogen following menopause: why hormone replacement therapy should be offered to postmenopausal women. Med J Aust. 2009;190(6):321–5. doi: 10.5694/j.1326-5377.2009.tb02423.x. [DOI] [PubMed] [Google Scholar]
- Yamashita Y, Shimada M. The release of EGF domain from EGF-like factors by a specific cleavage enzyme activates the EGFR-MAPK3/1 pathway in both granulosa cells and cumulus cells during the ovulation process. J Reprod Dev. 2012;58(5):510–4. doi: 10.1262/jrd.2012-056. [DOI] [PubMed] [Google Scholar]
- Yang FL, Hu KQ, Wang X, Liu ZM, Hu Q, Li JF, He H. Combination of raloxifene, aspirin and estrogen as novel paradigm of hormone replacement therapy in rabbit model of menopause. Acta Pharmacol Sin. 2011;32(8):1031–7. doi: 10.1038/aps.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Brigstock D, Besner GE. Heparin-binding EGF-like growth factor is a potent dilator of terminal mesenteric arterioles. Microvasc Res. 2009;78(1):78–85. doi: 10.1016/j.mvr.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]